
Abstract
Varroa mites, notorious for parasitizing honeybees, are generally classified as Varroidae. Their extremely modified morphologies and behaviors have led to debates regarding their phylogenetic position and classification as an independent family. In this study, two different datasets were employed to reconstruct the phylogenies of Varroa mites and related Laelapidae species: (1) 9257 bp from the whole 13 mitochondrial protein-coding genes of 24 taxa, (2) 3158 bp from 113 taxa using Sanger sequencing of four nuclear loci. Both mitochondrial and nuclear analyses consistently place Varroa mites within the Laelapidae. Here we propose to place Varroa mites in the subfamily Varroinae stat. nov., which represents a highly morphologically adapted group within the Laelapidae. Ancestral state reconstructions reveal that bee-associated lifestyles evolved independently at least three times within Laelapidae, with most phoretic traits originating from free-living ancestors. Our revised classification and evolutionary analyses will provide new insight into understanding the Varroa mites.
Similar content being viewed by others
Introduction
Varroa mites, represented by the notorious Varroa destructor Anderson and Trueman, 2000, are one of the most well-known honeybee pests in the world1,2. Varroa mites are defined as a family, Varroidae, consisting of six species in two genera3: Varroa jacobsoni Oudemans; Varroa underwoodi Delfinado-Baker and Aggarwal; Varroa rindereri Guzman and Delfinado-Baker; Varroa destructor Anderson and Trueman; Euvarra sinhai Delfinado and Baker; Euvarroa wongsirii Lekprayoon and Tangkanasing. All Varroa mites have remarkably adapted morphologically, physiologically, and ecologically to various species of honey bee4,5,6,7. Among them, V. destructor is the most widespread species in the group, posing a great threat to the overall apicultural and pollination industry after successfully adapting to the European honeybee (Apis mellifera Linnaeus) from its original host Asian honeybee (Apis cerana Fabricius)8,9. Currently, V. destructor is considered one of the major causes of colony collapse disorder (CCD) in European honeybees, serving as the primary vector for numerous contagious bee viruses such as Deformed Wing Virus (DWV)10. Likewise, the impact of Varroa mites extends beyond direct effects on bees and encompasses indirect consequences, including economic implications such as reduced pollinator populations, decreased crop yield, and increased expenditure on pest control11,12.
Despite numerous studies of Varroa mites, their phylogenetic relationship among related taxa remains largely unexplored. Currently, Varroa mites are widely accepted as a separate family, Varroidae, without much debate1,2,13,14,15,16 with only a few exceptions17,18. However, the classifications of Varroa mites had been the subject of prolonged controversy due to their unique external morphologies and parasitic life cycle. The genus Varroa was first established by Oudemans19 based on V. jacobsoni from the hive of Apis cerana indica Fabricius on Indonesian Java Island, by having both (i) broad ventral shields and (ii) reduced cheliceral digit. The genus Varroa was initially placed within Laelaptinae based on (i) metapodal and anal shield and (ii) numerous dorsal setae. Delfinado and Baker4 elevated Varroa to the family Varroidae based on densely dorsal setae, lack of fixed digit, looped stigma, and its unique leg-chaetotactic system (Supplementary Fig. 1). Casanueva20 asserted the tribe Varroini that belongs to the Hymenopteran-associated laelapid group, Melittiphidinae, by morphological phylogenetics using 83 characters. Casanueva20 suggested unique regressive apomorphies of Varroa also existed in some other laelapid genera, such as reduced gnathosomal setae in Tropilaelaps, Stevelus, Dinogamasus, and Urozercon, and reduced cheliceral digit in Myrmolaelaps. All the aforementioned morphological studies have in common that Varroa mites are closely related to Laelapidae but focused on whether to assign their taxonomic rank as a genus, tribe within Laelapidae, or a separate family.
Existing molecular phylogenies including Varroa mites, Laelapidae, and their relatives, have consistently indicated a close association between Varroa mites and Laelapidae, aligning with the findings from previous morphological analyses (Multi-locus phylogeny21,22,23; Mitogenome phylogeny24,25,26,27; Phylogenomics28). Varroa mites have been either placed within Laelapidae22,23,24, recovered as a sister group to Laelapidae28, or shown close affinity with Laelapidae + Blattisociidae27. However, most studies only included V. destructor22,23,24,27, utilized fewer genes below the current standard21,22,23, or lacked Varroa-related Laelapidae species in their taxon sampling, hindering the examination of whether Varroa mites constitute a separate family or are nested within other Laelapidae groups24,27,28. Therefore, an inclusive approach incorporating multiple Varroa mite species, broader coverage of Laelapid mites, and robust phylogenetic tree reconstruction with an extensive genetic dataset could elucidate their precise phylogenetic placement and aid in determining the most appropriate taxonomic rank for Varroa mites.
