
Abstract
Cisplatin is a widely used chemotherapeutic drug that can induce ovarian damage. Icariin (ICA), a natural antioxidant derived from Epimedium brevicornum Maxim., has been found to protect against organ injury. The aim of the present study was to investigate whether ICA can exert an ovarian-protective effect on cisplatin induced premature ovarian failure (POF) and the underlying mechanism involved. The preventive effect of ICA was evaluated using body weight, the oestrous cycle, ovarian histological analysis, and follicle counting. ICA treatment increased body weight, ovarian weight, and the number of follicles and improved the oestrous cycle in POF mice. ICA reduced cisplatin-induced oxidative damage and upregulated the protein expression levels of Nrf2, GPX4 and HO-1. Moreover, ICA reduced the expression levels of Bax and γH2AX and inhibited ovarian apoptosis. In addition, ICA activated the Nrf2 pathway in vitro and reversed changes in the viability of cisplatin-induced KGN cells, reactive oxygen species (ROS) levels, lipid peroxidation, and apoptosis, and these effects were abrogated when Nrf2 was knocked down or inhibited. Molecular docking confirmed that ICA promotes the release of Nrf2 by competing with Nrf2 for binding to Keap1. The inhibitory effects of ICA on cisplatin-induced oxidative stress, ferroptosis, and apoptosis may be mediated by its modulatory effects on the Nrf2 pathway, providing a novel perspective on the potential mechanisms by which ICA prevents POF.
Similar content being viewed by others

Introduction
Infertility, a range of symptoms associated with menopause, and lower oestrogen levels leading to long-term diseases such as osteoporosis, cardiovascular disease, and cognitive impairment are all key characteristics of premature ovarian failure (POF)1,2. Extensive research has indicated that POF can be influenced by various factors, including genetic, autoimmune, medical, and environmental factors3,4,5. Notably, chemotherapy-induced ovarian damage is the primary cause of ovarian reserve depletion and infertility in cancer survivors6. With the continued increase in cancer survival rates, it has become crucial to prioritize enhancing patients' quality of life. Impaired fertility, in particular, greatly impacts the well-being of patients of reproductive age and may even lead patients to choose not to receive chemotherapy7. However, there are currently no validated strategies available to effectively preserve and restore ovarian reproductive and hormonal functions.
Cisplatin, a nonspecific cell cycle inhibitor, is commonly used to treat solid tumours8. Cisplatin has been proven in studies to directly produce double-stranded DNA breaks, resulting in cell death9. However, while it has antitumour benefits, it also kills normal cells in the body, causing a variety of side effects, such as gastrointestinal dysfunction, nephrotoxicity, ototoxicity, and reproductive toxicity10,11,12. Clinical studies have reported that cisplatin can cause POF13,14. The studies in human ovarian cortical pieces or granulosa cells shown that cisplatin treatment reduced follicle numbers and steroidogenic activity11. However, the specific mechanism of cisplatin-induced POF is still unclear. An increasing number of studies have suggested that various mechanisms, including oxidative stress, inflammation, mitochondrial disruption, autophagy, and apoptosis, are involved in the pathophysiology of cisplatin-induced POF15,16,17. Notably, exposure to cisplatin increases the production of reactive oxygen species (ROS) in follicular and granulosa cells18. Intracellular ROS also attack organelles and biomolecules, causing varying degrees of oxidative stress in DNA, lipids, and proteins19. ROS can increase the acetylation and expression of p53, while decrease the expression of Sirtuin 1 (SIRT1), and promote cell apoptosis20. ROS is also able to up-regulate the expression of apoptosis genes by inducing FOXO1 overexpression19. Apoptosis induced follicular atresia has been hypothesized to be involved in the pathophysiology of POF21. Additionally, increased lipid-based ROS, particularly lipid hydroperoxides, may cause ferroptosis22. Ferroptosis is a novel type of nonapoptotic cell death distinguished by iron-dependent lipid peroxidation23. There is growing evidence that ferroptosis plays a crucial role in nonneoplastic diseases, such as ischaemic cardiomyopathy24, nonalcoholic fatty liver disease25 and acute kidney injury12. Our previous study revealed that it is associated with the pathological process of POF26. Thus, identifying the mechanism of cisplatin-induced ferroptosis and anti-ferroptosis drugs is critical for identifying new ways to prevent or protect against chemotherapy-induced ovarian damage.
One of the crucial transcription factors involved in the fight against oxidative stress is nuclear factor erythroid-2 related factor 2 (Nrf2), which not only restores redox homeostasis by activating genes containing antioxidant response elements (AREs), such as glutathione peroxidase 4 (GPX4) and haemoglobin oxygenase 1 (HO-1) but also plays a crucial role in the control of ferroptosis27,28. According to research by Sun et al., Nrf2/ARE pathway activation reduces the redox imbalance caused by hydrogen peroxide in primary cardiomyocytes and blocks ferroptosis, which delays cardiomyocyte death29. A recent study revealed that the Nrf2/ARE pathway plays an essential role in preventing chemotherapy-induced ferroptosis in ovaries30. As a result, activating Nrf2 to minimize lipid peroxidation and ferroptosis appears to be a potential therapy.
Icariin (ICA), a flavonoid derived from the Chinese medicinal plant Epimedium, has been demonstrated to have anti-inflammatory, antioxidative stress, antiaging, and antitumour effects31,32. For example, ICA decreases extracellular matrix buildup and ameliorates experimental diabetic nephropathy by inhibiting oxidative stress via GPER-mediated p62-dependent Keap1 degradation and Nrf2 activation33. Furthermore, earlier research revealed that ICA prevents hypoxia-reoxygenation-induced cardiomyocyte oxidation and ferroptosis via the Nrf2/HO-1 pathway24. In particular, ICA enhances ovarian endocrine function and successfully lessens ovarian damage caused by d-galactose34,35. However, it is worthwhile to determine whether the protective effect of ICA against cisplatin-induced POF improves oxidative stress and inhibits ferroptosis through activation of the Nrf2/ARE pathway, which would provide new insights into the management of cisplatin-induced POF.
Results
ICA improves ovarian function in cisplatin-induced POF
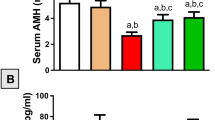
To evaluate whether ICA is able to protect against cisplatin-induced ovarian damage in vivo, we examined the body weights, oestrous cycles, ovarian appearances and follicle counts of the mice. We observed that the cisplatin group had considerably lower body weights, ovarian weights, and ovarian indices than did the control group (P < 0.0001) (Fig. 1A–D). These indices were restored in the cisplatin and ICA combination treatment group (P < 0.05) (Fig. 1A–D). Vaginal smears were subsequently obtained to assess the oestrous cycle of the mice. The results revealed that mice in the control and ICA groups presented regular oestrous cycles, and irregular oestrous cycles were observed in the mice in the cisplatin group, while cisplatin and ICA cotreatment altered oestrous cycle disorders in the mice (Fig. 1E,G). The number of follicles represents the ovarian reserve capacity, particularly the number of primordial follicles36. Therefore, we counted follicle numbers in each group of mice to assess the ovarian reserve. Compared to the control group, the cisplatin group exhibited a significant decrease in the number of follicles at all stages (P < 0.0001) and an increase in the percentage of growing follicles (P < 0.05), whereas the ICA and cisplatin cotreatment group showed an increase in the number of follicles at all levels and a decrease in the percentage of growing follicles (P < 0.05) (Fig. 1H–J). We also detected the Gdf9, Bmp15 and IL-1β mRNA level. As shown in figure S1, cisplatin treatment increased the expression level of Gdf9, Bmp15 and IL-1β, only IL-1β decreased after cisplatin and ICA cotreatment (P < 0.05) (Fig. S1A-C). The above results indicated that ICA has the capacity to alleviate cisplatin-induced POF.

