
Abstract
A bispecific antibody (bsAb) is a class of engineered antibody molecules that simultaneously binds to two different antigens by having two kinds of antigen-binding domains. One of the major obstacles for the bsAb production is the incorrect chain-pairing problem, wherein each heavy and light chain should form pairings with the correct counterpart’s chains, but the structural similarity of the incorrect partners also forms the incorrect pairings. This study aimed to demonstrate a bsAb construction method using intein-mediated protein trans-splicing to create IgG–Fab2–type bsAbs, which is a modified antibody with a structure in which two additional Fabs are linked to the N-terminus of the heavy chain of an IgG molecule. The chain-paring problem between a heavy chain and a light chain is circumvented by separate expression and purification of the IgG part and the Fab part. We found that the deletion of a possible glycosylation residue improved the reaction yield and side-reaction cleavage in the protein ligation step. The resulting bsAb, IgG–Fab2 (Her2/CD3), demonstrated target binding activity and cytotoxicity mediated by activated T cells. These results indicate that the use of the protein ligation to produce the IgG–Fab2 type bsAb will expand the bsAb production method.
Similar content being viewed by others


Introduction
A bispecific antibody (bsAb) is an engineered antibody having two different antigen-binding portions within one molecule, while general monoclonal antibodies (mAbs) target only one target antigen1,2,3,4. The dual binding ability of bsAbs has multiple applications, which cannot be achieved by general mAbs, including recruiting killer immune cells to cancer cells2 and activation of receptor molecules by co-cauterization5. Such ability makes bsAbs an emerging class of new antibody therapeutics. One difficulty for immunoglobulin G (IgG) bsAb development is a chain-pairing problem that four different polypeptide chains, consisting of two heavy chains and two light chains, should form correct pairings with each other, where only one combination out of 10 combinations is the correct pairing, although it has great potential. Several antibody engineering techniques have been developed to overcome this chain-pairing problem, such as knobs-into-holes mutation for heavy chain pairing, which introduces convex–concave mutations on the interface of the Fc dimer6 and CrossMab for heavy chain-light chain pairing, achieved by exchanging the order of domains in the Fab region7.
A split intein-mediated protein ligation can be used for generating bsAb molecules among such antibody engineering methods. The reaction, termed protein trans-splicing (PTS), is a widely used protein engineering technique to connect separately expressed two target proteins8,9,10. In the PTS reaction, the N-terminal and the C-terminal part of a split intein (intein-N and intein-C) are fused to the target proteins and ligated with each other to form a peptide bond, and the intein moiety is released without any structural trace at the ligation site. Connecting two single-domain nanobodies is the simplest usage of the ligation technique for the bsAb construction. We previously reported the construction of tandem VHHs in a bacterial cell11. Various combinations of tandem VHH bsAb can be created using this method. We further utilized the ligation technique to construct circularly connected VHH bsAb by ligating the N- and C-terminus12. The intein-mediated ligation between one Fab arm and the rest of the IgG molecule was also reported for constructing IgG-type bispecific antibodies13,14,15.
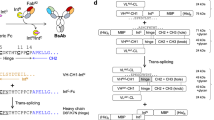
This study utilized the PTS reaction to construct the IgG–Fab2 bsAb (Fig. 1). The IgG–Fab2 format was initially developed to construct multivalent mono-specific antibodies16. The heavy chain-light chain-pairing problem, caused by the similarity of two different light chains, humpers its construction by the general recombinant expression method although IgG–Fab2 is an interesting format for bsAb. Thus, the use of a common light chain17 or exchanging one light chain with one of the VH-CH1 portions, FIT-Ig, was previously reported to overcome the mispairing issue18,19. Obtaining the common light chain is a cumbersome process and the FIT-Ig production potentially results in undesired Fab formation although these techniques are interesting. In this study, we report the PTS-based method for the IgG–Fab2 bsAb production by ligating the separately prepared IgG portion and the Fab portion. The heavy chain/light chain-pairing problem was avoided because the IgG part and Fab parts were separately expressed. Here, we demonstrate the construction of IgG–Fab2 bsAb, which binds to the Her2 and CD3 antigens.

Reaction scheme of IgG–Fab2 construction via PTS reaction.
