
Abstract
[4Fe–4S] clusters are essential cofactors in many proteins involved in biological redox-active processes. Density functional theory (DFT) methods are widely used to study these clusters. Previous investigations have indicated that there exist two local minima for these clusters in proteins. We perform a detailed study of these minima in five proteins and two oxidation states, using combined quantum mechanical and molecular mechanical (QM/MM) methods. We show that one local minimum (L state) has longer Fe–Fe distances than the other (S state), and that the L state is more stable for all cases studied. We also show that some DFT methods may only obtain the L state, while others may obtain both states. Our work provides new insights into the structural diversity and stability of [4Fe–4S] clusters in proteins, and highlights the importance of reliable DFT methods and geometry optimization. We recommend r2SCAN for optimizing [4Fe-4S] clusters in proteins, which gives the most accurate structures for the five proteins studied.
Similar content being viewed by others


Introduction
Direct electron transfer occurs in biological systems via iron–sulfur clusters, cytochromes, and blue copper proteins1. Iron–sulfur clusters were identified about 50 years ago in biological systems2. Since then, it has become evident that they play many important roles in biology. They are common in nature and are essential for electron transport3 and catalysis4. These roles often overlap with oxidoreductase proteins, which catalyze electron and proton transfers, as well as substrate binding and catalytic changes5,6,7,8.
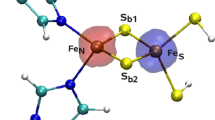
There are several types of FeS sites. Rubredoxins contain the simplest FeS site, which consists of a single Fe ion coordinated to four cysteine (Cys) residues9,10. The [2Fe–2S] ferredoxins have two Fe ions, two bridging sulfide ions, and two Cys residues coordinated to each Fe ion11,12. The Rieske site has another type of [2Fe–2S] cluster in which two histidine (His) residues coordinate one of the Fe ions in place of Cys13. Some proteins contain more complicated [4Fe–4S] clusters14,15,16, which comprise four Fe ions that are connected by four sulfide ions. In addition, each Fe ion is coordinated to a Cys residue17 (cf. Fig. 1). There are also ferredoxins with [3Fe–4S] clusters14,18, in which one Fe ion and one Cys residue are missing compared to [4Fe–4S] ferredoxins. More complicated iron–sulfur clusters are found in some proteins (such as the Fe8S7Cys6 P-cluster and the MoFe7S9C FeMo cluster in nitrogenase), are sometimes associated with catalytic functions19,20.

QM system in the QM/MM calculations.
The [4Fe–4S] clusters in ferredoxins use the \({\mathrm{Fe}}_{3}^{\mathrm{II}}{\mathrm{Fe}}_{1}^{\mathrm{III}}/{\mathrm{Fe}}_{2}^{\mathrm{II}}{\mathrm{Fe}}_{2}^{\mathrm{III}}\) redox couple, which results in redox potentials between − 0.7 and − 0.3 V21. High-potential iron–sulfur proteins (HiPIP) also contain [4Fe–4S] clusters, but in contrast to the [4Fe–4S] ferredoxins, they utilize \({\mathrm{Fe}}_{2}^{\mathrm{II}}{\mathrm{Fe}}_{2}^{\mathrm{III}}/{\mathrm{Fe}}_{1}^{\mathrm{II}}{\mathrm{Fe}}_{3}^{\mathrm{III}}\) redox pair, giving them a substantially more positive potential (+ 0.05 to + 0.5 V). Several spectroscopic, structural, and theoretical methods have been used to study the physical characteristics and electronic structures of FeS clusters22. Synthetic analogs of the FeS clusters have also been intensively studied23.
In recent years, computational chemistry calculations have been widely used to evaluate and predict structural and electrical properties of transition metal complexes22,24,25,26,27,28,29,30,31,32,33,34,35. Most of these studies have been performed with density functional theory (DFT).
Systems with several spin-coupled metal ions have complicated electronic structures. In the [4Fe–4S] clusters, the individual iron ions are typically in the high-spin state, but these spins are antiferromagnetically coupled to a lower total spin. It is crucial to characterize the antiferromagnetic coupling at the same level of theory as the strong metal–ligand bonding and the weaker metal–metal interactions to discuss trends in [4Fe–4S] clusters. The broken-symmetry (BS) approach36,37, which considers weakly interacting electrons physically, makes this possible.
