
Abstract
In order to further explore the effects of soil mercury pollution on soil microbial diversity and community structure, soil samples were randomly collected from 2 m, 20 m, 30 m, 500 m and 650 m periphery of Wanshan mining area, as 5 different treatments. Each treatment had 4 replicates. Soil microbial DNA was extracted from 20 soil samples, and then high-throughput sequencing technology was used to analyse the structure and distribution of bacterial and fungal communities. The results showed that the number of bacterial and fungal communities in T0–T30 treatments was significantly larger than that in T500–T650 treatments at order, family and genus level. Whatever, the number of uniquely distributed bacterial and fungal communities among 4 replicates soil samples was quite different at order, family and genus level. The results of the effect on the microbial community structure showed that there were both the same dominant bacterial and fungal communities, and the different dominant bacterial and fungal communities at any classification level, moreover, the number of same dominant bacterial and fungal communities was larger than that of different dominant bacterial and fungal communities. The results of relationship between soil environment factors and bacterial and fungal community structure showed that distance (Hg2+), EC and pH had a high correlation with community structure, especially the distance factor, that is, the content of mercury in soil had the highest effects on community structure. The internal heterogeneity of soil caused significant differences in bacterial and fungal community structure, and the emergence of dominant bacterial and fungal communities was a manifestation of better adaptability to long-term mercury stress and other stresses in soil, which will provide a scientific reference for further exploring the mechanism of mercury enrichment between microorganisms and plants.
Similar content being viewed by others

Introduction
Soil is an important link between water and atmosphere for material circulation. Soil carries about 90% of pollutants in the environment. Mercury was one of the main control pollutants in 129 priority control pollutants1,2,3. Mercury in soil was often enriched and transferred through crops, and then enriched and transferred to animals and humans through various food chains, resulting in serious ecological problems4,5. Soil bacteria, fungi and actinomycetes are the main components of the microflora in the soil ecosystem. When the soil is subjected to heavy metal stress, the soil microflora can reflect the change of soil environmental quality, so it can be used as one of the important biological indicators, and bacteria were more sensitive to heavy metal pollution than fungi6. In recent year, there have been many reports on the effects of mercury pollution on plants, and some plants with strong resistance to mercury stress have been found7,8, but there were few reports on the effects of mercury pollution on soil microorganisms9,10. Harris–Hellal and coworkers showed that when the mercury concentration was low (0–1 mg/kg), the microbial communities in the forest soil did not change significantly. When the mercury concentration increased to 20 mg/kg, the diversity and genetic structure of the soil microbial communities changed significantly11. The combined pollution of heavy metals cadmium and mercury could reduce the diversity of soil microbial communities, and the abundance ratio of gram-positive bacteria to gram-negative bacteria increases with the increase of pollution concentration12,13. Mercury in soil inhibited the growth of fungi, actinomycetes and bacteria, and seriously affected soil bacterial activity and bacterial community structure. In high-concentration mercury-contaminated environments, gemmatimonadetes were still active but Nitrospirae was declining. Fungi was prone to resistance in high-concentration mercury-contaminated environments14,15. Frossard and coworkers found that when the mercury concentration reached 32 mg/kg, it seriously affected the community structure, composition and diversity of bacteria and fungi in forest soil16. In order to further explore the effect of mercury on soil microorganisms, this experiment was carried out to collected soil samples at different distances from the abandoned mercury mining area in Wanshan, Tongren, Guizhou Province, China. Meanwhile, high-throughput sequencing was used to explore the effects of different concentrations of mercury in soils at different distances from the mercury mining area on soil microbial diversity and community structure, which provided theoretical support for further exploration of the synergistic mercury enrichment mechanism of mercury-resistant bacteria and plants.
Methods
General situation of soil sample plot
Wanshan mercury areas (E:109°07′–109°24′; N: 27°24′–27°38′) is located in the east of Tongren City, Guizhou Province, China (Fig. 1). It is 26 km wide from east to west and 22 km long from north to south. The total area of the region is 842 km2, and the mining area accounts for 45 km2. The terrain of the whole territory is low in the east and high in the west, and the central uplift, from the middle. The part tilts to the southeast on three sides. The altitude is 260–997 m, with an average of 850 m. It belongs to subtropical humid monsoon climate. The annual average temperature is 13.7 °C, the maximum temperature is 34.6 °C, and the minimum temperature is − 10.4 °C. Summer, The season is warm and rainy, and the precipitation is 1379 mm. Since the excavation of cinnabar in the Qin and Han Dynasties, the history of mercury mining and smelting in Wanshan has been more than two thousand years. In 2001, Wanshan Mercury Mine in Guizhou was closed due to resource depletion. The map of study area was created by ArcGIS software (Version 10.7 USA).