Results and discussion
Design of IgG and Fab parts for IgG–Fab2 bsAb
We designed intein-C fused to the N-terminus of the heavy chain of an IgG and intein-N fused to the C-terminus of the CH1 domain of a Fab to construct the IgG–Fab2 bsAb molecule by the split-intein–mediated ligation (Fig. 1). This process can separately express, purify, and connect these two portions by the split intein–mediated PTS reaction (Fig. 1). We chose Cfa DnaE split intein for this purpose, which is widely used for several protein engineering applications, including antibody labeling20 and bsAb constructions21. An anti-CD3 (M291) IgG22 and an anti-Her2 (trastuzumab)23 Fab were selected to construct IgG–Fab2. The anti-Her2/anti-CD3 bsAb is one of the popular constructs as a therapeutics for breast cancer treatment24,25,26. A VHH (Ia1; an anti-EGFR VHH)27 was fused to the N-terminus of intein-C of Cfa intein (CfaC) to improve the expression. The intein-N of Cfa intein (CfaN) was fused to the C-terminus of the CH1 domain of the trastuzumab Fab.
The PTS reaction for IgG–Fab2 (Her2/CD3)
CfaC-IgG (anti-CD3) and Fab-CfaN (anti-Her2) were expressed by a mammalian cell expression system and purified by protein A and Ni–NTA columns for CfaC-IgG and Fab-CfaN, respectively. Figure 2a, b show an SDS–polyacrylamide gel-electrophoresis (SDS-PAGE) image of protein purification. Non-reduced SDS-PAGE showed that interchain disulfide bonds were formed for both molecules shown by single bands at elute lanes (182.6 kDa for CfaC-IgG and 60.2 kDa for Fab-CfaN). The purified proteins were then mixed to perform the PTS reaction with a 1:2 molar ratio of CfaC-IgG and Fab-CfaN because there are two reaction sites and thus the reaction point for the PTS reaction is equimolar by mixing with a 1:2 molar ratio. A new band at high molecular weight appears after 17 h that corresponds to the ligated product of VH-CH1 of anti-Her2 and heavy chain of anti-CD3 (73.7 kDa; red arrow in Fig. 2c). Approximately 50% of the band of VHH-CfaC-Hchain (black arrow in Fig. 3a) remains after 24 h reaction although the ligation reaction proceeded. A band corresponds to the heavy chain cleaved at the C-terminal side of CfaC because the side reaction of the PTS reaction was observed in addition to the imperfect reaction. Such an undesired side reaction was commonly observed in the split intein-mediated PTS reaction. We examined the sequence around the intein reaction site to identify the causative factor for the low reaction yield and side cleavage reaction, and we revealed a possible glycosylation site at the extein portion at the C-terminal side of CfaC (Fig. 3a). The NheI cloning site introduced a “ ~ AS ~ ” sequence directly before the VH gene (anti-CD3), and the N-linked glycosylation consensus sequence formed, Asn-X-Ser/Thr28. Thus, we speculated that the possible N-linked glycosylation at the Ser portion caused the low reaction yield and side cleavage reaction.

Expression, purification, and ligation of CfaC-IgG and Fab-CfaN. (a), and (b) reduced and non-reduced SDS-PAGE for purification of CfaC-IgG and Fab-CfaN using a protein A column. (a) 1: culture medium, 2: pre-column, 3: flowthrough, 4: wash, 5: elute. M: maker. (b) 1: pre-column, 2: flowthrough, 3: wash, 4: elute. M: molecular-weight maker. (c) SDS-PAGE results for the PTS reactions between CfaC-IgG and Fab-CfaN. A top black arrow indicates unreacted CfaC-heavy chain. Red arrow indicates bands for the ligated product (VH-CH1-H chain). A blue arrow indicates heavy chain generated by the uncontrolled cleavage at the C-terminal side of CfaC.

Expression, purification, and ligation of CfaC-IgG-ΔS. (a) Domain order of heavy chain of VHH-CfaC-Hchain and VHH-CfaC-Hchain-ΔS. Amino acid sequence between CfaC and VH are shown. (b) Reduced and non-reduced SDS-PAGE for purification of CfaC-IgG-ΔS using Ni–NTA column. 1: pre-column, 2: flowthrough, 3: wash, 4: elute. M: molecular-weight maker. (c) SDS-PAGE result for the PTS reactions between CfaC-IgG-ΔS and Fab-CfaN. (d) SDS-PAGE result of the elution fraction (E1 and E2) of anion exchange column. The reaction mixture of the PTS reaction between CfaC-IgG-ΔS and Fab-CfaN was applied to the column. E1 and E2: Elution fractions, M: molecular-weight maker. Red arrows indicate bands for the ligated product (VH-CH1-H-chain). Blue arrows indicate heavy chain generated by the uncontrolled cleavage at the C-terminal side of CfaC.