ICA improves ovarian function in cisplatin-induced POF. (A) Gross appearance of ovaries between indicated groups. (B-D), Comparison of body weight (B), ovarian weight (C) and ovarian index (D) of mice after treatment in each group (n = 5). (E), Representative images of HE staining of mice at various stages of the estrous cycle (n = 5). Scale bars, 50 µm. (F), Percentage of estrous cycle in each stage. P, proestrus; E, estrus; M, metestrus; D, diestrus. (G), Representative graphs for monitoring the estrous cycle of each group of mice for 21 consecutive days. (H), HE staining of the ovaries in each group. Black arrows indicate primordial follicles; red arrows indicate primary follicles; green arrows indicate secondary follicle; blue arrows indicate antral follicles. Scale bars, 100 µm. (I), Ratio of number of growing follicles (primary follicle, secondary follicle and antral follicle) to primordial follicles in mice (n = 6). (J), Following therapy, the quantity of follicles at each stage in every mouse group (n = 6). PMF, primordial follicle; PF, primary follicle; SF, secondary follicle; AF, antral follicle. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, non-statistically significant.
ICA alleviates cisplatin-induced oxidative stress and inhibits apoptosis
Cisplatin-induced ROS can attack organelles and biomolecules, causing different degrees of oxidative damage to DNA, lipids, and proteins and triggering apoptosis19. To determine the level of oxidative damage, we used three commonly used biomarkers: 4-HNE (a marker of lipid peroxidation), 8-OHdG (a marker of DNA damage), and NTY (a marker of protein damage). IHC data revealed that cisplatin therapy significantly increased the levels of 4-HNE, 8-OHdG, and NTY (P < 0.01), whereas the combined treatment in the Cis + ICA group showed significantly decreased expression levels of these markers (P < 0.05) (Fig. 2A–D). Furthermore, ICA reduced the cisplatin-induced increase in Malondialdehyde (MDA) content in mouse ovaries (P < 0.05) (Fig. 2E). Numerous studies have revealed that the NRF2/ARE pathway is crucial for the regulation of oxidative stress and the induction of antioxidant damage by ICA24,33. As confirmed by both the IHC and WB results, the protein expression levels of Nrf2, HO-1, and GPX4 were significantly lower in the Cis group than in the control group (P < 0.01), and ICA reversed the inhibitory effect of cisplatin on the Nrf2, HO-1, and GPX4 proteins (P < 0.05) (Fig. 2F–J). Additionally, TUNEL analysis revealed an increase in the percentage of TUNEL-positive cells (particularly granulocytes) in the Cis group compared to the control group, as well as a decrease in the percentage of TUNEL-positive cells in the Cis + ICA cotreatment group (Fig. 3A). Moreover, we ascertained from the IHC data that cisplatin treatment with ICA considerably attenuated the cisplatin-induced increase in the levels of the pro-apoptotic gene Bax and the DNA double-strand break marker γH2AX (Fig. 3B). Moreover, WB data revealed that the Cis group had significantly higher expression levels of γH2AX and Bax than did the control group (P < 0.01) and that ICA intervention decreased the protein expression levels of γH2AX and Bax (P < 0.01) (Fig. 3C–E). Briefly, by accelerating the NRF2/ARE system, ICA decreases cisplatin-induced oxidative damage and prevents apoptosis in ovarian cells.

ICA alleviates cisplatin-induced oxidative stress. (A) Representative IHC images of 4-HNE, NTY and 8-OHdG in ovaries of each group (n = 6). Scale bars, 100 µm. (B-C), Quantification of positive area for 4-HNE (B), NTY (C) and 8-OHdG (D). (E), MDA levels in each group’s ovarian tissue were determined (n = 4). (F), Representative IHC images of Nrf2, HO-1, and Gpx4 in ovaries of each group (n = 6). Scale bars, 100 µm. (G), Nrf2, HO-1 and Gpx4 protein expression levels in ovarian tissues of each group were examined by WB (n ≥ 3). (H-J), Quantification of Nrf2 (H), HO-1 (I), and Gpx4 (J) protein levels. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, non-statistically significant.

ICA inhibits cisplatin-induced apoptosis in mouse ovaries. (A) TUNEL was performed on each group’s ovarian sections, and green fluorescent signals showed apoptosis. The nuclei of the cells were stained with Hoechst and fluoresced blue (n = 3). Scale bars, 100 µm. (B), Representative IHC images of Bax and γH2AX in each group of ovaries (n = 6). Scale bars, 100 µm. (C), Detection of γH2AX and Bax protein expression levels in ovarian tissues by WB (n ≥ 3). β-Actin was used as an internal control. (D,E), Quantitative analysis of γH2AX and Bax protein levels. Data are presented as mean ± SEM, **P < 0.01, ***P < 0.001, ns, non-statistically significant.
ICA protects KGN cells from cisplatin-induced damage by reducing oxidative stress and ferroptosis via the Nrf2/ARE pathway
To investigate whether ICA also reduces cisplatin-induced damage in vitro by activating the Nrf2/ARE pathway. First, the viability of KGN cells was determined using the CCK8 assay, and the results showed that 5 μg/ml or 10 μg/ml ICA did not affect the viability of KGN cells (Fig. 4A). We also observed that ICA alleviated the cisplatin-induced decrease in cell viability (P < 0.01) (Fig. 4B) and increase in cytotoxicity (P < 0.01) (Fig. 4C) by CCK-8 and lactate dehydrogenase (LDH) assays, respectively. Cisplatin exposure considerably elevated ROS levels in KGN cells, but ICA pretreatment significantly decreased cisplatin-induced ROS levels (P < 0.001) (Fig. 4D and 4E). In addition, the in vitro WB results were consistent with the in vivo findings. ICA elevated the protein levels of Nrf2, HO-1 and GPX4, which were decreased by cisplatin treatment (P < 0.05) (Fig. 4F–I). Moreover, the flow cytometry data of C11 BODIPY 581/591 support our hypothesis that lipid peroxidation accumulation was markedly elevated in the Cis group, while lipid peroxidation levels decreased when ICA was co-treated with cisplatin (P < 0.0001) (Fig. 4J–L). Comparable outcomes were noted for the capacity of ICA to prevent cisplatin-induced modifications to MDA levels. KGN cells treated with cisplatin showed high levels of lipid peroxidation; however, when both cisplatin and ICA were used for treatment, the level of lipid peroxidation decreased , as shown in Fig. 4J–L.