Location map of study area.
Sample collection
Five sampling points were selected at different distances around the abandoned area of Wanshan Mercury Mine in Tongren, Guizhou Province, China. Soil samples were collected at 16:44 p.m. on May 9, 2022, in summer. The T0 treatment soil samples, numbered MSO1, MSO2, MSO3 and MSO4, were collected at different points about 2 m from the sewage outlet of the mercury mining area. The T20 treatment soil samples, numbered CMSO21, CMSO22, CMSO23, and CMSO24 were collected from the wasteland of perennial ryegrass about 20 m away from the mining area. The T30 treatment soil samples, numbered GMSO301, GMSO302, GMSO303, and GMSO304 were collected from the wasteland of perennial ryegrass about 30 m away from the mining area. The T500 treatment soil samples, numbered GMSO500, GMSO501, GMSO502, and GMSO503 were collected from new cultivated land at different points about 500 m away from the mercury mining area. The T650 treatment soil samples, numbered GMSO651, GMSO652, GMSO653 and GMSO654 were collected from long-term cultivated land at different points about 650 m away from the mercury mining area, each treatment had 4 replicates.
All soil samples were taken from 0 to 20 cm soil under the surface, removed impurities, crushed, and sieved by 1 mm. A total of 20 soil samples were tested. In order to facilitate statistical analysis, all 20 soil samples were grouped. MSO1, MSO2, MSO3, MSO4, CMSO21, CMSO22, CMSO23, CMSO24, GMSO301, GMSO302, GMSO303 and GMSO304 soil samples were in the range of 2–30 m from the sewage outlet of the mercury mining area, and these soil samples were classified into the proximal group (CR). Similarly, GMSO500, GMSO501, GMSO502, GMSO503, GMSO651, GMSO652, GMSO653 and GMSO654 soil samples were collected 500–650 m away from the sewage outlet of the mercury mining area, and these soil samples were classified into the distal group (FBR). Two sets of soil samples were prepared, one for the determination of soil physical and chemical properties, and another for the determination of soil microbial DNA.
Mercury measurement
At first, 0.1–0.2 g over 200 mesh of dry soil was weighed accurately and placed in a 50 mL centrifuge tube, 5 mL deionized water was added, 5 mL freshly prepared aqua regia was added again, after being shaken well, and being heated in a 95 °C water bath in a fume hood 5 min, 1 mL BrCl was added to continue water bath for 30 min, after being cooled, being set the volume to 50 mL. Being placed for more than 24 h, BrCl oxidizes various forms of mercury to Hg2+, 0.4 mL hydroxylamine hydrochloride was added 30 min before the determination to remove the migration, an appropriate volume of supernatant was taken in a 10 mL borosilicate glass reaction flask, 0.4 mL SnCl2 was added, Determination of mercury by cold atomic absorption mercury analyzer (F732-V,China)3, The detection limit of the instrument was 0.05 μg/L.
The basic physical and chemical properties of the tested soils
The basic physical and chemical properties of soil samples were shown in Table 1. The content of basic nutrients in the soil samples was determined according to the standard analysis method17. In all 20 soil samples, pH was from 6.5 to 8.5; the content of available N was from 7.94 to 9.11 mg/kg, the mean of available N was T0 > T650 > T30 > T500 > T20; the content of available P was from 10.13 to 12.14 mg/kg, the mean of available P was T650 > T30 > T0 > T500 > T20; the content of available K was from 27.24 to 30.14 mg/kg, the mean of available P was T30 > T0 > T500 > T650 > T20; the content of Hg2+ was from 45.42 mg/kg to 167.14 mg/kg, the mean of Hg2+ was T20 > T0 > T650 > T30 > T500, Hg2+ content of T0 treatment and T20 treatment was higher than that of T30, T500, and T650 treatment (P < 0.05), Hg2+ content of T650 treatment was higher than that of T30 and T500 treatment (P < 0.05).
DNA extraction
Total community genomic DNA extraction was performed using a E.Z.N.A™ Mag-Bind Soil DNA Kit (Omega,M5635-02,USA), following the manufacturer's instructions. The concentration of the DNA was measured by using a Qubit 4.0 (Thermo,USA) to ensure that adequate amounts of high-quality genomic DNA had been extracted.
16S rRNA gene amplification by PCR
Our target was the V3-V4 hypervariable region of the bacterial 16S rRNA gene. PCR was started immediately after the DNA was extracted. The 16S rRNA V3-V4 amplicon was amplified using 2 × Hieff® Robust PCR Master Mix (Yeasen, 10105ES03, China). Two universal bacterial 16S rRNA gene amplicon PCR primers (PAGE purified) were used: the amplicon PCR forward primer (CCTACGGGNGGCWGCAG) and amplicon PCR reverse primer (GACTACHVGGGTATCTAATCC). The reaction was set up as follows: microbial DNA(10 ng/μl) 2 μl; amplicon PCR forward primer (10 μM)1 μl; amplicon PCR reverse primer (10 μM)1 μl; 2xHieff® Robust PCR Master Mix (Yeasen,10105ES03, China)(total 30 μl). The plate was sealed and PCR performed in a thermal instrument (Applied Biosystems9700, USA) using the following program: 1 cycle of denaturing at 95 °C for 3 min, first 5 cycles of denaturing at 95 °C for 30 s, annealing at 45 °C for 30 s, elongation at 72 °C for 30 s, then 20 cycles of denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s, elongation at 72 °C for 30 s and a final extension at 72 °C for 5 min. The PCR products were checked using electrophoresis in 2% (w/v) agarose gels in TBE buffer (Tris,boricacid,EDTA) stained with ethidium bromide (EB) and visualized under UV light.
ITS rRNA gene amplification by PCR
Our target was the ITS1-ITS2 hypervariable region of the fungal ITS rRNA gene. PCR was started immediately after the DNA was extracted. The ITS rRNA ITS1-ITS2 amplicon was amplified using 2 × Hieff® Robust PCR Master Mix (Yeasen, 10105ES03, China). Two universal fungal ITS rRNA gene amplicon PCR primers (PAGE purified) were used: the amplicon PCR forward primer (CTTGGTCATTTAGAGGAAGTAA) and amplicon PCR reverse primer (GCTGCGTTCTTCATCGATGC). The reaction was set up as follows: microbial DNA(10 ng/μl) 2 μl; amplicon PCR forward primer (10 μM)1 μl; amplicon PCR reverse primer (10 μM)1 μl; 2xHieff® Robust PCR Master Mix (Yeasen, 10105ES03, China) (total 30 μl). The plate was sealed and PCR performed in a thermal instrument (Applied Biosystems9700, USA) using the following program: 1 cycle of denaturing at 95 °C for 3 min, first 5 cycles of denaturing at 95 °C for 30 s, annealing at 45 °C for 30 s, elongation at 72 °C for 30 s, then 20 cycles of denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s, elongation at 72 °C for 30 s and a final extension at 72 °C for 5 min. The PCR products were checked using electrophoresis in 2% (w/v) agarose gels in TBE buffer (Tris, boricacid, EDTA) stained with ethidium bromide (EB) and visualized under UV light.
16S, ITS gene library preparation, quantification, and sequencing
Hieff NGSTM DNA Selection Beads (Yeasen, 10105ES03, China) were used to purify the free primers and primer dimer species in the amplicon product. Samples were delivered to Sangon BioTech (shanghai) for library preparation using universal lllumina adaptor and index. Before sequencing, the DNA concentration of each PCR product was determined using a Qubit® 4.0 Green double-stranded DNA assay and it was quality controlled using a bioanalyzer (Agilent 2100, USA). Depending on coverage needs, all libraries can be pooled for one run. The amplicons from each reaction mixture were pooled in equimolar ratios based on their concentration. Sequencing was performed using the lllumina MiSeq system (lllumina MiSeq, USA), according to the manufacturer's instructions.
Sequence processing, OTU clustering, Representative tags alignment and Biological classification
After sequencing, The two short lllumina readings were assembled by PEAR software (version 0.9.8) according to the overlap and fastq files were processed to generate individual fasta and qual files, which could then be analyzed by standard methods. The effective tags were clustered into operational taxonomic units (OTUs) of ≥ 97% similarity using Usearch software (version 11.0.667). Chimeric sequences and singleton OTUs (with only one read) were removed, after which the remaining sequences were sorted into each sample based on the OTUs. The tag sequence with the highest abundance was selected as a representative sequence within each cluster. Bacterial and fungal OTU representative sequences were classified taxonomically by blasting against the RDP Database and UNITE fungal ITS Database, respectively.
Results
The distribution of bacterial and fungal communities at the order level
At the order level, the distribution of bacterial communities in the 12 soil samples of the proximal group (CR) was shown in Fig. 2. Among them, the number of co-distributed bacterial communities in 12 soil samples was 72, and the number of uniquely distributed bacterial communities in the 12 soil samples was very different. Among them, the number of uniquely distributed bacterial communities in MSO1 was 5, only 1 in MSO3 and CMSO23 soil samples, no uniquely distributed bacterial communities in the other 9 soil samples. The bacterial community distribution of 8 soil samples in the distal group (FBR) was shown in Fig. 3. The number of co-distributed bacterial communities in the 8 soil samples was 56, of which 10 uniquely distributed bacterial communities in GMSO651 soil samples, 3 uniquely distributed bacterial communities in GMSO503 soil samples, 2 in GMSO653 soil samples, and 1 in GMSO500 soil samples. The results of bacterial community distribution of all 20 soil samples showed that the number of bacterial communities in each soil sample of 12 soil samples in the proximal group (CR) was about 72, and in each soil sample of 8 soil samples in the distal treatment group (FBR) was about 58. The number of bacterial communities in T0, T20, and T30 treatments was larger than that in T500 and T650 treatments.

