
Abstract
Fullerene free organic chromophores are widely utilized to improve the efficacy of photovoltaic materials. Herein, we designed D-π-A-π-D form chromophores (TAZD1-TAZD5) via end-capped redistribution of donor moieties by keeping the same π-bridge and central acceptor unit for organic solar cells (OSCs). To analyze the photovoltaic characteristics of these derivatives, DFT estimations were accomplished at B3LYP/6–311 G (d,p) functional. Different investigations like frontier molecular orbital (FMO), absorption spectra (UV–Vis), density of states (DOS), binding energy (Eb), open circuit voltage (Voc), and transition density matrix (TDMs) were performed to examine the optical, photophysical and electronic characteristics of afore-mentioned chromophores. A suitable band gap (∆E = 2.723–2.659 eV) with larger bathochromic shift (λmax = 554.218–543.261 nm in acetonitrile) was seen in TAZD1-TAZD5. An effective charge transference from donor to acceptor via spacer was observed by FMO analysis which further supported by DOS and TDM. Further, lower binding energy values also supported the higher exciton dissociation and greater CT in TAZD1-TAZD5. Among all the designed chromophores, TAZD5 exhibited the narrowest Egap (2.659 eV) and maximum red-shifted absorption in solvent as well as gas phase i.e. 554.218 nm and 533.219 nm, respectively which perhaps as a result of the phenothiazine-based donor group (MPT). In a nutshell, all the tailored chromophores can be considered as efficient compounds for promising OSCs with a good Voc response, interestingly, TAZD5 is found to be excellent chromophores as compared to all these designed compounds.
Similar content being viewed by others

Introduction
Solar energy has turn into a promising energy source which involve the phenomena of photoelectric effect and overcome the elevating power crisis as the sunlight is non-exhaustible, non-polluting and widely available1. Among promising and cost-effective substitutes for future sustainable energy are OSCs due to their exceptional advantages. OSCs have been proved as an effective scheme for light manipulation, which is capable of improving the light absorption process2. In this way, larger photocurrent is produced due to the light scattering. The photovoltaic innovation has gained consideration of academic along with industrial communities’ decades-long. The photovoltaic (PV) silicon-based solar cells were known as the foremost and popular energy gadgets owing to their remarkable eco-friendly nature, proficiency and low cost3. Crystalline silicon solar cells have the greatest manufacturing history with over 60 years of progress and their efficiency has increased to above 25% because of the improvements in their architecture4,5. However, they are fragile and possess non-tunable energy levels due to which the organic-based solar cells have become more popular in recent years. They possess various important characteristics such as; (i) light weight; (ii) low-cost materials; (iii) tunable energy levels; (iv) mechanical flexibility; (v) variety of structural modulations and (vi) compatibility with large manufacturing6,7,8,9. Furthermore, organic–inorganic perovskite photovoltaics have gained impressive power conversion efficiency (PCE) of 22.1% because of remarkable characteristics like intense absorption spectrum, greater charge mobility and long diffusion length of charges10,11,12. Another class of OSCs namely dye-sensitized solar cells (DSSC) has also captured significant attention owing to their stability besides tunable visual characteristics e.g. transparency and color13,14,15. Components in DSSCs, such as the dye catches special attention on because of light conversion capability to electricity supported by photoexcitation13. The DSSCs that are metal free are of great efficiency and advantageous because of their synthesis, purification and many optical properties by easy chemical modifications16. Although in beginning efforts to commercialize organic photovoltaics (OPVs) were difficult as a result of lower power conversion efficiency (PCE) than the approximated market viability of 15%17. Besides, the photovoltaic domain is developed with other very compelling substances like OSCs based on fullerene derivatives namely PC71BM, ICBA, and PC61BM. Organic solar cells (OSCs) keeping fascinating characteristics like simple processability, light weight, mechanical flexibility, high formulation area and ease made them significant alternative tools18,19,20,21. Due to the exceptional electronic and structural properties of fullerene-based OSCs, they have been widely examined since 198522. Low reorganization energy of excitons23,24, elevated electron affinity25, and high mobility of electrons26,27 are some fascinating and distinctive properties of fullerene based OSCs. There also exhibit certain drawbacks in fullerene acceptors (FAs) which include less absorption in visible and near IR regions, poor photochemical and thermal stability28, non-tunable LUMO energies29 and less sunshine assimilation which impelled the researchers to search for some more strongly absorbing analogues30. Therefore, non-fullerene acceptors (NFAs) are utilized in the OSCs in place of FA owing to their flexible nature, higher fabrication area and wide tunability of their energy levels31. The non-fullerene small molecule acceptors (NF-SMAs) are regarded as remarkable constituents for proficient OSCs32. A rapid improvement in PCE is observed (~ 18–19%) in NFAs benefiting from years of research on fullerene-based BHJ materials33,34. The obvious increase in fill factor (FF) as well as short-circuit current (Jsc) is also seen in NFA-based devices with greater open-circuit voltage (Voc) in comparison to their fullerene counterparts35. Literature is flooded with many examples in which fullerene free donor or acceptors are extensively utilized to improve the efficiencies of photovoltaic materials36,37,38,39. Keeping in view the importance of NF organic systems, herein, we have tried to design benzodithiophene based organic systems for high efficacy photovoltaic devices. For this purpose, we take a synthesized X94FIC40 fullerene free A-π-A-π-A architecture acceptor nature molecule and designed sequence of donor type D-π-A-π-D configured TAZD1-TAZD5 chromophores by structural modification of end capped acceptors with efficient donor moieties. Perhaps, it is first ever systematic comparative study of impact of NF chromophores with five-, six- and seven-membered rings on the electrochemical as well as photophysical characteristics. To check the influence of donor groups on photovoltaic characteristics, DFT method was employed and has anticipated their significance in OSCs.