ICA reduces oxidative stress and ferroptosis via the Nrf2/ARE pathway. (A) Cell viability of KGN cells after treatment with different concentrations of ICA for 24 h. (B), Detection of KGN cell viability treated by ICA or/with cisplatin. (C), Effect of ICA on Cis-induced toxicity in KGN cells validated by LDH assay. (D), Effect of ICA on cis-induced ROS levels in KGN cells detected by flow cytometry. (E), Geometric mean fluorescence intensity was employed for quantitative analysis of ROS levels. (F), The expression levels of Nrf2, HO-1 and Gpx4 proteins in KGN cells of each treatment group were detected by WB. (G–I), Quantification of Nrf2, HO-1, and Gpx4 protein levels.(J–K), Assessment of intracellular lipid peroxidation by flow cytometry using BODIPY C11 staining (J) and quantitative analysis (K). (L), Determination of the level of MDA in each group of KGN cells. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, non-statistically significant.
To investigate whether cisplatin and ICA cotreatment blocks the inhibitory effect of cisplatin on gynecological tumor cells, we examined the cell viability after treatment by different concentrations of cisplatin alone or in combination with ICA in gynecological cancer cell lines. The results showed that the viability of cervical cancer cell line SiHa cells, endometrial cancer cell line Ishikawa cells and ovarian cancer cell line A2780 cells decreased gradually with the increase of cisplatin concentration (Fig. 2SA–C). In each cell line, there was no significant difference between the cisplatin group and cisplatin + ICA group (Fig. 2SA–C).
To further validate the effect of ICA on cisplatin-induced ferroptosis in KGN cells, we treated KGN cells with the classical ferroptosis inducer RSL3 (a specific inhibitor of GPX4). A significant decrease in cell viability and a significant increase in LDH, MDA, ROS and lipid peroxidation levels were observed in the RSL3-treated group (P < 0.0001) (Fig. 5A–G). When ferroptosis was blocked with Fer-1, a ferroptosis inhibitor, these alterations were reversed (P < 0.0001) (Fig. 5A–G). Consistent with Fer-1, ferroptosis was suppressed by ICA administration (P < 0.0001) (Fig. 5A–G). These findings indicated that ICA activates the Nrf2/ARE signalling pathway to shield KGN cells from oxidative stress and ferroptosis induced by cisplatin.

ICA alleviates RSL3-induced oxidative damage. (A–C). Effects of ICA (5ug/ml) and Fer-1 (20 μM) on cell viability (A), cytotoxicity (B) and MDA levels (C) in KGN cells induced by RSL3 (300 nM). (D-G), ROS (D,E) and lipid oxidation levels (F,G) in KGN cells of different treatment groups were detected and quantified using DCFH-DA and C11-BODIPY 581/591 probes, respectively. Data are presented as mean ± SEM, ****P < 0.0001.
ICA suppressed cisplatin-induced apoptosis in KGN cells
We also investigated the potential impact of ICA on apoptosis induced by cisplatin treatment. The western blot results demonstrated that cis-treated cells had significantly higher levels of Bax and γH2AX expression (P < 0.0001) and that the increase in these proteins was significantly inhibited by ICA pretreatment (P < 0.01) (Fig. 6A-C). Apoptosis was also detected by flow cytometry using Annexin V and PI staining. We discovered that the cisplatin group exhibited more apoptosis than the control group and that ICA significantly protected against cisplatin-induced apoptosis (P < 0.01) (Fig. 6D,E). We also discovered that ICA and Fer-1 could block the apoptosis caused by RSL3 administration (P < 0.0001) (Fig. 6F,G). These results indicated that ICA can attenuate cisplatin- or ferroptosis-induced apoptosis.

ICA rescued cis-induced apoptosis in KGN cells. (A,B) Determination of γH2AX and Bax protein expression levels in KGN cells from different treatment groups by WB (A) and quantitative analysis (B,C). (D,E), Effect of ICA on the percentage of Cis-induced apoptosis in KGN cells using Annexin V/PI staining and flow cytometry. (F,G), Effect of ICA and Fer-1 on the percentage of RSL3-induced apoptosis in KGN cells using Annexin V/PI staining and flow cytometry. Data are presented as mean ± SEM, **P < 0.01, ****P < 0.0001, ns, non-statistically significant.
Knockdown or inhibition of Nrf2 reverses the protective effects of ICA on cisplatin-induced injury in KGN cells
To demonstrate that ICA protects KGN cells from cisplatin-induced injury by activating the Nrf2/ARE pathway, we transfected KGN cells with Nrf2 siRNA to knock down Nrf2, and the results were validated by WB. We observed that the Nrf2 protein level was significantly lower in the Nrf2 siRNA group than in the negative control group, and there was a significant reduction in HO-1 and GPX4 and an increase in γH2AX and Bax (P < 0.01) (Fig. 7A,B ). Moreover, CCK-8 and LDH assays confirmed that the protective effect of ICA against the cisplatin-induced decrease in cell viability (P < 0.01) and increase in cytotoxicity (P < 0.0001) was reversed by Nrf2 knockdown (Fig. 7C,D ). Furthermore, the ability of ICA to suppress ROS and lipid peroxidation, as well as apoptosis, was abolished in Nrf2-knockdown cells (Fig. 7E-J). Additionally, we utilized the Nrf2 inhibitor ML385 to block the activity of Nrf2 and examined the oxidation indices, apoptosis indices, and ferroptosis-related parameters with similar findings (Fig. 8).

Knockdown of Nrf2 reverses the protective effect of ICA on cis-induced KGN cell injury. (A) and (B), KGN cells were transfected with Nrf2 siRNA or negative control (NC) siRNA, and the protein levels of Nrf2, HO-1, Gpx4, γH2AX, and Bax were assessed by WB (A) and quantified (B). (C) and (D), Detection of cell viability and cytotoxicity of KGN cells in different treatment groups using CCK8 (C) and LDH (D), respectively. (E,F) , ROS were detected using DCFH-DA (E) and quantified (F). (G) and (H), Lipid peroxidation levels were detected using C11-BODIPY 581/591 probes (G) and quantified (H). (I) and (J), The percentage of apoptosis of KGN cells in different treatment groups was detected using Annexin V/PI staining and flow cytometry. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, non-statistically significant.

The Nrf2 inhibitor ML385 attenuates the protective effect of ICA on cis-induced KGN cell injury. (A–C) Effect of co-treatment of ICA with ML385 (2.5 μM) on cis-induced KGN cell viability (A), cytotoxicity (B) and MDA levels (C). (D) and (E), ROS were detected using DCFH-DA (D) and quantified (E). (F) and (G), Lipid peroxidation levels were detected using C11-BODIPY 581/591 probes (F) and quantified (G). (H) and (I), The percentage of apoptosis of KGN cells in different treatment groups was detected using Annexin V/PI staining and flow cytometry. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, non-statistically significant.