The distribution of bacterial communities in 12 soil samples of the proximal group (CR) at the order level.

The distribution of bacterial communities in 8 soil samples of the distal group (FBR) at the order level.
At the order level, the distribution of fungal communities in 12 soil samples of the proximal group (CR) was shown in Fig. 4. The total number of fungal communities in all 12 soil samples was 30, but the number of uniquely distributed fungal communities to each of the 12 soil samples was very different. Among them, the number of uniquely distributed fungal communities in MSO3 soil sample was 3, 2 uniquely distributed fungal communities in CMSO21, CMSO23, CMSO24, GMSO302, and GMSO303 soil samples, 1 uniquely distributed fungal communities in MSO2 and MSO4 soil samples, and 0 uniquely distributed fungal communities in MSO1, CMSO22, GMSO301, and GMSO304 soil samples. The distribution of fungal communities in 8 soil samples of the distal group (FBR) was shown in Fig. 5. The number of co-distributed fungi in 8 soil samples was 19, of which 9 uniquely distributed fungal communities in GMSO501 soil samples, 5 uniquely distributed fungal communities in GMSO503 soil sample, 3 uniquely distributed fungal communities in GMSO654 soil sample, 1 uniquely distributed fungal communities in GMSO500 and GMSO651 soil samples, and 0 uniquely distributed fungal communities in GMSO652 and GMSO653 soil samples. From the analysis of the number of fungal communities in all 20 soil samples, the number of fungal communities per sample in the 12 soil samples was about 31, and the number of fungal communities per soil sample in the 8 soil samples was about 22. The number of fungal communities in T0, T20, and T30 treatments was larger than that in T500 and T650 treatments. At the order level, the total number of bacterial communities in both the proximal group and the distal group was larger than that of fungal communities.

The distribution of fungal communities in 12 soil samples of the proximal group (CR) at the order level.

The distribution of fungal communities in 8 soil samples of the distal group (FBR) at the order level.
The distribution of soil bacterial and fungal communities at family level
At the family level, the distribution of bacterial communities in 12 soil samples in the proximal group (CR) was shown in Fig. 6. The total number of bacterial communities in 12 soil samples was 114, and the number of uniquely distributed bacterial communities to each of the 12 soil samples was quite different. Among them, the number of uniquely distributed bacterial communities in MSO1 soil sample was 10. 1 uniquely distributed bacterial communities in MSO2, MSO3, MSO4, CMSO21, CMSO22, CMSO23 and CMSO24 soil samples, while 0 uniquely distributed bacterial communities in GMSO301, GMSO302, GMSO303 and GMSO304 soil samples. The distribution of bacterial communities in 8 soil samples of the distal group (FBR) was shown in Fig. 7. There were 89 bacterial communities in 8 soil samples, and the number of uniquely distributed bacterial communities in 8 soil samples was quite different. Among them, the number of uniquely distributed bacterial communities in GMSO651 soil sample was 22, 7 uniquely distributed bacterial communities in GMSO653 soil sample, 3 uniquely distributed bacterial communities in GMSO500, GMSO503 and GMSO652 soil samples, 1 uniquely distributed bacterial communities in GMSO502 soil sample, 0 uniquely distributed bacterial communities in GMSO501 and GMSO654 soil samples. From the analysis of the number of bacterial communities in all 20 soil samples, the number of bacterial communities in each sample of 12 soil samples was about 115, and the number of bacterial communities in each sample of 8 soil samples was about 92. The number of bacterial communities in T0, T20, and T30 treatments was larger than that in T500 and T650 treatments. At the family level, the distribution of fungal communities in 12 soil samples of the proximal group (CR) was shown in Fig. 8. In the proximal group (CR), the number of co-distributed fungal communities in 12 soil samples was 36, and the number of uniquely distributed fungal communities in 12 soil samples was quite different. Among them, the number of uniquely distributed fungal communities in CMSO21 soil sample was 7, 6 uniquely distributed fungal communities in MSO4 and GMSO302 soil samples, 5 uniquely distributed fungal communities in MSO3 soil sample, 4 uniquely distributed fungal communities in MSO2 and CMSO24 soil samples, 3 uniquely distributed fungal communities in GMSO303, GMSO304, and CMSO23 soil samples. 1 uniquely distributed fungal communities in CMSO22 and GMSO301 soil samples, while 0 uniquely distributed fungal communities in MSO1soil sample. The distribution of fungal communities in 8 soil samples of the distal group (FBR) was shown in Fig. 9. The number of co-distributed fungal communities in 8 soil samples was 16, and the number of uniquely distributed fungal communities among the 8 soil samples was quite different. Among them, the number of uniquely distributed fungal communities in GMSO503, GMSO501 and GMSO502 soil samples was 22, 19 and 15 respectively, while 1 uniquely distributed fungal communities in GMSO500 soil sample. Among GMSO651, GMSO652, GMSO653 and GMSO654 soil samples, only GMSO654 soil sample had 6, GMSO651 and GMSO653 soil samples had 1 respectively, and GMSO652 soil sample had 0. From the distribution of fungal communities in all 20 soil samples, the number of fungal communities in each sample of 12 soil samples was about 39, and the number of fungal communities in each sample of 8 soil samples was about 22. The number of fungal communities distribution in T0, T20, and T30 treatments was larger than that in T500 and T650 treatments at the family level.

The distribution of bacterial communities in 12 soil samples of the proximal group (CR) at the family level.

The distribution of bacterial communities in 8 soil samples of the distal group (FBR) at the family level.

The distribution of fungal communities in 12 soil samples of the proximal group (CR) at the family level.