Computational procedure
Gaussian 09 package41 was exploited to understand the photovoltaic response of benzodithiophene based organic (TAZD1-TAZD5). To choose the suitable functional for current investigation, a relative investigation of X94FIC λmax outcomes among several TD-DFT functionals and experimental results was performed. For this purpose, the reference chromophore X94FIC was subjected to geometry optimization using four different functionals, including B3LYP42, M0643, MPW1PW9144 and ɷB97XD45 in acetonitrile solvent as range-separated functionals estimates HOMO–LUMO gaps and excited-state energies better46,47,48,49. Then these optimized geometries were applied to execute UV–Vis analysis in acetonitrile solvent and 818.864, 736.686, 676.142, and 495.461 nm values of λmax were obtained at aforesaid functionals, respectively. The λmax values of X94FIC obtained using these functionals were compared to the experimentally determined maximum absorption value of 783 nm39 of X94FIC chromophore. At B3LYP/6-311G (d,p) functional, closed harmony was seen with experimental results. Moreover, we also compared the band gap values of X94FIC calculated at aforesaid functional of TD-DFT (1.774, 2.142, 2.032 and 4.971 eV, respectively) with experimental ΔE value (1.41 eV)39 and interesting, good harmony with experimental results was seen at B3LYP, hence, this functional was selected for this study. First of all, structures of designed systems were optimized at B3LYP/6-311G(d,p) to get true minima geometries in acetonitrile solvent. The absence of any imaginary frequency specified that structures were at true minima potential energy surface. After the successful optimization of geometries, different analyses; FMOs, DOS, UV–Vis, Voc, Eb and TDMs were attained to inspect the optical, electronic and photophysical characteristics of afore-mentioned chromophores at B3LYP/6-311G (d,p) level of DFT/TDDFT in acetonitrile solvent. Nevertheless, in order to understand the effect of different media on UV–Vis properties, we performed absorption analysis in gas and acetonitrile at foresaid functional. For the extraction of data from output files, Gauss View 5.0 program50, Avogadro51, Chemcraft52, PyMOlyze 2.053, and Origin54 software were utilized and the data was recorded in the form of graphs and tables.
Results and discussion
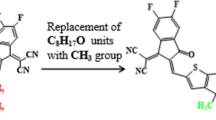
In current era, fullerene free-organic systems (FF-OSs) with some special architectures like D-π-A-π-D, A-π-A-π-A55, A-D-A56 and A-π-A gain significant importance in improving the efficiency of solar cell materials57,58,59. Therefore, in current study we formulated a range of donor nature chromophores (TAZD1-TAZD5) with D-π-A-π-D framework from a synthesized system X94FIC (A-π-A-π-A)40 by molecular replacement at the terminals with efficient azacycle donor moieties (see Fig. 2). First of all, we designed TAZD1 from X94FIC by replacing its terminal acceptors with four rings azacycle donor unit (9-phenyl-9H-carbazole) keeping the central ‘π-linker’ and ‘A’ same as shown in Fig. 1. After that TAZD2-TAZD5 are designed by replacing the four member azacycle donor rings unit with three, five and six member ring azacycle donor unit as exhibited in Fig. 2. The optimized structures of aforesaid systems are displayed in Fig. 3 while their Chemdraw structures are shown in Fig. S2, however, their IUPAC names are tabulated in Table S1. The utilized azacycle donor moieties: 9-phenyl-9H-carbazole (THC), 5-phenyl-10,11-dihydro-5H-dibenzo[b,f]azepine (THA), 5-phenyl-5H-dibenzo[b,f]azepine (TBA), 9,9,10-triphenyl-9,10-dihydroacridine (PTH), 3-methyl-10H-phenothiazine (MPT) and their structures can be seen in Fig. S1. We have calculated different parameters like FMOs, DOS, UV–Vis, Voc, Eb and TDMs of all the studied compounds. The modifications in derivatives with their respective donor moieties might prove as a significant step towards introducing efficient solar cells.

Modification of X94FIC into TAZD1 by replacing terminal acceptors with four ring azacycle donor unit, drawn utilizing ChemDraw software (https://chemistrydocs.com/chemdraw-pro-8-0/).

The sketch map of the TAZD1-TAZD5 compounds.

Optimized structures of TAZD1-TAZD5. Pictures are created by GaussView 5.0 and Gaussian 09 version D.01 (https://gaussian.com/g09citation/).
Frontier molecular orbitals (FMOs) analysis
The optoelectronic properties i.e. charge transfer, electronic features, reactivity, chemical stability and molecular interactions60 are investigated via utilizing FMOs61,62,63. The band gap of HOMO/LUMO orbitals is closely linked to these parameters64. As HOMO is the electronically filled highest orbital, so it is considered as an electron contributor, whereas, LUMO is considered to be an electron acceptor as it is an empty or unfilled orbital. Molecules having high energy gap (Egap) values are hard, because they resist changes in electronic configurations, resulting in lower reactivity and increased kinetic stability. Conversely, the compounds with low energy gap are attributed as soft molecules owing to their less stability and higher reactivity. These compounds reveal strong intramolecular charge transfer (ICT) possibilities due to their highly polarized nature and are extremely efficient molecules in the production of solar cell materials65. In addition, the HOMO–LUMO band difference is important in calculating a molecule’s total Voc and Eb64. So, FMO analysis is used to compute EHOMO, ELUMO and Egap of TAZD1-TAZD5 and the outcomes are exhibited in Table 1. The pictographs showing charge transference among orbitals are depicted in Fig. 4.

