
Abstract
A new series of 7-substituted coumarin scaffolds containing a methyl ester moiety at the C4-position were synthesized and tested for their in vitro anti-proliferative activity against MCF-7 and MDA-MB-231 breast cancer cell lines using Doxorubicin (DOX) as reference. Compounds 2 and 8 showed noticeable selectivity against MCF-7 with IC50 = 6.0 and 5.8 µM, respectively compared to DOX with IC50 = 5.6 µM. Compounds 10, 12, and 14 exhibited considerable selectivity against Estrogen Negative cells with IC50 = 2.3, 3.5, and 1.9 µM, respectively) compared to DOX with (IC50 = 7.3 µM). The most promising compounds were tested as epidermal growth factor receptor and aromatase (ARO) enzymes inhibitors using erlotinib and exemestane (EXM) as standards, respectively. Results proved that compound 8 elicited the highest inhibitory activity (94.73% of the potency of EXM), while compounds 10 and 12 displayed 97.67% and 81.92% of the potency of Erlotinib, respectively. Further investigation showed that the promising candidates 8, 10, and 12 caused cell cycle arrest at G0–G1 and S phases and induced apoptosis. The mechanistic pathway was confirmed by elevating caspases-9 and Bax/Bcl-2 ratio. A set of in silico methods was also performed including docking, bioavailability ADMET screening and QSAR study
Similar content being viewed by others

Introduction
One of the prominent causes of cancer mortality in women is breast cancer (BC), the second-most common cancer1. About 75% of BC is caused by the estrogen receptor (ER)2.
Inhibition of estrogen biosynthesis is one effective approach for hormone-dependent BC therapy in postmenopausal women3. The aromatase enzyme (ARO) is one of the enzymatic mechanisms that may regulate the increased estrogen levels found in BC cells. Exemestane (EXM) (Ia) is among the reported powerful aromatase inhibitors (AIs)4.
On the other hand, the most often overexpressed receptors in BC cells are tyrosine kinase receptors (TKRs), e.g., the epidermal growth factor receptor (EGFR)5. Thus, targeting those receptors is a promising way to develop novel anticancer drugs. In this context, the first reported EGFR inhibitor highly expressed in different forms of cancers is Erlotinib (Ib)6,7.
Moreover, targeting various apoptotic pathways is also an effective approach for all types of cancer8,9. Any pathway stage can be targeted for cancer treatment10.
In light of the above information, developing and synthesizing novel anticancer candidates with increased selectivity is essential. The varied biological characteristics of coumarin-containing drugs, especially anticancer activity, have generated much interest11,12,13. Investigations into the biological properties of coumarins have found that they can target multiple cancer pathways, including aromatase inhibition, kinase inhibition, cell cycle arrest, and angiogenesis suppression14,15.
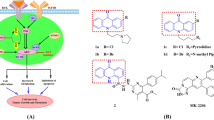
Studying the structures of EXM (Ia)16,17 and erlotinib (Ib)18 (Fig. 1a) has provided important structural information which helped us in the rational design of new potent ARO/EGFR inhibitors.

(a) Main structural features of EXM (Ia) and Erlotinib (Ib). (b) Design chart with reported coumarin derivatives as a potent anti-breast cancer agent.
Importantly, various anticancer candidates based upon coumarin skeletons (II-VII) and their structural characteristics were selected as the main pharmacophore in developing multitargeted anti-BC19,20,21,22,23. In this work, the molecular design was mostly based on two strategies:
In the first strategy, based on the previous evidence, we have selected EXM (Ia) and Erlotinib (Ib) as lead anti-BC compounds (Fig. 1a). Secondly, structural optimization of the lead compounds through the following (Fig. 1b):
-
1.
The planar coumarin (1,2-benzopyrone) scaffold is a bioisostere to the quinazoline scaffold in erlotinib and tetrahydronaphtalene-1,7-dione nucleus in EXM. Such a skeleton will be essential for anticancer activity24,25.
-
2.
The hydrophobic phenyl ring attached to the C4-position of the coumarin nucleus by introducing a spacer is previously considered a biologically active pharmacophore that is considered to be crucial for the activity26,27,28,29.
-
3.
The reported SAR research demonstrated that conjugates containing benzyl groups at the C7-position exhibited more activity than their alkyl analogs. Introducing F or Cl groups on the benzyl ring resulted in good activity19,23.
-
4.
The rotatable atoms of the linker enable the phenyl group to close the entrance cavity in the aromatase binding site30.
Based on the above findings, we worked on synthesizing 7-substituted coumarin derivatives with flexible C4-position ester functionality to examine their impact on two BC cell lines, MCF-7 and MDA-MB-231 cell lines, as well as normal breast cells MCF-10A. The synthetic compounds were tested in vitro for their ability to inhibit the EGFR and ARO enzymes.
The most potent compounds, such as cell cycle examination and apoptosis markers, were selected for additional studies to discover the molecular process behind their anticancer activity.
Moreover, molecular modeling studies were conducted to identify the two enzymes’ interaction patterns and investigate the relationship between their physicochemical features and inhibitory action. Additionally, QSAR analysis was performed.
Results
Chemistry
The steps in Figs. 2 and 3 were used to synthesize the intermediates and target molecules. The requisite 4-chloromethyl-7-hydroxycoumarin (1)31 was first synthesized through a Pechman cyclocondensation reaction involving acid-catalyzed condensation of resorcinol with ethyl 4-chloroacetoacetate in a readily scalable procedure32,33.

Synthetic pathways of the new coumarin derivatives (7–12).

Synthesis of new acetyl coumarin derivatives (13–15).
7-Hydroxycoumarin derivatives 2–4 were obtained through the nucleophilic substitution reaction from compound 1 with carboxylate ion bearing Na ion as the countercation in DMF (Fig. 2). The structures of new intermediates derivatives 2–4 were confirmed using spectral data. 1H NMR spectrum showed the appearance of exchangeable OH protons at 10.69–10.72 ppm and CH2 protons at 5.50–5.60 ppm. Besides, 13C NMR exhibited signals at 165.6–166.9 assigned to C=O of the carboxylate group. All the aromatic carbons appeared in the expected region. Moreover, the IR spectrum showed a characteristic broad band at 3100–3200 cm−1 corresponding to the OH group and a typical carbonyl group absorption at 1690–1720 cm−1 corresponding to two carbonyl groups.
The intermediates 5 and 6 were synthesized through benzoylation of the phenolic 7-OH group in 1 with the appropriate phenacyl bromide in the presence of acetone and K2CO3. Confirmation of structures by spectral data revealed the appearance of singlet 1H NMR signal at δ 5.77–5.79 characteristic for the newly formed –OCH2– and appearance of new C=O group 13C NMR signal at δ 193.3–194.8.
New Target coumarin derivatives 7–12 were obtained from 1 through two pathways: nucleophilic substitution of 1 and benzoylation of 2–4 (pathway A, Fig. 2)34,35. The order of these two steps can be reversed to produce the same target coumarin derivatives 7–12 (Pathway B, Fig. 2). It was observed that pathway B was less favorable than A in relation to the yield and purity of products. This may be due to competitive alkylation at the acidic methylenic group at position-4. The bulky benzoate group will hinder this side reaction sterically more than the chloride group, which goes along with the literature31. The reported yields (45–77%) and compound's characteristics of 7–12 were based on pathway A. It is worth reporting that the one-pot reaction was tried, but unfortunately, the yield and purity of products were poor (Pathway C, Fig. 2).
The structures of 7–12 were confirmed by spectral data in which the exchangeable proton OH disappeared in their 1H NMR. A singlet signal of COCH2 protons appeared at δ 5.78–5.81 ppm, and another singlet for CH2Cl appeared upfield at δ 5.60–5.64 ppm. The aromatic protons were found at the expected region and pattern. Moreover, 13C NMR spectra revealed the presence of three carbonyl groups at the ranges of 196.4–193.3, 165.6–164.8, and 162.9–161.6, corresponding to benzoyl C=O, benzoate C=O, and coumarin C=O; respectively.
As shown in Fig. 3, the acetylation reaction was carried out with a mixture of acetic anhydride and acetic acid to yield O-acetylated coumarins (13–15)36. The presence of the acetyl group was confirmed by a singlet peak at 2.33–2.34 ppm corresponding to COCH3 in 1H NMR and a signal of C=O at 169.2–169.3 in 13C NMR. In addition, the singlet signal of OCH2 in 1H NMR spectrum was found at 5.62–5.67 ppm. The aromatic protons were found at their expected aromatic regions. 13C NMR spectra proved the presence of three carbonyl groups at the ranges of 169.2–169.4, 165.4–165.6, and 158.9–159.8 corresponding to acetyl C=O, benzoate C=O, and coumarin C=O, respectively. All the chemical synthetic steps were found adequate and concise with appropriate yields.
In vitro biological evaluation
Measurement of in vitro cytotoxic activity by MTT assay
For the MCF-7 cell line, results shown in Table 1 revealed that the most powerful compounds were methyl benzoate derivatives 2 and 8 with IC50 = 6.0 µM and 5.8 µM, respectively, compared to IC50 = 5.6 µM for the standard drug DOX.