The distribution of fungal communities in 8 soil samples of the distal group (FBR) at the family level.
The distribution of bacterial and fungal communities at genus level
At the genus level, the bacterial community distribution of 12 soil samples in the proximal group (CR) was shown in Fig. 10. The number of co-distributed bacterial communities in 12 soil samples was 173, and the number of uniquely distributed bacterial communities to each of the 12 soil samples was very different. Among them, the number of uniquely distributed bacterial communities in MSO1 soil sample was the largest, reaching 29. 1, 4, and 1 uniquely distributed bacterial communities in MSO2, MSO3, and MSO4 soil samples respectively. 3, 5, 3, and 2 uniquely distributed bacterial communities in CMSO21, CMSO22, CMSO23, and CMSO24 soil samples respectively. 1, 1, 0, 1 uniquely distributed bacterial communities in GMSO301, GMSO302, GMSO303 and GMSO304 soil samples respectively. The distribution of bacterial communities of 8 soil samples in the distal (FBR) group was shown in Fig. 11. The number of co-distributed bacterial communities in 8 soil samples was 101, and the number of uniquely distributed bacterial communities to each soil sample varied greatly. Among them, the number of uniquely distributed bacterial communities in GMSO651 soil samples was 62, and 12, 14, 1 uniquely distributed bacterial communities in GMSO652, GMSO653, and GMSO654 soil samples respectively. 4, 6, 3 and 3 uniquely distributed bacterial communities in GMSO500, GMSO501, GMSO502 and GMSO503 soil samples respectively. According to the distribution of bacterial communities in all 20 soil samples, the number of bacterial community distribution in each sample of 12 soil samples was about 177, while the number of bacterial communities in each sample of 8 soil samples was about 114, the number of bacterial communities in T0, T20, and T30 treatments was larger than that in T500 and T650 treatments.

The distribution of bacterial communities in 12 soil samples of the proximal group (CR) at the genus level.

The distribution of bacterial communities in 8 soil samples of the distal group (FBR) at the genus level.
At the genus level, the distribution of fungal communities in 12 soil samples of the proximal group (CR) was shown in Fig. 12. The number of co-distributed fungal communities in 12 soil samples was 21, but there was a big difference in the number of uniquely distributed fungal communities among the 12 soil samples. The number of uniquely distributed fungal communities in MSO1, MSO2, MSO3 and MSO4 soil samples was 2, 13, 14, 21 respectively. 22, 5, 12, 16 uniquely distributed fungal communities in CMSO21, CMSO22, CMSO23 and CMSO24 soil samples respectively. 6, 10, 11, 12 uniquely distributed fungal communities in GMSO301, GMSO302, GMSO303 and GMSO304 soil samples respectively. The distribution of fungal communities in 8 soil samples of the distal group (FBR) was shown in Fig. 13. The total number of fungal communities in the 8 soil samples was 12, but the number of uniquely distributed fungal communities among the 8 soil samples varied greatly. Among them, the number of uniquely distributed fungal communities in GMSO500, GMSO501, GMSO502, GMSO503 soil samples was 8, 50, 44, 55, respectively, and 14, 6, 2, 11 uniquely distributed fungal communities in GMSO651, GMSO652, GMSO653, GMSO654 soil samples respectively. From the distribution of fungal communities in all 20 soil samples, the number of fungal communities each sample in 12 soil samples was about 33, and the number of fungal communities per sample in the 8 soil samples was about 35, The number of fungal communities in T0, T20, and T30 treatments was close to the number of fungal communities in T500 and T650 treatments.

The distribution of fungal communities in 12 soil samples of the proximal group (CR) at the genus level.

The distribution of fungal communities in 8 soil samples of the distal group (FBR) at the genus level.
Alpha diversity analysis
The alpha diversity indices of bacterial communities in 5 treatments were shown in the Table 2. The number of reads in T0–T650 treatments was 46,321, 42,898, 42,062, 44,585, 57,825, i.e. T650 > T0 > T500 > T20 > T300; the number of OUTs in T0–T650 treatments was 2558, 2267, 2088, 2151, 1541, i.e. T0 > T20 > T500 > T30 > T650; Shannon indices of T0–T650 treatments were 6.30911975, 6.256060, 6.0918122, 5.9032615, 5.103864 respectively, that is T0 > T20 > T30 > T500 > T650; Simpson indices of T0–T650 treatments were 0.00851625, 0.00630375, 0.0071275, 0.0122905, 0.0453070 respectively, that is T0 < T30 < T20 < T500 < T650. Comprehensive judgment based on the results of above 4 indices, the alpha diversity indices of bacterial communities in 5 treatments was T0 > T20 > T30 > T500 > T650.
The alpha diversity indices of fungal communities in 5 treatments were shown in Table 3. The number of reads in T0–T650 treatments was 69,062, 50,849, 89,767, 62,446, 51,248, i.e. T30 > T0 > T500 > T650 > T20; the number of OUTs in T0–T650 treatments was 559, 313, 610, 509, 159, i.e. T30 > T0 > T500 > T20 > T650; Shannon indices of T0–T650 treatments were 2.42374775, 2.63471725, 2.4174655, 3.260999, 4.02980475 respectively, i.e. T650 > T500 > T20 > T0 > T30, Simpson indices of T0–T650 treatments were 0.3465155, 0.2690235, 0.2884785, 0.1538875, 0.05412475 respectively, i.e. T650 < T500 < T20 < T30 < T0. Comprehensive judgment based on the results of above 4 indices, The alpha diversity indices of fungal communities in 5 treatments was T650 > T500 > T30 > T20 > T0.
Beta diversity analysis
The beta diversity analysis of bacterial communities among 20 soil samples was shown in Fig. 14 at the OTU level. From the bacterial community structure of 20 soil samples in T0–T650 treatments, except for the MSO3, CMS021, GMSO502 soil samples, the bacterial community composition of the other 18 soil samples was nearer to that of the soil samples in their respective treatment. Based on the similarity of bacterial communities of soil samples among T0–T650 treatments, from the perspective of phylogenetic distance, the results of the occurrence of bacterial communities in 5 treatments of soil samples from near to far was T20, T0, T500, T30, T650. By comparing the similarity of bacterial communities between CR and FBR group soil samples, the results showed that bacterial communities of T650 treatment in FBR group soil samples had lower similarity with T0, T20, T30 treatments in CR groups soil samples, but bacterial communities of T500 treatment in FBR group soil samples had higher similarity with T0, T20, T30 treatments in CR groups soil samples.

Weighted unifrac tree anaylsis of bacteria at OTU level.
The beta diversity analysis of fungal communities among 20 soil samples was shown in Fig. 15 at the OTU level. From the fungal community structure of 20 soil samples in T0–T650 treatments, except for the GMSO502 and GMSO503 soil samples, the fungal community composition of the other 18 samples was nearer to that of the soil samples in their respective treatments. Based on the similarity of fungal community structure of soil samples among T0–T650 treatments, From the perspective of phylogenetic distance, the results of the occurrence of fungal communities in 5 treatments of soil samples from near to far was T0, T30, T20, T650, T500. By comparing the similarity of fungal communities between CR and FBR group soil samples, the results showed that fungal communities of T500 and T650 treatments in FBR group soil samples had lower similarity with T0 and T30 treatments in CR groups soil samples.