Pictographic representation of HOMOs and LUMOs of TAZD1-TAZD5, units are in eV. Illustrations are made using Avogadro software, Version 1.2.0. (http://avogadro.cc/).
The above table reveals HOMO energy values for TAZD1, TAZD2, TAZD3, TAZD4 and TAZD5 as − 5.177, − 5.185, − 5.185, − 5.199 and − 5.104 eV while energies of LUMO are − 2.469, − 2.467, − 2.463, − 2.476 and − 2.445 eV, correspondingly. Egap is used to calculate molecules conductivity and net charge transfer66,67. The Egap values of designed chromophores (TAZD1-TAZD5) are revealed as 2.708, 2.718, 2.722, 2.723 and 2.659 eV, correspondingly. Highest energy difference between HOMO and LUMO (2.723 eV) is observed in TAZD4 among all the other derivatives which may be due to the 9,9-diphenyl-10-(p-tolyl)-9,10-dihydroacridine (PTH) donor moiety. The Egap value is abridged to 2.722 eV in TAZD3 due to the substitution of PTH with 5-(p-tolyl)-5H-dibenzo[b,f]azepine (TBA) donor moiety which may be due to the decreased in hindrance in charge transfer of TBA as compared to that of PTH. Furthermore, the replacement of donor of TAZD3 i.e. TBA with 5-(p-tolyl)-10,11-dihydro-5H-dibenzo[b,f]azepine (THA) in TAZD2 resulted in further reduction of bandgap to 2.718 eV owing to the enhancement in conjugation in the newly introduced donor moiety. TAZD1 is designed via replacing THA with carbazole containing donor moiety i.e. 9-(p-tolyl)-9H-carbazole (THC) in which nitrogen atom of carbazole exhibit the electron donating capability. As a result of this, the energy difference is lessen to 2.708 eV in TAZD1 because of the enhanced push pull mechanism. Moreover, TAZD5 has exhibited minimum energy gap as compared to all of the studied chromophores owing to the use of phenothiazine-based donor moiety such as 3-methyl-10H-phenothiazine (MPT) instead of THC in TAZD1. The extra electron-rich sulphur atom in phenothiazine might give an improved electron-donating capacity compared to donors that just include nitrogen atoms, like carbazole. Overall, the band gap descending order in the studied compounds is; TAZD4 > TAZD3 > TAZD2 > TAZD1 > TAZD5.
The electron density in HOMO of TAZD1-TAZD4 is predominantly located over the center ‘A’ and ‘π-spacer’ parts of the organic systems and minor over some atoms of donor, while in TAZD5 the electron density is dispersed on entire system. For LUMO, the electron density is majorly located over π-bridge and core acceptor in TAZD1-TAZD5. Among all the investigated compounds, TAZD5 is found to be the appropriate candidate for future OSCs with enhanced PV behavior due to less energy band gap and adequate charges transition from terminal donors to center acceptor (see Fig. 4).
UV–Vis analysis
UV–Vis analysis is significant to investigate the possibility of ICT, kind of configurations of transitions and electronic transitions in a compound. To calculate the absorption spectra of the excited states, the TD-DFT calculations are accomplished in gas and acetonitrile solvent. The observed oscillator strength (fos), transition energy (E), transition type and maximum absorption wavelength (λmax) are shown in Table 2 as well as Table 3 and other transitions are represented in Tables S2–S11, whereas the absorption spectra of studied compounds TAZD1-TAZD5 is displayed in Fig. 5.

Absorption spectra of TAZD1-TAZD5 in two different media. The UV–Vis graphs are illustrated utilizing Origin Pro 8.5 version (https://www.originlab.com/).
In solvent (acetonitrile), all the investigated compounds have revealed maximum absorbance in visible spectrum (Fig. 5). The designed molecules (TAZD1-TAZD5) exhibit absorption range from 543.361 to 554.218 nm in acetonitrile. In solvent phase, λmax values are found to be more red-shifted contrary to gas phase because of solvent effect. Furthermore, the absorption spectra of studied compounds (TAZD1-TAZD5) are dominated by π-π interactions68. The polar medium results in the stabilization of π-π* state associated with n-π* characteristics by the use of an efficient electronic state69. This indicates that, in the stabilization of first singlet state, hydrogen bonding and dipole interactions are imperative70 and the molecules exhibit red-shifted absorption as a result of enhancement of solvent polarity.