Another important aspect concerning the relationship between Varroa mites and Laelapidae pertains to the association between host use and phylogeny. Varroa mites are exclusively parasitic on bees, while Laelapidae exhibit a diverse array of parasitic behaviors. Previous studies have broadly categorized Laelapidae mites and their relatives based on their host types into three categories: (i) free-living, (ii) vertebrates, and (iii) arthropods29,30,31,32. Morphological analyses conducted by Casanueva20 revealed distinct monoclades sharing similar hosts among the included members, such as free-living forms, Hymenoptera parasites, beetles, and roach riders. Casanueva20 placed Varroa mites within Laelapidae, forming a monoclade alongside other bee-parasitic laelapids like Dinogamasus, Tropilaelaps, Urozercon, and Stevelus. However, Dowling and OConnor22 used two-loci multi-locus phylogenetics and identified multiple origins of both vertebrate and invertebrate parasitism. This contradicted Casanueva's findings, notably observing that Dinogamasus spp., a Carpenter bee parasite, located far apart from Varroa destructor on phylogeny. By employing a more reliable phylogenetic tree and a more refined host categorization than the existing ones, may unravel whether morphological traits of Varroa mites and Laelapidae are shaped by their hosts and how phoretic behavior aids in establishing a more robust classification.
In this study, we provide phylogenetic trees with the most up-to-date sample coverage that includes multiple species of Varroa mites and closely related groups such as Laelapidae, as well as other outgroup families. Based on our results, we were able to reach the Laelapidae s.lat. including Varroinae which is relegated from its family level. To confirm our phylogenetic hypothesis more robustly, we reconstructed phylogenetic trees using two different sampling strategies and types of molecular datasets widely employed in Acari phylogenies22,23,24,26: (i) mitochondrial genome sequences with a small number of terminal species and (ii) Sanger-based multi-locus data with a larger number of terminal species. We further utilized the resulting multigene phylogeny to re-examine the evolutionary history of the parasitic lifestyle within Laelapidae, an aspect that had not been extensively studied after Casanueva20 and Dowling and OConnor23. Our ancestral state reconstruction along with their host association suggests that the Varroa mite’s bee parasitic behavior evolved independently.
Result
Phylogenetic analysis of mitogenome sequences
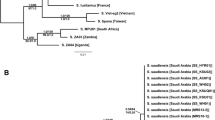
A total of 9257 bp (atp6: 651 bp, atp8: 174 bp, cox1: 1470 bp, cox2: 665 bp, cox3: 753 bp, cytb: 1008 bp, nad1: 768 bp, nad2: 783 bp, nad3: 327 bp, nad4: 939 bp, nad4L: 225 bp, nad5: 1143 bp, and nad6: 351 bp) were obtained from a combined dataset. This dataset included only the family Laelapidae and Varroidae among the superfamily Dermanyssoidea Laelapidae s.lat. with strong support (UFB = 100/BS = 100) (Fig. 1). Those laelapid mites were found to be sister to the clade containing superfamily Parasitoidea and Celaenopsoidea (UFB = 77/BS = 98) in both Maximum Likelihood (ML) and Bayesian Inference (BI) analyses (Supplementary Figs. 2, 3). Although the topologies of the constructed trees obtained from ML and BI analyses were not always congruent, Varroidae consistently belonged to Laelapidae s. lat. in both analyses. Varroidae was supported as a monophyletic group (UFB = 100/BS = 100) among the 11 ingroup taxa, which consisted of two genera and three species, including its type species V. jacobsoni. The other ingroups comprised the two broadest Laelapid superfamilies, Hypoaspidinae and Laelapinae. Hypoaspidinae (Coleolaelaps, Cosmolaelaps, Gaeolaelaps, Hypoaspis, and Stratiolaelaps) also exhibited non-monophyletic relationships in both ML and BI analyses. Furthermore, the genus Stratiolaelaps formed a sister group relationship with the clade of Laelaps with low support (UFB = 51/BS = –), as observed in the multi-locus phylogeny. Varroidae was formed to sister to Stratiolaelaps scimitus Berlese with low support (UFB = –/BS = 56) in the BI analysis, but this was not observed in the ML analysis.

Combined phylogenetic tree of varroa mites and relatives. (A) Mitogenome phylogeny. (B) Multi-locus phylogeny. The tree shown are resulted from Bayesian inference. Node colors represent posterior probabilities from MrBayes and ultrafast bootstrap values from IQ-tree. Monophyletic clades in the multi-locus phylogeny are shown as triangles for better visualization. Refer to Fig. 2A for comprehensive node details.
Phylogenetic analysis of multi-locus sequences
The dataset, encompassing 3,158 base pairs (28S: 798 bp, 18S: 1557 bp, ITS: 431 bp, and H3: 372 bp), led to the generation of topologies through BI and ML methods. These topologies were mostly congruent, though there were some divergences at certain nodes (Supplementary Figs. 4, 5). Notably, Varroa mites were accurately positioned within the Laelapidae family, exhibiting a close affinity with clade A, which itself demonstrated strong support, encompassing numerous species (UFB = 95/BS = 96). The topology of Varroa mites, Stratiolaelaps, and Laelaps mirrored that found in the Bayesian mitogenome tree. Among the included families, only the Macronyssidae (UFB = 88/BS = 76) and Varroidae (UFB = 100/BS = 100) formed monophyletic groups with moderate to strong support. However, the subfamilies Hypoaspidinae, Laelapinae, and Melittiphinae did not demonstrate monophyly, exhibiting varying levels of support. Hypoaspidinae fragmented into 12 clades with mostly non-monophyletic genera, except for the strongly supported monophyletic Stratiolaelaps (UFB = 100/BS = 100). Laelapinae subdivided into six clades, with Andreacarus being the only genus forming a monophyletic relationship, supported by strong values (UFB = 100/BS = 100). Lastly, Melittiphinae was divided into four disparate clades, with Gymnolaelaps and Tropilaelaps forming well-supported monophyletic clades (UFB = 100/BS = 100).