ICA promotes Nrf2 release by competing with Nrf2 for binding to Keap1
To investigate the mechanism of Nrf2 activation, we performed molecular docking studies between ICA and Keap1. We found that Asn414, Arg415, Arg380, and Ser363 of Keap1 formed hydrogen bonds with the ligand, and their binding energy was−7.6 kcal/mol (Fig. 9 and Table 1). Analysis of the UniProt database revealed that Arg380 and Ser363 are essential for the interaction between Keap1 and Nrf2, and a mutation at this locus resulted in the loss of this interaction. Thus, ICA may competitively bind to Keap1, promoting the dissociation of Nrf2 and Keap1 and allowing for Nrf2 to be released and activated.

ICA promotes Nrf2 release by competing with Nrf2 to bind Keap1. (A) Molecular docking of ICA to the key target KEAP1. (B), The KEAP1 protein is represented as a slate cartoon model, ICA is shown as a cyan stick, and their binding sites are shown as magentas stick structures. The hydrogen bond, ionic interactions, and hydrophobic interactions are depicted as yellow, magentas and green dashed lines, respectively.
Discussion
The long-term survival rates of women of reproductive age have increased in recent years due to considerable advances in cancer detection and treatment37. Maintaining fertility and eventual childbirth are crucial for these people38. Unfortunately, chemotherapy is a major cause of ovarian failure. POF leads not only to impaired fertility but also to many comorbidities. However, impaired fertility has a significant impact on the quality of life of patients of childbearing age and may lead to the abandonment of recommended treatments because of the risk of infertility39. Consequently, finding fertility-protective adjuvants to protect the ovaries from chemotherapy medicines is essential. Icariin is a flavonoid discovered in the Chinese medicinal plant Epimedium that has numerous pharmacological and biological properties31. Previous studies have shown that ICA ameliorates cisplatin-induced nephrotoxicity and cardiotoxicity by inhibiting oxidative stress, inflammation, and apoptosis through inhibition of the ROS-mediated PI3K/Akt pathway35,40. Furthermore, ICA has been shown to restore ovarian endocrine function and effectively reduce D-galactose-induced ovarian damage34,41.
Clinical studies have reported the toxicity of cisplatin to ovaries13,14. Cisplatin induced models of POF in rats and mice have been widely used to study the mechanism of ovarian injury and explore ways to alleviate ovarian injury15,17,18,26,42,43. The doses used in this study have been validated in our previous studies26 and others56. In this study, our results showed that cisplatin treatment significantly reduced ovarian volume and weight, disrupted oestrous cycle, and significantly reduced total follicles number and follicle number at all levels, suggesting that cisplatin caused POF. However, ICA treatment can alleviate these damage caused by cisplatin (Fig. 1). It is well known that the number of follicles at all levels reflects the reserve capacity of the ovary, especially the number of primordial follicles44. Cisplatin can directly cause damage and apoptosis in primordial follicular oocytes11. In women, the population of primordial follicle which serves as the reservoir for future fertility is determined at birth45. Excessive activation of primordial follicles can lead to premature ovarian insufficiency (POI), also known as POF36,45. Cisplatin treatment decreases the level of AMH which have an important inhibitory effect on primordial follicle activation42. Cytokines such as GDF9, BMP15 and IL-1β promote primordial follicle activation46,47. In addition, cisplatin has been shown to trigger the activation and maturation of primordial follicles via PI3K/AKT/FOXO3a activation, which eventually depletes primordial follicles48. We observed an increase in the proportion of growing follicles and increased expression levels of GDF9, BMP15, and IL-1β in the cisplatin group (Fig. S1A-C), which supports this activating impact on primordial follicles. ICA has been shown to alleviate osteoarthritis by down-regulating the AKT pathway49. Cotreatment with ICA and cisplatin decreased the proportion of growing follicles (Fig. 1I), possibly because ICA inhibited the PI3K/AKT pathway and IL-1β levels. The number of primordial follicles provides a more direct indication of chemotherapy-induced ovarian damage. The number of follicles at all stages in the cisplatin group significantly decreased, while the number of follicles in the ICA and cisplatin cotreatment group increased at all stages, as validated by our follicle counting results (Fig. 1J).
There is growing evidence that oxidative stress is one of the important causative factors of chemotherapeutic premature ovarian failure17,43. This is related to an imbalance between intracellular free radical products and cellular defence mechanisms50. Cisplatin has been shown to increase ROS production in follicular and granulosa cells18. In this study, we discovered that combining ICA with cisplatin decreased the levels of the oxidative damage products 4-HNE, 8-OHdG, and NTY (Fig. 2A–D). Similarly, ICA intervention significantly decreased the levels of the lipid peroxidation product MDA (Fig. 2E). Furthermore, we cocultured cisplatin-exposed KGN cells with ICA for in vitro treatment, and as expected, cisplatin exposure significantly increased ROS levels in KGN cells, whereas pretreatment with ICA significantly attenuated cisplatin-induced ROS accumulation (Fig. 4D–E). However, the accumulation of ROS will increases oxidative damage25. Nrf2/ARE has been demonstrated to be a crucial mechanism in the regulation of oxidative stress, and activation of this system promotes the production of antioxidant enzymes such as HO-1 and GPX4, which has an anti-oxidative stress effect27,28. There is evidence that ICA decreases oxidative stress and improves Escherichia coli lipopolysaccharide-mediated endometritis in mice by activating the NRF2 pathway51. Our findings showed that ICA activated the Nrf2/ARE pathway and reduced cisplatin-induced oxidative damage (Fig. 2F–J). Interestingly, multiple investigations have revealed that Nrf2 is a critical regulator of ferroptosis and has the ability to suppress ferroptosis by regulating GPX4 and HO-112,29. On the other hand, inhibition of GPX4 leads to an uncontrolled increase in lipid peroxidation, which results in ferroptosis. Conversely, there is disagreement concerning the involvement of HO-1 in ferroptosis. As mentioned above, HO-1 has been reported to inhibit ferroptosis, but in other studies HO-1 has been found to promote ferroptosis. For example, ferroptosis regulated by HO-1 is the main pathological process of sodium iodate induced retinal pigment epithelium degeneration52. Tagitinin C exerts anti-colorectal cancer effects by activating the NRF2/HO-1 pathway to promote ferroptosis53. In our study, we found that ICA upregulated the protein expression of Nrf2, GPX4 and HO-1. In addition, flow cytometry analysis of C11 BODIPY 581/591 further demonstrated that cotreatment with ICA and cisplatin reduced lipid peroxidation levels (Fig. 4D-L). Moreover, we found that ICA significantly increased cell viability and reduced the RSL3-induced increases in MDA, ROS and lipid peroxidation. Surprisingly, in cells with Nrf2 knockdown or treated with the Nrf2 inhibitor ML385, the ability of ICA to reduce ROS and lipid peroxidation generation, as well as apoptosis, was almost completely reversed. Our results confirmed that the Nrf2 pathway plays a role in the protective effect of ICA against cisplatin-induced oxidative stress, ferroptosis, and apoptosis in KGN cells(Fig. 7 and 8).