Accurate geometrical structures are important to predict the electronic properties of iron–sulfur clusters. Case et al.35 used DFT to calculate redox potentials for a few iron–sulfur clusters. Later, they extended their work using a similar method but with optimized structures of the clusters22 rather than assumed geometries. The results indicated that optimized structures tend to give longer bond lengths than the experimentally observed ones, e.g., by 0.03 Å for the [Fe4S4(SCH3)4]2− model cluster. This study also better reproduced the experimentally determined redox potentials, which they partly attributed to the geometry optimization.
Recently, we systematically investigated the redox potentials of iron–sulfur clusters with various quantum mechanics/molecular mechanics (QM/MM) and QM-cluster methods24. We then observed conspicuous differences in the Fe–Fe distances for some of the QM/MM structures for [4Fe–4S] clusters in proteins. Since the geometry plays a significant role in the accuracy of redox calculations, we decided to investigate this observation in depth. In this work, we investigate the occurrence and nature of two local minima for [4Fe–4S] clusters in proteins using QM/MM methods with various DFT functions and basis sets. We focus on five proteins that contain [4Fe–4S] clusters in different oxidation and spin states: three ferredoxins and two HiPIPs. We show that one local minimum (L state) has longer Fe–Fe distances than the other (S state). We also show that some DFT methods may only obtain the L state, while others may obtain both states. We compare the local minima and discuss their implications for computational models. Our work provides new insights into the structural diversity and stability of [4Fe–4S] clusters in proteins, and highlights the importance of using reliable DFT methods for accurate modeling of these systems. Additionally, we investigate how the local minima affect the BS states. The paper is organized as follows: “Methods” describes the computational methods and models used; “Result and discussion” presents the results and discussion; “Conclusions” summarizes the main conclusions and perspectives.
Methods
Studied systems
We have studied five [4Fe–4S] clusters in protein crystal structures. The proteins and the employed crystal structures are described in Table 1. Three [4Fe–4S] ferredoxins were studied from Bacillus thermoproteolyticus (1IQZ; 4Fd1)15, Desulfovibrio africanus (1FXR; 4Fd2)16, and Azotobacter vinelandii (5FD1; 4Fd3)14. In addition, two HiPIP sites were studied from Allochromatium vinosum (1CKU; Hip1)38 and Halorhodospira halophila (2HIP; Hip2). All QM/MM structures were taken from our recent study24, in which a description of the setup of the proteins, the protonation states, and the equilibration of the structure can be found.
QM calculations
QM calculations were performed using the Turbomole software40. We employed nine different DFT methods: four GGA functionals, with no admixture of HF exchange: PBE41, BP8642,43, BLYP42,44, and B97D45, two meta GGA functional with no HF exchange: TPSS46 and r2SCAN47, as well as three hybrid functionals: TPSSh (10% HF exchange)48, B3LYP (20% HF exchange42,44,49, and B3LYP* (15% HF exchange)42,44,49,50. All the functionals were combined with two different basis sets (def2-SV(P)51 or def2-TZVPD52,53). The calculations were sped up by the resolution-of-identity approximation54,55,56 which is a variational fitting of the electron density in an auxiliary basis set (we employed the built-in def2-SV(P)51 and universal57 auxiliary basis sets in Turbomole). Empirical dispersion corrections were included with the DFT-D3 approach58 and Becke–Johnson damping59, as implemented in Turbomole.
In some calculations, the QM system was immersed into a continuum solvent, employing the conductor-like screening model (COSMO)60,61. The default optimized COSMO atomic radii and a water solvent radius of 1.3 Å were employed to construct the solvent-accessible surface cavity62. For the Fe ions, a radius of 2.0 Å was used63. Structures for the QM + COSMO calculations were taken directly from the QM/MM calculations without further optimization. The dielectric constant of proteins has been much discussed, but values of 4–20 are typically used64,65. We tested three values for the dielectric constant (4, 20, and 80).
The QM system consisted of the Fe and S ions, as well as the directly coordinated Cys groups, modeled by CH3CH2S–, i.e., Fe4S4(SCH2CH3)4 for the whole cluster (cf. Fig. 1).
The electronic structures of the iron–sulfur clusters are complicated. Each Fe ion is in the high-spin state (five and four unpaired electrons for Fe(III) and Fe(II), respectively). However, these spins are coupled antiferromagnetically to a lower spin state in the polynuclear clusters, S = 0 or ½66,67, as is specified in Table 1. The BS approach in DFT calculations describes such antiferromagnetically coupled sites36,37. There are six possible BS states for the [4Fe–4S] clusters (two Fe ions with dominant beta spin can be selected among the four Fe ions in six different ways). We examined all possibilities and selected the one with the most favorable energy for the QM system of each protein and oxidation state with TPSS/def2-SV(P). This BS state was also used for the other calculations. In general, we discuss results only for the energetically lowest BS state.
