
Abstract
Long-term exposure to hyperglycemic conditions leads to β-cell dysfunction, particularly mitochondrial dysfunction, and inflammatory and oxidative stress responses, which are considered the primary causes of β-cell death and the hallmarks of diabetes. Plant-active ingredients may play a key role in glycemic control. Epigallocatechin gallate (EGCG) is a characteristic catechin derived from tea that possesses anti-diabetic properties. Nonetheless, its underlying mechanisms remain elusive. Herein, the protective role of EGCG on high glucose (33 mM)-induced pancreatic beta cell dysfunction and its possible molecular mechanisms were investigated. Briefly, MIN6 cells were treated with glucose and EGCG (10 µM, 20 µM, and 40 µM) for 48 h. Our results revealed that EGCG dose-dependently restored mitochondrial membrane potential and concomitantly alleviated cell apoptosis. Mechanistically, the expression level of apoptotic protein BAX and Dynamic related protein 1 (DRP1) was significantly downregulated following EGCG treatment, whereas that of the anti-apoptotic protein BCL-2 was significantly upregulated. Taken together, EGCG alleviated high glucose-induced pancreatic beta cell dysfunction by targeting the DRP1-related mitochondrial apoptosis pathway and thus can serve as a nutritional intervention for the preservation of beta cell dysfunction in patients with type 2 diabetes mellitus.
Similar content being viewed by others

Introduction
As a major public health concern worldwide, diabetes is severely threatening human health. In accordance with the International Diabetes Federation's report, the global prevalence of type 2 diabetes mellitus (T2DM) among adults aged 18–99 years is estimated to be 451 million, and this number will increase to 693 million by 20451. At present, China is the leading country in terms of the number of diabetics2. Earlier studies have reported that islet cell apoptosis plays a decisive role in islet cell dysfunction3,4. Consequently, preventing the apoptosis of islet cells is a candidate treatment option for T2DM. Programmed cell death, also referred to as apoptosis, is regulated by genes. Diabetes can induce apoptosis through three main pathways, namely the death receptor pathway, the apoptotic pathway mediated by endoplasmic reticulum stress, and the intrinsic mitochondrial pathway5,6. The intrinsic pathway initiates apoptosis with mitochondrial structural and functional abnormalities. Subsequently, cytochrome c (Cyt-C) is released into the cytosol and binds to activators of apoptotic proteins, thereby activating caspase-9 in the apoptosome. This is followed by the activation of the downstream caspase-37,8.
Recent studies have revealed that apoptosis is intimately associated with mitochondrial dynamics, both of which are involved in gene regulation9,10. Dynamic-related protein 1 (DRP1), a crucial regulator of mitochondrial division, can govern apoptosis by mediating mitochondrial division and fusion11. Meanwhile, the Bcl-2 family regulates the permeability of the mitochondrial membrane by modulating anti-apoptotic genes like Bcl-2 and pro-apoptotic genes such as Bax. The latter is typically found in the cytosol under physiological states. Nevertheless, following the activation of apoptosis, Bax relocates to the mitochondrial membrane. Of note, it has the ability to form homodimers or heterodimers with other pro-apoptotic members, resulting in the formation of mitochondrial pores12. By directly interacting with Bax, the antiapoptotic Bcl-2 protein impedes the ability of Bax to increase membrane potential, thereby inhibiting apoptosis and ultimately influencing cellular fate through the delicate equilibrium of these proteins13. Moreover, DRP1 stimulates truncated Bid (tBID)-triggered Bax oligomerization by driving the hemifusion of cardiolipin-containing membranes and then initiates apoptosis by releasing Cyt-C through a membrane hemifusion intermediate formed during mitochondrial division14. In the event of DRP1 deficiency, mitochondrial fragmentation is inhibited, and mitochondria become elongated11. Consequently, there is an urgent need to identify novel pharmacological agents that target DRP1 to protect mitochondrial function and mitigate apoptosis. Compelling evidence suggests that plant-based polyphenols possess significant potential in preserving the physiological functions of beta cells and attenuating the adverse impacts of glucolipotoxicity. Consequently, these polyphenols could potentially serve as a precise nutritional intervention for diabetes15.
Epigollatecatechin gallate (EGCG) is a characteristic catechin abundant in tea and a primary component of tea polyphenols. It has been documented to prevent several pathological disorders, encompassing cardiovascular disease, cancer, obesity, and diabetes16. Epidemiological studies have demonstrated that the ingestion of green tea can decrease body weight and fat accumulation by enhancing glucose and lipid metabolism. Additionally, earlier studies have described that green tea extracts and EGCG can alleviate hyperglycemia and insulin resistance in both human and animal subjects. However, the precise molecular mechanism underlying its anti-diabetic effect remains elusive17. Our previous study demonstrated that EGCG exerts a protective effect on hepatocytes and MIN6 pancreatic islet cells by modulating endoplasmic reticulum stress, decreasing reactive oxygen species (ROS) and UCP3 levels, and restoring mitochondrial membrane potential. This effect was observed following cellular exposure to glucotoxicity or IL-1β18,19,20. However, the mechanism by which EGCG confers a protective effect on islet cells through the regulation of the DRP1-related mitochondrial apoptosis pathway under hyperglycemic conditions remains to be elucidated. Therefore, the present study aimed to systemically explore the dose-dependent effect of EGCG on hyperglycemia-caused beta cell dysfunction and the molecular responses underlying the protective effect of EGCG on high glucose-induced pancreatic beta cell dysfunction via WB and RT-PCR.
Materials and methods
Cell culture
MIN6 cells were procured from Baili Company, China. They were incubated in a medium containing 15% fetal bovine serum (16000-044, GIBCO) supplemented with 100 µg/mL streptomycin (P1400-100, Solarbio) and 100U/mL penicillin (P1400-100, Solarbio) at a temperature of 37 °C and a humid atmosphere containing 5% carbon dioxide. The culture medium was renewed every 2–3 days, while the cells were transferred every 5–6 days. After being seeded in 24-well plates overnight, the cells were randomly divided into five ~ seven groups (n = 3) upon reaching 80% confluence. To construct a high glucose-induced model in vitro, MIN6 cells were incubated in RPMI-1640 supplemented with 33 mM glucose for 48 h (model group, HG). The EGCG (A-0159, YaJi Biological) treatment group was treated with a mixture of high glucose concentrations and EGCG (10 µM, 20 µM, 40 µM) for 48 h. Mdivi-1(S80654, MedMol) inhibitor groups include HG + inhibitor group, and HG + 40 µM EGCG + inhibitor group. The blank control group (blank group) consisted of MIN6 cells cultured under standard conditions and served as the baseline for subsequent experiments.
CCK-8 assay
The viability of MIN6 cells was assessed using a Cell Counting Kit-8 (CCK-8) (CP002, SAB). Briefly, the cells were centrifuged for 5 min at 500 rpm, and the supernatant was discarded. Afterward, the resuspended cells were incubated in a serum-free cell culture medium supplemented with 20 µL CCK-8 solution at 37 °C for 1 h. Absorbance was measured at 450 nm using a Microplate Reader (Bio-Rad, United States).
Insulin secretion assay
The insulin secretion by MIN6 cells was assessed using an ELISA Kit (CSB-E05071 m, CUSABIO). Briefly, removed particulates by centrifugation for 15 min at 1000g (4 °C) and assayed immediately according to the manufacturer’s instructions.