Weighted unifrac tree anaylsis of fungi at OTU level.
The community structure of bacteria and fungi at the order level
At the order level, the relative abundance of bacterial communities in 20 soil samples was shown in Fig. 16. Sphingomonadales(8.74%), Rhizobiales(8.38%), Sphingobacteriales(7.78%), Gp4(norank_Acidobacteria_Gp4)(5.16%) had higher average relative abundance in T0 treatment; Sphingobacteriales(9.10%), Gp6(norank_Acidobacteria_Gp6)(5.14%), Sphingomonadales(4.96%) in T20 treatment; Sphingomonadales(8.88%), Gp4(norank_Acidobacteria_Gp4)(8.83%), Sphingobacteriales(7.33%), Gp6(norank_Acidobacteria_Gp6)(7.12%) in T30 treatment; Sphingomonadales(11.31%), Sphingobacteriales(9.97%), Gp4(norank_Acidobacteria_Gp4)(6.90%), Gp6(norank_Acidobacteria_Gp6)(5.32%) in T500 treatment; Burkholderiales(20.01%), Bacteroidales(10.27%), Sphingomonadales(7.24%), Sphingobacteriales(4.60%) in T650 treatment. After comparison with bacterial community structure among 20 soil samples, the bacterial community structure had commonalities and differences at the order level. The commonality was that Sphingomonadales(4.96–11.31%) and Sphingobacteriales(4.60–9.97%) had higher average relative abundance in T0, T20, T30, T500, and T650 treatments, Gp4(norank_Acidobacteria_Gp4) in T0, T30, and T500 treatments, Gp6(norank_Acidobacteria_Gp6) in T20, T30, and T500 treatments; The differences was that Rhizobiales(8.38%) had higher average relative abundance in T0 treatment, Burkholderiales(20.01%) and Bacteroidales(10.27%) in T650 treatment.

The relative abundance of bacterial communities in 20 soil samples at the order level.
At the order level, the relative abundance of fungal communities in 20 soil samples was shown in Fig. 17. Agaricales(63.48%), Sebacinales(8.36%), and Hypocreales(5.59%) had higher average relative abundance in T0 treatment; Sebacinales(28.34%), Mortierellales(19.24%), and Trechisporales(6.94%) in T20 treatment; Agaricales(65.13%), Archaeorhizomycetales(15.27%), and Sebacinales(5.74%) in T30 treatment; Agaricales(23.93%), Geoglossales(14.46%), Sebacinales(12.62%), Archaeorhizomycetales(7.42%), and Corticiales(6.99%) in T500 treatment; Helotiales(16.03%), Mortierellales(8.88%), Sebacinales (8.17%), Saccharomycetales (6.06%), and Sordariales (5.54%) in T650 treatment. After comparison with fungal community structure among 20 soil samples, the fungal community structure had commonalities and differences at the order level. The commonality was that Agaricales had much higher average relative abundance in T0, T30, and T500 treatments(23.93–65.13%), Sebacinales in T0, T20, T30, T500 and T650 treatments(5.74–28.34%), Archaeorhizomycetales in T30 and T500 treatments(7.42–15.27%), Mortierellales in T20 and T650 treatments(8.88–19.24%). The differences was that Hypocreales(5.59%) only had higher average relative abundance in T0 treatment, Trechisporales(6.94%) in T20 treatment, Geoglossales(14.46%) and Corticiales(6.99%) in T500 treatment, Helotiales(16.03%), Saccharomycetales (6.06%) and Sordariales (5.54%) in T650 treatment.

The relative abundance of fungal communities in 20 soil samples at the order level.
The community structure of bacteria and fungi at the family level
At the family level, the relative abundance of bacterial communities in 20 soil samples was shown in Fig. 18. Sphingomonadaceae(8.01%), Chitinophagaceae(6.57%), and Gp4(norank_Acidobacteria_Gp4)(5.16%) had higher average relative abundance in T0 treatment; Chitinophagaceae(7.71%), Gp6(norank_Acidobacteria_Gp6)(5.14%), Anaerolineaceae(4.83%), and Sphingomonadaceae(4.74%) in T20 treatment; Gp4(norank_Acidobacteria_Gp4)(8.83%), Sphingomonadaceae(8.43%), Gp6 (norank_Acidobacteria_Gp6)(7.12%), and Chitinophagaceae(6.97%) in T30 treatment; Sphingomonadaceae(10.93%), Chitinophagaceae(9.42%) Gp4(norank_Acidobacteria_Gp4)(6.90%), and Gp6(norank_Acidobacteria_Gp6)(5.32%) in T500 treatment; Burkholderiaceae(13.81%), Porphyromonadaceae(8.40%), Sphingomonadaceae(7.16%), Oxalobacteraceae(4.75%), Gp1(norank_Acidobacteria_Gp1)(4.47%), Caulobacterales(4.37%), and Chitinophagaceae(4.26%) in T650 treatment. After comparison with bacterial community structure among 20 soil samples, the bacterial community structure had commonalities and differences at the family level. The commonality was that Sphingomonadales(4.74–8.43%) and Chitinophagaceae(4.26–7.71%) had higher average relative abundance in all 20 soil samples, Gp4(norank_Acidobacteria_Gp4)(5.16–8.83%) in T0, T30, T500 treatments, Gp6(norank_Acidobacteria_Gp6)(5.14–7.12%) in T20, T30, T500 treatments; The differences was that Anaerolineaceae(4.83%) only had higher average relative abundance in T20 treatment, Burkholderiaceae(13.81%), Porphyromonadaceae(8.40%), Oxalobacteraceae(4.75%), Gp1(norank_Acidobacteria_Gp1)(4.47%), and Caulobacterales(4.37%) in T650 treatment.