The BS states were generated either by the fragment approach of Szilagyi and Winslow33 or by obtaining one BS state by first optimizing the highest possible spin state (all unpaired electrons aligned), flipping the spins to the desired state, and then obtaining the other BS states by simply swapping coordinates of the Fe ions68. Spin densities, geometries and relative energies of the various BS states for the two oxidation states of 4Fd3 at the PBE/def2-SV(P) level of theory are shown in Supplementary Tables S12–S14 in the Supplementary Information.
QM/MM calculations
The QM/MM calculations were performed with the ComQum software69,70. In this approach, the protein and solvent are split into two subsystems: System 1 (the QM region) was relaxed by the QM method, whereas system 2 involved the remaining part of the protein and the solvent, and was kept fixed at the original coordinates (equilibrated crystal structure). In the QM calculations, system 1 was represented by a wavefunction (the functionals and basis sets are described in the previous section), whereas all the other atoms were represented by an array of partial point charges, one for each atom, taken from the MM setup. Thereby, the polarization of the QM system by the surroundings is included in a self-consistent manner. When there is a bond between systems 1 and 2, the hydrogen link-atom approach was employed: The QM system was capped with hydrogen atoms (hydrogen link atoms, HL), the positions of which are linearly related to the corresponding carbon atoms (carbon link atoms, CL) in the full system69,71. All atoms were included in the point-charge model, except the CL atoms72. Further details of the QM/MM calculations are given in the Supplementary Information.
Results and discussion
This study examines the structure of [4Fe–4S] clusters in proteins. The test set includes three [4Fe–4S] ferredoxins and two high-potential iron–sulfur proteins (cf. Table 1). In our previous study, we observed extensive differences in the Fe–Fe distances of the clusters in different proteins when optimizing the structures at the TPSS/def2-SV(P) level of theory. After some test calculations, we found out that the [4Fe–4S] clusters in all five proteins and all studied oxidation states could attain two local minima. Consequently, we have systematically studied the energies and electronic properties of these two minima. The two local minima were obtained with the QM/MM calculations, either by first restraining some Fe–Fe distances in initial optimizations and then removing all restraints and reoptimizing the structure or simply by starting from the other oxidation state (when they represent different minima). In the following, we analyze the geometry of the two local minima in both oxidation states, their spin states, and how their stability varies with the DFT functionals and basis sets. Furthermore, we examine how the environment (modeled either by point charges in QM/MM calculations or implicitly by a continuum solvent) affects the stability of the minima. We also test whether the minima can be obtained with other functionals and basis sets besides TPSS/def2-SV(P).
Geometries
Table 2 shows a statistical analysis of the calculated Fe–Fe and Fe–S distances for the five studied FeS clusters at the TPSS/def2-SV(P) level of theory (the raw data are given in Supplementary Tables S1–S5 in the Supplementary Information). It can be seen that the DFT calculations give two local minima for all structures and oxidation states. They differ primarily in the Fe–Fe distances. For example, for the Ox state of 4Fd1, one local minimum has Fe–Fe distances of 2.76–2.80 Å (2.77 Å on average), whereas for the other, the distances are 2.56–2.66 Å (2.64 Å on average), i.e. a difference of 0.13 Å on average. In the following, these local minima will be referred to as L and S (long and short). From Table 2, it can be seen that the average Fe–Fe distances are the same for all ferredoxin sites: 2.76–2.78 vs. 2.63–2.65 Å for L and S, respectively (i.e. a difference of 0.12–0.13 Å between L and S). The reduced sites have only 0.01 Å shorter average Fe–Fe distances. The reduced HiPiPs, also have similar Fe–Fe distances, but for the oxidized HiPIPs the Fe–Fe distances are 2.80–2.81 vs. 2.58 Å, with an appreciably larger difference between L and S (0.22–0.23 Å) and also a larger difference between the two oxidation states (0.02–0.04 Å; in fact, the average Fe–Fe distance increase upon reduction for the S states). This is caused by the differing \({\mathrm{Fe}}_{1}^{\mathrm{II}}{\mathrm{Fe}}_{3}^{\mathrm{III}}\) oxidation state.