Mitochondrial activity
The JC-1 dye is commonly employed as a fluorescent probe to detect mitochondrial membrane potential. Specifically, this dye exhibits a potential-dependent accumulation pattern within mitochondria. Red fluorescence denotes the accumulation of JC-1 in the mitochondrial matrix. Following apoptosis, the mitochondrial membrane potential diminishes, leading to a concomitant reduction in the intensity of red fluorescence21. To assess the mitochondrial membrane potential in MIN6 cells, a kit containing JC-1 (C2006, Beyotime) was employed. Following the resuspension of cells in 0.5 mL PBS, an additional 0.5 mL of JC-1 staining solution was introduced, and the resulting mixture was incubated for twenty minutes at 37 °C in the dark. A flow cytometer (Accuri C6, BD) was then used to detect cells after resuspension in a buffer.
Transmission electron microscopy (TEM)
Following treatment, the cells were rinsed twice with PBS and then fixed for 4 h using 2.5% glutaraldehyde. After washing three times with PBS, 1% osmium tetroxide was subsequently added, followed by serial dehydration using 50, 70, 80, 90, and 100% acetone. Next, the cells were infiltrated with acetone embedding liquid (1:1), buried liquid (1:3), and finally embedded in pure embedded liquid, which was sequentially polymerized at 37 °C overnight at 60 °C for 48 h. A Leica ultramicrotome EM UC7 from Germany was utilized to section the samples, which were then stained with uranyl acetate and alkaline lead citrate for 30 min and scanned using an H-7500 scanner.
Apoptosis assay
Flow cytometry and TUNEL staining were utilized to investigate the apoptotic rate of MIN6 cells. After the cells were washed twice with cold PBS and suspended in a binding buffer, they were stained with Propidium iodide and Annexin V/FITC (C1056, Beyotime). After 20 min of incubation in the dark (37 °C), fluorescence intensity was detected within 1 h using a FACSCalibur flow cytometer (Acuuri, USA). Annexin V-FITC + PI −, Annexin V-FITC − PI +, Annexin V-FITC + PI +, and Annexin V-FITC − PI − represent early apoptotic cells, necrotic cells, late apoptotic cells, and living cells, respectively. Regarding TUNEL staining, the apoptosis levels were measured using an in situ cell death detection kit (11-684-817-910, Roche) following the manufacturer's instructions. After incubation with 50 µL of the TUNEL reaction mixture solution at 37 C for 1 h, fluorescence-positive cells were captured using an IX71 fluorescence microscope (Olympus, Japan).
Immunohistochemistry staining
Immunostaining was performed by fixing MIN6 cells with 4% paraformaldehyde for 30 min at room temperature, washing three times with PBS before permeabilization for 20 min using 0.5% Triton X-100. After blocking with normal goat serum at room temperature for 20 min, MIN6 cells were incubated overnight at 4 °C with the (1:100) primary antibodies against BCL-2 (ab7973, Abcam), BAX (ab32503, Abcam), and DRP1 (ab56788, Abcam). After three washes with PBS, they were incubated with the Fluorescein-Conjugated AffiniPure goat anti-rabbit IgG secondary antibody (ZF-0311, ZSGB-BIO) or the goat anti-mouse IgG (ZB-2305, ZSGB-BIO) in the dark for 20 min. After removing the excess liquid, the slides were subsequently exposed to a DAPI solution at room temperature for 10 min. Following drying and sealing with anti-fluorescence quenching sealing tablets, the slides were visualized under a fluorescence microscope (IX71, Olympus).
Quantitative real-time PCR
TRIzol reagent (15596-026, Invitrogen) was used to extract total RNA. After quantification, the IANScript RT Kit (KR104-02, Tiangen) was used for reverse transcription in accordance with the manufacturer's instructions. A Real-Time PCR System (Connect CFXTM, USA) was used for qPCR, using SYBR green (FP205, Tiangen). The thermal cycles consisted of 10 min at 95 °C, followed by 45 cycles of 15 s at 95 °C, and lastly 60 s at 60 °C. The primers used in this study are as follows: Bax-F: 5ʹ-GGCGAATTGGAGATGAAC-3ʹ; R: 5ʹ-CCAGTTGAAGTTGCCATC-3ʹ. Bcl-2-F: 5ʹ-ACTTGCGTGAAGGCTTGAGA-3ʹ; R: 5ʹ-ATCCAGAGACAAGCGAGCTG-3ʹ. Drp1-F: 5ʹ-AACACCCCGACTTTGCTGAT-3ʹ; R: 5ʹ-ATGGGACCTTGTCCCGTGAT-3ʹ. Gapdh-F: 5ʹ -TGATGGGTGTGAACCACGAGA-3ʹ; R: 5ʹ -TGGTCATGAGCCCTTCCACAA-3ʹ. Gene expression was normalized to Gapdh, and relative quantitative calculation analysis was conducted using the 2 − ΔΔct approach.
Western blotting
To identify differences in the protein expression levels of BCL-2, BAX, and DRP1 between the experimental groups, MIN6 cells were collected and lysed with 200 mL of lysis buffer containing PMSF (ST505, Beyotime) after two PBS washes. Coomassie blue staining (P0017B, Beyotime) was used to determine protein concentrations for each sample, which was denatured at 95 °C for 5 min. After separation using 12% SDS-PAGE (80 V), the protein samples were then transferred to PVDF membranes (Millipore, USA), which were incubated overnight with the following primary antibodies after blocking for an hour at room temperature: Anti-BCL-2 rabbit polyclonal antibody (ab7973, Abcam), Anti-BAX rabbit monoclonal antibody (ab32503, Abcam), Anti-DRP1 mouse monoclonal (ab56788, Abcam) (1:500) and mouse mAb-actin (AA128, Beyotime) (1:1000). Afterward, after washing with TBST, the membranes were incubated with HRP-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies (1:1000) for one hour at room temperature. Finally, Western Lighting ECL (Fusion solos, China) was used to develop the chemiluminescent HRP substrate (Merk Millipore, USA) and to visualize the blots. Finally, the absorbance values for target proteins were calculated using Image J 180 for visualization.
Statistical analysis
Statistical analyses were performed using the SPSS 15.0 statistical software. Variables were expressed as mean and standard deviation. The F-test was used to compare multiple samples. Multiple sample means were compared using the Dunnet t-test and SNK-q test. p < 0.05 was considered statistically significant.