The relative abundance of bacterial communities in 20 soil samples at the family level.
At the family level, the relative abundance of fungal communities in 20 soil samples was shown in Fig. 19. Hygrophoraceae(58.45%) and Clavariaceae(4.03%) had higher average relative abundance in T0 treatment; Sebacinaceae(28.02%), Mortierellaceae(19.24%), and Hygrophoraceae(6.94%) in T20 treatment; Hygrophoraceae(26.34%), Tricholomataceae(21.18%), Clavariaceae(17.10%), Archaeorhizomycetace(15.27%), and Serendipitaceae(4.85%) in T30 treatment; Geoglossaceae(14.37%), Clavariaceae(12.94%), Archaeorhizomycetace(7.42%), Corticiaceae(6.99%), Serendipitaceae(6.30%), Sebacinaceae(5.58%), Entolomataceae(5.14%), and Hygrophoraceae(4.55%) in T500 treatment; Helotiaceae(10.16%), Mortierellaceae(8.88%), Sebacinaceae(6.17%), and Nectriaceae(4.15%) in T650 treatment, After comparison with fungal community structure among 20 soil samples, the fungal community structure had commonalities and differences at the family level. The commonality was that Hygrophoraceae had very higher average relative abundance in T0, T20, T30, and T500 treatment(4.55–58.45%), especially in T0 treatment, the relative abundance reached 58.45%, Clavariaceae(4.03–17.10%) in T0, T30, T500 treatments, Archaeorhizomycetace(7.42–15.27%) in T30, T500 treatments, Sebacinaceae(5.58–28.02%) in T20, T500, T650 treatments, Mortierellales(8.88–19.24%) in T20 and T650 treatments, Serendipitaceae(4.85–6.30%) in T30 and T500 treatments; The differences was that Clavariaceae(4.03%) only had higher average relative abundance in T0 treatment, Tricholomataceae(21.18%) in T30 treatment, Geoglossaceae (14.37%), Corticiaceae (6.99%), and Entolomataceae(5.14%) in T500 treatment, Helotiaceae(10.16%) and Nectriaceae (4.15%) in T650 treatment.

The relative abundance of fungal communities in 20 soil samples at the family level.
The community structure of bacteria and fungi at the genus level
At the genus level, the relative abundance of bacterial communities in 20 soil samples was shown in Fig. 20. Gp6(norank_Acidobacteria_Gp6)(4.14%), Flavobacterium(4.12%), Sphingomonas(3.77%), Gp4(norank_Acidobacteria_Gp4)(3.49%) had higher average relative abundance in T0 treatment; Gp6(norank_Acidobacteria_Gp6)(5.14%), Flavobacterium(3.37%), Sphingomonas(3.30%), Gemmatimonas(3.19%) in T20 treatment; Gp4(norank_Acidobacteria_Gp4)(7.20%), Gp6(7.12%), Sphingomonas(4.11%), Spartobacteria_genera_incertae_sedis(3.82%) in T30 treatment; Gp6(norank_Acidobacteria_Gp6)(5.32%), Sphingomonas(4.70%), Gp4(4.40%), and Gemmatimonas(4.21%) in T500 treatment; Ralstonia(13.49%), Herbaspirillum (4.43%), and Phenylobacterium(4.33%) in T650 treatment. After comparison with bacterial community structure among 20 soil samples, the bacterial community structure had commonalities and differences at the genus level. The commonality was that Sphingomonas(3.30–4.70%) had higher average relative abundance in T0, T20, T30, and T500 treatments, Gp6(norank_Acidobacteria_Gp6)(4.14–7.12%) in T0, T20, T30, T500 treatments, Flavobacterium(3.37–4.12%) in T0 and T20 treatments, Gp4(norank_Acidobacteria_Gp4)(3.49–7.20%) in T0, T30, and T500 treatments, Gemmatimonas(3.19–4.21%) in T20 and T500 treatments; The differences was that Spartobacteria_genera_incertae_sedis(3.82%) only had higher average relative abundance in T30 treatment, Ralstonia (13.49%), Herbaspirillum(4.43%), and Phenylobacterium(4.33%) in T650 treatment.

The relative abundance of bacterial communities in 20 soil samples at the genus level.
At the genus level, the relative abundance of fungal communities in 20 soil sample was shown in Fig. 21. Hygrocybe(58.45%), Clavaria(4.03%), Archaeorhizomyces(2.98%), and Fusarium(2.88%) had higher average relative abundance in T0 treatment; Helvellosebacina(27.85%), Mortierella(19.24%), Trechispora(6.94%) in T20 treatment; Hygrocybe(26.34%), Dermoloma(21.04%), Clavaria(17.10%), Archaeorhizomyces (15.27%), and Serendipita (4.85%) in T30 treatment; Trichoglossum(13.9%), Clavaria(12.94%), Archaeorhizomyces(7.42%), Serendipita(6.30%), Entoloma(5.06%), Hygrocybe(4.55%), Sebacina(3.76%) in T500 treatment; Collophora(9.74%), Mortierella(8.88%), Sebacina(3.84%), Archaeorhizomyces(3.14%) in T650 treatment. After comparison with fungal community structure among 20 soil samples, the fungal community structure had commonalities and differences at the genus level. The commonality was that Archaeorhizomyces(2.98–15.27%) had very higher average relative abundance in T0, T30, T500, and T650 treatments, Hygrocybe(4.55–58.45%) in T0, T30, and T500 treatments, Clavaria(4.03–17.10%) in T0, T30, T500 treatments, Mortierella(8.88–19.24%) in T20 and T650 treatments; The differences was that Fusarium(2.88%) only had higher average relative abundance in T0 treatment, Helvellosebacina(27.85%) and Trechispora (6.94%) in T20 treatment, Dermoloma(21.04%) and Serendipita(4.85%) in T30 treatment, Trichoglossum(13.9%), Serendipita(6.30%), Entoloma(5.06%), and Sebacina(3.76%) in T500 treatment, Collophora(9.74%), Sebacina(3.84%) in T650 treatment.

The relative abundance of fungal communities in 20 soil samples at the genus level.
The relationship between soil environment factors with bacterial and fungal abundance
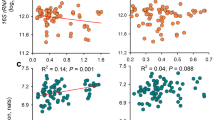
The relationship between soil environment factors and bacterial community abundance of CR and FBR group soil samples was shown in the Fig. 22. The results showed that among 6 soil environmental factors, distance, pH and EC had very important effects on the distribution of bacterial communities, In particular, the distance had the greatest effect, indicating that mercury in the soil played a dominant role in the distribution of bacterial communities. The bacterial community abundance of FBR group soil samples was more affected by distance than that in CR group soil samples.

Redundancy analysis (db-RDA) between soil environment factors and bacterial abundance of CR and FBR group at OTU level.
The relationship between soil environment factors and fungal community abundance of CR and FBR group soil samples was shown in the Fig. 23. The results showed that among the 6 soil environmental factors, distance, EC and pH had very important effects on the distribution of fungal communities, In particular, the distance had the greatest effect, indicating that mercury in the soil played a dominant role in the distribution of fungal communities. The fungal community abundance of FBR group soil samples was more affected by distance than that of CR group soil samples. While the fungal community abundance of CR group soil samples was more affected by pH, and EC than that of FBR group soil samples.