Table 2 shows also the average Fe–S distances. They show appreciably smaller differences between the L and S local minima: 0.01–0.02 Å for the ferredoxins and reduced HiPIPs, but 0.04 Å for the oxidized HiPIPs (L always gives longer average Fe–S distances). The average Fe–S distances increase upon reduction by 0.02–0.05 Å for all sites and minima (most for S of the HiPIPs).
Naturally, it is interesting to know which of the two local minima agrees best with the crystal structures of the corresponding proteins. In Table 2, we show the average Fe–Fe and Fe–S distances in the crystal structures, as well as the mean absolute difference of the Fe–Fe (MADFe–Fe) and Fe–S distances (MADFe–S) from the crystal values for the four optimized structures of each protein. It can be seen that the crystallographic Fe–Fe distances are always in between those obtained for the two minima, 2.66–2.73 Å. For three of the proteins, the reduced L minimum gives the smallest MADFe–Fe (0.03–0.06 Å) among the four optimized structures, whereas for Hip2, instead Ox–S gives the best results and for 4Fd3, all four structures give the same MAD. For MADFe–S, instead, Ox–S gives the best results for four of the structures (0.03–0.07 Å) and Red–S for Hip1. However, the differences are small and in two cases, other structures (Ox–L or Red–S) give the same MAD. Thus, it seems hard to point out which of the two minima correspond to the crystal structures, probably owing to the mediocre accuracy of the crystal structures and the risk that they are partly photoreduced during crystallography and therefore may represent a mixture of oxidation states.
Stability of the local minima with different methods
Next, we calculated the relative stability of the two local minima in the two oxidation states and the five proteins. From the results in Table 3, it can be seen that the L state is always more stable than the S state, by 26–30 kJ/mol for the ferredoxins and by 19–24 kJ/mol for the HiPIPs (3 kJ/mol less for the Ox state than for the Red state).
We also performed single-point energy calculations on the TPSS/def2-SV(P)-optimized structures of the two local minima for all systems, changing the functional to B3LYP (with the def2-SV(P) basis set), changing the basis set to def2-TZVPD (with the TPSS functional), and changing the explicit QM/MM environment of the [4Fe–4S] cluster to an implicit one (COSMO continuum solvation model with dielectric constants of 4, 20, or 80; TPSS/def2-SV(P) calculations). The results presented in Table 3 show that the description of the surroundings has a very small effect, changing the relative stabilities by less than 5 kJ/mol. Likewise, the three dielectric constants give the same relative energies within 2 kJ/mol. On the other hand, the basis set gives a larger effect, up to 20 kJ/mol. Moreover, B3LYP increases the stability of the L minimum (to 86–94 kJ/mol, but 151–159 for the Ox state of the HiPIPs, compared to 19–30 kJ/mol with the TPSS).
Spin states of the local minima
Mulliken spin populations of the iron ions in the various oxidation states and local minima at the TPSS/def2-SV(P) level of theory are collected in Table 4. It can be seen that for the sites with the \({\mathrm{Fe}}_{2}^{\mathrm{II}}{\mathrm{Fe}}_{2}^{\mathrm{III}}\) charge state (Ox 4Fd and Red HiPIP), the Fe spin populations are the same on all four Fe ions (within 0.03–0.09 e), 3.4–3.5 e for the L minimum and 3.3–3.4 e for the S minimum (in absolute terms). Thus, the Fe spin populations of the S local minimum are always slightly lower than those of the L minimum, by 0.13 e on average. For the other two charge states, two of the Fe ions (those with a surplus of β spin, i.e. a negative spin population in Table 4) have a larger spin population, and the other two have a lower spin population, with a difference of 0.2–0.5 e. For the Red 4Fd sites, the spin populations are 2.9–3.6 e, still with a difference of 0.1–0.2 e between the two minima. However, for the oxidized state of the two HiPIPs, the difference is much larger, 0.4–0.8 e with an average of 0.53 e, because the S minimum has smaller spin populations, 3.1 e for those with negative populations and 2.6–2.9 e, for those with positive populations. It can also be seen that the L and S local minima do not depend on the BS states (i.e., which Fe ions have negative spin populations): The various proteins have different preferred BS states, but both local minima are found for all proteins, obtained for the same BS state for each protein. Enlarging the basis set from def2-SV(P) to def2-TZVPD has a small effect on the spin populations. Further details of spin populations with the B3LYP functional and the def2-TZVPD basis set are given in the Supplementary Information.