It is seen that, \(\lambda\)max values are controlled efficiently by end-capped donor moieties which successively drive the red shifted absorption spectra71,72. The absorption band of TAZD4 is noticed at 543.361 nm having 2.282 eV energy of transition, 1.031 fos by exhibiting 98% molecular orbital contribution from HOMO to LUMO. The computed λmax value is shifted towards bathochromic shift in TAZD3 due to the replacement of PTH donor of TAZD4 by TBA so, TAZD3 has exhibited λmax at 544.101 nm, 2.279 eV transition energy, and 0.964 oscillator strength via showing HOMO → LUMO MO contribution of 98%. Furthermore, the substitution of TBA with THA donor moiety resulted in red-shifted absorption of 544.483 nm in TAZD2 along with lower transition energy (2.277 eV) and 1.006 oscillator strength via same MO contributions. Additionally, TAZD1 absorption spectra further shifted towards bathochromic shift (546.427 nm), owing to the deposition of another donor moiety i.e., THC in TAZD1 in the replacement of THA in TAZD2. Finally, the substitution of TTC with MPT donor moiety resulted in maximum red shift of 554.218 nm in TAZD5 due to phenothiazine group in MPT by showing 96% HOMO → LUMO and 2% HOMO-2 → LUMO molecular orbital contribution at 1.005 oscillator strength and minimum transition energy (2.237 eV) owing to its lowest band gap. \(\lambda\)max of all the compounds in acetonitrile solvent is found to be in increasing order as TAZD4 < TAZD3 < TAZD2 < TAZD1 < TAZD5.
In gaseous phase (Table 3), all the entitled compounds have almost exhibited equivalent order as well as characteristics as in solvent phase. The absorption spectrum shifts towards the red shift as the dielectric constant of media enhanced. Therefore, greater bathochromic shift is seen in acetonitrile due to its higher dielectric constant than that of gas phase. Nevertheless, it can be concluded from above discussion that, TAZD5 compound has exhibited maximum absorption wavelength, the lowest transition energy and minimum band gap which implies that, it can be used as an efficient material for photophysical characteristics in non-fullerene OSC materials.
Open circuit voltage
Open circuit voltage (Voc) is another significant study that provides insights into the performance of OSCs i.e. their maximum working capability. The total current that can be produced via any optical system can be estimated by Voc31,55. So, Voc shows direct relation with EHOMO and ELUMO of donor and acceptor molecules, correspondingly. Thus, Voc outcomes of TAZD1-TAZD5 are determined via Eq. (1) proposed by Scharber and his coworkers.
The major purpose of the calculation of Voc is to associate HOMO of the investigated donors with LUMO of PC61BM acceptor which is a well-known acceptor having energy of HOMO = − 6.10 eV and energy of LUMO = − 3.70 eV73 and the results obtained are represented in Table 4.
As Table 4 reveals that the values of Voc for TAZD1-TAZD5 by considering the energy gap of HOMOdonor –LUMOPC61BM are found to be 1.446, 1.454, 1.454, 1.468, and 1.373 V, respectively. Among all, TAZD4 shows maximum results of Voc. The descending order of open circuit voltage of all the studied molecules is: TAZD4 > TAZD2 = TAZD3 > TAZD1 > TAZD5. From literature, we have found that for a significant transference from DHOMO towards ALUMO, the LUMO of acceptor should be at lesser energy level than that of the LUMO of donor molecules46,71. Interestingly, the LUMO of our compounds is higher than the PC61BM. These higher values of Voc elucidate the higher ICT from donor HOMO TAZD1-TAZD5 towards PC61BM which shows highly efficient donating capability of all the studied donors as shown in Fig. 6.

Pictographic representation of Voc of TAZD1-TAZD5 with respect to PC61BM.
Density of states (DOS)
DOS was accomplished to assist FMO analysis and their comparative evaluation demonstrates that both of these are analogs to one another. The DOS graphs are displayed in Fig. 7. DOS are performed to disclose the dissemination of electron density on FMOs with the analysis of percentage composition for TAZD1-TAZD5. It provides useful data related to contribution of donor as well as acceptor in the development of FMOs. The DOS pictographs illustrate the bonding, non-bonding, and antibonding interactions amongst HOMO and LUMO74. The FMO diagrams in the Fig. 7 signify the electronic transitions that demonstrate the intramolecular charge transfer (ICT). In DOS pictographs, the negative values characterize HOMOs while, the positively charged outcomes depict the LUMOs and the difference among their values represents the energy gap on x-axis75.

DOS around HOMO and LUMO of TAZD1-TAZD5. The DOS pictographs are drawn utilizing PyMOlyze 1.1 version.
The maximum density on LUMO is noticed at − 2.5 to 4 eV in all the investigated chromophores (TAZD1-TAZD5), while on HOMO highest density is observed from − 7 to − 12 eV as shown in Fig. 7. In TAZD2, TAZD4 and TAZD5 both HOMO and LUMO have comparable charge densities which depict their equal contribution toward FMOs. In TAZD1-TAZD5, the donor contributes 7.7, 5.3, 4.8, 4.6 and 22.0% to HOMO, whereas to LUMO its participation is 5.5, 5.5, 5.2, 5.9 and 4.0%, respectively. In the same fashion, π-spacer participates 72.5, 74.4, 74.8, 74.8 and 61.7% to HOMO, while 35.0, 35.2, 34.2, 35.9 and 32.6% to LUMO in TAZD1-TAZD5, respectively. Likewise, acceptor shows its percentage participation 19.8, 20.3, 20.4, 20.5 and 16.2% to HOMO in TAZD1-TAZD5, respectively, while 59.5, 59.3, 60.6, 58.2 and 63.4% to LUMO, respectively. Overall, the pattern of electronic charge distribution elucidates the delocalization of charges and large amount of charge transfer has taken place in all the modulated chromophores. Interestingly, all the designed derivatives portray almost alike contributions and the electron density is more prominent on the central unit (π-spacer and A units).