Redundancy analysis (db-RDA) between soil environment factors and fungal abundance of CR and FBR group at OTU level.
Discussion
Mercury is one of the most common heavy metal pollutants in the environment. Soil mercury pollution is often closely related to the mining of surrounding mercury-related metal mines and the smelting of mercury-related metals. Once soil is polluted by mercury, it will be transferred to plant roots, stems, leaves and other parts through plant enrichment. When the soil mercury content continues to rise, it will seriously affect the normal growth and development of plants18,19. Meanwhile, it will have adverse effects on the soil ecosystem, especially on soil microorganisms and soil animals20. Previous studies have confirmed that the concentration of Cd, Zn, Pb and Cu in soil had a strong correlation with soil microbial biomass21, and different microbial groups had different tolerance to different heavy metals22. The reason why heavy metals had adverse effects on microbial diversity could be that high concentration heavy metals had a certain restrictive effect on microbial cell metabolism and other functions, which ultimately led to a decrease in microbial diversity23.
In this work, the results of bacterial and fungal community composition in soils with different mercury contents at different distances from mercury mining areas showed that the number of bacterial and fungal communities in 20 soil samples increased significantly from the order level to the genus level. The number of uniquely distributed bacterial and fungal communities to each soil sample of the 20 soil samples showed a significant increase trend from the order level to the genus level. This result showed that even in a certain mercury stress environment, the number of bacterial and fungal communities in the soil increased with the decline of the grade level, which was not significantly affected by soil mercury and other factors. Meanwhile, the alpha diversity of bacterial and fungal communities were analyzed by using reads, OTUs, Shannon index and Simpson index (showed in Tables 2 and 3), our results indicated that there were large differences in the diversity of bacterial and fungal communities among 4 replicates soil samples, from T0 to T650 treatments, the diversity of bacterial communities decreased gradually, however, the diversity of fungal communities increased gradually. This results indicated that in the outdoor natural environment, the diversity distribution of bacterial and fungal communities at different points within the same range was affected by the internal factors of the soil. The diversity of bacterial and fungal communities was affected by many factors, resulting in the difference in the number of uniquely distributed bacterial and fungal communities to individual soil samples24. It also indicated that there was a certain heterogeneity in the soil around the mercury mining area. Therefore, it can be speculated that mercury press will lead to a decline in soil microbial diversity to a certain extent, but the relationship between the two was not a simple linear relationship, also affected by other factors within the soil. Meanwhile, the results on beta diversity analysis of bacterial and fungal communities showed that there were some differences between bacterial and fungal communities, the similarity of bacterial communities from far to near was T20, T0, T500, T30, and T650 treatment soil samples, the similarity of fungal communities from near to far was T0, T30, T20, T650, and T500 treatment soil samples. The similarity of bacterial communities and fungal communities in different soil samples was significantly different, and the main reason for this result was related to the internal heterogeneity of soil.
The results of this study showed that the number of bacterial communities in T0 and T20 treatments was much larger than that in T30, T500, and T650 treatments at order, family and genus level. Also lying in fungal communities. That is, the number of bacteria and fungal communities decreased with decline of mercury concentration at order, family and genus level. This result was inconsistent with the research results of Shan and coworkers25. This could be caused by the difference of mercury concentration or experimental environment. Previous studies belonged to the artificial simulation laboratory soil environment. This study was the natural soil environment of mercury mining area, various bacterial and fungal communities reached an equilibrium state after a long period of comprehensive influence of various factors inside the soil. The real existence of these bacterial and fungal communities was a good adaptation to mercury-containing soil environment for a long time. The reason for this result could be related to the soils of different types. Maliszewska and coworkers26found that the compounds of Cu and Hg showed the strongest inhibitory effect on microbial proliferation, and the order of toxicity of HgCl2 to microorganisms was bacteria > fungi > actinomycetes. The results showed that the inhibitory effect of mercury on microbial proliferation was fluctuated, and the inhibition of microorganisms decreased with the prolongation of culture time26. In this work, the soil around the mercury mining area had been subjected to high concentration mercury pollution stress for a long time. The increase in the number of bacterial and fungal communities under higher concentration mercury environment could be the best evidence that the inhibition of bacterial and fungal communities decreased with time.
The structure of bacterial, fungal and communities of actinomycetes in soil was easily affected by environmental variables around the soil, such as nutrient cycling and heavy metal concentration changed27,28. In this work, the effects of mercury concentration on the structure of bacterial and fungal communities at the order, family and genus levels were analyzed. The results indicated that mercury had a certain effect on the structure of bacterial and fungal communities, and the tolerance of different bacterial and fungal communities to mercury pollution was also very different, which was consistent with the results of Prasad29. Our results indicated that the dominant bacterial communities in T0–T650 treatments were different at order, family and genus level. At the order level, Sphingomonadales and Sphingobacteriales were the dominant bacterial communities, the relative abundance remained at about 10%; at the family level, for Sphingomonadaceae and Chitinophagaceae; at the genus level, for Gp6(norank_Acidobacteria_Gp6), Sphingomonas,Gp4 (norank_Acidobacteria_Gp4). Similar results were also found in fungal communities, at the order level, Agaricales and Sebacinales were the dominant fungal communities; at the family level, for Sebacinaceae and Hygrophoraceae; at the genus level, for Hygrocybe and Sebacina. Regardless of the classification level, bacterial and fungal communities with higher relative abundance were the performance of long-term adaptation to higher mercury soil environment, indicating that they had strong tolerance to heavy mercury stress. This was similar to the conclusion of Feris24 and Gillan et al.30. Meantime, at order, family and genus level, the dominant bacterial and fungal communities in different positions of mercury mining area were quite different, which was closely related to the heterogeneity of many factors in the internal environment of soil. The relationship between soil environment factors and bacterial and fungal community abundance was analyzed by using db-RDA method at OTU level, the results indicated that distance(Hg2+concentration), EC, and pH three soil factors had greater effects on the relative abundance of bacterial and fungal communities than available N, P, K. Among distance(Hg2+concentration), EC, and pH, distance(Hg2+concentration) had the greatest effects on the relative abundance of bacterial and fungal communities. This results fully demonstrated that these dominant bacterial and fungal communities had much stronger resistance to mercury pollution stress.