Therefore, we constructed an expression vector for the Ser deletion mutant at the glycosylation site, termed VHH-CfaC-Hchain-ΔS (Fig. 3a). We expressed and purified CfaC-IgG-ΔS (anti-CD3) similarly with CfaC-IgG (Fig. 3b). SDS-PAGE showed that the CfaC-IgG-ΔS was purified after the protein A column purification with a single band in the non-reduced condition (Fig. 3b; 182.4 kDa).
Then, we performed the PTS reaction between CfaC-IgG-ΔS and Fab-CfaN (Fig. 3c). The concentration effect of reducing reagent TCEP (Tris(2-carboxyethyl)phosphine)) during the PTS reaction was evaluated by performing 1 mM and 10 mM TCEP conditions (Fig. 3c). Both TCEP conditions demonstrated no significant difference of the reaction yield. The use of the serine deletion mutant (CfaC-IgG-ΔS) showed the reduction of the side-reaction product of the uncontrolled cleavage and the improvement of the ligation yield compared with the original CfaC-IgG and Fab-CfaN reaction. Although we set the reaction stoichiometry 1:2 for CfaC-IgG-ΔS and Fab-CfaN, the band for the Fab-CfaN remained after the ligation reaction while the band for CfaC-H-chain mostly converted to the ligated product, VH-CH1-H-chain (Fig. 3c). We speculated that the reason for the residual reactant of Fab-CfaN after the ligation reaction could be a result of the amount excess of the Fab-CfaN compared with CfaC-IgG-ΔS because of the inaccuracy of concentration determination using UV. The reaction mixture was applied to the Ni–NTA column to remove the unreacted Fab-CfaN then the flowthrough fraction was applied to an anion-exchange chromatography column. The SDS-PAGE showed the purified target bsAb molecule with a trace amount of the cleaved side product (Fig. 3d). Hereafter, we termed the target bsAb molecule with the ΔS mutation as IgG–Fab2(Her2/CD3).
Binding activity of IgG–Fab2(Her2/CD3)
The binding activity of the constructed IgG–Fab2(Her2/CD3) was evaluated. First, we observed the IgG–Fab2 binding to Her2-positive cells by fluorescent microscopy (Fig. 4a). A clear fluorescence was obtained on the cell surface for the sample treated with bsAb compared with the fluorescent microscopy image without adding bsAb. The fluorescence is emitted from a FITC-labeled anti-Fc antibody, and the anti-Her2 Fab portion locates the N-terminal side of IgG–Fab2. Thus, the fluorescent image indicates not only the binding activity to the Her2 antigen but also the correct structure formation of the Fab (Her2)-Fab (CD3)-Fc structure (Figs. 1, 4a). Then, we evaluated the binding activity by flow cytometry using Her2-positive cells and CD3-positive cells, and the IgG–Fab2(Her2/CD3) bsAb demonstrated a binding activity toward both cells (Fig. 4b). These results revealed the binding activities of IgG–Fab2(Her2/CD3) toward Her2 and CD3.

(a) Fluorescent microscopy image of SK-BR-3 cells. Top panels: fluorescent images. Bottom panel: bright field images of the same observation areas of the fluorescent images. The pictures indicate 100-μm scale bars. Antibody combinations for fluorescence imaging is illustrated on the right. (b) Flowcytometry analyses using Her2 positive (SK-BR-3) or CD3 positive (HPB-ALL) cells. Black: negative control (w/o antibody). Blue: positive control using anti-Her2 or anti-CD3 antibodies. Red: IgG–Fab2. (c) In vitro cytotoxicity assay. Target cell: Her2+ SK-BR-3 cell. Effector cell: LAK T-cell. E/T ratio = 5. Standard deviations of the determined cytotoxicity values are indicated with error bars (n = 4).