Results
EGCG treatment improved activity and function in high glucose-treated MIN6 cells and partially restored mitochondrial membrane potential
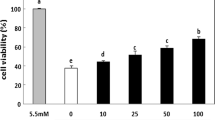
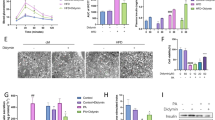
Cell viability was evaluated via the CCK8 assay following treatment with glucose and pre-defined concentrations of EGCG. As illustrated in Fig. 1A, the cell viability of MIN6 cells was significantly decreased (P < 0.0001) following exposure to a high glucose concentration. However, MIN6 cells treated with a combination of high glucose levels and varying concentrations of EGCG (10 µM, 20 µM, 40 µM) for 48 h displayed a concentration-dependent enhancement in cell viability compared to the group treated solely with high levels of glucose. To investigate the dose–effect and duration-effect of EGCG on cell viability, different concentrations of EGCG (0, 10, 20, 40, 80, 160, and 320 μM) were added to culture media for varying durations (0, 12, 24, and 48 h) in the selected groups. As anticipated, cell viability increased with increasing initial EGCG concentrations and peaked at 80 μM, after which the proliferative ability progressively decreased (Supplementary Fig. S1A). Conversely, the administration of high levels of glucose to MIN6 cells inhibited the proliferative abilities of MIN6 cells at 12, 24, and 48 h. Meanwhile, EGCG partially restored the viability of MIN6 cells at different time points, particularly after 48 h of culture. Moreover, treatment with a DRP1 inhibitor (mdivi-1) attenuated the inhibitory effects of high glucose, which was similar to the effects of 10 μM EGCG (Supplementary Fig. S1B). Consistent with the findings of previous studies, exposure to high glucose (33.3 mmol/L) impaired glucose-stimulated insulin secretion and insulin synthesis in MIN6 cells compared with the control group. However, EGCG treatment significantly increased insulin secretion in high glucose-stimulated MIN6 cells in a dose-dependent manner. Likewise, DRP1 inhibitor treatment significantly upregulated the expression of insulin, but its effect was inferior to that of 20 and 40 μM EGCG (Fig. 1B). The JC-1 fluorochrome is extensively employed for the determination of mitochondrial membrane potentials and emits a red fluorescence in healthy cells due to its aggregated structure in the mitochondrial matrix. As is well documented, mitochondrial depolarization leads to an increase in the permeability of the mitochondrial membrane, causing the dispersion of the JC-1 probe into the cytoplasm. Consequently, the probe disaggregates into monomers, resulting in a weakening of red fluorescence. In the current study, flow cytometry was used to measure fluorescence intensity. The results revealed a significant reduction (P < 0.0001) in red fluorescence in the high glucose group, suggesting a decline in mitochondrial membrane potential and an elevation in the proportion of early mitochondrial apoptotic cells compared to the control group. Nonetheless, EGCG reversed this effect in a dose-dependent manner (Fig. 1C,D), indicating that it minimized the loss in mitochondrial membrane potential compared to high glucose treatment alone.

Effect of EGCG on cell viability, function, and mitochondrial membrane potential in MIN6 cells treated with high glucose concentrations (HG). MIN6 cells were treated with high levels of glucose and with or without different doses of EGCG (10 µM, 20 µM, 40 µM) and DRP1 inhibitor for 48 h. (A) The CCK-8 assay was used to assess cell viability. (B) Insulin levels in the cell supernatants were determined using an ELISA kit. (C,D) Flow cytometry was utilized to determine mitochondrial membrane potential in each group of MIN6 cells; the bar chart (C) displays a summary of the results of (D). The data are representative of two independent experiments and are presented as mean ± standard deviation; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. A post hoc test was conducted to examine differences using one-way ANOVA.
Effect of EGCG on the mitochondrial ultrastructure of high glucose-treated MIN6 cells
Transmission electron microscopy was used to visualize cellular ultrastructures, revealing that mitochondria in the control group exhibited an elliptic or elongated morphology. Furthermore, these mitochondria displayed an intact membrane structure, longitudinal cristae that were complete and evenly arranged, and a dense interstitium without evidence of swelling. In the high glucose group, alterations in mitochondrial morphology were observed, characterized by vacuolized swelling and the absence of cristae. Additionally, mitochondrial outer membrane rupture and collapse, cytoplasmic and nucleoplasmic swelling, and cell membrane impairment were noted (Fig. 2). On the other hand, mitochondria were swollen in the low concentration (10 μM) EGCG group, accompanied by cristae disruption, disorganized cristae structure, and blurred internal structures. Interestingly, the degree of mitochondrial damage was slightly lower compared to the high glucose group. In the middle concentration (20 μM) EGCG group, mitochondria were marginally swollen; nonetheless, cristae breaks were visible. In the high-concentration (40 μM) EGCG group, there was no significant mitochondrial swelling, and the mitochondrial cristae and membrane were fused and blurred.

Effect of EGCG on the mitochondrial ultrastructure of high glucose-treated MIN6 Cells. MIN6 cells, with or without EGCG treatment for 48 h, were examined under a transmission electron microscope to visualize the mitochondrial ultrastructure. Representative images (× 4000, × 10,000) are presented. The yellow arrows represent mitochondria, and the green arrows denote the location of the nucleus.
EGCG treatment significantly attenuated apoptosis of high glucose-treated MIN6 cells
The TUNEL assay revealed a significant increase in the proportion of TUNEL-positive cells in the high glucose group compared to the control group in MIN6 cell samples, Notably, EGCG (Fig. 3A,B) alleviated high glucose-induced cell death in a concentration-dependent manner. In order to further investigate the anti-apoptotic effect of EGCG in high glucose-treated MIN6 cells, alterations in apoptotic rates were analyzed through flow cytometry. As anticipated, the rate of early apoptosis and late apoptosis was significantly increased after 48 h of treatment with high glucose compared to the control group. Besides, EGCG yielded a concentration-dependent reduction in apoptotic rates. In comparison to the high glucose group, the administration of 10 μM EGCG for 48 h resulted in a significant reduction in the percentage of cells undergoing early apoptosis from 17.7 ± 2.13% to 14.1 ± 2.63%. Additionally, the percentage of cells undergoing late apoptosis also decreased from 20.0 ± 3.18% to 16.2 ± 2.26%. Moreover, this impact was more pronounced in the EGCG 20 μM and 40 μM groups. Finally, treatment with the DRP1 inhibitor could also partially inhibit high glucose-induced cell death, but its effect was inferior to that of 40 μM EGCG, as depicted in Fig. 3C,D.

Effect of EGCG on apoptosis in MIN6 cells treated with high glucose. MIN6 cells were treated with high glucose and in the presence or absence of EGCG (10 µM, 20 µM, 40 µM) and DRP1 inhibitor for 48 h. (A) TUNEL staining was employed to observe alterations in the apoptosis level of MIN6 cells. Representative images (× 400) are displayed. Apoptotic cells are denoted by yellow arrows. (B) Quantification of TUNEL-positive cells. (C) Representative flow cytometry plots depicting the proportion of cells in the early apoptotic, necrotic, late apoptotic, and living states. (D) The summary of the statistical findings in (C) is presented in the bar charts. The data are representative of two independent experiments and are presented as mean ± standard deviation; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. A post hoc test was conducted to assess differences variations using a one-way ANOVA.
EGCG regulates Bax, Bcl-2, and Drp1 mRNA expression in high glucose-treated MIN6 cells
RT-qPCR was performed to detect the mRNA expression levels of Bax, Bcl-2, and Drp1. The expression of Drp1 and proapoptosis-related gene Bax was significantly upregulated in the high glucose group compared with the blank control (p < 0.0001). Conversely, exposure to high glucose resulted in a decrease in the expression level of the anti-apoptotic gene Bcl-2 (Fig. 4A–C). However, the up-regulation in the mRNA expression of Drp1 and Bax mRNA and the down-regulation in that of Bcl-2 was reversed by EGCG in a dose-dependent manner. In other words, the expression levels of Drp1 and Bax in the high-dose EGCG group were restored to a level comparable to the blank control, which was approximately twice as high as those in the blank group.