In summary, the Nrf2 pathway may be a crucial link between oxidative stress and ferroptosis12,29,53, and activated ICA protects against early ovarian failure. However, the exact mechanism by which ICA upregulates Nrf2 is still unclear. Previous research has demonstrated that in the absence of stress, Nrf2 binds to Keap1 in the cytoplasm and is thus ubiquitinated and destroyed50. Once exposed to oxidative stress or electrophilic chemicals, the conformation of Keap1 changes, resulting in inhibited ubiquitination and degradation of Nrf2, which can translocate to the nucleus and activate target genes such as HO-1, NQO1, and GPX454. As a result, we examined the molecular docking model of the ICA molecule with Keap1 and revealed that ICA binds to the Asn414, Arg415, Arg380, and Ser363 residues of Keap1 (Fig. 9). Whereas Arg380 and Ser363 are more critical for Keap1 to interact with Nrf2, mutation of this site results in the loss of the interaction between the two. Thus, ICA may competitively bind to the Nrf2 binding site of Keap1 and promote the dissociation of Nrf2 from Keap1, allowing for Nrf2 to be released and activated. Therefore, we hypothesized that ICA could competitively bind to Keap1 and release NRF2, thus exerting an antioxidant effect.
Overall, we discovered that ICA alleviated cisplatin-induced POF. Mechanistically, ICA prevented cisplatin-induced POF by competitively binding to Keap-1 and activating the Nrf2/ARE pathway, which inhibited oxidative stress, ferroptosis, and apoptosis. Therefore, icariin is a candidate for the treatment of chemotherapeutic premature ovarian failure. However, there are some limitations to our study. First, the protective effect of ICA found in this study was not evaluated in a tumor mouse model. The effect of ICA on tumor and whether it can play a protective role in tumor bearing mouse model are unclear. In addition, we found in this study that different types of tumor cell lines have different sensitivity to cisplatin. The protective effect of ICA at higher treatment-appropriate concentrations of cisplatin still needs to be further clarified.
Methods
Reagents and antibodies
Icariin (C13319079) was purchased from MACKLIN (Shanghai, China). Cisplatin (HY-17394), ferrostatin-1 (HY-100579), ML385 (HY-100523), RSL3 (HY-100218A), and BODIPY 581/591 C11 (HY-D1301) were purchased from MCE (Monmouth Junction, NJ, USA). An Annexin V-FITC/PI Apoptosis Kit (40302ES60), foetal bovine serum (FBS; 40130ES76), Dulbecco’s modified Eagle’s medium/Ham's F-12 (DMEM/F12; 41420ES76), penicillin–streptomycin (PS; 60162ES76), trypsin–EDTA (0.25%; 40127ES60), Hieff Trans® siRNA/miRNA in vitro transfection reagents (40806ES03), and a 1st Strand cDNA Synthesis Kit (11141ES60) were purchased from Yeasen (Shanghai, China). An LDH Cytotoxicity Assay Kit (C0017), One Step TUNEL Apoptosis Assay Kit (C1086) and Reactive Oxygen Species Assay Kit (S0033S) were purchased from Beyotime (Shanghai, China). A Cell Counting Kit-8 (CT0001) was purchased from SparkJade (Shandong, China). A malondialdehyde (MDA) content kit (JL-T0761) was purchased from Jianglai (Shanghai, China).
The primary antibodies used in this study were as follows: anti-GPX4 (381958), anti-HO-1 (R24541), anti-Bax (380709), anti-p-γH2AX (381558), anti-beta actin (250136), goat anti-mouse IgG H&L (HRP) (511103) and goat anti-rabbit IgG H&L (HRP) (511203), which were purchased from Zen BioScience (Chengdu, China); and anti-4-HNE (bs-6313R), anti-NTY (bs-8551R), and anti-8-OHdG (bs-1278R), which were purchased from Bioss (Beijing, China). The anti-Nrf2 antibody (16396-1-AP) was purchased from Proteintech (USA).
Animal grouping and treatment
Female ICR mice (6–8 weeks) were purchased from Anhui Medical University's Animal Centre. All mice were housed in a favourable experimental setting (temperature, 25 ℃; humidity, 55%; 12 h light/dark cycle) with free access to water and food. After three days of acclimatization, a total of 88 mice were randomly assigned to four groups (22 mice were randomly selected for each group): the control (Ctr), cisplatin (Cis), cisplatin + Icariin (Cis + ICA), and icariin (ICA) groups. In the Ctr and ICA groups, saline containing 1% dimethyl sulfoxide and ICA (30 mg/kg)55 was injected intraperitoneally daily; Cis (5 mg/kg)56 was injected intraperitoneally every 5 days in the Cis group; and ICA (30 mg/kg) was injected intraperitoneally in the Cis + ICA group, followed by an interval of 3 h before an intraperitoneal injection of cisplatin (5 mg/kg). In each group, 4 mice were used for western blotting, 6 mice for RT-PCR, 6 mice for follicle counting and 6 mice were used for IHC and other staining. After 21 days, all the mice were sacrificed, and the ovaries were collected and weighed for further experiments. The individual mouse was considered the experimental unit within the studies. All the animal procedures used in this study were in accordance with the protocols of the National Institute of Health Guide for the Care and Use of Laboratory Animals and ARRIVE guidelines. The permission (LLSC20220164) was obtained from the Laboratory Animal Ethics Committee of Anhui Medical University.
Oestrous cycle analysis
During the course of the 21 days of therapy, vaginal swabs were collected every day at 8 a.m. to evaluate the oestrous cycle of the mice. Briefly, 20 μl of saline was used to clean the vagina, and then the collected vaginal secretions were placed on slides, dried, and stained with HE. The oestrus cycle was differentiated according to the predominant cell type in the vaginal smear. Preoestrus is predominantly nucleated epithelial cells; oestrus consists of anucleate, keratinized epithelial cells; postoestrus consists of equal proportions of anucleate keratinized epithelial cells and neutrophils; and interoestrus is predominantly composed of neutrophils.
HE staining and follicle count
Mouse ovaries were submerged in 4% paraformaldehyde, fixed for at least 24 h, dehydrated in gradient alcohol and embedded in paraffin. One side of the ovary from each group was cut into consecutive sections at a thickness of 5 µm, and the sections were stained with haematoxylin and eosin solution at intervals of 5th for follicle counting and morphological observation. To eliminate double counting, only follicles containing oocytes were assessed, and the number of follicles at each level was counted by two independent experienced technicians using predefined criteria. We used the recognized criteria developed by Pedersen and Peters for follicular staging57.