Transition density matrix (TDM) analysis
TDM is considerably utilized for the evaluation of electronic transitions along with their nature for TAZD1-TAZD5 in solvent phase. The study of the charge carriers localization along with delocalization and the interaction between donor and acceptor groups followed by electron–hole delocalization as calculated by TDM analysis76. The role of hydrogen atoms has been neglected because of their small contribution. The TDM heat maps of all the formulated molecules (TAZD1-TAZD5) are presented in Fig. S3. To study the transition of electrons within molecules in detail, we split our compound into fragments i.e. donor (D), π-spacer and acceptor (A).
It has been seen that adequate charge is transmitted out of donor towards acceptor moieties as the electron–hole pair is constituted diagonally on the entire TDM plot and represented by clear red and green spots near the acceptor portion. The electron delocalization is seen in the diagonals of A and π-spacers and very little in the D region. Moreover, the charge coherence and electron–hole pair generation are similarly observed in the off-diagonal portions of TDM heat maps (see Fig. S3).
Hole-electron analysis
Hole-electron analysis is popularly accomplished by utilizing the Multiwfn 3.8 software. It is a very useful method for revealing the nature of electron excitations. Moreover, it offers a deep understanding of all different electron transfer properties77, 78. In this study, hole-electron analysis is performed at B3LYP/6-311G (d,p) to understand the charge transmission in our studied molecules. Figure 8 shows that hole intensity is found maximum at sulphur atom (S15 and S16) of the thiophene ring of π-linker in parent compound (X94FIC) while, the hole intensity is observed at C36 and C38 of the acceptor region. Furthermore, it is also clear from Fig. 8 that electron intensity is found at its peak at sulphur atom (S9) of the acceptor region in compounds TAZD2, TAZD3 and TAZD4 whereas, hole intensity is observed to maximum at sulphur atoms (S15 and S16) of the π-spacer. However, in TAZD5 hole intensity is higher at methyl group of the π-spacer and electron density is intense at nitrogen atoms (N7 and N8) of the acceptor portion. Moreover, electronic cloud is observed to be thick at nitrogen (N7 and N8) and sulphur (S9) atoms of the acceptor in TAZD6 however, hole intensity is maximum at carbon atoms (C12 and C14) of the π-linker. The labeled structures of entitled chromophores without hydrogen atoms are illustrated in Fig. S5. In conclusion, all the designed molecules except TAZD5, are electron type materials as electronic cloud is observed thick at electronic band in contrast to hole intensity at hole band. In TAZD5, hole intensity is found higher at hole band therefore, it is a hole type material.

Graphical representation of hole-electron analysis of investigated compounds.
Exciton binding energy (Eb)
One more consideration to estimate the working proficiency, optoelectronic characteristics, and separation potential is binding energy (Eb)79. The Eq. (2) is used to compute the binding energy of the studied systems.
Here, Eopt represents the least energy that is obtained when an electron moves from S0 (ground state) to S1 (excited state) during the first electronic transition. EH-L signifies the energy difference among HOMO and LUMO whereas, Eb is the binding energy that is obtained by the difference in band gap between molecular orbitals and first singlet exciton energy. Usually, the lower the value of Eb, greater would be the charge separation and current charge density Jsc which results in higher PCE80. The outcomes for the studied compounds calculated in acetonitrile are formulated in Table 5.
According to our obtained results, the values of Eopt decreases progressively in all the designed compounds and is found as minimum in TAZD5 and the same behavior is observed in the HOMO and LUMO energy gap. The binding energy values in TAZD1-TAZD5 are found to be 0.439, 0.441, 0.443, 0.441 and 0.422 eV, correspondingly. The lowermost Eb value of TAZD5 depicts that it has excessive charges that can be separated into isolated charges. It is noted that TAZD5 exhibits high segregation of charges along with high Jsc which indicates that it is the leading candidate to improve the efficiency of organic photovoltaics. Furthermore, Eb data unveil a good agreement with TDM outcomes.
Charge transfer analysis
In order to understand the intermolecular charge transfer between donor and acceptor, a complex is developed between a donor molecule (TAZD5) and acceptor (PC16BM) polymer and FMO is investigated as shown in Fig. S4. For charge transfer analysis, we selected TAZD5 due to its unique properties such as reduced band gap and greater UV–Vis absorption spectra etc. among all fabricated chromophores. According to Fig. S4, in HOMO the charge is located over the TAZD5 donor chromophore and significantly transferred towards the acceptor polymer in LUMO which elucidates the significantly charge transfer from donor towards acceptor.
Conclusion
In a nutshell, through the molecular engineering with azacycle donor moieties in an organic system (X94FIC) fullerene free donor based chromophores (TAZD1-TAZD5) were designed. To comprehend their photophysical properties, the behavior of charge transfer, and structure–activity relationship, various analyses were performed at quantum chemical approach. A reasonable energy gap between LUMO/HOMO (∆E = 2.723–2.659 eV) and significant charge transfer with wider absorption spectra (λmax = 554.218–543.261 nm in acetonitrile) was examined in all non-fullerene donor chromophores. Additionally, the less binding energy outcomes (Eb = 0.422–0.411 eV) in formulated compounds specified higher rate of exciton dissociation that also reinforce the tremendous charge transition out of HOMO towards LUMO as shown by FMOs, DOS and TDMs analyses. Moreover, the Voc values are also determined with regarding to \({\mathrm{HOMO}}_{\mathrm{donor}}-{\mathrm{LUMO}}_{\mathrm{PC}61\mathrm{BM}}\) and interesting data was found with this order; TAZD4 (1.199 V) > TAZD2 (1.185 V) = TAZD3 (1.185 V) > TAZD1(1.177 V) > TAZD5(1.104 V). Consequently, significant photovoltaic materials can be developed by structural tailoring with efficient azacycle donor moieties. Moreover. this study also encourages the experimentalist to synthesize these efficient materials for practical use.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Nozik, A. J. Photoelectrochemistry: Applications to solar energy conversion. Annu. Rev. Phys. Chem. 29, 189–222 (1978).