Evolution of Laelapidae phoretic traits
The ancestral states of phoretic traits reconstructed by Mesquite and Bayestraits generally showed congruent results (Fig. 2). The most probable common ancestral trait within Laelapidae s.lat. was 'free-living', with an estimated probability of approximately 98% (node 1). This trait also appeared as the most probable ancestral trait at various deeper nodes on the trees (nodes 2, 3, 4). Phoretic behavior on ants and bees is estimated to have originated at least three times independently within Laelapidae s.lat., all likely evolving from a free-living ancestry (ants: nodes 2, 4, 10; bees: nodes 7, 8, 5). Beetle phoretic behavior evolved at least twice within closely related clades from a free-living ancestry (node 3). The parasitic behavior of Varroa mites also appears to have evolved from a free-living ancestry. Reverse transitions from invertebrate hosts to free-living habits were seldom observed. Rodent parasites arose from at least three clades, with multiple terminal species within the subfamily. In the most species-rich rodent phoretic clade (node 6) of our phylogenetic tree, phoresy of roaches (node 9) and diploids (node 11) evolved at least once within the family from rodent phoresy.

(A) Multi-locus phylogeny resulting from MrBayes. (B) Summary of Parsimony and Bayesian Ancestral State Reconstructions for Phoretic Hosts. Branch colors depict the results of ancestral state reconstruction via parsimony analysis. The pie charts display mean posterior probability values computed through RJ-MCMC analysis using BayesTraits.
Discussion
Our study has revealed significant challenges in the current classification of Laelapidae. These include seriously non-monophyletic Laelapidae subfamilies and paraphyletic Laelapidae under multi-locus phylogenetics, when Macronyssidae is included. The monophyly of Laelapidae under mitochondrial genome phylogeny (Fig. 1) can be interpreted as a result of better and larger genetic data, but it may also be due to a simple sampling bias, with missing Macronyssidae in the sampling. Despite these challenges, it is clear that conventional morphology and molecular phylogenetic analyses are inconsistent, and a new perspective not bound by traditional morphology is needed to develop a more stable phylogeny. While our sampling of Laelapidae is limited and lacks many representative taxa, including type genera and species of each subfamily, and a more densely sampled multi-locus analysis showed low supports on the deeper nodes, we have not made significant changes to the taxonomic ranks. One major discovery of our study is that the Varroa mites have a close affinity to Laelaps, the type genus of the Laelapidae family. This was identified by both four molecular phylogenetic results, namely multi-locus and mitochondrial phylogenetic results, under both ML and BI frames (Fig. 1). This discovery reasonably relegates the disreputable Varroa mites, currently known as a family Varroidae, to a subfamily within Laelapidae. Based on our molecular analysis and prior morphological studies31,32,33,34,35,36,37,38,39, additional morphological evidence was identified regarding crucial characteristics that play a significant role in establishing the family Varroidae. (i) cheliceral structure: Oudemans19 noted the similarity of the chelicerae of female Berlesia to those of Varroa and their modifications for parasitism; he also noted that the involution of the cheliceral digit is dissimilar. Looking for comparisons with Varroa chelicerae in other parasitic Acari, Oudemans19 found similarities with those of Berlesia and ticks in terms of their construction for piercing the host cuticle, although Varroa lacks retrospective anchoring structures for attachment to its host as in the latter two taxa. In particular, a similar reduction in the size of the fixed digit was observed in the females of Varroa and Berlesia. The chelicerae of Varroa tend to have less robust movable digits with a vertical cutting plane typical of mesostigmatid mites, and the cutting actions of the right and left digits remain functionally independent40 and appear to be restricted to the tips of the cheliceral shafts. (ii) internal hypostomal setae (h3): The subcapitulum generally bears a pair of palp coxal setae, and the hypostome bears three pairs of setae (h1–3) arranged in a triangular pattern. The h3 is lost in some genera of Laelapidae, such as Dicrocheles and some Jacobsonia32. (iii) hypertrichy of idiosoma: This character can be easily found in the family Laelapidae, as Oudemans19 rightly pointed out the similarity of V. jacobsoni with Pogonolaelaps canestrinii (mentioned as H. canestrinii) with regard to the hypertrichy of the dorsal shield. Also, in the genera such as Eulaelaps, Haemogamasus41, Dinogamasus, Neohypoaspis, Sphaeroseius, Suracarus, and Tropilaelaps, the hypertrichy character can be observed32,38,42. (iv) extra setae on genital shield: The genital shield generally only bears the st5 setae. However, if the shield is posterolaterally expanded, it bears 1–5 additional setae pairs (Jv1–3, Zv1–2) or more in cases of hypertrichy, as in the genera Eulaelaps, Haemogamasus, or Laelaps41 or in the myrmecophilous genus Sphaeroseius32. (v) enlarged metapodal plates: The family Laelapidae generally have one or two pairs of small metapodal plates, but sometimes greatly enlarged, as in the genera Eulaelaps41, and Neohypoaspis32. (vi) conspicuously short legs: found in some arthropod-related genera such as Conolaelaps, Dynatochela, Scorpionyssus, and some Myrmozercon species. Their legs are shortened compared to the idiosoma. Additionally, according to Burkett-Cadena43, the most notable characteristics of parasitic arthropods are their body shape, mouthparts, and leg shape. These specifically modified features can be found in other parasitic arthropods such as Myrmecophilidae (ant cricket), Mystacinobiidae (bat fly), and other nest-dwelling mites like Tropilaelaps, Melittiphis, and Myrmozercon. These inquilines have undergone changes in their morphology to adapt to specialized environments. Thus, we can assume the modified appearance of Varroa mites is a result of the optimal adaption to the limited space of the bee hive, and Varroidae should be demoted and transferred to Varroinae stat.nov., with the specific unique morphological features previously assigned to Varroidae.