Immunohistochemistry
The steps for fixation, paraffin embedding, and tissue sectioning were the same as those for H&E staining described in Section “Oestrous cycle analysis”. Tissue sections were deparaffinized with xylene, rehydrated with gradient alcohol and heated for antigen repair using sodium eurycomonate. After blocking with endogenous peroxidase for 10 min, diluted primary antibody was added, and the membrane was incubated at 4 ℃ overnight. Subsequently, the sections were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody at 37 ℃ for 40 min, stained with DAB, restained with haematoxylin, air-dried, and viewed under a microscope, and positive expression was indicated by dark brown-stained areas.
Western blot analysis
Ovarian tissue and KGN cells were lysed with precooled RIPA buffer to extract total protein (n ≥ 3). Next, the protein samples were subjected to SDS‒PAGE and then transferred to a PVDF membrane. After being blocked with 5% skim milk powder for 1 h, the membranes were cut prior to hybridisation with primary antibodies overnight at 4 ℃. The next day, the membranes were incubated with anti-rabbit or murine IgG secondary antibody at 1:10,000 for 1 h, and then chemiluminescence detection was performed using a CS analysis system (5200, Tanon, Shanghai, China). The following antibodies were used: anti-Nrf2 (1:1000), anti-HO-1 (1:1000), anti-GPX4 (1:1000), anti-Bax (1:1000), anti-γH2AX (1:1000), and anti-beta actin (1:1000). Protein bands were quantified using ImageJ software and normalized to each control.
TUNEL staining
A terminal deoxynucleotidyl transferase (TdT) dUTP end labelling (TUNEL) staining kit was used to measure ovarian tissue cell death. After dewaxing, rehydration, and proteinase K permeabilization, we treated the tissue slices with 50 μl of TDT buffer solution and incubated them for 1 h at 37 ℃ in a dark and humid setting. Cell nuclei were stained with Hoechst 33342 (1:500) for 5 min at room temperature and observed and photographed under a fluorescence microscope.
Cell culture and treatment
SiHa cells and Ishikawa cells were cultured in DMEM medium and A2780 cells were cultured in DMEM medium. KGN cells were cultured in DMEM/F-12 medium. All medium supplemented with 10% foetal bovine serum and 1% penicillin/streptomycin.
The cells were kept at 37 ℃ and 5% CO2 in a humid environment. We generated the following groups: the control group, in which KGN cells were cultured in DMEM/F-12 medium supplemented with 1% DMSO for 48 h; the cisplatin group, in which KGN cells were cultured in DMEM/F-12 medium supplemented with cisplatin (1 μg/ml) for 48 h; the Cis + ICA group, in which KGN cells were treated with ICA (5 μg/ml) for 6 h, after which cisplatin (1 μg/ml) was added for 48 h; the ICA group, in which KGN cells were cultivated for 48 h with ICA (5 μg/ml) alone; and the Cis + ICA + ML385 group, in which ICA (5 μg/ml) and ML385 were cotreated for 6 h, after which cisplatin (1 μg/ml) was added for 48 h.
Cell transfection
The Nrf2 siRNA sequences were purchased from Universal, and the sequences were as follows: forwards 5′- CCGGCAUUUCACUAAACACAATT-3′; reverse, 5′-UUGUGUUUAGUGAAAUGCCGGTT-3′. Briefly, KGN cells were transiently transfected with Nrf2 siRNA or negative control siRNA when they reached 50–80% confluence according to the manufacturer's protocol, and the transfection efficiency was assessed by WB 24 h later.
Detection of cell viability
Cell viability was determined using the Cell Counting Kit-8 (CCK-8) assay. Cells were inoculated in 96-well plates, and cell processing was completed. Ten microlitres of CCK8 solution was added to each well, and the plates were incubated at 37 ℃ for 2 h. Then, the OD value at 450 nm was measured by an enzyme marker, and cell viability was calculated.
Lactate dehydrogenase (LDH) assay
After cell treatment, LDH release reagent was added to untreated KGN cells, which were then incubated for 1 h. Subsequently, 120 μl of supernatant from each group was removed and transferred to a new 96-well plate, 60 µl of LDH assay working solution was added, the cells were incubated for 30 min at room temperature on a shaker protected from light, and the OD at 490 nm was measured by an enzyme marker to determine the cytotoxicity.
Determination of MDA levels
Fresh ovarian tissue was lysed using RIPA lysis buffer, MDA working solution was added, mixed and heated at 100 ℃ for 15 min, centrifuged, and the supernatant was removed and transferred to a new 96-well plate. The OD value at 532 nm was measured, and the amount of MDA in the sample solution was calculated. In addition, we inoculated KGN cells into 6-cm dishes, and after the cells had attached to the wall for 24 h, they were processed and cellular MDA measurements were performed using a kit according to the manufacturer's instructions.
Lipid peroxidation assay
C11 BODIPY 581/591 (HY-D1301, MedChemExpress, Shanghai, China) was dissolved in DMSO to 1 mM for storage. To assess intracellular lipid peroxidation levels, KGN cells were seeded in 12-well plates. After drug treatment, the cells were incubated with 1 ml of serum-free medium containing 2 μM BODIPY 581/591C11 dye for 30 min at 37 ℃ in the dark. Then, the cells were washed three times with PBS after digestion with trypsin. Finally, the cells were resuspended in PBS, and the lipid peroxidation signal results were acquired by flow cytometry and analysed with FlowJo V10 software.
Detection of ROS
The fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) was used to detect cellular ROS levels. Cells were inoculated in 12-well plates, and after the completion of drug treatment, the cells were washed with serum-free DMEM/F-12, followed by incubation with serum-free medium containing 10 μM DCFH-DA for 30 min at 37 ℃ in the dark. Then, the KGN cells were washed three times with cold PBS before being collected and resuspended in PBS. The results were acquired by flow cytometry and analysed using FlowJo V10 software.
Detection of apoptotic cells
An Annexin V-FITC Apoptosis Kit was used to calculate the number of apoptotic cells. KGN cells were collected and washed twice with precooled PBS before being resuspended in binding buffer containing 5 μl of Annexin V-FITC and 10 μl of propidium iodide (PI). Then, the cells were incubated at room temperature in the dark for 15 min. Flow cytometry was used to determine the rate of apoptosis. Cells that were positive for Annexin V were considered early apoptotic cells, and cells that were positive for both Annexin V and PI were considered late apoptotic cells. We calculated the sum of the proportions of early- and late-stage apoptotic cells for statistical analysis.
Quantitative real-time PCR (qRT-PCR)
RNA extraction kit (YEASEN, 19221ES50) was used to extract total RNA. RNA concentration was measured by Nanodrop Spectrophotometer (Thermo Scientific, Nanodrop2000, USA). After removing the genomic DNA, a total of 2.5 μg RNA was reverse-transcripted into cDNA using the 1st Strand cDNA Synthesis Kit (YEASEN, 11141ES40). The cDNA was amplified using the Hieff qPCR SYBR Green Master Mix (YEASEN, 11201ES08) on the Real-time PCR Detection System (Roche LightCycler480, Switzerland). The primers purchased from GENERAL BIOL (Anhui, China) are listed in Table 1. All target genes were standardized with β-Actin. The relative gene expression levels were calculated using the 2−ΔΔCT method.