Yang, D. et al. Tailoring morphology compatibility and device stability by adding PBDTTPD-COOH as third component to fullerene-based polymer solar cells. ACS Appl. Energy Mater. 3, 2604–2613 (2020).
Dusastre, V. Materials for Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group (World Scientific, 2010).
Saga, T. Advances in crystalline silicon solar cell technology for industrial mass production. NPG Asia Mater. 2, 96–102 (2010).
Battaglia, C., Cuevas, A. & De Wolf, S. High-efficiency crystalline silicon solar cells: Status and perspectives. Energy Environ. Sci. 9, 1552–1576 (2016).
Sharma, S., Jain, K. K. & Sharma, A. Solar cells: In research and applications—A review. Mater. Sci. Appl. 6, 1145 (2015).
Li, Y. et al. High efficiency near-infrared and semitransparent non-fullerene acceptor organic photovoltaic cells. J. Am. Chem. Soc. 139, 17114–17119 (2017).
Xu, C. et al. Ternary small molecules organic photovoltaics exhibiting 12.84% efficiency. Nano Energy 66, 104119 (2019).
Zhao, F. et al. Single-junction binary-blend nonfullerene polymer solar cells with 12.1% efficiency. Adv. Mater. 29, 1700144 (2017).
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Burschka, J. et al. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 499, 316–319 (2013).
Snaith, H. J. Perovskites: The emergence of a new era for low-cost, high-efficiency solar cells. J. Phys. Chem. Lett. 4, 3623–3630 (2013).
Hagfeldt, A., Boschloo, G., Sun, L., Kloo, L. & Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 110, 6595–6663 (2010).
Grätzel, M. Conversion of sunlight to electric power by nanocrystalline dye-sensitized solar cells. J. Photochem. Photobiol. A 164, 3–14 (2004).
Mathew, S. et al. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 6, 242–247 (2014).
Madili, N. Theoretical and Experimental Studies of Combination of Synthetic Dye (Indoline d131) and Natural Dye (Lawsone) for Dye Sensitized Solar Cells Applications (NM-AIST, 2019).
Cui, Y. et al. Achieving over 15% efficiency in organic photovoltaic cells via copolymer design. Adv. Mater. 31, 1808356 (2019).
Chen, H.-Y. et al. Polymer solar cells with enhanced open-circuit voltage and efficiency. Nat. Photonics 3, 649–653 (2009).
Liang, Y. et al. Development of new semiconducting polymers for high performance solar cells. J. Am. Chem. Soc. 131, 56–57 (2009).
Zhao, J. et al. Efficient organic solar cells processed from hydrocarbon solvents. Nat. Energy 1, 1–7 (2016).
Li, M. et al. Solution-processed organic tandem solar cells with power conversion efficiencies> 12%. Nat. Photonics 11, 85–90 (2017).
Kroto, H. W., Heath, J. R., O’Brien, S. C., Curl, R. F. & Smalley, R. E. C60: Buckminsterfullerene. Nature 318, 162–163 (1985).
Guldi, D. M., Neta, P. & Asmus, K.-D. Electron-transfer reactions between C60 and radical ions of metalloporphyrins and arenes. J. Phys. Chem. 98, 4617–4621 (1994).
Hiroshi, I. et al. The small reorganization energy of C60 in electron transfer. Chem. Phys. Lett. 263, 545–550 (1996).
Reed, C. A. & Bolskar, R. D. Discrete fulleride anions and fullerenium cations. Chem. Rev. 100, 1075–1120 (2000).
Frankevich, E., Maruyama, Y. & Ogata, H. Mobility of charge carriers in vapor-phase grown C60 single crystal. Chem. Phys. Lett. 214, 39–44 (1993).
Gudaev, O. A., Malinovsky, V. K., Okotrub, A. V. & Shevtsov, Y. V. Charge transfer in fullerene films. Fuller. Sci. Technol. 6, 433–443 (1998).
Distler, A. et al. The effect of PCBM dimerization on the performance of bulk heterojunction solar cells. Adv. Energy Mater. 4, 1300693 (2014).
Bloking, J. T. et al. Comparing the device physics and morphology of polymer solar cells employing fullerenes and non-fullerene acceptors. Adv. Energy Mater. 4, 1301426 (2014).
Khalid, M. et al. First theoretical framework for highly efficient photovoltaic parameters by structural modification with benzothiophene-incorporated acceptors in dithiophene based chromophores. Sci. Rep. 12, 1–15 (2022).
Mehboob, M. Y. et al. Designing N-phenylaniline-triazol configured donor materials with promising optoelectronic properties for high-efficiency solar cells. Comput. Theor. Chem. 1186, 112908 (2020).