Furthermore, at the viewpoint of host traits, our findings revealed that parasitic traits tend to evolve from a free-living state to various invertebrates and vertebrates independently, with a few reverse transitions to invertebrates that share similar habitats with closely related mammal host species (Fig. 2). Similarly, the parasitic behavior observed in Varroa mites most likely evolved from a history of free-living (69%). However, a significant challenge arose at Node 5 due to insufficient support, rendering uncertain their origin from a free-living state. Minor alterations in topology significantly impact the reconstruction of ancestral states, leaving open the possibility that the evolution of bee parasitic behavior in Varroa mites might be linked to ant phoretic behavior. To address this, it is necessary to add more terminal species that are possibly related to Varroa mites (e.g., Myrmozercon) into our molecular tree or use markers with better resolution to enhance support across nodes. Our results indicate the host traits may play a significant role in the classification of the family Laelapidae, as host specificity is frequently observed in numerous groups within the subclass Acari30,32,44,45,46. In particular, Casanueva20, which, although not widely used today, seems to be reevaluated positively which integrated host and morphology in its classifications. We found that the highly supported clades tended to be in slightly better concordance with paraphyletic characters rather than traditional morphological classifications. Examples include the relegation of Varroa from an independent family to Laelapidae, and the formation of a highly supported monoclade (node 4) of Melittiphinae (Laelaspis, Holostaspis) and Hypoaspidinae (Cosmolaelaps partim) found in ants, Laelapinae (Andreacarus, Laelaps, Mysolaelaps, Echinolaelaps, Ondatralaelaps), Hypoaspidinae (Blaberolaelaps, Androlaelaps), and Ipiopsidinae (Julolaelaps), which primarily host rodents and some of the organisms that live around them, form a high-supporting monoclade. In this context, the genus Androlaelaps may be included in the subfamily Laelapinae a subfamily reliant on mammals (node 6), as their close affinity has been the subject of occasional debate in several prior studies23,47. Given the independent evolution of similar phoretic behaviors across diverse clades, a comprehensive analysis is required, extending beyond host associations, and encompassing morphological and phylogenetic analyses. For example, bee-associated behavior has independently evolved at least three times (node 5, 7, and 8) within Laelapidae s.lat., paralleling the independent occurrences of ant phoretic behavior (node 2, 4), also observed at least three times. This independent evolution of traits is supported by highly robust MRCA nodes across nearly all clades. The initial step toward refining classification in line with phylogenetic relationships entails identifying the morphological characteristics at each node. In the future, we will need to revise not only Varroinae but also other subfamily classifications based on trees that include more terminal species, type species, and genera using more powerful markers, and in the process, we learned that the role of phoretic behavior should not be underestimated.
Systematic account of Varroinae
Hyporder (Subcohort) Dermanyssiae Evans and Till, 1979, sensu Lindquist et al. 2009.
Superfamily Dermanyssoidea Kolenati, 1859, sensu Beaulieu et al. 2011.
Subfamily Varroinae subfam. nov.
Type genus Varroa Oudemans, 1904.
Genera included. Varroa Oudemans, 1904; Euvarroa Delfinado and Baker, 1974.
Diagnosis. strongly flattened, hypertrichous idiosoma, with extra setae present on the genital shield, peritremes looped and shifted to a position lateral to the stigma, subcapitulum with three pairs hypostomal setae arranged in a line (internal hypostomal setae (h3) absent), fixed digit of chelicera reduced and legs conspicuously shorter than idiosoma.