Dependence of the S and L local minima on the DFT functional and basis set
The results presented above are based on structures optimized at the TPSS/def2-SV(P) level of theory. In this section, we consider structures optimized with other DFT functionals or basis sets, testing also the larger def2-TZVPD basis set52,53. We used the PBE41, BP8642,43, BLYP42,44, and B97-D45, functionals with no HF exchange, as well as the meta GGA TPSS46 and r2SCAN47 functionals, and the hybrid TPSSh (10% HF exchange)48, B3LYP (20% HF exchange)42,44,49, B3LYP* (15% HF exchange)42,44,49,50 and PBE073,74 (25% HF exchange) functionals. Interestingly, we were able to reproduce the two local minima for some combinations of the functionals and basis sets, but for many combinations, the L local minimum was only obtained and S local minimum disappeared (structures optimized starting from the S minimum converged to the L minimum, indicating that S is converted from a local minimum to a shoulder on the potential energy surface). Table 5 summarizes the difference between AvFe–Fe values in the L and S states (ΔDFe–Fe) as well as the energy difference between the L and S states (ΔE = ES – EL) for all combinations of the functionals and the basis sets. As can be seen, the TPSS, BLYP, PBE, and BP86 pure functionals with the def2-SV(P) basis set give both local minima for all proteins in both the Red and Ox states. On the other hand, B97-D/def2-SV(P) gives only the L local minima, except for the Red states of Hip1 and Hip2. Moreover, the r2SCAN, TPSSh, B3LYP, and B3LYP* functionals give only the L minima for all proteins and redox states. As the basis set is expanded to def2-TZVPD, the S local minimum disappears for most functionals. Only some of the pure functionals (especially BP86 and PBE) give both local minima for some states, whereas the hybrid functionals and r2SCAN never show the S local minima with the larger basis set.
It can be seen that the four functionals with results for all proteins with the small basis set give quite similar ΔDFe–Fe (slightly lower for BLYP than for the other three functionals) and also similar trends among the five proteins. For the proteins and functionals that give both minima for the large basis set, the increase in the basis set typically leads to a slightly smaller ΔDFe–Fe (by up to 0.04 Å). However, for ∆E the variation is larger. BLYP gives the largest values (52–66 kJ/mol), whereas PBE gives the smallest values (up to 19 kJ/mol), actually suggesting that for the two Ox HiPiP sites, the two minima are essentially degenerate. Still, the trends among the five proteins are the same. Increasing the basis sets typically increases ∆E, but the effect is small (− 4 to 11 kJ/mol).
Since the L local minimum is more stable than the S local minimum in both oxidation states for all enzymes and in all combinations of the functionals and basis sets, as well as being the only possible minimum for some functionals, we compared the calculated structures of L minima with the crystal structures. The raw data are shown in Tables S7–S11 in the Supplementary Information and the results are summarized in Tables 6 and 7, showing MADFe–Fe and MADFe–S for all proteins and oxidation states and all functionals and basis sets. To compare the various approaches, we averaged the lowest MAD value for the two oxidation states for each protein (the oxidation state of all crystal structures is not always reported and it may change during data collection owing to photoreduction). Based on these values for the Fe–Fe distances, it can be seen that r2SCAN/def2-SV(P) performs best with an average deviation of only 0.04 Å. The same functional with the larger basis set and TPSS, BLYP and PBE with the small basis set all have average deviations of 0.06 Å. Increasing the basis set deteriorates the results for all functionals (by 0.01 Å on average), indicating some cancellation of errors. B3LYP and B3LYP* give appreciably worse results than the other functionals (0.12–0.13 Å), whereas TPSSh gives results similar to those of the worst pure functionals. It is notable that Hip2 (the protein with the lowest resolution) gives twice as high MADFe–Fe as the other proteins, 0.13 Å, compared to 0.06–0.08 Å (averaged over all functionals and basis sets).
The results for the Fe–S bond lengths are quite different. In this case, optimizations with the larger basis set give the best results (improving the average MADs by 0.01 Å on average). The lowest average MAD (0.04 Å) is obtained for PBE, TPSSh and r2SCAN. B3LYP and B3LYP* still give the worst results (0.06–0.07 Å), but the differences are small. Again, Hip2 gives appreciably worse MADs than the other four structures (0.10 compared to 0.03–0.06 Å, averaged over all DFT methods). Averaging the results for the two sets of distances indicates that r2SCAN with both basis sets gives the best result (0.05 Å), but the differences are small (all the other methods, except B3LYP and B3LYP* give an average MAD of 0.06 Å).