Cytotoxic activity of IgG–Fab2(Her2/CD3) mediated by activated T cells
An in vitro cytotoxicity assay measured the cancer cell-killing activity of IgG–Fab2(Her2/CD3) mediated by activated T cells (lymphokine-activated killer cells with the T-cell phenotype [T-LAK]). The Her2-positive SK-BR-3 cells were incubated with activated T cells containing different IgG–Fab2(Her2/CD3) concentrations (Fig. 4c). The effector cells/target cells (E/T) ratio was set to 5. Over 80% cell killing was observed at a 1 pM concentration of bsAb. This indicates that the constructed IgG–Fab2(Her2/CD3) molecule simultaneously bridges an SK-BR-3 cell and an activated T cell through the binding of the Her2 and CD3 antigens. It is known that the Her2/CD3 antibody induced T-cell activation through cytokine production, and we expected a similar mechanism should be induced by IgG–Fab2(Her2/CD3) in our cytotoxicity experiment.
Conclusions
An advantage of the IgG–Fab2 bsAb over the general IgG type bsAb is the bivalency for the two targets. Because of the bivalency, the IgG–Fab2 bsAb can bind to each target strongly through the avidity effect, which may contribute to better therapeutic outcomes. Here, we demonstrated the novel bsAb construction method with the format of IgG–Fab2 using the PTS reaction to achieve a specific pairing between the heavy chain and the light chain of two Fab parts. Previous studies used the common light chain17 or connecting light chain to the N-terminus of IgG heavy chain, FIT-Ig18,19, to construct IgG–Fab2 type bsAb. Obtaining the common light chain is a cumbersome process and FIT-Ig production potentially generates mispaired byproduct Fab. Our method can prevent such production problems by separating the IgG part and the Fab part productions. Additionally, our method can be utilized for screening tasks to evaluate several IgG and Fab part combinations. Any combinations can be generated by mixing CfaC-IgG and Fab-CfaN, once several clones of CfaC-IgG and Fab-CfaN are constructed. The use of the split intein–based protein ligation method for bsAb production will expand our bsAb construction strategy, and highly efficient cancer-treating drugs will be obtained based on this technology.
Materials and methods
Expression and purification of proteins
The Expi293 mammalian cell expression system (Thermo Fisher Scientific, MA USA) with HE400 medium (GMEP, Fukuoka, Japan) was used to produce recombinant proteins. Expression vectors of VHH-CfaC fused heavy chain gene and light chain gene of M291 antibody were simultaneously transfected to the cell using PEI MAX reagent (Polyscience Inc., PA USA) for CfaC-IgG expression. Expression vectors of CfaN fused VH-CH1 gene and the light chain gene of trastuzumab for Fab-CfaN were transfected similarly for CfaC-IgG expression. The supernatants were collected and applied to the purification columns 7 days after culture. Protein A column (UNOsphere SUPrA, Biorad, CA USA) was used for the CfaC-IgG purification and Ni–NTA column (FUJIFILM Wako chemicals, Osaka, Japan) for the Fab-CfaN purification.
PTS reaction
Purified CfaC-IgG and Fab-CfaN samples were mixed in a PTS ligation buffer (50 mM HEPES, pH: 7.0, 200 mM of NaCl, 1 or 10 mM TCEP) at 37 °C. The final concentrations of CfaC-IgG and Fab-CfaN were 2 μM and 4 μM, respectively. The reaction mixture was subjected to the Ni–NTA column and the flowthrough fraction was collected after 3 h of the ligation reaction. AKTA start chromatography system with a 5 mL of HiPrep Q HP column (Cytiva, MA USA) was used for anion-exchange chromatography.
Flow cytometry
For 15 min on ice, 106 cells of SK-BR-3 (Her2+) (obtained from ATCC) or HPB-ALL (CD3+) (obtained from Cell resource center for biomedical research, Tohoku university; ID: TKG 0199) were incubated with 30 nM of IgG–Fab2 in phosphate-buffered saline (PBS). An anti-human Fc FITC conjugate antibody (Sigma-Aldrich, MO USA) was added after washing the cells with PBS twice. PBS-suspended cells were used for the analysis using an RF-500 flow cytometry machine (Sysmex, Kobe, Japan) after washing the cells with PBS twice. The analyses used 100 μL of cell suspensions for each measurement. FCSalyzer software (Souceforge; https://sourceforge.net/projects/fcsalyzer/) was used to analyze the obtained data. As positive controls, trastuzumab (Chugai Pharmaceutical Co., Ltd, Tokyo, Japan) and anti-CD3 FITC (Proteintech Group, Inc., IL, USA) were used for SK-BR3 and HPB-ALL, respectively.