Molecular docking
The X-ray crystal structures of Keap1 (PDB: 2FLU) were retrieved from the Protein Data Bank. The protonation state of all the compounds was set at pH = 7.4, and the compounds were expanded to 3D structures using Open Babel58. AutoDock Tools (ADT3) were applied to prepare and parametrize the receptor protein and ligands. The docking grid documents were generated by AutoGrid of sitemap, and AutoDock Vina (1.2.0) was used for docking simulation59,60. The optimal pose was selected for interaction analysis. Finally, the protein‒ligand interaction was generated by PyMOL. The Keap1 protein is represented as a slate cartoon model, the ligand is shown as a cyan stick, and its binding sites are shown as magenta stick structures. (Table 2) Nonpolar hydrogen atoms are omitted. The hydrogen bond, ionic interactions, and hydrophobic interactions are depicted as yellow, magenta and green dashed lines, respectively.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9.0. Each experiment was repeated at least three times, and all the data are expressed as the mean ± sem. A t test was used to compare differences between two groups, one-way ANOVA was used to analyse differences between three or more groups, and post hoc analyses were carried out with the Bonferroni correction. p < 0.05 was considered to indicate a statistically significant difference.
Ethics approval and consent to participate
This study was approved (LLSC20220164) by t Laboratory Animal Ethics Committee of Anhui Medical University.
Data availability
Data is provided within the manuscript or supplementary information files.
Abbreviations
- ICA:
-
Icariin
- POF:
-
Premature ovarian failure
- ROS:
-
Reactive oxygen species
- Nrf2:
-
Nuclear factor erythroid-2 related factor 2
- ARE:
-
Antioxidant response element
- Gpx4:
-
Glutathione peroxidase 4
- HO-1:
-
Hemoglobin oxygenase 1
- HRP:
-
Horseradish peroxidase
- qRT-PCR:
-
Quantitative real-time PCR
- MDA:
-
Malondialdehyde
- LDH:
-
Lactate dehydrogenase
- POI:
-
Premature ovarian insufficiency
References
Webber, L. et al. ESHRE guideline: Management of women with premature ovarian insufficiency. Hum. Reprod. 31, 926–937 (2016).
Rebar, R. W. Premature ovarian failure. Obstet. Gynecol. 113, 1355–1363 (2009).
Qin, Y., Jiao, X., Simpson, J. L. & Chen, Z. J. Genetics of primary ovarian insufficiency: New developments and opportunities. Hum. Reprod. Update 21, 787–808 (2015).
Szeliga, A. et al. Autoimmune diseases in patients with premature ovarian insufficiency-our current state of knowledge. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22052594 (2021).
Goswami, D. & Conway, G. S. Premature ovarian failure. Hum. Reprod. Update 11, 391–410 (2005).
Levine, J. M. et al. Nonsurgical premature menopause and reproductive implications in survivors of childhood cancer: A report from the childhood cancer survivor study. Cancer-Am. Cancer Soc. 124, 1044–1052 (2018).
Vitale, S. G., La Rosa, V. L., Rapisarda, A. & Lagana, A. S. Fertility preservation in women with gynaecologic cancer: The impact on quality of life and psychological well-being. Hum. Fertil. (Camb) 21, 35–38 (2018).
Oberoi, H. S. et al. Cisplatin-loaded core cross-linked micelles: Comparative pharmacokinetics, antitumor activity, and toxicity in mice. Int J Nanomed. 7, 2557–2571 (2012).
Maccio, A. & Madeddu, C. Cisplatin: An old drug with a newfound efficacy— from mechanisms of action to cytotoxicity. Expert. Opin. Pharmacother. 14, 1839–1857 (2013).
Morse, H. et al. Acute onset of ovarian dysfunction in young females after start of cancer treatment. Pediatr. Blood Cancer 60, 676–681 (2013).
Spears, N. et al. Ovarian damage from chemotherapy and current approaches to its protection. Hum. Reprod. Update 25, 673–693 (2019).
Hu, J., Gu, W., Ma, N., Fan, X. & Ci, X. Leonurine alleviates ferroptosis in cisplatin-induced acute kidney injury by activating the Nrf2 signalling pathway. Br. J. Pharmacol. 179, 3991–4009 (2022).
Maneschi, F. et al. Menstrual and hormone patterns in women treated with high-dose cisplatin and bleomycin. Gynecol. Oncol. 54, 345–348 (1994).
Sklar, C. A. et al. Premature menopause in survivors of childhood cancer: A report from the childhood cancer survivor study. J. Natl. Cancer Inst. 98, 890–896 (2006).
Tang, X. et al. Ubiquitin-like modifier 1 ligating enzyme 1 relieves cisplatin-induced premature ovarian failure by reducing endoplasmic reticulum stress in granulosa cells. Reprod. Biol. Endocrinol. 20, 84 (2022).
Wang, R. et al. FGF2 is protective towards cisplatin-induced KGN cell toxicity by promoting FTO expression and autophagy. Front. Endocrinol. (Lausanne) 13, 890623 (2022).
Ibrahim, M. A. et al. Resveratrol protects against cisplatin-induced ovarian and uterine toxicity in female rats by attenuating oxidative stress, inflammation and apoptosis. Chem. Biol. Interact. 338, 109402 (2021).
Nguyen, Q. N. et al. Cisplatin- and cyclophosphamide-induced primordial follicle depletion is caused by direct damage to oocytes. Mol. Hum. Reprod. 25, 433–444 (2019).
Shen, M. et al. Involvement of the up-regulated FoxO1 expression in follicular granulosa cell apoptosis induced by oxidative stress. J. Biol. Chem. 287, 25727–25740 (2012).
Park, J. H. et al. TOPK inhibition accelerates oxidative stress-induced granulosa cell apoptosis via the p53/SIRT1 axis. Int. J. Mol. Med. 46, 1923–1937 (2020).
Stringer, J. M., Alesi, L. R., Winship, A. L. & Hutt, K. J. Beyond apoptosis: Evidence of other regulated cell death pathways in the ovary throughout development and life. Hum. Reprod. Update. 29, 434–456 (2023).
Dixon, S. J. et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Liu, X. J. et al. Icariin inhibits hypoxia/reoxygenation-induced ferroptosis of cardiomyocytes via regulation of the Nrf2/HO-1 signaling pathway. Febs Open Bio. 11, 2966–2976 (2021).
Gao, G. et al. Dehydroabietic acid improves nonalcoholic fatty liver disease through activating the Keap1/Nrf2-ARE signaling pathway to reduce ferroptosis. J. Nat. Med. 75, 540–552 (2021).
Wang, S. et al. Inhibition of cisplatin-induced Acsl4-mediated ferroptosis alleviated ovarian injury. Chem. Biol. Interact. 387, 110825 (2024).
Kinowaki, Y. et al. Glutathione peroxidase 4 overexpression inhibits ROS-induced cell death in diffuse large B-cell lymphoma. Lab. Invest. 98, 609–619 (2018).
Dodson, M., Castro-Portuguez, R. & Zhang, D. D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107 (2019).