Effect of EGCG on the mRNA expression levels of Bax, Bcl-2, and Drp1 in MIN6 cells treated with high glucose. MIN6 cells were treated with high glucose in the presence or absence of EGCG (10 µM, 20 µM, 40 µM) for 48 h. Total RNA was extracted from cells and subjected to qRT-PCR to determine the levels of Bax (A), Bcl-2 (B), and Drp1 (C). The data are representative of two independent experiments and are presented as mean ± standard deviation; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. A post hoc test was conducted to assess differences using one-way ANOVA.
EGCG regulates DRP1 and downstream effector proteins in high glucose-treated MIN6 cells
DRP1 may influence cellular outcomes via its regulation of the balance between the apoptotic proteins BAX/BCL-2. To investigate the impact of EGCG on apoptosis via the DRP1-BAX/BCL-2 pathway in high glucose-stimulated MIN6 cells, western blotting, and immunofluorescence assays were conducted to detect the protein expression levels of BAX, BCL-2, and DRP1. The findings were consistent with those of qRT-PCR (Fig. 4). Moreover, compared with control cells, high glucose elicited a downregulation in the expression of the anti-apoptotic BCL-2 protein (Figs. 5B,D, 6B,F). In contrast, the expression of the pro-apoptotic BAX protein (Figs. 5A,C, 6B,E) and DRP1 (Fig. 6A,C,D) was upregulated. Notably, high glucose conditions also disrupted the BAX/BCL-2 balance and increased the BAX/BCL-2 ratio (Figs. 5E, 6G). Importantly, alterations in the expression level of BAX, BCL-2, and DRP1 were reversed by EGCG in a dose-dependent manner. Furthermore, a DRP1 inhibitor was introduced to explore the effects of hyperglycemia on mitochondrial apoptosis. Western blot analysis demonstrated that compared with the high glucose group, the expression level of DRP1 and BAX decreased following the concurrent administration of the inhibitor mdivi-1, whereas the expression level of BCL-2 was restored. Furthermore, the effect was more pronounced following DRP1 down-regulation and treatment with 40 μM EGCG (Fig. 6).

Immunofluorescence staining and expression levels of BAX and BCL-2. MIN6 cells were treated with high glucose in the presence or absence of EGCG (10 µM, 20 µM, 40 µM) for 48 h. Immunocytochemical analysis was conducted to evaluate the protein level of BAX and BCL-2. The blue fluorescence represents the nucleus, whereas the green fluorescence represents the protein of interest (× 400), (A) representative images for BAX, (B) Representative images for BCL-2, (C) presents a bar chart summarizing the findings from (A). (D) Bar chart delineating a summary of results from (B), (E) BAX/BCL-2 ration. The data are representative of two independent experiments and are presented as mean ± standard deviation; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. A post hoc test was conducted to assess differences using one-way ANOVA.

Immunofluorescence staining of DRP1 and Western blot analysis. MIN6 cells were treated with high glucose in the presence or absence of EGCG (10 µM, 20 µM, 40 µM) and DRP1 inhibitor for 48 h. Immunocytochemical analysis was conducted to evaluate the level of DRP1. The blue fluorescence represents the nucleus, while the green fluorescence represents the protein of interest (× 400), (A) representative images for DRP1, (C) Bar graph portraying a summary of the findings in (A). (B) Western blot bands and quantitative analysis of the expression of DRP1 (D), BAX (E), and BCL-2 (F). (G) The BAX/BCL-2 ratio. The data are representative of two independent experiments and are presented as mean ± standard deviation; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. A post hoc test was conducted to assess differences using one-way ANOVA.
Discussion
The MIN6 cell line, originating from mouse pancreatic β-cells, demonstrates a comparable degree of insulin secretion to that of isolated islets when stimulated by glucose. Thus, this particular cell line is a valuable model for investigating pancreatic islet development, cell differentiation, and functional characteristics22. β-cells within an islet can synthesize and release insulin in response to variations in glucose levels within the bloodstream and surrounding environment. A wide range of chemicals, biological agents, and ions have the capacity to exert both direct and indirect effects on biological functions. The impact of glucose, amino acids, vitamins, and Ca2+ on the survival and insulin release of pancreatic islets has been well-documented23. Glucose toxicity is widely recognized as a significant contributor to mitochondrial apoptosis in pancreatic β-cells. Islets exposed to a glucose concentration exceeding 24.4 mmol/L for 48 h exhibited a higher incidence of apoptotic events compared to islets exposed to a glucose concentration ranging from 5.5 to 11.1 mmol/L24. In other words, the persistent exposure to high glucose levels induces oxidative stress, thereby limiting ATP production and mitochondrial membrane potential. This, in turn, triggers the mitochondrial apoptosis pathway, ultimately resulting in apoptosis25. Herein, high glucose levels (33 mM) were used to stimulate MIN6 cells, and our results corroborated that increased glucose concentrations can suppress growth and trigger the apoptosis of MIN6 cells along with a decrease in insulin secretion.
The prevalence of metabolic diseases is on the rise among younger populations due to the popularization of contemporary diets rich in sugar and fat, coupled with sedentary behaviors. EGCG, a prominent active component derived from green tea, has demonstrated outstanding efficacy in the treatment of metabolic disorders, including obesity, type 2 diabetes, cardiovascular disease, kidney disease, and gout26. Specifically, its anti-diabetic and anti-obesity properties are intricately linked to mechanisms involving insulin resistance, inflammation, and metabolic dysregulation. Human experiments demonstrated that continuous supplementation of EGCG or green tea was effective in improving body weight, blood lipids, and so on27,28. Furthermore, findings from animal experiments indicated that the anti-obesity mechanism of EGCG may be ascribed to its modulation of inflammatory factor release, IRS-1, and the AMPK signaling pathway29,30,31. Diabetes mellitus comprises a collection of metabolic disorders characterized by elevated blood glucose levels stemming from deficiencies in insulin secretion, insulin action, or a combination of both factors31. At present, antidiabetic medications are associated with a wide range of adverse effects and may lead to complications32,33. The consumption of an appropriate dosage of EGCG has been shown to effectively and consistently regulate blood glucose levels, enhance insulin sensitivity, and decrease blood lipid concentrations34. These effects can be primarily attributed to its ability to reverse pancreatic β-cell damage and apoptosis, mitigate oxidative stress, and reduce the levels of inflammatory factors such as IL-1β and IL-1835,36. In a mouse model of partial liver ischemia–reperfusion injury, the application of EGCG (50 mg/kg) successfully decreased oxidative stress and apoptosis37. Xie et al. described that EGCG can attenuate nephrotoxicity induced by gentamicin, an aminoglycoside antibiotic, by suppressing apoptosis and ferroptosis. Therefore, EGCG holds significant potential for further research and development owing to its well-documented anti-apoptotic properties38.