Conclusions
Geometries are important for predicting accurate electronic properties of molecular systems and iron-sulfur clusters, and today DFT methods are widely used to evaluate and predict the geometrical and electronic properties of transition metal complexes. For example, DFT-optimized structures for a [Fe4S4(SCH3)4]2– model cluster reproduce experimentally determined redox potentials more accurately than experimental structures22. In this study, we examined the recent observation that QM/MM calculations for [4Fe–4S] clusters in proteins may give two local minima at the differing in the Fe–Fe distances24. Accordingly, we have studied five [4Fe–4S] clusters in protein crystal structures shown in Table 1. We examined the geometry of local minima in both oxidation states, their spin states, and how their stability is affected by the choice of DFT functionals and basis sets.
The results indicate that the crystallographic Fe–Fe distances are always in between those obtained for the two minima. Nevertheless, it is difficult to determine which of the two minima corresponds to the crystal structure, probably owing to the mediocre accuracy of the crystal structures and the possibility that they are partially photoreduced during crystallography and therefore may represent a mixture of oxidation states due to the photoreduction process. However, the calculations indicate that the L state is always more stable than the S state.
Moreover, we investigated whether the two local minima could also be obtained with other DFT functionals, as well as the dependence of the results on the basis set. Using seven other functionals (both pure and hybrid functionals) along with two basis sets, we optimized the cluster structures. Interestingly, while all methods could give the L local minimum, the S minimum was only obtained with some pure functionals. In particular, the S minimum was never found with the r2SCAN, TPSSh, B3LYP, and B3LYP*. Increasing the basis set often led to the disappearance of the S minimum, also with the pure functionals. Considering that the S minimum is less stable than the L minimum and that is found mainly with the small basis set and with the older DFT functionals, it is likely that it represents a spurious artifact, rather than a real alternative that could be observed experimentally.
Therefore, we finally compared the structures of the L local minimum optimized with the nine DFT methods and the two basis sets with the starting crystal structures. The results in Tables 6 and 7 show that r2SCAN/def2-SV(P) gives the best structures. Considering that this method is quite fast and it does not give the spurious S local minimum, we recommend this approach for optimizing a [4Fe–4S] cluster in proteins.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information file.
References
Liu, J. et al. Metalloproteins containing cytochrome, iron-sulfur, or copper redox centers. Chem. Rev. 114, 4366–4469 (2014).
Beinert, H. Recent developments in the field of iron-sulfur proteins. FASEB J. 4, 2483–2491 (1990).
Han, A. L., Yagi, T. & Hatefi, Y. Studies on the structure of NADH:ubiquinone oxidoreductase complex: Topography of the subunits of the iron-sulfur protein component. Arch. Biochem. Biophys. 275, 166–173 (1989).
Beinert, H., Kennedy, M. C. & Stout, C. D. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 96, 2335–2373 (1996).
Lindahl, P. A. & Kovacs, J. A. Reactivities and biological functions of iron-sulfur clusters. J. Cluster Sci. 1, 29–73. https://doi.org/10.1007/BF00703585 (1990).
Qiu, D., Kumar, M., Ragsdale, S. W. & Spiro, T. G. Nature’s carbonylation catalyst: Raman spectroscopic evidence that carbon monoxide binds to iron, not nickel, in CO dehydrogenase. Science 264, 817–819 (1994).
Lindahl, P. A., Ragsdale, S. W. & Munck, E. Mössbauer study of CO dehydrogenase from Clostridium thermoaceticum. J. Biol. Chem. 265, 3880–3888 (1990).
Howard, J. B. & Rees, D. C. Structural basis of biological nitrogen fixation. Chem. Rev. 96, 2965–2982 (1996).
Dauter, Z., Wilson, K. S., Sieker, L. C., Moulis, J. M. & Meyer, J. Zinc- and iron-rubredoxins from Clostridium pasteurianum at atomic resolution: A high-precision model of a ZnS4 coordination unit in a protein. Proc. Natl. Acad. Sci. U. S. A. 93, 8836–8840 (1996).
Cuypers, M. G., Mason, S. A., Mossou, E., Haertlein, M. & Forsyth, V. T. RCSB PDB-5NW3: The cryofrozen atomic resolution X-ray crystal structure of perdeuterated Pyrococcus furiosus Rubredoxin (100K, 0.59A resolution). https://doi.org/10.2210/pdb5nw3/pdb (2017).