Fluorescent microscopy
SK-BR-3 cells were cultured on a 24-well cell culture dish in Dulbecco’s Modified Eagle Medium medium with 10% fetal bovine serum. IgG–Fab2 at 30 nM in PBS was added and incubated for 20 min after removing the medium. Then, an anti-human Fc FITC conjugate antibody (Sigma-Aldrich, MO USA) was added after washing the wells with PBS twice. Cells were observed by ZOE fluorescent imager (Biorad, CA USA) after washing the wells. No antibody was added for the control cells.
In vitro cytotoxicity assay
T-LAK cells were induced as described in the literature29. Peripheral blood mononuclear cells (CTL-UP1 uncharacterized PBMC) were cultured for 48 h at a density of 1 × 106 cells/mL in a medium supplemented with 100 IU/mL of recombinant human interleukin 2 (Shionogi Pharmaceutical Co.) in a culture flask (A/S Nunc) pre-coated with anti-CD3 monoclonal antibody (10 μg/mL). The in vitro growth inhibition of cancer cells using Her2 positive SK-BR-3 cells was assayed using an MTS assay kit (CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay; Promega) with the E/T ratio set to 5, as reported in the literature29.
Data availability
All raw image data used for SDS-PAGE and fluorescent microscopy are included in the supplementary information file. The other datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Ma, J. et al. Bispecific antibodies: from research to clinical application. Front. Immunol. https://doi.org/10.3389/fimmu.2021.626616 (2021).
Wu, Y., Yi, M., Zhu, S., Wang, H. & Wu, K. Recent advances and challenges of bispecific antibodies in solid tumors. Exp. Hematol. Oncol. 10, 56. https://doi.org/10.1186/s40164-021-00250-1 (2021).
Kontermann, R. E. & Brinkmann, U. Bispecific antibodies. Drug Discov. Today. 20, 838–847. https://doi.org/10.1016/j.drudis.2015.02.008 (2015).
Brinkmann, U. & Kontermann, R. E. The making of bispecific antibodies. MAbs 9, 182–212. https://doi.org/10.1080/19420862.2016.1268307 (2017).
Kitazawa, T. et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat. Med. 18, 1570–1574. https://doi.org/10.1038/nm.2942 (2012).
Ridgway, J. B. B., Presta, L. G. & Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. Des. Sel. 9, 617–621. https://doi.org/10.1093/protein/9.7.617 (1996).
Schaefer, W. et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. 108, 11187–11192. https://doi.org/10.1073/pnas.1019002108 (2011).
Wang, H., Wang, L., Zhong, B. & Dai, Z. Protein splicing of inteins: a powerful tool in synthetic biology. Front. Bioeng. Biotechnol. https://doi.org/10.3389/fbioe.2022.810180 (2022).
Shah, N. H. & Muir, T. W. Inteins: Nature’s gift to protein chemists. Chem Sci. 5, 446–461. https://doi.org/10.1039/C3SC52951G (2014).
Li, Y. Split-inteins and their bioapplications. Biotechnol. Lett. 37, 2121–2137. https://doi.org/10.1007/s10529-015-1905-2 (2015).
Shibuya, Y. et al. Generation of camelid VHH bispecific constructs via in-cell intein-mediated protein trans-splicing. Protein Eng. Des. Sel. 30, 1–7. https://doi.org/10.1093/protein/gzw057 (2016).
Hemmi, S. et al. Construction of a circularly connected VHH bispecific antibody (cyclobody) for the desirable positioning of antigen-binding sites. Biochem. Biophys. Res. Commun. https://doi.org/10.1016/j.bbrc.2019.12.018 (2020).
Han, L. et al. Efficient generation of bispecific IgG antibodies by split intein mediated protein trans-splicing system. Sci. Rep. 7, 1–11. https://doi.org/10.1038/s41598-017-08641-3 (2017).
Han, L. et al. Naturally split intein Npu DnaE mediated rapid generation of bispecific IgG antibodies. Methods 154, 32–37. https://doi.org/10.1016/j.ymeth.2018.10.001 (2019).
Sun, R. et al. A rational designed novel bispecific antibody for the treatment of GBM. Biomedicines. https://doi.org/10.3390/biomedicines9060640 (2021).
Miller, K. et al. Design, construction, and in vitro analyses of multivalent antibodies. J. Immunol. 170, 4854–4861. https://doi.org/10.4049/jimmunol.170.9.4854 (2003).