For MDA-MB-231, the most promising action was reported for compounds 4, 5, methyl benzoate derivatives 10, 12, and compound 14 with IC50 values of 6.4, 5.8, 2.3, 3.5, and 1.9 µM, respectively, compared to the standard drug DOX which had IC50 value of 7.3 µM.
Structure–activity relationship (SAR) study
Starting compound 1 was devoid of activity against both types of cell lines.
The introduction of benzoate substituent at position-4 in the coumarin scaffold was observed to potentiate the activity. This is possibly explained by specific hydrophobic interactions of the phenyl group.
Evidently, the compound bearing unsubstituted benzoate 2 exhibited superior selectivity against MCF-7 than against MDA-MB-231 with IC50 = 6 µM versus IC50 = 51.9 µM, respectively, compared to the reference drug.
Incorporation of the electron-withdrawing group at position-4 on the aromatic moiety in compound 3 showed moderate selectivity towards MCF-7 than MDA-MB-231. Conversely, the substitution of an electron-donating methoxy group at position-2 as in compound 4 resulted in a beneficial selectivity against MDA-MB-231 than MCF-7 cell lines was IC50 values = 6.4 and 17.2 µM, respectively.
The activity of 7-(2-oxo-2-phenylethoxy)-4-methyl benzoate coumarin series (7–12) varied according to the type of substituents compared to intermediates 2–4. Incorporation of 2-oxo-2-phenylethoxy at position-7 of coumarin moiety in compounds 7, 9, and 11 diminished the activity against the MCF-7 cell line while resulting in poor anti-proliferative activity against the MDA-MB-231 cell line. Compound 9 bearing halogen atom at p-position was identified as the most significant cytotoxic candidate among the series revealing potent selectivity against the MDA-MB-231 cell line with IC50 value of 10.4 µM.
In general, mono substitution with an electron-withdrawing group on phenyl moiety such as p-chloro atom located at C7- of coumarin ring in compounds 8, 10, 12 and 14 dramatically improved the activity against both cell lines, in comparison to the unsubstituted derivatives. In this context, it could be stressed that the target compound 8 showed high selectivity against MCF-7 than MDA-MB-231 (IC50 = 5.8 µM versus IC50 = 27.5 µM), while compounds 10, 12 and 14 showed superior selectivity against MDA-MB-231 with IC50 = 2.3, 3.5 and 1.9 µM, respectively. Fortunately, compound 12 was the most potent among this series against both MCF-7 and MDA-MB-231 with IC50 = 13.2 µM and 3.5 µM, respectively.
Surprisingly, compound 6 with an electron-withdrawing chlorine atom exhibited a lower activity level against both cell lines, which was assumed to involve H-bonds with amino acids in the ligand-binding domain.
Concerning coumarins 13–15 carrying 7-acetyloxy group, derivative 14 showed superior selectivity against MDA-MB-231 than MCF-7 by 150 times with (IC50 = 1.9 µM). In contrast, compound 15 showed moderate anti-proliferative activity against MCF-7 (IC50 = 17.1 µM).
In vitro cytotoxicity against nonmalignant human cells
The compounds were also examined against the human nonmalignant cells MCF10A (breast) to investigate their selectivity between nonmalignant cells and cancerous cells. Compared to DOX (IC50 = 12.4 µM), the most active compounds 8, 10, 12 and 14 were less toxic to MCF10A cells (IC50 = 79.9, 76.8, 72.1 and 263.1 µM, respectively) with good SI values (more than 10) (Table 1).
In vitro enzyme inhibition assays
The most active compounds were subjected further to in vitro enzyme inhibition via ELISA enzyme assay.
-
EGFR enzyme inhibition assay
MDA-MB-231 cell lines could express mainly EGFR enzyme, whereas MCF-7 cell lines only produced aromatase enzyme23. So, an enzyme assay study was performed for ARO and EGFR enzymes in MCF-7 and MDA-MB-231 cell lines, respectively.
It was observed that compounds 10 and 12 showed the highest inhibition effect towards EGFR, displaying 97.67% and 81.92% of the potency of erlotinib, respectively (Table 2, Supplementary Fig. S1). Therefore, their potent selectivity against MDA-MB-231 might be due to EFGR inhibition pathway.
-
(ARO) enzyme inhibition assay
To convert androgens into estrogens, the MCF-7 cell line was shown to possess enough aromatase enzyme activity37. Results are presented in Table 3, Supplementary Fig. S2, and revealed that target compound 8 is considered the most potent AIs with (IC50 value = 0.114 µM), compared to EXM (IC50 value = 0.108 µM), and this described the elevated value of IC50 in MCF-7 (5.8 µM). Furthermore, target compound 2 exhibited half ARO inhibitory activity as that of reference EXM.
Cell cycle arrest
Results are presented in Table 4, Supplementary Fig. S3. Regarding the MCF-7 cell line, the percentage of apoptotic cells was increased significantly in the pre-G1 phase by 18.67 folds with significant arrest in G0-G1 and S phases by 1.508 and 1.33 folds, respectively compared to the untreated control cells for compound 8. For MDA-MB-231 cell lines, when treated with compounds 10, 12 and 14, the percentage of apoptotic cells in the pre-G1 phase increased by 16.22, 22.46 and 19.9 folds with consequential cell cycle arrest at the S phase by 1.59, 1.08 and 1.38 folds, respectively. Results indicated that compound 8 tested against the MCF-7 cell line and compounds 10, 12 and 14 tested against the MDA-MB-231 cell line showed pre-G1 apoptosis and cell cycle arrest at the S phase in addition to G0-G1 by compound 8.
Apoptosis determination using annexin V-FITC/PI dual staining assay.
To further assure the apoptotic ability of compounds 8, 10, 12 and 14 using dual staining with propidium iodide (PI) and annexin-V-FITC, a flow cytometric analysis was carried out to distinguish between living, early, late apoptotic and necrotic cells. Propidium iodide stains late apoptotic and necrotic cells’ DNA, while annexin-V binds powerfully and distinctively to phosphatidylserine (PS) on their surfaces and fluoresces green38. The assay results are provided in Table 5 and (Supplementary Figs. S4 and S5) as fluorescence-activated cell sorter (FACS) and cytometry profiles with PI on the Y-axis and annexin V-FITC on the X-axis. In addition, each profile’s quadrant has four sections: necrosis, late and early apoptosis, and living cells, clockwise from the upper left. Results are demonstrated in Table 5, indicating that compound 8 showed % apoptosis 31.19 for MCF-7 cells while % apoptosis for MDA-MB-231 cells of compounds 10, 12 and 14 was found to be 33.43, 46.28 and 41.02, respectively; compared to 1.67 and 2.06 for control untreated MCF-7 and MDA-MB-231.
Effect on the levels of Bax/Bcl-2
Apoptosis is provoked in a cell through two major apoptotic pathways, the extrinsic and intrinsic pathways. By overexpressing anti-apoptotic proteins like Bcl-2 or downregulating pro-apoptotic proteins like Bax through the intrinsic apoptotic pathway, cancer cells can develop apoptotic resistance39. In this study, we measured the levels of Bax and Bcl2 to evaluate the effects of compounds 8, 10, 12 and 14, which showed promising apoptosis-inducing activity on the intrinsic apoptotic pathway. As shown in (Supplementary Table S6 and Supplementary Fig. S7), the tested compounds 8, 10, 12 and 14 significantly increased the expression of the pro-apoptotic protein Bax by 5.27, 5.27, 5.22 and 4.9 folds, respectively, with a consequent decline in the anti-apoptotic protein Bcl-2 levels by 0.44, 0.377, 0.271 and 0.329 folds, respectively compared to the control. As a result, a significant increase in the Bax/Bcl-2 ratio was observed, supporting the claim that the tested compounds can improve the therapeutic response in breast cancer.
Caspase-9 activation
Caspase-9, activated in apoptotic cells by intrinsic pathways, can be used as a marker for the induction of apoptosis by anticancer agents.40. In the present study, the levels of caspase-9 were then measured in the compounds that showed a noticeable increase in the Bax/Bcl-2 ratio to identify a possible pathway for the antiproliferative activity of the most promising compounds. Significantly elevated levels of caspase-9 protein level in treated samples were observed in (Supplementary Table S8 and Supplementary Fig. S9) by 6.71 folds for compound 8, while compounds 10, 12 and 14 resulted in 6.42-, 7.33- and 4.37-folds elevation, respectively, compared to control cells. These results suggested that all tested compounds could induce a significant overexpression in caspase-9 protein level higher than that elicited by control, assuming that apoptosis was probably due to the activation of caspase-9 protein.
Statistical analysis and data interpretation
Data analysis was performed by SPSS software, version 25 (SPSS Inc., PASW Statistics for Windows version 25. Chicago: SPSS Inc.). Apoptosis was described using numbers and percentages. Quantitative data were described using mean ± Standard deviation for normally distributed data after testing normality using Shapiro Wilk test. The significance of the obtained results was judged at the (≤ 0.05) level. Chi-Square and Z-tests were used to compare qualitative data between groups as appropriate. One Way ANOVA test was used to compare more than 2 independent groups with Post Hoc Tukey test to detect pair-wise comparison.