Sun, C. et al. Ferroptosis-specific inhibitor Ferrostatin-1 relieves H(2)O(2)-induced redox imbalance in primary Cardiomyocytes through the Nrf2/ARE pathway. Dis. Markers 2022, 4539932 (2022).
Zhang, S. et al. Chemotherapy impairs ovarian function through excessive ROS-induced ferroptosis. Cell Death Dis. 14, 340 (2023).
Chen, X. J. et al. Chemical constituents, quality control, and bioactivity of Epimedii folium (Yinyanghuo). Am. J. Chin. Med. 43, 783–834 (2015).
Kong, L. et al. Icariin inhibits TNF-alpha/IFN-gamma induced inflammatory response via inhibition of the substance P and p38-MAPK signaling pathway in human keratinocytes. Int. Immunopharmacol. 29, 401–407 (2015).
Wang, K. et al. Icariin prevents extracellular matrix accumulation and ameliorates experimental diabetic kidney disease by inhibiting oxidative stress via GPER mediated p62-dependent keap1 degradation and Nrf2 activation. Front. Cell. Dev.Biol. 8, 559 (2020).
Li, N., Wang, J., Wang, X., Sun, J. & Li, Z. Icariin exerts a protective effect against d-galactose induced premature ovarian failure via promoting DNA damage repair. Biomed. Pharmacother. 118, 109218 (2019).
Xia, J. et al. Icariin exhibits protective effects on cisplatin-induced cardiotoxicity via ROS-mediated oxidative stress injury in vivo and in vitro. Phytomedicine 104, 154331 (2022).
Persani, L., Rossetti, R. & Cacciatore, C. Genes involved in human premature ovarian failure. J. Mol. Endocrinol. 45, 257–279 (2010).
Kim, S. S. et al. Recommendations for fertility preservation in patients with lymphoma, leukemia, and breast cancer. J. Assist. Reprod. Genet. 29, 465–468 (2012).
Ruddy, K. J. et al. Prospective study of fertility concerns and preservation strategies in young women with breast cancer. J. Clin. Oncol. 32, 1151–1156 (2014).
Sella, T. et al. Impact of fertility concerns on endocrine therapy decisions in young breast cancer survivors. Cancer. Am. Cancer Soc. 127, 2888–2894 (2021).
Zhou, Y. D. et al. Icariin ameliorates cisplatin-induced cytotoxicity in human embryonic kidney 293 cells by suppressing ROS-mediated PI3K/Akt pathway. Biomed. Pharmacother. 109, 2309–2317 (2019).
Nie, X. et al. Effect of Hyperin and Icariin on steroid hormone secretion in rat ovarian granulosa cells. Clin. Chim. Acta. 495, 646–651 (2019).
Liu, J. et al. Erxian decoction alleviates cisplatin-induced premature ovarian failure in rats by reducing oxidation levels in ovarian granulosa cells. J. Ethnopharmacol. 304, 116046 (2023).
Barberino, R. S. et al. Melatonin protects against cisplatin-induced ovarian damage in mice via the MT1 receptor and antioxidant activity. Biol. Reprod. 96, 1244–1255 (2017).
Findlay, J. K., Hutt, K. J., Hickey, M. & Anderson, R. A. How is the number of primordial follicles in the ovarian reserve established?. Biol. Reprod. 93, 111 (2015).
Ford, E. A., Beckett, E. L., Roman, S. D., McLaughlin, E. A. & Sutherland, J. M. Advances in human primordial follicle activation and premature ovarian insufficiency. Reproduction 159, R15–R29 (2020).
Field, S. L., Dasgupta, T., Cummings, M. & Orsi, N. M. Cytokines in ovarian folliculogenesis, oocyte maturation and luteinisation. Mol. Reprod. Dev. 81, 284–314 (2014).
Passos, J. R. et al. Protein and messenger RNA expression of interleukin 1 system members in bovine ovarian follicles and effects of interleukin 1beta on primordial follicle activation and survival in vitro. Domest. Anim. Endocrinol. 54, 48–59 (2016).
Chang, E. M. et al. Cisplatin induces overactivation of the dormant primordial follicle through PTEN/AKT/FOXO3a pathway which leads to loss of ovarian reserve in mice. PLoS ONE 10, e144245 (2015).
Chen, Y. et al. Icariin alleviates osteoarthritis through PI3K/Akt/mTOR/ULK1 signaling pathway. Eur. J. Med. Res. 27, 204 (2022).
Gopoju, R., Panangipalli, S. & Kotamraju, S. Metformin treatment prevents SREBP2-mediated cholesterol uptake and improves lipid homeostasis during oxidative stress-induced atherosclerosis. Free Radic. Biol. Med. 118, 85–97 (2018).
Shaukat, A. et al. Icariin alleviates Escherichia coli lipopolysaccharide-mediated endometritis in mice by inhibiting inflammation and oxidative stress. Int. J. Mol. Sci. https://doi.org/10.3390/ijms231810219 (2022).
Tang, Z. et al. HO-1-mediated ferroptosis as a target for protection against retinal pigment epithelium degeneration. Redox Biol. 43, 101971 (2021).
Wei, R. et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int. J. Biol. Sci. 17, 2703–2717 (2021).
Zhao, Y. et al. Dexmedetomidine alleviates hepatic injury via the inhibition of oxidative stress and activation of the Nrf2/HO-1 signaling pathway. Eur. Cytokine Netw. 30, 88–97 (2019).
Jia, J. et al. Icariin improves cardiac function and remodeling via the TGF-beta1/Smad signaling pathway in rats following myocardial infarction. Eur. J. Med. Res. 28, 607 (2023).
Biyik, I. et al. The effects of recombinant klotho in cisplatin-induced ovarian failure in mice. J. Obstet. Gynaecol. Res. 47, 1817–1824 (2021).
Pedersen, T. & Peters, H. Proposal for a classification of oocytes and follicles in the mouse ovary. J. Reprod. Fertil. 17, 555–557 (1968).
O’Boyle, N. M. et al. Open babel: An open chemical toolbox. J. Cheminform. 3, 33 (2011).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model 61, 3891–3898 (2021).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Funding
This work is financially supported by Anhui Provincial Key Research and Development Project (2022e07020050); Science Research Project of Anhui Health Commission (AHWJ2021b097).
Author information
Authors and Affiliations
Contributions
FL, FZ, SW contributed equally to this work. The study was designed and wrote by FZ, XL, LC. FZ, FL, HQ, SW performed research; DZ, JC, ZH analyzed the data. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, F., Zhu, F., Wang, S. et al. Icariin alleviates cisplatin-induced premature ovarian failure by inhibiting ferroptosis through activation of the Nrf2/ARE pathway. Sci Rep 14, 17318 (2024). https://doi.org/10.1038/s41598-024-67557-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67557-x
- Springer Nature Limited
Keywords
This article is cited by
-
Icariin inhibits cisplatin-induced ovarian toxicity via modulating NF-κB and PTEN/AKT/mTOR/AMPK axis
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)