However, EGCG typically displays a biphasic bioactivity pattern, with high doses exerting pro-oxidative and pro-apoptotic effects and low doses demonstrating neuroprotective characteristics39. In vivo and in vitro studies have reported that EGCG is well tolerated and non-toxic to cells within a specific concentration range. According to an earlier study, the oral bioavailability of EGCG is estimated to range from 0.1% to 0.3% in rats and humans40. Administering a high concentration of EGCG (50 mg kg−1 day−1) alleviated the adverse impacts of a high-fat diet on well-being, chronic ailments, and longevity. By applying the specific surface area method and assuming an average weight of 60 kg for humans, the estimated dosage would be around 500 mg per kilogram per day30. Furthermore, the oral administration of EGCG to wild-type mice at a daily dosage of 100 μg day−1 g−1 of body weight for 2 months led to a notable decrease in the level of serum alanine transferase and alkaline phosphatase, which were initially elevated due to the consumption of a Western diet. This observation infers that EGCG did not induce hepatotoxicity41. In a research conducted on healthy women, a dosage of 752 mg of EGCG, which is equivalent to 9 cups of green tea, was well tolerated, with only a few participants experiencing mild nausea or vomiting after 90 min of capsule ingestion. In addition, a study enrolling healthy individuals assessed the safety of 800 mg/day of pure EGCG for 4 weeks. The results revealed no significant symptoms or adverse events, suggesting that a daily intake of 752 mg to 800 mg is safe42,43,44. The cytotoxicity of EGCG at elevated concentrations has been investigated in multiple in vitro experiments. Specifically, the function of BEAS-2B cells was considerably suppressed compared to the control group following treatment with 100 μg/mL of EGCG for 48 h. Conversely, cellular activity was not significantly impacted following exposure to 50 μg/mL of EGCG45. Additionally, another study demonstrated that EGCG showed no signs of toxicity towards the normal embryonic fibroblast cell line (3T3), even at a concentration of 1000 μM46. In conclusion, EGCG exerts differential cytotoxicity based on cell species and tissue origin. Herein, MIN6 cells were treated with varying concentrations of EGCG in order to identify a safe and effective therapeutic dose. Compared to high EGCG concentrations (> 80 μM), medium and low concentrations (10–40 μM) resulted in significantly higher cell viability. On the other hand, the protective effects of EGCG persisted over time, given that high glucose loads fully inhibited cell proliferation with prolonged exposure. Moreover, cell proliferation initially increased after co-cultivation for 12 h with the DRP1 inhibitor or EGCG and was significantly higher at 24 h. Noteworthily, this protective effect was more prominent at 48 h. Based on the results of the cell viability test, a 48-h duration was selected for EGCG treatment in the subsequent experiments. As anticipated, EGCG partially reversed the decrease in cell viability induced by high glucose levels. Additionally, flow cytometry demonstrated that EGCG exerted a protective effect against high glucose-induced apoptosis in MIN6 cells, consistent with the results of TUNEL staining.
As is well documented, hyperglycemia exacerbates the impairment of insulin secretion, a phenomenon commonly referred to as glucotoxicity47. This reduction in insulin secretion may be ascribed to direct damage to pancreatic islets by high glucose levels. In addition, signaling pathways, such as the nodal signaling pathway in islet β-cells, AKT signaling pathway, and PPAR signaling pathway, may also play a role under hyperglycemic conditions48,49,50. According to the results of this study, EGCG increased the viability of MIN6 cells by inhibiting apoptosis in a dose-dependent manner. Importantly, the surviving cells remained functional and were capable of synthesizing and secreting insulin. Moreover, although EGCG only partially restored cell viability, it significantly promoted insulin secretion. Nevertheless, the mechanism by which EGCG influences insulin sensitivity remains elusive. Besides, EGCG exerted a significant effect in restoring the mitochondrial membrane potential, particularly at a concentration of 40 μM. At this concentration, both cellular viability and mitochondrial membrane potential were comparable to those of the control group. Therefore, within the specified concentration range, EGCG partially stimulated cellular proliferation, alleviated cell apoptosis, and restored insulin secretion and mitochondrial membrane potential under hyperglycemic conditions.
Mitochondria undergo morphological alterations through fusion and division processes to adapt to the demands of cellular metabolism. In mammals, mitochondrial fusion is facilitated by the fusion proteins mitofusin 1, mitofusin 2, and optic atrophy 1, whilst mitochondrial fission is regulated by Drp1. It is crucial for essential mitochondrial functions, such as energy metabolism, cellular specialization, and growth. On the other hand, mitochondrial division plays a pivotal role in the generation of new mitochondria during cell division and in the removal of impaired mitochondria via mitophagy51,52. DRP1, a soluble protein predominantly located in the cytoplasm, typically remains in an inactive state through dephosphorylation. However, DRP1 can be activated through dephosphorylation during apoptosis. Additionally, both DRP1 and Endophilin B1 can translocate from the cytoplasm to the region of mitochondrial division, thereby driving the segregation of mitochondria into two distinct daughter mitochondria25,53. Besides, the delicate balance between mitochondrial fusion and fission is considerably influenced by DRP1. Suppressing DRP1 function has been hypothesized to hinder not only mitochondrial fission but also the activation of caspases and subsequent cell death54. Conversely, an excessive up-regulation in the expression of DRP1 can promote mitochondrial fragmentation and disrupt the BAX/BCL-2 balance, thereby regulating the Caspase-9 pathway and contributing to cell apoptosis via the mitochondrial pathway55,56. However, the mechanism by which DRP1 interacts with BAX/BCL-2 during apoptosis remains unclear. DRP1-dependent mitochondrial autophagy may serve as a regulatory mechanism preceding apoptosis, with DRP1 down-regulation resulting in partial mitigation of intrinsic apoptosis57. Zhao et al. concluded that DRP1 can induce the oligomerization of BAX and the release of cytochrome C, processes essential for increasing mitochondrial membrane permeability and the release of apoptotic factors58. Several previous studies have demonstrated that co-localization and interaction between DRP1 and BAX facilitate mitochondrial membrane permeabilization and fragmentation59,60. The findings of our study indicated that high glucose levels decreased the mitochondrial membrane potential and compromised mitochondrial integrity through DRP1-mediated mitochondrial apoptosis. The introduction of a DRP1 inhibitor further supports our hypothesis, given the enhanced viability of MIN6 cells and reduced number of apoptotic cells following DRP1 inhibition compared to the control group. Furthermore, a decrease in downstream BAX protein expression levels was observed, accompanied by an elevation in BCL-2 levels. These findings collectively indicated that high glucose levels may activate the DRP1-BAX/BCL-2 axis, ultimately initiating the mitochondrial apoptotic pathway in MIN6 cells.