In silico computational study
Docking into EGFR crystal structure
Re-docking results of the co-crystallized ligand (Erlotinib) and its binding mode18,30,41 were described in (Table 6, Supplementary Figs. S10, and S11).
Docking results showed that compounds 10 and 12 exhibited good binding interactions and docking scores similar to Erlotinib (Table 6). It is suggested that the planarity of quinazoline moiety in erlotinib seems important for stabilizing the EGFR binding site. Similarly, the planar nature of the coumarin scaffold in compounds 10 and 12 allowed them to be embedded into the active site of EGFR, as shown in their 2D interactions (Supplementary Fig. S12) and 3D binding modes as in Fig. 5. It was found that the C=O group of the coumarin skeleton overlapped with the N1 atom of the quinazoline moiety of erlotinib, which equivalently served to anchor with Met 769, which was considered the key interaction improving the stabilization. By investigating the binding site of the receptor, the presence of o-methoxy or p-chloro phenyl moiety at position 4-methyl carboxylate of coumarin nucleus in compounds 10 and 12, respectively, were observed to be positioned in the hydrophobic pocket, which included side chains Leu 764, Leu 753, Met 742, Ile 720 and Ile 765 as for the ethynyl-phenyl group in erlotinib42 (Supplementary Fig. S13).
Moreover, additional halogen bond interactions were shown with the amino acid His 781 with the chloride atom on C4 of the phenyloxy side chain at C7 of the coumarin nucleus with bond length (2.22 Å for compound 10 and 2.32 Å for compound 12). This interaction appears to help their immobilization; in the same way, the ether side chain in erlotinib as illustrated in (Fig. 4).

(a) Binding mode of compound 10 (blue) and erlotinib (green), (b) Binding mode of compound 12 (blue) and erlotinib (green), (c) Binding mode of compound 10 (red), compound 12 (green) and erlotinib (blue) occupying the active site. The 2 compounds had the same binding mode as the native ligand. They bound with the key residue Met 769. The presence of p-chloro atom improved their stability. The hydrophobic side chain in both compounds changed their orientation to embed in hydrophobic pocket. The coumarin moiety was buried in the pocket in parallel to quinazoline nucleus in erlotinib.
In order to rationalize the biological outcomes, it was believed that the poor EGFR inhibitory action of compounds 11 and 14 was related to their failure to bind with the essential amino acid and their low docking score of -5.7394 and -5.4982 kcal/ mol, respectively (Supplementary Fig. S14). Hence, the acquired data revealed that the binding modes of the investigated compounds were consistent with their EGFR inhibitory action.
On the other hand, despite compound 8 showing the same binding features as the most potent EGFR inhibitors 10 and 12 into the active site with a docking score of -6.7717 kcal/ mol and RMSD = 1.9314 Å, it unexpectedly possessed weak EGFR inhibitory activity. However, it could be stated that other factors that affect activity related to lipophilicity and solubility may be involved.
Docking into ARO crystal structure
Results of re-docked ligands (ASD and EXM) are summarized in (Table 6) and their binding modes are described in (Supplementary Fig. S15). Aromatase enzymes comprise a heme prosthetic group containing iron at the active reaction center43.
Regarding compound 8, the most potent AI showed an acceptable binding mode (Table 6, Fig. 5). When compared to the binding mode of ligands, it was found that the coumarin scaffold was stabilized by arene-arene interaction with the 5-membered ring of the heme moiety, which is considered the binding driving interaction.

3D binding modes of compounds 2 and 8 inside ARO binding pocket (PDB ID: 3EQM) and compounds 10 and 12 into the active site of EGFR (PDB ID: 1 M17).
Unsubstituted benzoate at position-4 of coumarin is essential for activity, binding with Met 374 by H-bond (2.84 Å) and providing a convenient allocation in the hydrophobic pocket, constituted of Val 373, Met 374, Leu 372, Ile 398 residues.
Introducing a substituent on the phenyl moiety (p-chloro or o-methoxy) resulted in a dramatic decrease in activity due to either a steric effect or a change in lipophilicity.
In particular, the distance between the benzene ring placed in the hydrophobic pocket and the coumarin moiety is likely the same as the entire length of ASD (6.97 Å, 6.69 Å, respectively) and plays a significant role in the control of affinity. The substitution at position-4 on the methylene bridge could be responsible for the activity.
In addition, the phenyl ethoxy side chain at position-7 slightly twisted to be embedded into the binding site and seemed to induce additional weak H-bond with Thr 310 (3.64 Å) to close the entrance cavity between Asp 309 and Ser 47820. The longer the substituent at position-7 could justify the better affinity to the binding site than the free OH moiety. These three binding site interactions were fulfilled when the coumarin ring carried benzyl carboxylate methyl group at position-4 and p-chlorophenyl ethoxy substituent at position-7. Therefore, the better binding mode of target compound 8 when compared to compound 2 can be explained, in which it showed the same binding manner with the backbone amide group of Met 374 (3.42 Å) and the heme moiety through arene–H interaction. Although, the free hydroxyl group at position-7 exhibited a decrease in activity due to the inability to close the entrance side of the pocket (Fig. 5). The 2D binding poses of both compounds are shown in (Supplementary Fig. S16).
On the other hand, because of the correlation between the bulkiness of the position-4 substituent and the activity, it may be presumed that the o-methoxy substituent in compound 11 (IC50 = 5.98 µM) generated steric hindrance in the rear side of the pocket and unstable binding mode with score (S) = − 0.1435 kcal/mol. In addition, compound 6 (IC50 = 3.93 µM) did not illustrate the three-site binding hypothesis since its molecular volume is smaller than the pocket site. They both shared the lack of essential interaction in either the rear side or the entrance side and unacceptable values of RMSD of more than 2 (Supplementary Fig. S17).
Bioavailability and ADMET properties screening
To understand the pharmacokinetic behavior of the most active compounds 8, 10, 12 and 14, their physicochemical characters were calculated with the aid of the online application ADMETSAR server http://lmmd.ecust.edu.cn/admetsar1 by applying various qualitative ADMET models as described in (Table 7). The results revealed that all compounds possessed good intestinal absorption and could penetrate the blood–brain barrier. Besides, the candidate compounds illustrated good oral bioavailability, making them promising candidates as anti-BC agents.
QSAR study
The QSAR analysis was carried out on the synthesized compounds against MDA-MB-231 cancer cells, where most compounds showed good selectivity using MOE software. The synthesized compounds were used as a training set with their experimental pIC50 (- log IC50). Various physicochemical descriptors were calculated.
Vsurf descriptors explained the hydrophilic and hydrophobic interactions, while the hydrophobic bindings were described by the vsurf_D descriptors (vsurf_D5 and vsurf_D3)44,45.
The following equation represents the best QSAR model is: pIC50 = 7.49008 + 0.41537 log P (o/w)—0.01752 E_vdw + 0.26664 opr_nrot—0.02428 vsurf_D3—0.00142 Weight + 0.01549 vsurf_D5—0.26814 logS (with root mean square error (RMSE) = 0.39205 and squared correlation coefficient (r2) = 0.21403. According to the previous equation, the anticancer activity was enhanced by log P (lipophilicity), vsurf D5 (hydrophobic bindings), and Opr nrot (number of rotatable bonds), while the activity is negatively correlated with the following: van der Waal energy, polar interactions, molecular weight, and log S (aqueous solubility).
The reliability of the built model was confirmed by excellent linearity [$PRED = 0.2094 (pIC50) + 4.215], with R2 = 0.326 (Fig. 6) and the relative values between the predicted and experimental activities (Table 8). It was found that the predicted values are close to those experimentally investigated, indicating that the QSAR model is reliable and can be safely applied to predict more effective compounds.

Experimental versus predicted pIC50 of the tested compounds against MCF-7 human cancer cell line.
Discussion
In the current study, a new series of nonsteroidal 4,7-disubstituted coumarin (1,2-benzopyrone) derivatives bearing methyl ester moiety at the C4-position were designed as anti-BC agents. Easy and good-yielding synthetic pathways were adopted to prepare the designed compounds. MTT assay method was used to evaluate their in vitro anti-BC activity against MCF-7, MDA-MB-231, and MCF-10A cell lines using Doxorubicin (DOX) as a reference drug. Generally, most of the tested compounds revealed MDA-MB-231 selectivity more than MCF-7. Particularly, compounds 2 and 8 exhibited obvious selectivity against MCF-7, while compounds 4, 5, 10, 12, and 14 exhibited considerable selectivity against the MDA-MB-231 cell line.
In conclusion, it might be emphasized that "mono substitution with electron-withdrawing groups as chlorine on phenyl moiety is crucial for the anti-BC activity against both cell lines as it could be involved in electrostatic interactions.