Correll, C. C., Batie, C. J., Ballou, D. P. & Ludwig, M. L. Phthalate dioxygenase reductase: A modular structure for electron transfer from pyridine nucleotides to [2Fe–2S]. Science (80-). 258, 1604–1610 (1992).
Morales, R. et al. Refined X-ray structures of the oxidized, at 1.3 Å, and reduced, at 1.17 Å, [2Fe−2S] Ferredoxin from the cyanobacterium anabaena PCC7119 show redox-linked conformational changes. Biochemistry 38, 15764–15773 (1999).
Kolling, D. J., Brunzelle, J. S., Lhee, S. M., Crofts, A. R. & Nair, S. K. Atomic resolution structures of rieske iron-sulfur protein: Role of hydrogen bonds in tuning the redox potential of iron-sulfur clusters. Structure 15, 29–38 (2007).
Stout, C. D. Crystal structures of oxidized and reduced Azotobacter vinelandii ferredoxin at pH 8 and 6. J. Biol. Chem. 268, 25920–25927 (1993).
Fukuyama, K., Okada, T., Kakuta, Y. & Takahashi, Y. Atomic resolution structures of oxidized [4Fe-4S] ferredoxin from Bacillus thermoproteolyticus in two crystal forms: Systematic distortion of [4Fe-4S] cluster in the protein. J. Mol. Biol. 315, 1155–1166 (2002).
Séry, A. et al. Crystal structure of the ferredoxin I from Desulfovibrio africanus at 2.3 Å resolution. Biochemistry 33, 15408–15417 (1994).
Fukuyama, K., Matsubara, H., Tsukihara, T. & Katsube, Y. Structure of [4Fe-4S] ferredoxin from Bacillus thermoproteolyticus refined at 2.3 Å resolution. Structural comparisons of bacterial ferredoxins. J. Mol. Biol. 210, 383–398 (1989).
Kissinger, C. R., Sieker, L. C., Adman, E. T. & Jensen, L. H. Refined crystal structure of ferredoxin II from Desulfovibrio gigas at 1.7 Å. J. Mol. Biol. 219, 693–715 (1991).
Roth, L. E. & Tezcan, F. A. X-ray Crystallography BT—Nitrogen Fixation: Methods and Protocols (ed. Ribbe, M. W.) 147–164 (Humana Press, 2011). https://doi.org/10.1007/978-1-61779-194-9_10.
Einsle, O. Nitrogenase FeMo cofactor: An atomic structure in three simple steps. J. Biol. Inorg. Chem. 19, 737–745. https://doi.org/10.1007/s00775-014-1116-7 (2014).
Sticht, H. & Rösch, P. The structure of iron-sulfur proteins. Prog. Biophys. Mol. Biol. 70, 95–136 (1998).
Torres, R. A., Lovell, T., Noodleman, L. & Case, D. A. Density functional and reduction potential calculations of Fe4S4 clusters. J. Am. Chem. Soc. 125, 1923–1936 (2003).
Holm, R. H. Synthetic approaches to the active sites of iron-sulfur proteins. Acc. Chem. Res. 10, 427–434 (1977).
Jafari, S., Tavares Santos, Y. A., Bergmann, J., Irani, M. & Ryde, U. Benchmark study of redox potential calculations for iron-sulfur clusters in proteins. Inorg. Chem. 61, 5991–6007 (2022).
Siegbahn, P. E. M. A quantum chemical approach for the mechanisms of redox-active metalloenzymes. RSC Adv. 11, 3495–3508 (2021).
Kurniawan, I. et al. A theoretical study on redox potential and pKa of [2Fe-2S] cluster model from iron-sulfur proteins. Bull. Chem. Soc. Jpn. 91, 1451–1456 (2018).
Bruschi, M., Breglia, R., Arrigoni, F., Fantucci, P. & De Gioia, L. Computational approaches to the prediction of the redox potentials of iron and copper bioinorganic systems. Int. J. Quantum Chem. 116, 1695–1705 (2016).
Arumugam, K. & Becker, U. Computational redox potential predictions: Applications to inorganic and organic aqueous complexes, and complexes adsorbed to mineral surfaces. Minerals. 4, 345–387. https://doi.org/10.3390/min4020345 (2014).
Cheng, J., Liu, X., VandeVondele, J., Sulpizi, M. & Sprik, M. Redox potentials and acidity constants from density functional theory based molecular dynamics. Acc. Chem. Res. 47, 3522–3529 (2014).