Numerous chemicals and active constituents can modify cellular outcomes by modulating DRP1 expression. Recent investigations have revealed that the cardioprotective impact of carbon monoxide (CO) on cardiomyocytes following cardiopulmonary resuscitation (CPR) is attributed to a reduction in the expression levels of DRP1 and the inhibition of mitochondria-mediated apoptosis61. Furthermore, the downregulation of dynamin DRP1 and the preservation of mitochondrial homeostasis during ischemia/reperfusion injury is closely linked to anti-apoptotic effects in brain neurons and cardiac cells62,63. The aforestated protective effect was also observed in studies on diabetes. Makoto Tagaya et al.64 demonstrated that the inhibition of mitochondrial division promoted podocytes to relieve albumin-induced cell damage in a diabetic nephropathy model. Additionally, the modulation of mitochondrial dynamics through optoprotein D and rapamycin was found to ameliorate diabetes-induced myocardial injury and concurrently enhance brain injury recovery in rats with middle cerebral artery occlusion65,66. It is worthwhile acknowledging that tea polyphenols mitigate injury inflicted upon diabetic oocytes through a mechanism linked to a reduction of ROS levels and the expression of mitochondrial DRP167. Nevertheless, the underlying mechanism by which EGCG protects against high glucose stress remains poorly understood. Herein, co-cultivation with EGCG successfully reversed the decline in mitochondrial membrane potential and mitigated mitochondrial swelling and fragmentation induced by high glucose levels. Furthermore, EGCG markedly reduced the levels of DRP1, an essential protein participating in mitochondrial fission, as well as downstream BAX, while simultaneously up-regulating the expression of BCL-2. Taken together, these results suggest that EGCG may protect MIN6 cells from high glucose-induced mitochondrial apoptosis by modulating the DRP1-BAX/BCL-2 signaling pathway. It is worthwhile emphasizing that EGCG yielded a greater decrease in apoptosis level and down-regulating DRP1 expression compared to the high glucose group with the DRP1 inhibition, particularly at higher EGCG concentrations. This phenomenon may be attributed to the diverse biological effects exerted by EGCG. There are numerous pathways for cell death, each with its own morphological and biochemical characteristics, including apoptosis, necroptosis, autophagy, ferroptosis, pyroptosis, necrosis, etc.68. Diabetes mellitus is hallmarked by an intricate pathogenesis involving several mechanisms leading to complications69. Indeed, multiple forms of programmed cell death play a decisive role in the development and progression of diabetes mellitus70. Recent research implied that EGCG can inhibit ferroptosis-induced cell death, thereby attenuating liver injury in mice71. Additionally, EGCG has been found to enhance the restoration of sensory function following dorsal root crush injury in rats by up-regulating KAT6A and suppressing pyroptosis72. Thus, the current study validated that EGCG may preserve MIN6 cell viability by protecting against various forms of programmed cell death, especially by modulating the DRP1-mediated mitochondrial apoptotic pathway.
Furthermore, our study uncovered that EGCG possesses the capacity to restore cell viability, inhibit apoptosis, and increase mitochondrial membrane potential in MIN6 cells induced by high glucose loads, potentially by regulating DRP1-induced mitochondrial apoptosis. Nevertheless, it is imperative to acknowledge that previous research has demonstrated that EGCG can modulate multiple signaling pathways, including those related to inflammation, oxidative stress, endoplasmic reticulum stress, and other metabolic processes73, ultimately leading to apoptosis. Our study indicated that EGCG plays a role in the synthesis and secretion of insulin, thereby expanding our understanding of the mechanisms underlying its anti-diabetic properties. Type 2 diabetes is a chronic condition that lacks a definitive cure. In recent years, the therapeutic potential of traditional Chinese medicine, particularly tea polyphenols, as agents for the management of diabetes has garnered extensive attention74,75. The findings of this study signaled that EGCG exerts anti-apoptotic effects in islet cells and enhances insulin secretion, highlighting its potential as a promising supplement for obese individuals or those with a genetic predisposition to diabetes.
Conclusions
One of the limitations inherent in this study is the exclusive reliance on MIN6 cells for the analysis of EGCG. While MIN6 cells secrete insulin levels comparable to those of cultured normal islet cells, it is important to acknowledge that the MIN6 cell line may not fully encompass all the physiological characteristics of pancreatic beta cells. Additionally, the effective concentration of EGCG within pancreatic islets in vivo remains unknown, warranting further investigations to comprehensively elucidate the in vivo role of EGCG in pancreatic beta cells. In summary, EGCG played a protective role in β-cell apoptosis by regulating DRP1-mediated mitochondrial apoptosis pathways. Our research offers empirical evidence of the protective impact of EGCG on pancreatic beta-cell dysfunction caused by high glucose stress, thereby expanding our understanding of the anti-diabetic effects of EGCG and positioning it as a nutritional intervention aimed at mitigating beta-cell glucotoxicity in diabetes.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Q&A: Key points for IDF Diabetes Atlas 2017. Diabetes Res. Clin. Pr. 135 (2018).
Yu, X. et al. Prevalence and control of diabetes in Chinese adults. JAMA 310, 1 (2013).
Daisuke, A., Hiderou, Y. & Yukihiro, H. Endoplasmic reticulum (ER) stress and endocrine disorders. Int. J. Mol. Sci. 18, 1 (2017).
Fukunaka, A. & Fujitani, Y. Role of zinc homeostasis in the pathogenesis of diabetes and obesity. Int. J. Mol. Sci. 19, 1. https://doi.org/10.3390/ijms19020476 (2018).
Qiao, L. & Wong, B. C. Targeting apoptosis as an approach for gastrointestinal cancer therapy. Drug Resist. Updat 12, 55–64. https://doi.org/10.1016/j.drup.2009.02.002 (2009).
Shoshan-Barmatz, V. et al. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 1848, 2547–2575. https://doi.org/10.1016/j.bbamem.2014.10.040 (2015).
Guo, H. et al. Research advances on pathways of nickel-induced apoptosis. Int. J. Mol. Sci. 17, 1. https://doi.org/10.3390/ijms17010010 (2015).
Liu, D. M. et al. Emodin induces apoptosis in HepG2 cells via mitochondrial pathway [J]. Chin. J. Exp. Tradition. Med. Form. 24(03), 104–108 (2018).
Ansari, M. Y., Novak, K. & Haqqi, T. M. ERK1/2-mediated activation of DRP1 regulates mitochondrial dynamics and apoptosis in chondrocytes. Osteoarth. Cartil. 30, 315–328. https://doi.org/10.1016/j.joca.2021.11.003 (2022).
Scaini, G. et al. Dysregulation of mitochondrial dynamics, mitophagy and apoptosis in major depressive disorder: Does inflammation play a role?. Mol. Psychiatry 27, 1095–1102. https://doi.org/10.1038/s41380-021-01312-w (2022).
Kim, D., Sesaki, H. & Roy, S. Reduced levels of Drp1 protect against development of retinal vascular lesions in diabetic retinopathy. Cells-Basel 10, 1. https://doi.org/10.3390/cells10061379 (2021).
Vyssokikh, M. Y. et al. Bax releases cytochrome c preferentially from a complex between porin and adenine nucleotide translocator: Hexokinase activity suppresses this effect. Mol. Biol. Rep. 29, 93–96. https://doi.org/10.1023/a:1020383108620 (2002).
Seid, A. N., Khodarahmi, P. & Naseh, V. The effect of cadmium on apoptotic genes mRNA expression of Bax and Bcl-2 in small intestine of rats. Iran J. Pathol. 13, 408–414 (2018).
Chen, F. Y. et al. BIK ubiquitination by the E3 ligase Cul5-ASB11 determines cell fate during cellular stress. J. Cell Biol. 218, 3002–3018. https://doi.org/10.1083/jcb.201901156 (2019).
Chen, Y. et al. Cyanidin-3-O-glucoside ameliorates palmitic-acid-induced pancreatic beta cell dysfunction by modulating CHOP-mediated endoplasmic reticulum stress pathways. Nutrients 14, 1. https://doi.org/10.3390/nu14091835 (2022).
Olech, M. et al. Polyphenol composition and antioxidant potential of instant gruels enriched with Lycium barbarum L. Fruit. Mol. 25, 1. https://doi.org/10.3390/molecules25194538 (2020).
Ueda-Wakagi, M. et al. Green tea ameliorates hyperglycemia by promoting the translocation of glucose transporter 4 in the skeletal muscle of diabetic rodents. Int. J. Mol. Sci. 20, 1. https://doi.org/10.3390/ijms20102436 (2019).
Luo, Z. R. et al. Experimental study on the effect of epigallocatechin gallate on hepatocyte apoptosis and the expression of endoplasmic reticulum stress protein PERK and GRP78 in type 2 diabetic rats [J]. J. Tea Commun. 47(04), 665–674 (2020).