The cytotoxic activity of compounds 10 and 12 against MDA-MB-231 could be due to its potent EGFR inhibitory activity, while compound 8 against MCF-7 might be attributed to its ARO inhibitory activity. Regarding compound 14, it possessed weak inhibitory activity toward EGFR despite its potent anticancer activity in MDA-MB-231 cell lines. Consequently, further investigations for the most active compounds 8, 10, 12 and 14 were done to discover more mechanistic pathways for their antiproliferative activity, such as cell cycle analysis and apoptosis assay using flow cytometry. It was found that they were able to induce cell cycle arrest at G0-G1 and S phases. Their apoptotic mechanism was proved by a significant increase in both Bax/Bcl-2 ratio and caspase-9. The inhibitory activities of compounds 2 and 8 on ARO enzymes and 10 and 12 on EGFR were confirmed by docking studies, highlighting significant binding interactions. ADMET study was also carried out to further describe the synthesized drugs' pharmacokinetics. The findings of the QSAR model revealed that there should be a balance between the compounds' hydrophilic and hydrophobic substituents, which was confirmed by the docking results. Therefore, the observed results showed that these compounds were susceptible to being modified further to serve as promising multitargeted anti-BC agents (Supplementary Fig. S18).
Methods
Chemistry
Melting points were determined on Stuart melting point apparatus and are uncorrected. Microanalyses were performed at Cairo University and performed on a Perkin-Elmer 240 elemental analyzer for C, H, and N elements, and the results were within the acceptable range of the theoretical values. 1H and 13C NMR were performed at Mansoura University and recorded on Brucker 400 MHz spectrometer and 100 MHz spectrometer, respectively. Chemical shifts are expressed in δ ppm with reference to TMS. Using a Nicolet iS10 infrared spectrometer, IR spectra were recorded at Mansoura University. Mass spectral analyses were performed on Thermo SCIENTIFIC DCQII at Azhar University. All the used chemicals and reagents were purchased from Aldrich Chemicals Co, USA.
General procedure for the preparation of (7-hydroxy-2-oxo-2H-chromen-4-yl) methyl benzoate derivatives (2–4)
To a solution of 1 (0.45 g, 2 mmol) in DMF (8 mL), the appropriate sodium salt of (un)substituted benzoic acid (2 mmol) was added. The reaction mixture was heated at 90 °C overnight, then cooled and poured over ice-water. The separated solid products were filtered off, dried and then recrystallized from EtOAc/MeOH (4:1).
(7-Hydroxy-2-oxo-2H-chromen-4-yl)methyl benzoate (2).
White solid; Yield 72%; mp 248–250 °C. IR (KBr) υmax (cm−1): 3154 (OH); 2943 and 2840 (CH aliphatic); 1720 and 1689 (C=O); 1614 and 1568 (C=C); 1136 (C–O–C). 1H NMR (400 MHz, DMSO-d6): δ 10.72 (s, 1H, OH, D2O exchangeable), 8.08 (d, 2H, J = 7.5 Hz, Phenyl-C2-H and C6-H), 7.73 (t, J = 7.2 Hz, 1H, Phenyl-C4-H), 7.68 (d, J = 8.6 Hz, 1H, C5-H), 7.59 (t, 2H, J = 7.6 Hz, Phenyl-C3-H and C5-H), 6.85 (dd, 1H, J1 = 8.6, J2 = 1.5 Hz, C6-H), 6.79 (d, 1H, J = 1.5Hz, C8-H), 6.29 (s, 1H, C3-H), 5.60 (s, 2H, OCH2). 13C NMR (100 Hz, DMSO-d6), δ (ppm): δ 165.6 (C=O), 162.0 (C2=O), 160.6 (C7), 155.6 (C4), 150.9 (C8a), 134.3 (Phenyl-C4), 130.0 (two Phenyl-C2 and C6), 129.5 (two Phenyl-C3 and -C5), 129.4 (Phenyl-C1), 126.6 (C5), 113.7 (C6), 110.0 (C4a), 108.8 (C3), 103.0 (C8), 62.5 (CH2). MS (m/z %): 296.22 (1.39, M+), 294.85 (M-H+), 105.18 (100). Elemental analysis for C17H12O5, calcd.: C, 68.92; H, 4.08; Found: C, 68.78; H, 4.26.
(7-Hydroxy-2-oxo-2H-chromen-4-yl)methyl 4-chlorobenzoate (3).
White solid; Yield 80%; mp 173–175 °C. IR (KBr) υmax (cm−1): 3174 (OH); 2930 and 2846 (CH aliphatic); 1721 and 1690 (C=O); 1599 (C=C); 1130 (C–O–C). 1H NMR (400 MHz, DMSO-d6): δ 10.69 (s, 1H, OH, D2O exchangeable), 7.95 (d, 2H, J = 8.3 Hz, Phenyl-C2-H and C6-H), 7.58 (d, 2H, J = 8.3 Hz, Phenyl-C3-H and C5-H), 7.69 (d, 1H, J = 8.7 Hz, C5-H), 6.86 (dd, 1H, J1 = 8.7, J2 = 1.3 Hz, C6-H), 6.77 (d, 1H, J = 1.3 Hz, C8-H), 6.43 (s, 1H, C3–H), 5.50 (s, 2H, OCH2). 13C NMR (100 Hz, DMSO-d6), δ (ppm) 166.9 (C=O), 161.7 (C2=O), 160.8 (C7), 155.7 (C4), 151.5 (C8a), 138.4 (Phenyl-C4), 131.6 (Phenyl-C2), 131.6 (Phenyl-C6), 129.9 (Phenyl-C1), 129.3 (two Phenyl-C3 and C5), 127.1 (C5), 113.6 (C6), 111.5 (C4a), 109.9 (C3), 102.9 (C8), 62.5 (CH2). MS (m/z %): 332.03 (22.66, M + 2), 330.50 (24.62, M+), 252.22 (100). Elemental analysis for C17H11ClO5, calcd.: C, 61.74; H, 3.35; Found: C, 61.97; H, 3.51.
(7-Hydroxy-2-oxo-2H-chromen-4-yl)methyl 2-methoxybenzoate (4).
White solid; Yield 64%; mp 268–270 °C. IR (KBr) υmax (cm−1): 3260 (OH); 3097 (CH Aromatic), 2999, 2941 and 2836 (CH aliphatic); 1721 and 1693 (C=O); 1612 and 1567 (C=C); 1130 (C–O–C). 1H NMR (400 MHz, DMSO-d6): δ 10.69 (s, 1H, C7–OH, D2O exchangeable), 7.77 (d, 1H, J = 7.5 Hz, Phenyl-C6-H), 7.69 (d, 1H J = 8.7 Hz, C5–H), 7.62 (t, 1H, J = 7.9 Hz, Phenyl-C4-H),7.22 (d, 1H, J = 8.4 Hz, Phenyl-C3-H), 7.08 (t, 1H, J = 7.5 Hz, Phenyl-C5-H), 6.85 (dd, 1H, J1 = 8.7, J2 = 1.6 Hz, C6-H), 6.78 (d, 1H, J = 1.6 Hz, C8-H), 6.37 (s, 1H, C3-H), 5.55 (s, 2H, OCH2), 3.88 (s, 3H, CH3). 13C NMR (100 Hz, DMSO-d6), δ (ppm) δ 165.6 (C=O), 162.7 (C2=O), 161.8 (C7), 160.6 (Phenyl-C2), 158.8 (C4), 155.5 (C8a), 151.1 (Phenyl-C4), 134.7 (Phenyl-C6), 131.6 (C5), 126.5 (Phenyl-C1), 120.7 (Phenyl-C5), 119.3 (C6), 113.5 ((Phenyl-C3), 113.1 (C4a), 109.6 (C3), 108.6 (C8), 62.3 (CH2), 56.2 (OCH3). MS (m/z %): 326.24 (4.06, M+), 325.61 (10.6, M-H+), 44.22 (100). Elemental analysis for C18H14O6, calcd.: C, 66.26; H, 4.32; Found: C, 66.45; H, 4.59.
General procedure for the preparation of 4-(chloromethyl)-7-(2-oxo-2-phenylethoxy)-2H-chromen-2-one derivatives (5, 6)
To a solution of 1 (0.32 g, 1.5 mmol) in acetone (10 mL), anhydrous K2CO3 (0.29 g, 3 mmol) and the appropriate phenacyl bromide (1.5 mmol) were added. The reaction mixture was kept under stirring at room temperature for about 4 h and was monitored by TLC (petroleum ether/ethyl acetate, 7:3 v/v). After filtration, the solution was concentrated under vacuum. The obtained residue was recrystallized from a mixture of EtOAc/acetone (4:1).
4-(Chloromethyl)-7-(2-oxo-2-phenylethoxy)-2H-chromen-2-one (5).
White solid; Yield 63%; mp 180–182 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.06 (d, 2H, J = 7.2 Hz, Phenyl-C2-H and C6-H), 7.79 (d, 1H, J = 8.9 Hz, C5-H), 7.73 (t, 1H, J = 7.4 Hz, Phenyl-C4-H), 7.60 (t, 2H, J = 7.6 Hz, Phenyl-C3-H and C5-H), 7.16 (d, 1H, J = 2.5 Hz, C8-H), 7.10 (dd, 1H, J1 = 8.9, J2 = 2.5 Hz, C6-H), 6.52 (s, 1H, C3-H), 5.79 (s, 2H, OCH2), 5.02 (s, 2H, CH2Cl). 13C NMR (100 Hz, DMSO-d6), δ (ppm) 194.8 (C=O), 161.8 (C2=O), 160.4 (C7), 155.5(C8a), 151.2 (C4), 134.6 (Phenyl-C4), 134.4 (Phenyl-C1), 129.3 (two Phenyl-C2 and-C6), 128.3 (two Phenyl-C3 and -C5), 126.8 (C5), 113.2 (C6), 112.6 (C3), 111.2 (C4a), 102.45 (C8), 71.07 (CH2), 41.8 (CH2). MS (m/z %): 328.88 (19.45, M+), 330.20 (15.51, M+ + 2), 329.80 (10.36, M+ + 1), 125.26 (100). Elemental analysis for C18H13ClO4, calcd.: C, 65.76; H, 3.99; Found: C, 66.02; H, 4.13.