Zhao, J. et al. A strategy for the efficient construction of anti-PD1-based bispecific antibodies with desired IgG-like properties. MAbs 14, 2044435. https://doi.org/10.1080/19420862.2022.2044435 (2022).
Gong, S., Ren, F., Wu, D., Wu, X. & Wu, C. Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets. MAbs 9, 1118–1128. https://doi.org/10.1080/19420862.2017.1345401 (2017).
Gong, S. & Wu, C. Generation of Fabs-in-tandem immunoglobulin molecules for dual-specific targeting. Methods 154, 87–92. https://doi.org/10.1016/j.ymeth.2018.07.014 (2019).
Stevens, A. J. et al. Design of a split intein with exceptional protein splicing activity. J. Am. Chem. Soc. 138, 2162–2165. https://doi.org/10.1021/jacs.5b13528 (2016).
Akiba, H. et al. Production of IgG1-based bispecific antibody without extra cysteine residue via intein-mediated protein trans-splicing. Sci. Rep. 11, 19411. https://doi.org/10.1038/s41598-021-98855-3 (2021).
Cole, M. S., Stellrecht, K. E., Shi, J. D., Homola, M., Hsu, D.-H., Anasetti, C., Vasquez, M., Tso, J. Y. HuM291, A humanized anti-cd3 antibody, is immunosuppressive to t cells while exhibiting reduced mitogenicity in VITRO1, Transplantation. 68 (1999). https://journals.lww.com/transplantjournal/Fulltext/1999/08270/HuM291,_A_HUMANIZED_ANTI_CD3_ANTIBODY,_IS.20.aspx. 563–571.
Carter, P. et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. 89, 4285–4289. https://doi.org/10.1073/pnas.89.10.4285 (1992).
Yu, S. et al. A novel asymmetrical anti-HER2/CD3 bispecific antibody exhibits potent cytotoxicity for HER2-positive tumor cells. J. Exp. Clin. Cancer Res. 38, 355. https://doi.org/10.1186/s13046-019-1354-1 (2019).
Zhou, Y. et al. Fully human HER2/cluster of differentiation 3 bispecific antibody triggers potent and specific cytotoxicity of T lymphocytes against breast cancer. Mol Med Rep. 12, 147–154. https://doi.org/10.3892/mmr.2015.3441 (2015).
Staflin, K. et al. Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI Insight. https://doi.org/10.1172/jci.insight.133757 (2020).
Roovers, R. C. et al. Efficient inhibition of EGFR signalling and of tumour growth by antagonistic anti-EGFR Nanobodies. Cancer Immunol. Immunother. 56, 303–317. https://doi.org/10.1007/s00262-006-0180-4 (2006).
Mellquist, J. L., Kasturi, L., Spitalnik, S. L. & Shakin-Eshleman, S. H. The amino acid following an Asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37, 6833–6837. https://doi.org/10.1021/bi972217k (1998).
Asano, R. et al. Highly effective recombinant format of a humanized IgG-like bispecific antibody for cancer immunotherapy with retargeting of lymphocytes to tumor cells*. J. Biol. Chem. 282, 27659–27665. https://doi.org/10.1074/jbc.M704719200 (2007).
Acknowledgements
This research was supported by AMED under Grant Number 21ak0101143h0002, the research fund from Yamagata University Research Institute, Grant-in-Aid from JSPS for Scientific Research (KAKENHI Category B 19H03511 and 22H02915), JST PRESTO Grant Number JPMJPR21AD, Takahashi Industrial and Economic Research Foundation, Hokuto Foundation for Bioscience, Japan Foundation for Applied Enzymology, and Astellas Foundation for Research on Metabolic Disorders. We thank Dr. Shota Shiga for the support of protein expression. The cell illustration used in the Fig. 4a was obtained from IRASUTOYA website (https://www.irasutoya.com).
Author information
Authors and Affiliations
Contributions
K.M. and R.Y. wrote the main manuscript text. K.M. designed the experiments. R.Y., I.N., I.K., R.A., and T.N. performed the experiments. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamada, R., Nakahara, I., Kumagai, I. et al. Construction of IgG–Fab2 bispecific antibody via intein-mediated protein trans-splicing reaction. Sci Rep 13, 15961 (2023). https://doi.org/10.1038/s41598-023-43110-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-43110-0
- Springer Nature Limited