Liu, H. et al. Effects of Epigallocatechin gallate on IL-lβ induced MIN6 cells apoptosis[J]. Chongqing Med. 44(23), 3183–3186 (2015).
Jia, X. et al. EGCG upregulates UCP(3) levels to protect MIN(6) pancreatic islet cells from interleukin-1beta-induced apoptosis. Drug Des. Dev. Ther. 14, 4251–4261. https://doi.org/10.2147/DDDT.S270345 (2020).
Dressler, C. et al. Fluorescence imaging of heat-stress induced mitochondrial long-term depolarization in breast cancer cells. J. Fluoresc. 16, 689–695. https://doi.org/10.1007/s10895-006-0110-z (2006).
Nakashima, K. et al. MIN6 is not a pure beta cell line but a mixed cell line with other pancreatic endocrine hormones. Endocr. J. 56, 45–53. https://doi.org/10.1507/endocrj.K08E-172 (2009).
Kang, H. C. & Bae, Y. H. Transfection of rat pancreatic islet tissue by polymeric gene vectors. Diabetes Technol. 11, 443–449. https://doi.org/10.1089/dia.2008.0117 (2009).
Mellado-Gil, J. M. & Aguilar-Diosdado, M. Assay for high glucose-mediated islet cell sensitization to apoptosis induced by streptozotocin and cytokines. Biol. Proced. Online 7, 162–171. https://doi.org/10.1251/bpo113 (2005).
Senft, D. & Ronai, Z. A. Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 39, 43–52. https://doi.org/10.1016/j.ceb.2016.02.001 (2016).
James, A., Wang, K. & Wang, Y. Therapeutic activity of green tea epigallocatechin-3-gallate on metabolic diseases and non-alcoholic fatty liver diseases: The current updates. Nutrients 15, 1. https://doi.org/10.3390/nu15133022 (2023).
Li, F. et al. Epigallocatechin gallate reduces uric acid levels by regulating xanthine oxidase activity and uric acid excretion in vitro and in vivo. Ann. Palliat. Med. 9, 331–338. https://doi.org/10.21037/apm.2019.11.28 (2020).
Xu, X. Y. et al. Effects and mechanisms of tea on obesity. Crit. Rev. Food Sci. Nutr. 63, 3716–3733. https://doi.org/10.1080/10408398.2021.1992748 (2023).
Wu, G. et al. Tea polyphenol EGCG ameliorates obesity-related complications by regulating lipidomic pathway in leptin receptor knockout rats. J. Nutr. Biochem. 118, 109349. https://doi.org/10.1016/j.jnutbio.2023.109349 (2023).
Yuan, H. et al. The phytochemical epigallocatechin gallate prolongs the lifespan by improving lipid metabolism, reducing inflammation and oxidative stress in high-fat diet-fed obese rats. Aging Cell 19, e13199. https://doi.org/10.1111/acel.13199 (2020).
Rocha, A. et al. Green tea extract activates AMPK and ameliorates white adipose tissue metabolic dysfunction induced by obesity. Eur. J. Nutr. 55, 2231–2244. https://doi.org/10.1007/s00394-015-1033-8 (2016).
Wan, C. et al. Effects of green tea polyphenol extract and epigallocatechin-3-O-gallate on diabetes mellitus and diabetic complications: Recent advances. Crit. Rev. Food Sci. Nutr. 1, 1–29. https://doi.org/10.1080/10408398.2022.2157372 (2022).
Marin-Penalver, J. J. et al. Update on the treatment of type 2 diabetes mellitus. World J. Diabetes 7, 354–395. https://doi.org/10.4239/wjd.v7.i17.354 (2016).
Asbaghi, O. et al. Effect of green tea extract on lipid profile in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetes Metab. Syndr 14, 293–301. https://doi.org/10.1016/j.dsx.2020.03.018 (2020).
Dinarello, C. A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550. https://doi.org/10.1146/annurev.immunol.021908.132612 (2009).
Zhang, C. et al. Epigallocatechin-3-gallate prevents inflammation and diabetes -Induced glucose tolerance through inhibition of NLRP3 inflammasome activation. Int. Immunopharmacol. 93, 107412. https://doi.org/10.1016/j.intimp.2021.107412 (2021).
Tak, E. et al. Epigallocatechin-3-gallate protects against hepatic ischaemia-reperfusion injury by reducing oxidative stress and apoptotic cell death. J. Int. Med. Res. 44, 1248–1262. https://doi.org/10.1177/0300060516662735 (2016).
Zhou, D. et al. Epigallocatechin-3-gallate protects porcine oocytes against post-ovulatory aging through inhibition of oxidative stress. Aging (Albany NY) 14, 8633–8644. https://doi.org/10.18632/aging.204368 (2022).
Feng, S. et al. (-)-Epigallocatechin-3-gallate protects PC12 cells against corticosterone-induced neurotoxicity via the hedgehog signaling pathway. Exp. Ther. Med. 15, 4284–4290. https://doi.org/10.3892/etm.2018.5936 (2018).
Noguchi-Shinohara, M. et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. Plos One 9, e96013. https://doi.org/10.1371/journal.pone.0096013 (2014).
Sheng, L. et al. Obesity treatment by epigallocatechin-3-gallate-regulated bile acid signaling and its enriched Akkermansia muciniphila. Faseb J. 32, j201800370R. https://doi.org/10.1096/fj.201800370R (2018).
Fernandes, R. C. et al. Acute epigallocatechin 3 gallate (EGCG) supplementation delays gastric emptying in healthy women: A randomized, double-blind placebo-controlled crossover study. Nutrients 10, 1122. https://doi.org/10.3390/nu10081122 (2018).
Sherry Chow, H. H. et al. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin. Cancer Res. 9, 3312–3319 (2003).
Siblini, H. et al. Assessing the hepatic safety of epigallocatechin gallate (EGCG) in reproductive-aged women. Nutrients 15, 1. https://doi.org/10.3390/nu15020320 (2023).
Mou, Q. et al. EGCG induces beta-defensin 3 against influenza A virus H1N1 by the MAPK signaling pathway. Exp. Ther. Med. 20, 3017–3024. https://doi.org/10.3892/etm.2020.9047 (2020).
Md, N. Z. et al. Induction of endoplasmic reticulum stress pathway by green tea epigallocatechin-3-gallate (EGCG) in colorectal cancer cells: Activation of PERK/p-eIF2alpha/ATF4 and IRE1alpha. Biomed. Res. Int. 2019, 3480569. https://doi.org/10.1155/2019/3480569 (2019).
Kato, T. et al. Palmitate impairs and eicosapentaenoate restores insulin secretion through regulation of SREBP-1c in pancreatic islets. Diabetes 57, 2382–2392. https://doi.org/10.2337/db06-1806 (2008).
Li, J. et al. Antagonistic interaction between Nodal and insulin modulates pancreatic beta-cell proliferation and survival. Cell Commun. Signal 16, 79. https://doi.org/10.1186/s12964-018-0288-0 (2018).
Li, Y. et al. hMSCs migrate under the chemotaxis of CXCL-13 and enhance Islet B cell activity through p-AKT signaling pathway in high-glucose environment. J. Healthc Eng. 2022, 5430175. https://doi.org/10.1155/2022/5430175 (2022).