Conclusion
From the perspective of the effects of mercury stress on bacterial and fungal communities at different distances from mercury mining areas, Based on the experimental results, we could infer the following conclusions. 1. Under the same classification, the diversity of bacterial and fungal communities in the proximal soil was much higher than that in the distal soil; 2. At the same classification level, among five different distance soils sample, there were both the same dominant bacterial and fungal communities and different dominant bacterial and fungal communities; 3. The structure and distribution of bacterial and fungal communities were highly correlated with soil environment factors, and the mercury content in soil had more important effect on the structure and distribution of bacterial and fungal communities.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA011955) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. Please cite the following required publications: [1] The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics, Proteomics & Bioinformatics 2021, 19(4):578–583. https://doi.org/10.1016/j.gpb.2021.08.001 [PMID = 34400360]. [2] Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res 2022, 50(D1):D27–D38. https://doi.org/10.1093/nar/gkab951 [PMID = 34718731].
References
Hui, T. et al. Bioaccumulation of mercury in plants collected from Wanshan mercury mining areas in Guizhou province. Environ. Chem. 41(12), 4047–4056 (2022) (Chinese with English abstract).
Jiating, Z. et al. Study of mercury resistant wild plants growing in the mercury mine area of Wanshan district, Guizhou Province. Asian J. Ecotoxicol. 9(5), 881–887 (2014) (in Chinese with English abstract).
Xiaoli, Q. Tolerant Plants and Their Accumulation Mechanism of Mercury in Typical Mercury Mining Areas (Guizhou University, 2018) (in Chinese with English abstract).
Jianxiong, D. et al. Effects of Hg stress on growth characteristics and Hg enrichment ability of 4 turfgrass, forage varieties seedlings. Southwest China J. Agricult. Sci. 34(09), 1969–1976. https://doi.org/10.16213/j.cnki.scjas.2021.9.021 (2021) (in Chinese with English abstract).
Shijia, M. et al. Effects of low-temperature pyrolysis of mercury-contaminated soil on the accumulation of total mercury and methylmercury in vegetables. Earth Environ. https://doi.org/10.14050/j.cnki.1672-9250.2023.51.010 (2023) (in Chinese with English abstract).
Mei, L., Yanlin, H. & Guangjie, P. Microbial populations and bacterial physiological groups in purple soils polluted by mercury. J. Agro-Environ. Sci. 04, 668–673 (2004) (in Chinese with English abstract).
Youdan, L. Screening and Perfoemance Evaluation of Mercury-Rich Plant (Beijing University of Chemical Technology, 2015) (in Chinese with English abstract).
Xiaorong, X. The Selection of Hg-Tolerance Plants and the Study of the Tolerance Mechanisms in the WanShan Mine (Guizhou Normal University, 2008) (in Chinese with English abstract).
Zhenjing, L. Biological Effects of Soil Mercury on Plant Growth and Microbial Community (Beijing University of Chemical Technology, 2019). https://doi.org/10.26939/d.cnki.gbhgu.2019.000461 (in Chinese with English abstract).
Taotao, F. Study on the Screening, Identification and Mercury Conversion Mechanism of Mercury Tolerant Strains (Lanzhou Jiaotong University, 2019). https://doi.org/10.27205/d.cnki.gltec.2019.000826 (in Chinese with English abstract).
Harris, H. J. et al. Effects of mercury on soil microbial communities in tropical soils of French Guyana. Appl. Soil Ecol. 41(1), 59–68 (2009).
Xie, X. et al. Effects of contamination of single and combined cadmium and mercury on the soil microbial community structural diversity and functional diversity. Chin. J. Geochem. 30(3), 366–374 (2011) (in Chinese with English abstract).
Liao, M. et al. Effects of Cadmium and Mercury Alone and in Combination on the Soil Microbial Community Structural Diversity. Molecular Environmental Soil Science at the Interfaces in the Earth’s Critical Zone 337–341 (Springer, 2010).
Frey, B. & Rieder, S. R. Response of forest soil bacterial communities to mercury chloride application. Soil Biol. Biochem. 65(5), 329–337 (2013).
Yurong, L. et al. Patterns of bacterial diversity along a long-term mercury-contaminated gradient in the paddy soils. Microbial Ecol. 68(3), 575–583 (2014).
Frossard, A., Hartmann, M. & Frey, B. Tolerance of the forest soil microbiome to increasing mercury concentrations. Soil Biol. Biochem. 105, 162–176 (2017).
Johnson, S. W. Agricultural chemistry-soil-analysis. York (2010).
Yu, Z. et al. Enrichment research of several aquatic plants on arsenic and mercury combined pollution in water. Metal World 4, 15–19 (2018) (in Chinese with English abstract).
Fangqun, G. et al. Accumulation of mercury in rice grown in pot experimental soils and its impact factors. J. Ecol. Rural Environ. 34(7), 630–635 (2018) (in Chinese with English abstract).
Lei, W. Research on the Abiotic Methylation of Mercury and Its Effect on the Microbial Community in Mercury-Contaminated Soil (Chongqing University, 2017) (in Chinese with English abstract).
Xiuli, W. et al. Effects of Cu, Zn, Cd and Pb compound contamination on soil microbial community. Acta Scientiae Circumstantiae 23(1), 22–27 (2003) (in Chinese with English abstract).
Jun, Z. et al. Diversity change of microbial communities responding to zinc and arsenic pollution in a river of northeastern China. J. Zhejiang Univ.-Sci. B 15(7), 670–680 (2014) (in Chinese with English abstract).
Huaqun, Y. Development and Application of the Microarray Technology During the Research on the Structure and Function of Microbial Communities for Acid Mine Drainages from the Copper Mines (Central South University, 2007).
Feris, K. et al. Differences in hyporheic-zone microbial community structure along a heavy-metal contamination gradient. Appl. Environ. Microbiol. 69(9), 5563–5573 (2003).
Ping, Sh., Zhenwei, W. & Gaofei, G. Effect of exogenous mercury addition on soil microbial populations. J. Anhui Agricult. Univ. 43(2), 248–251 (2016).
Maliszewska, W. The influence of various heavy metal compounds on the development and activity of soil microorganism. Environ. Pollut. 37(3), 195–215 (1985).
Mbuthia, L. W. et al. Long term tillage, cover crop, and fertilization effects on microbial community structure, activity: implications for soil quality. Soil Biol. Biochem. 89, 24–34 (2015).
Aždajińc, M. et al. The effect of legacy gold mining on methyl-mercury cycling and microbial community structure in northern freshwater lakes. Environ. Sci. Process. Impacts 23(8), 1220–1230 (2021).
Prasad, M. N. V. Metals in the environment: Analysis by biodiversity. Met. Environ. Anal. Biodivers. (2001).
Gillan, D. C. et al. Structure of sediment associated microbial communities along a heavy-metal contamination gradient in the marine environment. Appl. Environ. Microbiol. 71(2), 679–690 (2005).
Acknowledgements
Thanks to the Science and Technology Plan Project of Guizhou Province (Qiankehe foundation-ZK [2021] General 229 and [2020] 1Z077) and Guizhou Outstanding Young Scientific Talents Program ([2021] 5625) for supporting this project with funds.
Author information
Authors and Affiliations
Contributions
J.D. and J.L. performed experiments. J.D. and Y.R. wrote the manuscript. S.Z. searched and verified the formula of the manuscript. Y.R. polished the manuscript, H.H. and J.L. carried out data collection on the device in the manuscript, all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, J., Ren, Y., Li, J. et al. The study on the effect of mercury pollution on soil microorganisms around mercury mining area. Sci Rep 13, 21605 (2023). https://doi.org/10.1038/s41598-023-48932-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48932-6
- Springer Nature Limited