4-(Chloromethyl)-7-(2-(4-chlorophenyl)-2-oxoethoxy)-2H-chromen-2-one (6)
Buff solid; Yield 66%; mp 195–197 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.07 (d, 2H, J = 8.4 Hz, Phenyl-C2-H and C6–H), 7.79 (d, 1H, J = 8.8 Hz, C5–H), 7.69 (d, 2H, J = 8.4 Hz, Phenyl-C3-H and C5-H), 7.17 (d, 1H, J = 2.2 Hz, C8–H), 7.11 (dd, 1H, J1 = 8.8 Hz, J2 = 2.2 Hz, C6-H), 6.53 (s, 1H, C3-H), 5.77 (s, 2H, OCH2), 5.02 (s, 2H, CH2Cl). 13C NMR (100 Hz, DMSO-d6): δ (ppm) 193.3 (C=O), 161.7 (C2=O), 160.4 (C7), 155.5 (C8a), 151.2 (C4), 139.2 (Phenyl-C4), 133.3 (Phenyl-C1), 130.3 (two phenyl-C2 and-C6), 129.4 (two phenyl-C3 and -C5), 126.8 (C5), 113.2 (C6), 112.6 (C3), 111.2 (C4a), 102.4 (C8), 71.1 (CH2), 41.8 (CH2). MS (m/z %): 362.08 (2.37, M+), 363.81 (4.51, M+ + 2), 139.18 (100). Elemental analysis for C18H12Cl2O4, calcd.: C, 59.53; H, 3.33; Found: C, 59.74.; H, 3.58.
General Procedure for the Preparation of 7-[(2-oxo-2-phenylethoxy)-2H-chromen-4-yl] methyl benzoate derivatives (7–12)
Pathway (A)
The same procedure used for the synthesis of 5 and 6 was adopted, except that 7-hydroxycoumarin derivatives 2–4 were used instead of 1.
Pathway (B)
The same procedure used for the synthesis of derivatives 2–4 was adopted, except that coumarin derivatives 5 and 6 were used instead of 1.
Pathway (C)
To a solution of 1 (0.32 g, 1.5 mmol) in DMF (10 mL), the appropriate sodium salt of (un)substituted benzoic acid (1.5 mmol), anhydrous K2CO3 (0.29 g, 3 mmol) and the appropriate phenacyl bromide (1.5 mmol) were added. The reaction mixture was kept under stirring at room temperature for about 4 h, heated at 90 °C overnight, then cooled and poured over ice-water. The separated solid products were found to be an impure mixture of different products with very low yields.
[2-Oxo-7-(2-oxo-2-phenylethoxy)-2H-chromen-4-yl]methyl benzoate (7)
Buff solid; Yield 66%; mp 242–244 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.07 (d, 2H, J = 7.3 Hz, Phenyl C2–H and C6–H), 7.96 (s, 2H, Phenyl C2`-H and C6’–H), 7.73 (t, 2H, J = 7.4 Hz, Phenyl C4 -H and C4’-H) 7.68 (d, 1H, J = 8.7 Hz, C5-H), 7.59–7.57 (m, 4H, Phenyl C3–H, C5–H, C3’–H and C5’–H), 6.85 (dd, 1H, J1 = 8.7 Hz, J2 = 2.0 Hz, C6-H), 6.78 (d, 1H, J = 2.0 Hz, C8–H), 6.28 (s, 1H, C3–H), 5.79 (s, 2H, COCH2), 5.60 (s, 2H, OCH2). 13C NMR (100 Hz, DMSO-d6), δ (ppm) 196.4 (C=O), 165.6 (C=O), 162.9 (C2=O), 160,6 (C7), 155.6 (C4), 150.9 (C8a), 134.3 (two Phenyl-C4’ and Phenyl-C1’), 133.10 (two Phenyl-C1 and Phenyl-C4), 129.9 (two Phenyl-C2 and C6), 129.5 (two Phenyl-C2` and C6`), 129.3 (two Phenyl-C3’ and C5’), 128.4 (two Phenyl-C3 and C5), 126.6 (C5), 113.7 (C6), 109.7 (C4a), 108.8 (C3), 102.9 (C8), 70.0 (COCH2), 62.5 (OCH2). MS (m/z %): 414.14 (23.61, M+), 413.24 (6.94, M-H+), 105.12 (100). Elemental analysis for C25H18O6, calcd.: C, 72.46; H, 4.38; Found: C, 72.23; H, 4.57.
{7-[2-(4-Chlorophenyl)-2-oxoethoxy]-2-oxo-2H-chromen-4-yl}methyl benzoate (8)
White solid; Yield 51%; mp 245–247 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.08 (d, 2H, J = 8.7 Hz, Phenyl C2–H and C6–H), 8.05 (d, 2H, J = 8.9 Hz, Phenyl C2’–H and C6’–H), 7.78 (d, 1H, J = 8.9 Hz, C5–H), 7.73 (t, 1H, J = 7.4 Hz, Phenyl C4–H), 7.66 (d, 2H, J = 8.6 Hz, Phenyl C3’–H, C5’–H), 7.61 (t, 2H, J = 7.7 Hz, Phenyl C3–H, C5–H,), 7.18 (d, 1H, J = 2.3 Hz, C8–H), 7.09 (dd, 1H, J1 = 8.6 Hz, J2 = 2.3 Hz, C6-H), 6.41 (s, 1H, C3-H), 5.81 (s, 2H, COCH2), 5.64 (s, 2H, OCH2). 13C NMR (100 Hz, DMSO-d6): δ (ppm): δ 194.2 (C=O), 164.8 (C=O), 161.8 (C2=O), 160.4 (C7), 155.4 (C4), 150.5 (C8a), 136.3 (Phenyl-C4`), 134.7 (Phenyl-C1`), 134.4 (Phenyl-C1), 131.8 (Phenyl-C4), 129.7 (two Phenyl-C2`and C6`), 129.3 (two Phenyl-C2 and C6), 128.4 (two Phenyl-C3`and C5`), 128.3 (two Phenyl-C3 and C5), 126.4 (C5), 113.4 (C6), 111.1 (C4a), 110.1 (C3), 102.4 (C8), 71.1 (COCH2), 62.8 (OCH2). MS (m/z %): 447.97 (12.78, M+), 450.63 (32.38, M+ + 2), 43.19 (100). Elemental analysis for C25H17ClO6, calcd.: C, 66.90; H, 3.82; Found: C, 67.12; H, 3.97.
[2-Oxo-7-(2-oxo-2-phenylethoxy)-2H-chromen-4-yl]methyl 4-chlorobenzoate (9)
White crystals; Yield 77%; mp 217–219 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.10–8.07 (m, 4H, Phenyl C2–H, C6–H, C2’–H and C6’–H), 7.78 (d, 1H, J = 8.9 Hz, C5–H), 7.73 (t, 1H, J = 7.4 Hz, Phenyl C4’–H), 7.66 (d, 2H, J = 8.5 Hz, Phenyl C3–H and C5–H), 7.61 (t, 2H, J = 7.6 Hz, Phenyl C3’–H and C5’–H), 7.18 (d, 1H, J = 2.4 Hz, C8-H), 7.09 (dd, 1H, J1 = 8.9 Hz, J2 = 2.4 Hz, C6-H), 6.41 (s, 1H, C3–H), 5.81 (s, 2H, COCH2), 5.64 (s, 2H, OCH2). 13C NMR (100 Hz, DMSO-d6): δ 194.2 (C=O), 164.8 (C=O), 161.8 (C2=O), 160.4 (C7), 155.3 (C4), 150.5 (C8a), 139.2 (Phenyl-C4), 134.6 (Phenyl-C4’), 134.5 (Phenyl-C1’), 131.8 (Phenyl-C2), 131.8 (Phenyl-C6), 129.7 (Phenyl-C2’), 129.7 (Phenyl-C6’), 129.4 (Phenyl-C3’), 129.4 (Phenyl-C5’), 128.4 (Phenyl-C3), 128.4 (Phenyl-C5), 128.2 (Phenyl-C1), 126.4 (C5), 113.4 (C6), 111.1 (C4a), 110.1 (C3), 102.4 (C8), 71.1 (CH2), 62.8 (CH2). MS (m/z %): 448.41 (38.92, M+), 450.14 (22.31, M+ + 2), 447.68 (28.44, M+-1), 384.02 (100). Elemental analysis for C25H17ClO6, calcd.: C, 66.90; H, 3.82; Found: C, 66.83; H, 4.05.