Methods
Mitochondrial genome phylogeny
The major goal of sampling strategy was to determine the phylogenetic position of Varroa mites by using a wide range of Dermanyssoidea mites. Therefore, 47 sequences and 25 novel sequences were obtained including three Varroa mites from two genera: Varroa destructor, V. jacobsoni, and Euvarroa sinhai. In total, 11 species of Dermanyssodea and 13 outgroup species (Celaenopsoidea, Eviphidoidea, Parasitoidea, Phytoseioidea, and Rhodacaroidea) were used (Supplementary Table 1). For the novel sequences, DNA libraries were prepared using the Nextera DNA Flex Library Preparation Kit (Illumina Inc., Cambridge, UK) and sequenced on an Illumina Novaseq 6000S4 (Illumina Inc.). The raw reads from the whole-genome sequences were assembled into contigs using Velvet 1.2.10. Among the contigs, those with high-GC ratio contents were screened, and ambiguous contigs were manually generated. Additionally, 77 data belonging to parasitiformes served as mitogenome reference sequences and were downloaded from NCBI Genbank (https://www.ncbi.nlm.nih.gov/genbank/). The public sequences were aligned using MAFFT 7.149 and extracted separately through Geneious Prime 2023.02.02 (https://www.geneious.com). All 13 protein-coding genes (atp6, atp8, COX1, COX2, COX3, CYTB, NAD1, NAD2, NAD3, NAD4, NAD4L, NAD5, NAD6) annotated with the MITOS Web Server48 and used. For three important taxa that lack full-mitogenome data, only COI sequences were used: Euvarroa sinhai, Laelaps agilis, and L. clethrionomydis. These data were partitioned using PARTITION-FINDER2 software, and the best-fit partition schemes were applied to Maximum Likelihood (ML) and Bayesian Inference (BI) analyses. Prior to obtaining the final results, species with lower Taxonomic Instability Index (Tii) > 0.4 or Leaf Stability Index (Lsi) < 0.9 were removed using the RoughNaRok web server49.
Phylogenetic analyses were conducted using both ML and BI methods. The ML analysis was carried out using IQ-TREE50 with 1,000 replicates of ultrafast bootstrap approximation (UFB) with the best partition scheme and the best-fit substitution models found by the PARTITION-FINDER251. Bootstrap values were designated as moderate (≥ 85) and strong (≥ 95). The Bayesian Inference analysis was conducted using MRBAYES v.3.2.752, the analysis was performed using 20 million Markov Chain Monte Carlo (MCMC) generations, and trees were sampled every 1000 generations. A burnin of 25% of the sampled trees was applied to ensure adequate mixing of the MCMC chain. Both analyses results were visualized using FIGTREE v.1.4.4.53.
Multi-locus phylogeny
Two species of Varroa mites were included in multi-locus phylogeny: V. destructor and V. jacobsoni. A total of 108 species of Dermanyssoidea were used for the ingroup, with 94 species belonging to the family Laelapidae. The outgroup consisted of two species of Ologamasidae (rhodacaroidea) and three species of Eviphididae (eviphidoidea), which were selected based on previous studies20,21,22. Public sequences of 73 species were obtained from Genbank and those of 128 sequences from 40 species were newly added to the database (Supplementary Table 2). Newly sequenced mites were collected using Berlese-Tullgren funnel traps (60w/48 h) from various regions in Korea. Voucher specimens for identification were made after the experiment by Poly Vinyl Alcohol (PVA) mounting fluid54 and DNAs were stored in − 20 °C conditions. All the DNA vouchers and voucher specimens were deposited in the Insect Biosystematics Laboratory at Seoul National University.
Total genomic DNAs were extracted from Dermanyssoidea mites using DNeasy Blood & Tissue kit (Quiagen, Inc.) following the manufacturer's protocol. Four genes commonly used in Araneae phylogenetics were selected, including one nuclear protein-coding gene (H3) and three ribosomal genes (28S rDNA, 18S rDNA, ITS) which were used in previous studies22,23,55,56,57. The sequences of primers and PCR conditions are provided (Supplementary Table 3). PCR products were purified and sequenced at Bionics Inc. The raw sequence data were assembled and checked using SEQMAN PRO v.7.1.0 (DNASTAR, Madison, WI, USA). The sequences were aligned using MAFFT version 758 and adjusted by MUSCLE in MEGA v.7.0.26. The aligned sequences were combined using SEQUENCEMATRIX v.1.7.859. ML and BI analysis were performed the same as in Mitogenome phylogenetics.
Ancestral character states reconstruction of host
The phoretic behavior was designated into seven states: free-living and phoretic to beetle, bee, ant, vertebrates, roaches, and diplopod. Mites that were observed on the soil surface, within animal nests, or were never found attached to the bodies of other organisms under any circumstances were categorized as 'free-living.' This classification aligns with the common lifestyle observed in many acarine species, as suggested by Krantz and Walter60. In contrast to the free-living category, mites with previous records of phoretic behavior or detachment from other animals' bodies. The 'ant' state includes mites detaching from ant bodies directly, within the nest, or even close to nest entrances. In total, our dataset includes 110 host designation records, excluding those marked with the '?' symbol (Supplementary Table 4).
The hosts of Laelapidae were coded as seven discrete states by their phoretic preference: (A) free-living, (B) beetle, (C) bee, (D) ant, (E) vertebrates, (F) roaches, (G) diplopod. The probability of the ancestral state of each node was calculated by Bayestraits V.4.0.061 using reversible jump Markov Chain Monte Carlo (RJ-MCMC). An exponential distribution was implemented, seeding from a uniform prior in an interval of 0–100. 50 million iterations were done, sampling every 1000th iteration. The first million iterations were discarded as burn-in. Acceptance rates were automatically adjusted and achieved in the preferred range of nearly 35%. Parsimony-based ancestral character state reconstruction was conducted using the trace character over trees option in Mesquite 3.8162 on a single Bayesian Inference (BI) based tree.