Liao, C. et al. Green solvent-processed efficient non-fullerene organic solar cells enabled by low-bandgap copolymer donors with EDOT side chains. J. Mater. Chem. A 7, 716–726 (2019).
Chen, H. et al. Central unit fluorination of non-fullerene acceptors enables highly efficient organic solar cells with over 18% efficiency. Angew. Chem. Int. Ed. 61, e202209580 (2022).
Li, D. et al. Fibrillization of non-fullerene acceptors enables 19% efficiency pseudo-bulk heterojunction organic solar cells. Adv. Mater. 35, 2208211 (2023).
Lin, Y. & Zhan, X. Non-fullerene acceptors for organic photovoltaics: An emerging horizon. Mater. Horiz. 1, 470–488 (2014).
Janjua, M. R. S. A. Deciphering the role of invited guest bridges in non-fullerene acceptor materials for high performance organic solar cells. Synth. Met. 279, 116865 (2021).
Mehboob, M. Y., Adnan, M., Hussain, R. & Irshad, Z. Quantum chemical designing of banana-shaped acceptor materials with outstanding photovoltaic properties for high-performance non-fullerene organic solar cells. Synth. Met. 277, 116800 (2021).
Afzal, Q. Q. et al. Designing benzothiadiazole based highly efficient non-fullerene acceptor molecules for organic solar cells. Polymer 238, 124405 (2022).
Saeed, M. U. et al. End-capped modification of Y-Shaped dithienothiophen [3, 2-b]-pyrrolobenzothiadiazole (TPBT) based non-fullerene acceptors for high performance organic solar cells by using DFT approach. Surf. Interfaces 30, 101875 (2022).
Xiao, J. et al. Organic solar cells based on non-fullerene acceptors of nine fused-ring by modifying end groups. Org. Electron. 81, 105662 (2020).
Frisch, M. et al. Gaussian 09, revision D. 01 (2009).
Civalleri, B., Zicovich-Wilson, C. M., Valenzano, L. & Ugliengo, P. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 10, 405–410 (2008).
Bryantsev, V. S., Diallo, M. S., Van Duin, A. C. & Goddard, W. A. III. Evaluation of B3LYP, X3LYP, and M06-class density functionals for predicting the binding energies of neutral, protonated, and deprotonated water clusters. J. Chem. Theory Comput. 5, 1016–1026 (2009).
Adamo, C. & Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. J. Chem. Phys. 108, 664–675 (1998).
Farrokhpour, H. & Jouypazadeh, H. Description of adenine and cytosine on Au (111) nano surface using different DFT functionals (PW91PW91, ɷB97XD, M06–2X, M06-L and CAM-B3LYP) in the framework of ONIOM scheme: Non-periodic calculations. Chem. Phys. 488, 1–10 (2017).
Foster, M. E. & Wong, B. M. Nonempirically tuned range-separated DFT accurately predicts both fundamental and excitation gaps in DNA and RNA nucleobases. J. Chem. Theory Comput. 8, 2682–2687 (2012).
Komjáti, B. et al. Systematic study on the TD-DFT calculated electronic circular dichroism spectra of chiral aromatic nitro compounds: A comparison of B3LYP and CAM-B3LYP. Spectrochim. Acta A Mol. Biomol. Spectrosc. 155, 95–102 (2016).
Gündüz, B. & Kurban, M. Photonic, spectroscopic properties and electronic structure of PTCDI-C8 organic nanostructure. Vib. Spectrosc. 96, 46–51 (2018).
Kurban, M., Gündüz, B. & Göktaş, F. Experimental and theoretical studies of the structural, electronic and optical properties of BCzVB organic material. Optik 182, 611–617 (2019).
Dennington, R., Keith, T. & Millam, J. Gauss view, version 5 (2009).
Hanwell, M. D. et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 1–17 (2012).
Zhurko, G. A. Chemcraft: http://www.chemcraftprog.com. Received 22 October (2014).
O’boyle, N. M., Tenderholt, A. L. & Langner, K. M. Cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 29, 839–845 (2008).
May, R. A. & Stevenson, K. J. Software review of Origin 8. J. Am. Chem. Soc. https://doi.org/10.1021/ja809638x (2009).
Khalid, M. et al. Efficient tuning of small acceptor chromophores with A1-π-A2-π-A1 configuration for high efficacy of organic solar cells via end group manipulation. J. Saudi Chem. Soc. 25, 101305 (2021).
Arshad, M. N., Shafiq, I., Khalid, M. & Asiri, A. M. Exploration of the intriguing photovoltaic behavior for fused indacenodithiophene-based A-D–A conjugated systems: A DFT model study. ACS Omega 7, 11606–11617 (2022).
Adnan, M., Mehboob, M. Y., Hussain, R. & Irshad, Z. In silico designing of efficient C-shape non-fullerene acceptor molecules having quinoid structure with remarkable photovoltaic properties for high-performance organic solar cells. Optik 241, 166839 (2021).
Khan, M. I. et al. End-capped group modification on cyclopentadithiophene based non-fullerene small molecule acceptors for efficient organic solar cells; a DFT approach. J. Mol. Graph. Model. 113, 108162 (2022).
Janjua, M. R. S. A. Photovoltaic properties and enhancement in near-infrared light absorption capabilities of acceptor materials for organic solar cell applications: A quantum chemical perspective via DFT. J. Phys. Chem. Solids 171, 110996 (2022).