{7-[2-(4-Chlorophenyl)-2-oxoethoxy]-2-oxo-2H-chromen-4-yl}methyl 4-chlorobenzoate (10)
Buff crystals; Yield 51%; mp 228–230 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.09–8.07 (m, 4H, Phenyl C2–H, C6–H, C2`-H and C6`-H), 7.78 (d, 1H, J = 8.9 Hz, C5–H), 7.68–7.65 (m, 4H, Phenyl C3–H, C5–H, C3’–H and C5’–H), 7.20 (d, 1H, J = 2 Hz, C8–H), 7.09 (dd, 1H, J1 = 8.9, J2 = 2.0 Hz, C6–H), 6.41 (s, 1H, C3–H), 5.78 (s, 2H, COCH2), 5.64 (s, 2H, OCH2). 13C NMR (100 Hz, DMSO-d6): δ 193.3 (C=O), 164.8 (C=O), 161.8 (C2=O), 160.4 (C7), 155.6 (C4), 150.7 (C8a), 139.2 (two Phenyl-C4 and C4’), 131.8 (two Phenyl-C2 and C6), 130.4 (two Phenyl- C2’ and C6’), 129.6 (two Phenyl-C3’ and C5’), 129.4 (two Phenyl-C3 and C5), 128.3 (two Phenyl-C1 and C1’), 126.5 (C5), 113.9 (C6), 113.4 (C4a), 103.1 (C8), 102.4 (C3), 71.56 (CH2), 62.8 (CH2). MS (m/z %): 483.13 (35.54, M+), 485.01 (15.28, M+ + 2), 141.69 (100). Elemental analysis for C25H16Cl2O6, calcd.: C, 62.13; H, 3.34; Found: C, 61.96; H, 3.56.
[2-Oxo-7-(2-oxo-2-phenylethoxy)-2H-chromen-4-yl]methyl 2-methoxybenzoate (11)
White solid; Yield 45%; mp 220–222 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.06 (d, 2H, J = 7.7 Hz, Phenyl C2’–H and C6’–H), 7.78 (d, 1H, J = 6.4 Hz, C5–H), 7.76 (d, 1H, J = 8.7 Hz, Phenyl C6–H), 7.73 (m, 1H, Phenyl C4’–H), 7.64–7.62 (m, 3H, Phenyl C3’–H and C5’–H and C4’H), 7.23 (d, 1H, J = 7.2 Hz, C6–H), 7.18 (s, 1H, C8-H), 7.09 (dd, 2H, J1 = 13.7, J2 = 6.5 Hz, Phenyl C3-H and C5-H), 6.46 (s, 1H, C3-H), 5.81 (s, 2H, COCH2), 5.60 (s, 2H, OCH2), 3.88 (s, 3H, OCH3). 13C NMR (100 Hz, DMSO-d6): δ (ppm) 194.2 (C=O), 165.6 (C=O), 161.7 (C2=O), 160.4 (C7), 158.9 (Phenyl-C2), 155.3 (C4), 151.0 (C8a), 134.7 (Phenyl-C4), 134.6 (Phenyl-C4`), 134.4 (phenyl-C1`), 131.6 (phenyl-C6), 129.3 (Phenyl-C2`), 129.3 (Phenyl-C6`), 128.4 (Phenyl-C3`), 128.4 (Phenyl-C5`), 126.4 (C5), 120.8 (Phenyl-C1), 119.4 (Phenyl-C5), 113.2 (Phenyl-C3), 113.1 (C6), 111.1 (C4a), 109.8 (C3), 102.4 (C8), 71 (COCH2), 62.4 (OCH2), 56.28 (OCH3). MS (m/z %): 444.46 (9.60, M+), 77.32 (100). Elemental analysis for C26H20O7, calcd.: C, 70.27; H, 4.54; Found: C, 70.44; H, 4.63.
{7-[2-(4-Chlorophenyl)-2-oxoethoxy]-2-oxo-2H-chromen-4-yl}methyl 2-methoxybenzoate (12)
White solid; Yield 51%; mp 218–220 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.07 (d, 2H, J = 8.2 Hz, Phenyl C2’–H and C6’–H), 7.78 (d, 1H, J = 6.8 Hz, C5–H), 7.74 (d, 1H, J = 7.7 Hz, Phenyl C6–H), 7.69 (d, 2H, J = 8.3 Hz, Phenyl C3’–H and C5’–H), 7.62 (t, 1H, J = 8.0 Hz, Phenyl C4–H), 7.22 (d, 1H, J = 7.7 Hz, C6–H), 7.19 (s, 1H, C8–H), 7.09 (dd, 2H, J1 = 11.3 Hz, J2 = 7.7 Hz, Phenyl C3–H and C5–H), 6.46 (s, 1H, C3–H), 5.78 (s, 2H, COCH2), 5.60 (s, 2H, OCH2), 3.87 (s, 3H, OCH3). 13C NMR (100 Hz, DMSO-d6): δ (ppm) 193.3 (C=O), 165.6 (C=O), 161.6 (C2=O), 160.4 (C7), 158.9 (Phenyl-C2`), 155.3 (C4), 150.9 (C8a), 139.2 (Phenyl-C4`), 134.7 (Phenyl-C4), 133.3 (Phenyl-C1`), 131.7 (Phenyl-C6), 131.6 (Phenyl-C2`), 130.3 (Phenyl-C6`), 129.4 (Phenyl-C3), 126.5 (Phenyl-C3`), 126.4 (Phenyl-C5`), 120.8 (C5), 119.4 (Phenyl-C1), 113.2 (Phenyl-C5), 113.1 (C6), 111.1 (C4a), 109.8 (C3), 102.3 (C8), 71.1 (COCH2), 62.3 (OCH2), 56.2 (OCH3). MS (m/z %): 478.24 (8.62, M+), 479.23 (18.46, M+ + 1), 480.20 (12.17, M+ + 2), 235.15 (100). Elemental analysis for C26H19ClO7, calcd.: C, 65.21; H, 4.00; Found: C, 65.03; H, 4.19.
General procedure for the preparation of (7-acetoxy-2-oxo-2H-chromen-4-yl)methyl benzoate derivatives (13–15)
The appropriate 2–4 derivative (1.5 mmol) was added to a mixture of acetic anhydride and acetic acid (1:1, 10 mL). The reaction mixture was refluxed for 12 h, cooled and poured onto ice. The crude off-white solid was then filtered, washed with water, and dried. The obtained solid was recrystallized from ethanol.
(7-Acetoxy-2-oxo-2H-chromen-4-yl)methyl benzoate (13)
White solid; Yield 71%; mp 192–194 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.09 (d, 2H, J = 7.3 Hz, Phenyl C2–H and C6–H), 7.92 (d, 1H, J = 8.6 Hz, C5–H), 7.73 (t, 1H, J = 7.1 Hz, Phenyl C4–H), 7.59 (t, 2H, J = 7.4 Hz, Phenyl C3–H and C5–H), 7.37 (s, 1H, C8–H), 7.25 (d, 1H, J = 8.5 Hz, C6–H), 6.56 (s, 1H, C3–H), 5.66 (s, 2H, OCH2), 2.33 (s, 3H, COCH3). 13C NMR (100 Hz, DMSO-d6): δ (ppm) 169.2 (C=O), 165.5 (C=O), 159.8 (C2=O), 154.2 (C7), 153.6 (C4), 150.2 (C8a), 134.3 (phenyl-C4), 129.9 (two Phenyl-C2 and C6), 129.4 (two Phenyl-C3 and C5), 129.3 (phenyl-C1), 126.4 (C5), 119.2 (C6), 115.3 (C4a), 112.7 (C8), 110.9 (C3), 62.5 (OCH2), 21.3 (COCH3). MS (m/z %): 338.55 (24.52, M+), 337.37 (18.27, M+-1), 280.45 (100). Elemental analysis for C19H14O6, calcd.: C, 67.45; H, 4.17; Found: C, 67.63; H, 4.39.
(7-Acetoxy-2-oxo-2H-chromen-4-yl)methyl 4-chlorobenzoate (14).
Off-White solid; Yield 88%; mp 216–218 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.09 (d, 2H, J = 8.2 Hz, Phenyl C2–H and C6-H), 7.92 (d, 1H, J = 7.0 Hz, C5–H), 7.67 (d, 2H, J = 6.6 Hz, Phenyl C3–H and C5–H), 7.37 (s, 1H, C8–H), 7.25 (d, 1H, J = 5.2 Hz, C6–H), 6.60 (s, 1H, C3–H), 5.67 (s, 2H, OCH2), 2.34 (s, 3H, COCH3). 13C NMR (100 Hz, DMSO-d6): δ (ppm) 169.4 (C=O), 165.4 (C=O), 159.4 (C2=O), 154.3 (C7), 153.6 (C4), 149.9 (C8a), 134.6 (phenyl-C4), 131.8 (two Phenyl-C2 and C6), 129.6 (two Phenyl-C3 and C5), 128.2 (phenyl-C1), 126.4 (C5), 119.2 (C6), 115.5 (C4a), 112.8 (C8), 110.9 (C3), 62.7 (OCH2), 21.4 (COCH3). MS (m/z %): 372.14 (43.76, M+), 374.07 (63.76, M+ + 2), 312.09 (100). Elemental analysis for C19H13ClO6, calcd C, 61.22; H, 3.52; Found C, 61.39; H, 3.68.