Data availability
The authors declare that all other data supporting the findings of this study are available within the supplementary information files.
References
Ramsey, S. D. et al. Varroa destructor feeds primarily on honey bee fat body tissue and not hemolymph. PNAS 116, 1792–1801. https://doi.org/10.1073/pnas.1818371116 (2019).
Reams, T. & Rangel, J. Understanding the enemy: A review of the genetics, behavior and chemical ecology of Varroa destructor, the parasitic mite of Apis mellifera. J. Insect Sci. 22, 1–10. https://doi.org/10.1093/jisesa/ieab101 (2022).
Chantawannakul, P., de Guzman, L. I., Li, J. & Williams, G. R. Parasites, pathogens, and pests of honeybees in Asia. Apidologie. 47, 301–324. https://doi.org/10.1007/s13592-015-0407-5 (2016).
Delfinado, M. D. & Baker, E. W. Varroidae, A new family of mites on honey bees (Mesostigmata: Acarina). J. Wash. Acad. Sci. 64, 4–10 (1974).
Delfinado-Baker, M. & Aggarwal, K. A new Varroa (Acari: Varroidae) from the nest of Apis cerena (Apidae). Int. J. Acarol. 13, 233–237. https://doi.org/10.1080/01647958708683777 (1987).
Lekprayoon, C. & Tangkanasing, P. Euvarroa wongsirii, a new species of bee mite from Thailand. Int. J. Acarol. 17, 255–258. https://doi.org/10.1080/01647959108683915 (1991).
de Guzman, L. I. & Delfinado-Baker, M. A new species of Varroa (Acari: Varroidae) associated with Apis koschevnikovi (Apididae: Hymenoptera) in Borneo. Int. J. Acarol. 22, 23–27. https://doi.org/10.1080/01647959608684077 (1996).
Wilfert, L. et al. Deformed wing virus is a recent global epidemic in honeybees driven by Varroa mites. Science 354, 594–597. https://doi.org/10.1126/science.aac9976 (2016).
Traynor, K. S. et al. Varroa destructor: A complex parasite, crippling honey bees worldwide. Trends Parasitol. 36, 592–606. https://doi.org/10.1016/j.pt.2020.04.004 (2020).
Francis, R. M., Nielsen, S. L. & Kryger, P. Varroa-virus interaction in collapsing honey bee colonies. PLos ONE 8, e57540. https://doi.org/10.1371/journal.pone.0057540 (2013).
Cook, D. C. et al. Predicting the economic impact of an invasive species on an ecosystem service. Ecol. Appl. 17, 1832–1840. https://doi.org/10.1890/06-1632.1 (2007).
PHA (Plant Health Australia). Varroa mite preparedness of pollination dependent industries. Rural Industries Research and Development Corporation (RIRDC), 29–39 (2013).
Anderson, D. L., Halliday, R. B. & Otis, G. W. The occurrence of Varroa underwoodi (Acarina: Varroidae) in Papua New Guinea and Indonesia. Apidologie 28, 143–147. https://doi.org/10.1051/apido:19970305 (1997).
Bruce, W. A., Delfinado-Baker, M. & Vincent, D. L. Comparative morphology of the peritremes of Varroa and Euvarroa (Varroidae), parasites of honey bees (Apidae). Int. J. Acarol. 2, 13–20. https://doi.org/10.1080/01647959708684114 (1997).
Anderson, D. L. & Trueman, J. W. H. Varroa jacobsoni (Acari: Varroidae) is more than one species. Exp. Appl. Acarol. 24, 165–189. https://doi.org/10.1023/A:1006456720416 (2000).
Beaulieu, F. et al. Superorder parasitiformes Reuter, 1909. In: Zhang, Z.-Q. (Ed.). Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness. Zootaxa 3148, 123–128. https://doi.org/10.11646/zootaxa.3148.1.23 (2011).
Sammataro, D., Gerson, U. & Needham, G. Parasitic mites of honey bees: Life history, Implications, and impact. Annu. Rev. Entomol. 45, 519–548. https://doi.org/10.1146/annurev.ento.45.1.519 (2000).
Khanjani, M. & Ueckermann, E. A. Hypoaspis (Hypoaspis) polyphyllae N. SP. (Mesostigmata: Laelapidae) parasitic on larvae of Polyphylla olivieri Castelnau (Coleoptera: Scarabaeidae) in Iran. Int. J. Acarol. 31, 119–122. https://doi.org/10.1080/01647950508683661 (2005).
Oudemans, A. C. Note VIII. On a new genus and species of parasitic acari. Notes Leyden Mus. 24, 216–222 (1904).
Casanueva, M. E. Phylogenetic studies of the free-living and arthropod associated Laelapidae (Acari: Mesostigmata). Gayana Zool. 57, 21–46 (1993).
Klompen, H., Lekveishvili, M. & Black, W. C. IV. Phylogeny of parasitiform mites (Acari) based on rRNA. Mol. Phylogenet. Evol. 43, 936–951. https://doi.org/10.1016/j.ympev.2006.10.024 (2007).