Zhou, Y. et al. Comparative evaluation of chiglitazar and sitagliptin on the levels of retinol-binding protein 4 and its correlation with insulin resistance in patients with type 2 diabetes. Front. Endocrinol. (Lausanne) 13, 801271. https://doi.org/10.3389/fendo.2022.801271 (2022).
Yang, X. et al. Inhibition of Drp1 protects against senecionine-induced mitochondria-mediated apoptosis in primary hepatocytes and in mice. Redox. Biol. 12, 264–273. https://doi.org/10.1016/j.redox.2017.02.020 (2017).
Pernas, L. & Scorrano, L. Mito-morphosis: Mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 78, 505–531. https://doi.org/10.1146/annurev-physiol-021115-105011 (2016).
Hoppins, S. & Nunnari, J. Mitochondrial dynamics and apoptosis—the ER connection. Science 337, 1052–1054. https://doi.org/10.1126/science.1224709 (2012).
Suen, D. F., Norris, K. L. & Youle, R. J. Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590. https://doi.org/10.1101/gad.1658508 (2008).
Edwards, J. L. et al. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 53, 160–169. https://doi.org/10.1007/s00125-009-1553-y (2010).
Chen, S. D. et al. Roles of PTEN-induced putative kinase 1 and dynamin-related protein 1 in transient global ischemia-induced hippocampal neuronal injury. Biochem. Biophys. Res. Commun. 460, 397–403. https://doi.org/10.1016/j.bbrc.2015.03.045 (2015).
Denton, K. et al. Impaired mitochondrial dynamics underlie axonal defects in hereditary spastic paraplegias. Hum. Mol. Genet. 27, 2517–2530. https://doi.org/10.1093/hmg/ddy156 (2018).
Zhao, Y. X. et al. Amelioration of ischemic mitochondrial injury and Bax-dependent outer membrane permeabilization by Mdivi-1. CNS Neurosci. Ther. 20, 528–538. https://doi.org/10.1111/cns.12266 (2014).
Jenner, A. et al. DRP1 interacts directly with BAX to induce its activation and apoptosis. EMBO J. 41, e108587. https://doi.org/10.15252/embj.2021108587 (2022).
Maes, M. E. et al. Completion of BAX recruitment correlates with mitochondrial fission during apoptosis. Sci. Rep. 9, 16565. https://doi.org/10.1038/s41598-019-53049-w (2019).
Liu, M. et al. Carbon monoxide-releasing molecule-2 ameliorates postresuscitation myocardial dysfunction in rat via mitochondrial-mediated apoptosis pathway and the regulation of mitochondrial dynamics. Eur. J. Pharmacol. 927, 175038. https://doi.org/10.1016/j.ejphar.2022.175038 (2022).
Zhao, J. et al. O-GlcNAc transferase (OGT) protects cerebral neurons from death during ischemia/reperfusion (I/R) injury by modulating Drp1 in mice. Neuromol. Med. 24, 299–310. https://doi.org/10.1007/s12017-021-08688-6 (2022).
Sun, S. et al. TBC1D15-Drp1 interaction-mediated mitochondrial homeostasis confers cardioprotection against myocardial ischemia/reperfusion injury. Metabolism 134, 155239. https://doi.org/10.1016/j.metabol.2022.155239 (2022).
Tagaya, M. et al. Inhibition of mitochondrial fission protects podocytes from albumin-induced cell damage in diabetic kidney disease. Biochim. Biophys. Acta Mol. Basis Dis. 1868, 166368. https://doi.org/10.1016/j.bbadis.2022.166368 (2022).
Li, W. et al. Ophiopogonin D alleviates diabetic myocardial injuries by regulating mitochondrial dynamics. J. Ethnopharmacol. 271, 113853. https://doi.org/10.1016/j.jep.2021.113853 (2021).
Hei, C. et al. Rapamycin ameliorates brain damage and maintains mitochondrial dynamic balance in diabetic rats subjected to middle cerebral artery occlusion. Metab. Brain Dis. 38, 409–418. https://doi.org/10.1007/s11011-022-01020-6 (2023).
Lu, J. et al. Tea polyphenols alleviate the adverse effects of diabetes on oocyte quality. Food Funct. 13, 5396–5405. https://doi.org/10.1039/d1fo03770f (2022).
Shi, Y. X. et al. Identifies KCTD5 as a novel cancer biomarker associated with programmed cell death and chemotherapy drug sensitivity. BMC Cancer 23, 408. https://doi.org/10.1186/s12885-023-10895-2 (2023).
Antu, K. A. et al. Antidiabetic property of Symplocos cochinchinensis is mediated by inhibition of alpha glucosidase and enhanced insulin sensitivity. Plos One 9, e105829. https://doi.org/10.1371/journal.pone.0105829 (2014).
Yang, X. D. & Yang, Y. Y. Ferroptosis as a novel therapeutic target for diabetes and its complications. Front. Endocrinol. (Lausanne) 13, 853822. https://doi.org/10.3389/fendo.2022.853822 (2022).
Yang, C. et al. Epigallocatechin-3-gallate alleviates liver oxidative damage caused by iron overload in mice through inhibiting ferroptosis. Nutrients 15, 1. https://doi.org/10.3390/nu15081993 (2023).
Wang, J. et al. EGCG promotes the sensory function recovery in rats after dorsal root crush injury by upregulating KAT6A and inhibiting pyroptosis. Transl. Neurosci. 14, 20220326. https://doi.org/10.1515/tnsci-2022-0326 (2023).
Luo, Y. et al. Exploring the absorption mechanisms of imidazolium-based ionic liquids to epigallocatechin gallate. Int. J. Mol. Sci. 23, 1. https://doi.org/10.3390/ijms232012600 (2022).
Mohd, N. N. et al. The role of polyphenol in modulating associated genes in diabetes-induced vascular disorders. Int. J. Mol. Sci. 23, 1. https://doi.org/10.3390/ijms23126396 (2022).
Rasouli, H. et al. Differential alpha-amylase/alpha-glucosidase inhibitory activities of plant-derived phenolic compounds: a virtual screening perspective for the treatment of obesity and diabetes. Food Funct. 8, 1942–1954. https://doi.org/10.1039/c7fo00220c (2017).
Funding
This study was supported by the Sichuan Provincial Administration of Traditional Chinese Medicine Scientific and Technological Research Special Project (2023MS563), Nanchong City and University Strategic Cooperation Science and Technology Project (20SXQT0003, 22SXQTO132), National college students' Innovation and entrepreneurship Projects (202210634053), Innovation and Entrepreneurship Projects for Sichuan University Students (S20211063009, S202210634181), Science and technology development project of Affiliated Hospital of North Sichuan Medical College (2022JC027) and Scientific Research and Development Fund project of North Sichuan Medical College (CBY21-QA12).
Author information
Authors and Affiliations
Contributions
Conceptualization, Z.Q. and L.H.T.; methodology and investigation, K.J.Y., Z.Y.L., Z.X.Y., L.Z.R., L.X.H., T.R. and H.R.H.; writing and editing, J.X., M.D.T. and G.J.W.; funding acquisition, Z.Q. and J.X.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jia, X., Mao, D., Guo, J. et al. Epigallocatechin gallate attenuated high glucose-induced pancreatic beta cell dysfunction by modulating DRP1-mediated mitochondrial apoptosis pathways. Sci Rep 14, 16809 (2024). https://doi.org/10.1038/s41598-024-67867-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67867-0
- Springer Nature Limited