(7-Acetoxy-2-oxo-2H-chromen-4-yl)methyl 2-methoxybenzoate (15).
White solid; Yield 74%. mp 186–188 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.89 (d, 1H, J = 8.5 Hz, Phenyl C6–H), 7.79 (d, 1H, J = 6.7 Hz, C5–H), 7.62 (t, 1H, J = 7.5 Hz, Phenyl C4–H), 7.35 (s, 1H, C8–H), 7.25 (d, 1H, J = 7.9 Hz, C6–H), 7.22 (d, 1H, J = 8.8 Hz, Phenyl C3–H), 7.08 (t, 1H, J = 7.4 Hz, Phenyl C5–H), 6.63 (s, 1H, C3–H), 5.62 (s, 2H, OCH2), 3.88 (s, 3H, OCH3), 2.33 (s, 3H, COCH3). 13C NMR (100 Hz, DMSO-d6): δ (ppm) 169.3 (C=O), 165.6 (C=O), 158.9 (C2=O), 155.5 (phenyl-C2), 154.2 (C7), 153.5 (C4), 150.2 (C8a), 134.7 (phenyl-C4), 131.6 (Phenyl C6), 126.5 (Phenyl-C1), 126.4 (phenyl-C5), 120.7 (C5), 119.3 (C6), 115.2 (C4a), 113.5 (phenyl-C3), 113.1 (C8), 109.6 (C3), 62.5 (OCH2), 56.2 (OCH3), 21.3 (COCH3). MS (m/z %): 368.36 (6.80, M+), 44.13 (100). Elemental analysis for C20H16O7, calcd.: C, 65.22; H, 4.38; Found C, 65.41; H, 4.45.
Biological tests
In vitro cytotoxic activity using MTT assay
In this study, all synthesized compounds were initially screened for their in vitro antitumor activities against two human BC cell lines, namely, MCF-7 (estrogen positive) and MDA-MB-231(triple negative), to select the most promising ones for further screenings via MTT assay using Doxorubicin, as a reference drug as described in the reported method46,47.
In vitro (EGFR) enzyme inhibition assay
Fourteen compounds that showed good cytotoxic activity against the TNBC cell line (MDA-MB-231) were examined for in vitro EGFR inhibition assay relative to erlotinib as a reference drug according to the reported protocol48.
In vitro (ARO) enzyme inhibition assay
Eleven compounds that showed good cytotoxic activity against the ER + cell line (MCF-7) were examined for in vitro ARO inhibition assay relative to EXM as a reference drug according to the reported method49.
Cell cycle arrest
DNA flow cytometric cell cycle analysis was performed on treated MCF-7 cells with IC50 concentration of compound 8 and treated MDA-MB-231 cells with IC50 concentration of compounds 10, 12 and 14 according to the reported procedure50.
Apoptosis assay
Annexin V-FITC/PI dual staining assay was performed according to the reported method51 to further study the ability of compounds 8, 10, 12 and 14 to induce apoptosis by introducing flow cytometry-based analysis.
Measurement of the level of Bax Bcl-2 and Caspase-9
MCF-7 cells were treated with compound 8, while MDA-MB-231 cells were exposed to compounds 10, 12 and 14 according to the manufacturer's instructions52.
Molecular docking study with both EGFR and human ARO enzymes
Crystal structures of EGFR (PDB ID: 1M17)18 and ARO (PDB ID: 3EQM)53 were obtained from the PDB. Regarding enzyme inhibition assay, the most potent compounds were selected for docking study using Molecular Operating Environment (MOE) software.
Data availability
All study data are presented in this article and its supplementary information files.
References
Schick, J., Ritchie, R. P. & Restini, C. Breast cancer therapeutics and biomarkers: Past, present, and future approaches. Breast Cancer Basic Clin. Res. 15, 1178223421995854. https://doi.org/10.1177/1178223421995854 (2021).
Murphy, C. G. & Dickler, M. N. Endocrine resistance in hormone-responsive breast cancer: Mechanisms and therapeutic strategies. Endocr.-Relat. Cancer 23(8), R337–R352. https://doi.org/10.1530/erc-16-0121 (2016).
Johnston, S. R. D. & Dowsett, M. Aromatase inhibitors for breast cancer: Lessons from the laboratory. Nat. Rev. Cancer 3(11), 821–831. https://doi.org/10.1038/nrc1211 (2003).
Samara, N. & Casper, R. F. Aromatase Inhibitors. Infertility in Women with Polycystic Ovary Syndrome 119–133 (Springer, 2018).
Nuciforo, P., Radosevic-Robin, N., Ng, T. & Scaltriti, M. Quantification of HER family receptors in breast cancer. Breast Cancer Res. BCR 17, 53. https://doi.org/10.1186/s13058-015-0561-8 (2015).
Noolvi, M. N., Patel, H. M. & Kaur, M. Benzothiazoles: Search for anticancer agents. Eur. J. Med. Chem. 54, 447–462. https://doi.org/10.1016/j.ejmech.2012.05.028 (2012).
El-Helby, A. A. et al. Design, synthesis, molecular modeling, in vivo studies and anticancer activity evaluation of new phthalazine derivatives as potential DNA intercalators and topoisomerase II inhibitors. Bioorg. Chem. 103, 104233. https://doi.org/10.1016/j.bioorg.2020.104233 (2020).
Liu, Y. & Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener 6, 21. https://doi.org/10.1186/s40035-017-0092-6 (2017).
Bao, H. et al. BHX, a novel pyrazoline derivative, inhibits breast cancer cell invasion by reversing the epithelial-mesenchymal transition and down-regulating Wnt/β-catenin signalling. Sci. Rep. 7(1), 9153. https://doi.org/10.1038/s41598-017-09655-7 (2017).
Hassan, M., Watari, H., AbuAlmaaty, A., Ohba, Y. & Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. BioMed. Res. Int. 2014, 150845. https://doi.org/10.1155/2014/150845 (2014).
Batran, R. Z. et al. Coumarinyl pyranopyrimidines as new neuropeptide S receptor antagonists; Design, synthesis, homology and molecular docking. Bioorgan. Chem. 75, 274–290. https://doi.org/10.1016/j.bioorg.2017.09.017 (2017).
Batran, R. Z., Kassem, A. F., Abbas, E. M. H., Elseginy, S. A. & Mounier, M. M. Design, synthesis and molecular modeling of new 4-phenylcoumarin derivatives as tubulin polymerization inhibitors targeting MCF-7 breast cancer cells. Bioorgan. Med. Chem 26(12), 3474–3490. https://doi.org/10.1016/j.bmc.2018.05.022 (2018).
Abdel Latif, N. A., Batran, R. Z., Khedr, M. A. & Abdalla, M. M. 3-Substituted-4-hydroxycoumarin as a new scaffold with potent CDK inhibition and promising anticancer effect: Synthesis, molecular modeling and QSAR studies. Bioorgan. Chem. 67, 116–129. https://doi.org/10.1016/j.bioorg.2016.06.005 (2016).
Lin, M. H. et al. Induction of ROS-independent JNK-activation-mediated apoptosis by a novel coumarin-derivative, DMAC, in human colon cancer cells. Chem.-Biol. Interact. 218, 42–49. https://doi.org/10.1016/j.cbi.2014.04.015 (2014).
Saidu, N. E. et al. Coumarin polysulfides inhibit cell growth and induce apoptosis in HCT116 colon cancer cells. Bioorgan. Med. Chem. 20(4), 1584–1593. https://doi.org/10.1016/j.bmc.2011.12.032 (2012).
Colozza, M., Califano, R., Minenza, E., Dinh, P. & Azambuja, E. Aromatase inhibitors: A new reality for the adjuvant endocrine treatment of early-stage breast cancer in postmenopausal women. Mini Rev. Med. Chem. 8(6), 564–574. https://doi.org/10.2174/138955708784534472 (2008).
Gobbi, S. et al. Novel highly potent and selective nonsteroidal aromatase inhibitors: Synthesis, biological evaluation and structure− activity relationships investigation. J. Med. Chem. 53(14), 5347–5351. https://doi.org/10.1021/jm100319h (2010).
Stamos, J., Sliwkowski, M. X. & Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 277(48), 46265–46272. https://doi.org/10.1074/jbc.M207135200 (2002).
Stefanachi, A. et al. Design, synthesis, and biological evaluation of imidazolyl derivatives of 4,7-disubstituted coumarins as aromatase inhibitors selective over 17-α-hydroxylase/C17−20 lyase. J. Med. Chem. 54, 1613–1625. https://doi.org/10.1021/jm101120u (2011).
Yamaguchi, Y. et al. Evaluation of synthesized coumarin derivatives on aromatase inhibitory activity. Bioorgan. Med. Chem. Lett. 27(12), 2645–2649. https://doi.org/10.1016/j.bmcl.2017.01.062 (2017).
Chen, S., Cho, M., Karlsberg, K., Zhou, D. & Yuan, Y.-C. Biochemical and biological characterization of a novel anti-aromatase coumarin derivative*. J. Biol. Chem. 279(46), 48071–48078. https://doi.org/10.1074/jbc.M406847200 (2004).