Dowling, A. P. G. & OConnor, B. M. Phylogenetic relationships within the suborder Dermanyssina (Acari: Parasitiformes) and a test of dermanyssoid monophyly. Int. J. Acarol. 36, 299–314. https://doi.org/10.1080/01647951003604569 (2010).
Dowling, A. P. G. & OConnor, B. M. Phylogeny of Dermanyssoidea (Acari: Parasitiformes) suggests multiple origins of parasitism. Acarologia 50, 113–129. https://doi.org/10.1051/acarologia/20101957 (2010).
Li, W. N. et al. Mitochondrial genome reorganization characterizes various lineages of mesostigmatid mites (Acari: Parasitiformes). Zool. Scr. 48, 679–689. https://doi.org/10.1111/zsc.12369 (2019).
Yan, Y. et al. Characterization of the complete mitochondrial genome of the predatory mites Stratiolaelaps scimitus (Acari: Laelapidae). Mitochondrial DNA B 5, 885–886. https://doi.org/10.1080/23802359.2020.1717393 (2020).
Yan, Y. et al. A highly contiguous genome assembly of a polyphagous predatory mite Stratiolaelaps scimitus (Womersley) (Acari: Laelapidae). Genome Biol. Evol. 13, 1–7. https://doi.org/10.1093/gbe/evab011 (2021).
Yang, H., Chen, T. & Dong, W. The complete mitochondrial genome of Parasitus fimetorum (Berlese, 1904) (Arachnida: Parasitiformes: Parasitidae). Mitochondrial DNA B 7, 1044–1045. https://doi.org/10.1080/23802359.2022.2081944 (2022).
Techer, M. A. et al. Divergent evolutionary trajectories following speciation in two ectoparasitic honey bee mites. Commun. Biol. 2, 357. https://doi.org/10.1038/s42003-019-0606-0 (2019).
Tipton, V. J. The genus Laelaps: with a review of the Laelaptinae and a new subfamily Alphalaelaptinae, Acarina, Laelaptidae. Univ. Calif. Publ. Entomol. 16, 233–356 (1960).
Radovsky, F. J. The Macronyssidae and Laelapidae (Acarina: mesostigmata) Parasitic on Bats Vol. 46 (University of California Publications in Entomology, 1967).
Fain, A. & Rack, G. Scorpionyssus heterometrus gen. n., sp. n. (Acari, Laelapidae) parasitic on a scorpion from Sri Lanka. Ent. Mitt. Zool. Mus. Hamburg 9, 99–108 (1988).
Moraes, G. J. D. et al. Catalogue of the free-living and arthropod-associated Laelapidae Canestrini (Acari: Mesostigmata), with revised generic concepts and a key to genera. Zootaxa 5184, 001–509. https://doi.org/10.11646/zootaxa.5184.1.1 (2022).
Evans, G. O. A review of the Laelaptid paraphages of the Myriapoda with descriptions of three new species (Acarina: Laelaptidae). Parasitology 45, 352–368. https://doi.org/10.1017/S0031182000027694 (1954).
Moss, W. W. & Funk, R. C. Studies on the developmental chaetotaxy of Dyscinetonyssus hystricosus n. g., n. sp. (Acari: Mesostigmata: Laelaptoidea). Acarologia 7, 235–267 (1965).
Kethley, J. B. & Narceolaelaps, N. G. (acari: laelapidae) with four new species parasitizing spiroboloid millipeds. Int. J. Acarol. 4, 195–210. https://doi.org/10.1080/01647957808683117 (1978).
Fain, A. & Lukoschus, F. S. Katydiseius nadchatrami n.g., n.sp. (Acari, Otopheidomenidae) from the tracheae of a Malaysian katydid Chloracris brullei Pictet & Saussure, 1892 (Orthoptera, Pseudophyllidae). Int. J. Acarol. 9, 173–178. https://doi.org/10.1080/01647958308683333 (1983).
Walter, D. E. A new mite from an arboreal and (Formicidae: Polyrachis sp.): Myrmozercon iainkayi n sp. (Mesostigmata: Laelapidae). Int. J. Acarol. 29, 81–85. https://doi.org/10.1080/01647950308684325 (2003).
Joharchi, O., Khodaparast, R. & Moghadam, S. G. First report of the genus Dinogamasus Kramer (Acari: Mesostigmata: Laelapidae) from the Middle East Region, with the description of a new species. Syst. Appl. Acarol. 21, 791–799. https://doi.org/10.11158/saa.21.6.6 (2016).
Lindquist, E. E., OConnor, B. M., Shaw, M. D. & Sidorchuk, E. A. Review of the genera Berlesia Canestrini, 1884, and Katydiseius Fain & Lukoschus, 1983, the subfamily Katydiseiinae Fain & Lukoschus, 1983, and their family group relationships (Acari: Mesostigmata: Gamasina), with description of three new species parasitic on gryllacridid crickets (Orthoptera). Zootaxa 4857, 005–070. https://doi.org/10.11646/zootaxa.4857.1.4 (2020).
de Lillo, E., Di Palma, A. & Nuzzaci, G. Morphological adaptations of mite chelicerae to different trophic activities (Acari). Entomologica 35, 125–180 (2002).