Khan, M. U. et al. First theoretical framework of triphenylamine–dicyanovinylene-based nonlinear optical dyes: Structural modification of π-linkers. J. Phys. Chem. C 122, 4009–4018 (2018).
Srnec, M. & Solomon, E. I. Frontier molecular orbital contributions to chlorination versus hydroxylation selectivity in the non-heme iron halogenase SyrB2. J. Am. Chem. Soc. 139, 2396–2407 (2017).
Kandemirli, F. & Sagdinc, S. Theoretical study of corrosion inhibition of amides and thiosemicarbazones. Corros. Sci. 49, 2118–2130 (2007).
Khan, M. U. et al. Prediction of second-order nonlinear optical properties of D–π–A compounds containing novel fluorene derivatives: A promising route to giant hyperpolarizabilities. J. Clust. Sci. 30, 415–430 (2019).
Khan, M. U. et al. First theoretical framework of Z-shaped acceptor materials with fused-chrysene core for high performance organic solar cells. Spectrochim. Acta A Mol. Biomol. Spectrosc. 245, 118938 (2021).
Chemla, D. S. Nonlinear Optical Properties of Organic Molecules and Crystals V1 Vol. 1 (Elsevier, 2012).
Janjua, M. R. S. A. Theoretical and conceptual framework to design efficient dye-sensitized solar cells (DSSCs): Molecular engineering by DFT method. J. Clust. Sci. 32, 243–253 (2021).
Hussain, S. et al. Zinc-doped boron phosphide nanocluster as efficient sensor for SO2. J. Chem. 2020, 1–12 (2020).
Rafiq, M. et al. Synthesis, XRD, spectral (IR, UV–Vis, NMR) characterization and quantum chemical exploration of benzoimidazole-based hydrazones: A synergistic experimental-computational analysis. Appl. Organomet. Chem. 33, e5182 (2019).
Rahmalia, W., Fabre, J.-F., Usman, T. & Mouloungui, Z. Aprotic solvents effect on the UV–visible absorption spectra of bixin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 131, 455–460 (2014).
Adeoye, M. D. et al. Effect of solvents on the electronic absorption spectra of 9, 14 dibenzo (a, c) phenazine and tribenzo (a, c, i) phenazine. Sci. Res. Essays 4, 107–111 (2009).
Khalid, M. First principles study of electronic and nonlinear optical properties of A-D–π–A and D–A–D–π–A configured compounds containing novel quinoline–carbazole derivatives. RSC Adv. 10, 22273–22283 (2020).
Ans, M., Ayub, K., Muhammad, S. & Iqbal, J. Development of fullerene free acceptors molecules for organic solar cells: A step way forward toward efficient organic solar cells. Comput. Theor. Chem. 1161, 26–38 (2019).
Deibel, C. & Dyakonov, V. Polymer–fullerene bulk heterojunction solar cells. Rep. Prog. Phys. 73, 096401 (2010).
Jabeen, S., Khera, R. A., Iqbal, J. & Asgher, M. Design, synthesis and application of triazole ligands in suzuki miyaura cross coupling reaction of aryl chlorides. J. Mol. Struct. 1206, 127753 (2020).
Lu, T. & Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Ans, M. et al. Designing three-dimensional (3D) non-fullerene small molecule acceptors with efficient photovoltaic parameters. ChemistrySelect 3, 12797–12804 (2018).
Wang, X. et al. Photophysical properties and optical nonlinearity of cyclo [18] carbon (C 18) precursors, C 18–(CO) n (n = 2, 4, and 6): Focusing on the effect of the carbonyl groups. Phys. Chem. Chem. Phys. 24, 7466–7473 (2022).
Liu, Z., Wang, X., Lu, T., Yuan, A. & Yan, X. Potential optical molecular switch: Lithium@ cyclo [18] carbon complex transforming between two stable configurations. Carbon 187, 78–85 (2022).
Köse, M. E. Evaluation of acceptor strength in thiophene coupled donor–acceptor chromophores for optimal design of organic photovoltaic materials. J. Phys. Chem. A 116, 12503–12509 (2012).
Dkhissi, A. Excitons in organic semiconductors. Synth. Metals 161, 1441–1443 (2011).
Acknowledgements
Dr. Muhammad Khalid gratefully acknowledges the financial support of HEC Pakistan (project no. 20-14703/NRPU/R&D/HEC/2021). Authors are thankful for cooperation and collaboration of A.A.C.B from IQ-USP, Brazil especially for his continuous support and providing computational lab facilities. A.A.C.B. acknowledges the financial support of the São Paulo Research Foundation (FAPESP) (Grants 2014/25770-6 and 2015/01491-3), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) of Brazil for academic support (Grant 309715/2017-2), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) that partially supported this work (Finance Code 001). The authors thank the Researchers Supporting Project number (RSP2023R29), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
I.S.: Conceptualization; methodology. M.K.: Methodology; software; project administration. M.A.A.: Data curation; formal analysis. R.B.: Conceptualization; methodology; software. A.A.C.B.: Conceptualization; resources. S.M.A.: Data curation; formal analysis; validation, S.A.: Data curation; formal analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shafiq, I., Khalid, M., Asghar, M.A. et al. Influence of azacycle donor moieties on the photovoltaic properties of benzo[c][1,2,5]thiadiazole based organic systems: a DFT study. Sci Rep 13, 14630 (2023). https://doi.org/10.1038/s41598-023-41679-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41679-0
- Springer Nature Limited