Nasr, T., Bondock, S. & Youns, M. Anticancer activity of new coumarin substituted hydrazide-hydrazone derivatives. Eur. J. Med. Chem. 76, 539–548. https://doi.org/10.1016/j.ejmech.2014.02.026 (2014).
Dhawan, S. et al. Synthesis, computational studies and antiproliferative activities of coumarin-tagged 1,3,4-oxadiazole conjugates against MDA-MB-231 and MCF-7 human breast cancer cells. Bioorgan. Med. Chem. 26(21), 5612–5623. https://doi.org/10.1016/j.bmc.2018.10.006 (2018).
Ragab, F. A. et al. Synthesis and biological evaluation of new coumarin derivatives as cytotoxic agents. Arch. der Pharm. 354(8), 2100029. https://doi.org/10.1002/ardp.202100029 (2021).
Hussain, M. K. et al. Design and synthesis of ERα/ERβ selective coumarin and chromene derivatives as potential anti-breast cancer and anti-osteoporotic agents. RSC Adv. 4(17), 8828–8845. https://doi.org/10.1039/C3RA45749D (2014).
Zhu, W. et al. Design, synthesis and 3D-QSAR analysis of novel 2-hydrazinyl-4-morpholinothieno [3, 2-d] pyrimidine derivatives as potential antitumor agents. Eur. J. Med. Chem. 57, 162–175. https://doi.org/10.1016/j.ejmech.2012.09.002 (2012).
Liu, Z. et al. Design, synthesis and biological evaluation of novel thieno [3, 2-d] pyrimidine derivatives possessing diaryl semicarbazone scaffolds as potent antitumor agents. Eur. J. Med. Chem. 87, 782–793. https://doi.org/10.1016/j.ejmech.2014.10.022.(2014) (2014).
Kini, S. G., Choudhary, S. & Mubeen, M. Synthesis, docking study and anticancer activity of coumarin substituted derivatives of benzothiazole. J. Comput. Methods Mol. Des. 2(1), 51–60 (2012).
Bana, E. et al. A novel coumarin-quinone derivative SV37 inhibits CDC25 phosphatases, impairs proliferation, and induces cell death. Mol. Carcinog. 54(3), 229–241. https://doi.org/10.1002/mc.22094 (2015).
Farghaly, A. M. et al. New thieno[3,2-d]pyrimidine-based derivatives: Design, synthesis and biological evaluation as antiproliferative agents, EGFR and ARO inhibitors inducing apoptosis in breast cancer cells. Bioorgan. Chem. 115, 105208. https://doi.org/10.1016/j.bioorg.2021.105208 (2021).
Pisani, L. et al. Discovery of a novel class of potent coumarin monoamine oxidase B inhibitors: Development and biopharmacological profiling of 7-[(3-chlorobenzyl)oxy]-4-[(methylamino)methyl]-2H-chromen-2-one methanesulfonate (NW-1772) as a highly potent, selective, reversible, and orally active monoamine oxidase B inhibitor. J. Med. Chem. 52(21), 6685–6706. https://doi.org/10.1021/jm9010127 (2009).
Chen, H. M., Armstrong, Z., Hallam, S. J., Withers, S. & G,. Synthesis and evaluation of a series of 6-chloro-4-methylumbelliferyl glycosides as fluorogenic reagents for screening metagenomic libraries for glycosidase activity. Carbohydr. Res. 421, 33–39. https://doi.org/10.1016/j.carres.2015.12.010 (2016).
Ragab, F. A. et al. Synthesis and biological evaluation of new coumarin derivatives as cytotoxic agents. Arch. der Pharm. 354(8), e2100029. https://doi.org/10.1002/ardp.202100029 (2021).
Stefanachi, A. et al. Design, synthesis, and biological evaluation of imidazolyl derivatives of 4,7-disubstituted coumarins as aromatase inhibitors selective over 17-α-hydroxylase/C17–20 lyase. J. Med. Chem. 54(6), 1613–1625. https://doi.org/10.1021/jm101120u (2011).
Frasinyuk, M., Bondarenko, S. & Khilya, V. Synthesis and properties of 4-chloromethyl-6-hydroxy-coumarins and 4-(2-benzofuryl)-6-hydroxycoumarins. Chem. Heterocycl. Compd. 45, 290–296. https://doi.org/10.1007/s10593-009-0275-x (2009).
Kathuria, A. et al. Substrate specificity of acetoxy derivatives of coumarins and quinolones towards Calreticulin mediated transacetylation: Investigations on antiplatelet function. Bioorgan. Med. Chem. 20(4), 1624–1638. https://doi.org/10.1016/j.bmc.2011.11.016 (2012).
Sonne-Hansen, K. & Lykkesfeldt, A. E. Endogenous aromatization of testosterone results in growth stimulation of the human MCF-7 breast cancer cell line. J. Steroid Biochem. Mol. Biol. 93(1), 25–34. https://doi.org/10.1016/j.jsbmb.2004.11.005 (2005).
Vermes, I., Haanen, C., Steffens-Nakken, H. & Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184(1), 39–51. https://doi.org/10.1016/0022-1759(95)00072-i (1995).
Zimmermann, K. C. & Green, D. R. How cells die: Apoptosis pathways. J. Allergy Clin. Immunol. 108(4 Suppl), S99-103. https://doi.org/10.1067/mai.2001.117819 (2001).
Ghavami, S. et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 46, 497–510. https://doi.org/10.1136/jmg.2009.066944 (2009).
Wu, P., Nielsen, T. E. & Clausen, M. H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 36(7), 422–439. https://doi.org/10.1016/j.tips.2015.04.005 (2015).
Verma, G. et al. Pharmacophore modeling, 3D-QSAR, docking and ADME prediction of quinazoline based EGFR inhibitors. Arab. J. Chem. 12(8), 4815–4839. https://doi.org/10.1016/j.arabjc.2016.09.019 (2019).
Favia, A. D., Cavalli, A., Masetti, M., Carotti, A. & Recanatini, M. Three-dimensional model of the human aromatase enzyme and density functional parameterization of the iron-containing protoporphyrin IX for a molecular dynamics study of heme-cysteinato cytochromes. Proteins 62(4), 1074–1087. https://doi.org/10.1002/prot.20829 (2006).
Mohamed, T. K., Batran, R. Z., Elseginy, S. A., Ali, M. M. & Mahmoud, A. E. Synthesis, anticancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis. Bioorgan. Chem. 85, 253–273. https://doi.org/10.1016/j.bioorg.2018.12.040 (2019).
Cruciani, G., Crivori, P., Carrupt, P. A. & Testa, B. Molecular fields in quantitative structure–permeation relationships: The VolSurf approach. J. Mol. Struct. THEOCHEM 503(1), 17–30. https://doi.org/10.1016/S0166-1280(99)00360-7 (2000).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 65(1), 55–63. https://doi.org/10.1016/0022-1759(83)90303-4 (1983).
Grela, E., Kozłowska, J. & Grabowiecka, A. Current methodology of MTT assay in bacteria—a review. Acta Histochem. 120(4), 303–311. https://doi.org/10.1016/j.acthis.2018.03.007 (2018).
Nakamura, J. L. The epidermal growth factor receptor in malignant gliomas: Pathogenesis and therapeutic implications. Expert Opin. Ther. Targets 11(4), 463–472. https://doi.org/10.1517/14728222.11.4.463 (2007).
Fantacuzzi, M. et al. Synthesis, biological evaluation, and docking study of indole aryl sulfonamides as aromatase inhibitors. Eur. J. Med. Chem. 185, 111815. https://doi.org/10.1016/j.ejmech.2019.111815 (2020).
Calvert, M. E., Lannigan, J. A. & Pemberton, L. F. Optimization of yeast cell cycle analysis and morphological characterization by multispectral imaging flow cytometry. Cytom. Part A J. Int. Soc. Anal. Cytol. 73(9), 825–833. https://doi.org/10.1002/cyto.a.20609 (2008).
Lieschke, E. et al. Flow cytometric single cell-based assay to simultaneously detect cell death, cell cycling, DNA content and cell senescence. Cell Death Differ. 29(5), 1004–1012. https://doi.org/10.1038/s41418-022-00964-7 (2022).
Abo-Salem, H. M. et al. Synthesis and bioactivity assessment of novel spiro pyrazole-oxindole congeners exhibiting potent and selective in vitro anticancer effects. Mol. (Basel Switz.) 25, 5. https://doi.org/10.3390/molecules25051124 (2020).
Ghosh, D., Griswold, J., Erman, M. & Pangborn, W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 457(7226), 219–223. https://doi.org/10.1038/nature07614 (2009).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
F.T. conducted the experimental part. All authors have substanial and equal contributions in the conception, design, data analysis, writing and reviewing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Takla, F.N., Bayoumi, W.A., El-Messery, S.M. et al. Developing multitarget coumarin based anti-breast cancer agents: synthesis and molecular modeling study. Sci Rep 13, 13370 (2023). https://doi.org/10.1038/s41598-023-40232-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-40232-3
- Springer Nature Limited