Evans, G. O. & Till, W. M. Studies on the British Dermanyssidae (Acari: Mesostigmata). Part II. Classification. Bull. Br. Museum (Natl. Hist.) Zool. 14, 1–370 (1966).
Lundqvist, L. Taxonomic revision of the genus Dinogamasus Kramer (Acari: Mesostigmata: Laelapidae). Entomol. Scand. Suppl. 54, 1–109 (1999).
Burkett-Cadena, N. D. Chapter 2—Morphological adaptations of parasitic arthropods. In Medical and Veterinary Entomology (Third Edition) (eds Mullen, G. R. & Durden, L. A.) 17–22 https://doi.org/10.1016/B978-0-12-814043-7.00002-9 (Academic Press, 2019).
Costa, M. Cerambylaelaps nadchatrami, N. gen., N. sp., An unusual Mesostigmatic mite (Acari) associated with a cerambycid beetle in Malaysia. Acarologia t. XX. fasc 2, 188–195 (1978).
OConnor, B. M. The mite community associated with Xylocopa latipes (Hymenoptera: Anthophoridae: Xylocopinae) with description of a new type of acarinarim. Int. J. Acarol. 19, 159–166. https://doi.org/10.1080/01647959308683975 (1993).
Metwally, A. M., Al-Azazzy, M. M. & Abd El-Hady, M. A. H. Mites associated with Coleoptera. Acarines 8, 55–58. https://doi.org/10.21608/ajesa.2014.4910 (2014).
Radovsky, F. J. Adaptive radiation in the parasitic Mesostigmata. Acarologia t. XI, fasc. 3, 450–483 (1969).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. https://doi.org/10.1016/j.ympev.2012.08.023 (2013).
Aberer, A. J., Krompass, D. & Stamatakis, A. Pruning rogue taxa improves phylogenetic accuracy: An efficient algorithm and webservice. Syst. Biol. 62, 162–166 (2013).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. https://doi.org/10.1093/molbev/msu300 (2015).
Lanfear, R. et al. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. https://doi.org/10.1093/molbev/msw260 (2016).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Rambaut, A. FigTree, a Graphical Viewer of Phylogenetic Trees. Institute of Evolutionary Biology, The University of Edinburgh. http://tree.bio.ed.ac.uk/software/figtree/ (2009).
Downs, W. G. Polyvinyl alcohol: A medium for mounting and clearing biological specimens. Science 97, 539–540. https://doi.org/10.1126/science.97.2528.539 (1943).
Colgan, D. J. et al. Histone H3 and U2 snRNA DNA sequences and arthropod molecular evolution. Aust. J. Zool. 46, 419–437. https://doi.org/10.1071/ZO98048 (1998).
Lekveishvili, M. & Klompen, H. Phylogeny of infraorder Sejina (Acari: Mesostigmata). Zootaxa 629, 1–19. https://doi.org/10.11646/zootaxa.629.1.1 (2004).
Anderson, D. L. & Morgan, M. J. Genetic and morphological variation of bee-parasitic Tropilaelaps mites (Acari: Laelapidae): New and re-defined species. Exp. Appl. Acarol. 43, 1–24. https://doi.org/10.1007/s10493-007-9103-0 (2007).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Vaidya, G., Lohman, D. J. & Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. https://doi.org/10.1111/j.1096-0031.2010.00329.x (2011).
Krantz, G. W. & Walter, D. E. A Manual of Acarology. 3rd ed. Lubbock Texas, viii + 807 pp (Texas Tech University Press, 2009).
Pagel, M., O’Donovan, C. & Meade, A. Bayesian estimation of ancestral character states on phylogenies. Syst. Biol. 53, 673–684. https://doi.org/10.1080/10635150490522232 (2004).
Maddison, W. P. & Maddison, D. R. Mesquite: A modular system for evolutionary analysis. Version 3.2. Mesquite. http://www.mesquite project.org (2017).
Acknowledgements
We appreciate Kyeong-Yeoll Lee (Kyungpook National University), Seongjun Choe (Chungbuk National University), and Sehyeon Bang (KUMDOMI INSECT BRAND) for providing DNA materials. This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF2020R1I1A2069484, RS-2023-00237795). Omid Joharchi’s study was also supported by the Russian Science Foundation, grant No. 24–46–00024 (https://rscf.ru/en/project/24-46-00024/).
Author information
Authors and Affiliations
Contributions
J.O., S.L.1, and S.K. conceived and designed the research. J.O., S.L.1, and O.J. wrote the first draft of the manuscript, O.J. confirmed and wrote the morphological information. W.K. had a part in the raw data for the mitochondrial genome analyses. S.L.2 supervised the experiment, S.L.2 and S.K. critically revised the manuscript. All authors read and approved the final manuscript. S.L.1: Seunghyun Lee S.L.2: Seunghwan Lee.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oh, J., Lee, S., Kwon, W. et al. Molecular phylogeny reveals Varroa mites are not a separate family but a subfamily of Laelapidae. Sci Rep 14, 13994 (2024). https://doi.org/10.1038/s41598-024-63991-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-63991-z
- Springer Nature Limited