Perrin, B. S. Jr. et al. Web-based computational chemistry education with CHARMMing III: Reduction potentials of electron transfer proteins. PLoS Comput. Biol. 10, e1003739 (2014).
Perrin, B. S. Jr., Niu, S. & Ichiye, T. Calculating standard reduction potentials of [4Fe–4S] proteins. J. Comput. Chem. 34, 576–582 (2013).
Niu, S. & Ichiye, T. Density functional theory calculations of redox properties of iron–sulphur protein analogues. Mol. Simul. 37, 572–590 (2011).
Szilagyi, R. K. & Winslow, M. A. On the accuracy of density functional theory for iron–sulfur clusters. J. Comput. Chem. 27, 1385–1397 (2006).
Higashi, M. & Kato, S. Theoretical study on electronic and spin structures of [Fe2S2]2+,+ cluster: Reference interaction site model self-consistent field (RISM-SCF) and multireference second-order Møller−Plesset Perturbation Theory (MRMP) approach. J. Phys. Chem. A 109, 9867–9874 (2005).
Mouesca, J.-M., Chen, J. L., Noodleman, L., Bashford, D. & Case, D. A. Density functional/Poisson-Boltzmann calculations of redox potentials for iron-sulfur clusters. J. Am. Chem. Soc. 116, 11898–11914 (1994).
Noodleman, L. Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 74, 5737–5743 (1981).
Noodleman, L., Lovell, T., Liu, T., Himo, F. & Torres, R. A. Insights into properties and energetics of iron–sulfur proteins from simple clusters to nitrogenase. Curr. Opin. Chem. Biol. 6, 259–273 (2002).
Parisini, E. et al. Ab initio solution and refinement of two high-potential iron protein structures at atomic resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 55, 1773–1784 (1999).
Breiter, D. R., Meyer, T. E., Rayment, I. & Holden, H. M. The molecular structure of the high potential iron-sulfur protein isolated from Ectothiorhodospira halophila determined at 2.5-Å resolution. J. Biol. Chem. 266, 18660–18667 (1991).
Furche, F. et al. Turbomole. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 91–100 (2014).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 38, 3098–3100 (1988).
Perdew, J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 33, 8822 (1986).
Lee, C., Yang, W. & Parr, G. R. Development of the Colic–Salvetti correlation-energy into a functional of the electron density. Am. Phys. Soc. 37, 785–789 (1988).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Tao, J., Perdew, J. P., Staroverov, V. N. & Scuseria, G. E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 91, 146401 (2003).
Furness, J. W., Kaplan, A. D., Ning, J., Perdew, J. P. & Sun, J. Accurate and numerically efficient r2SCAN meta-generalized gradient approximation. J. Phys. Chem. Lett. 11, 8208–8215 (2020).
Staroverov, V. N., Scuseria, G. E., Tao, J. & Perdew, J. P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 119, 12129–12137 (2003).
Becke, A. D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 96, 2155–2160 (1992).
Vosko, S. H., Wilk, L. & Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 58, 1200–1211 (1980).
Schäfer, A., Horn, H. & Ahlrichs, R. Fully optimized contracted Gaussian-basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577 (1992).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297 (2005).
Rappoport, D. & Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 133, 134105 (2010).
Lehtola, S. Straightforward and accurate automatic auxiliary basis set generation for molecular calculations with atomic orbital basis sets. J. Chem. Theory Comput. 17, 6886–6900 (2021).
Eichkorn, K., Treutler, O., Öhm, H., Häser, M. & Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 240, 283–289 (1995).
Eichkorn, K., Weigend, F., Treutler, O. & Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem Acc. Theory Comput. Model. Theor. Chim. Acta. 97, 119–124 (1997).
Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 8, 1057–1065 (2006).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Klamt, A. & Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2, 799–805. https://doi.org/10.1039/P29930000799 (1993).
Schäfer, A., Klamt, A., Sattel, D., Lohrenz, J. C. W. & Eckert, F. COSMO implementation in TURBOMOLE: Extension of an efficient quantum chemical code towards liquid systems. Phys. Chem. Chem. Phys. 2, 2187–2193 (2000).
Klamt, A., Jonas, V., Bürger, T. & Lohrenz, J. C. W. Refinement and parametrization of COSMO-RS. J. Phys. Chem. A 102, 5074–5085 (1998).
Sigfridsson, E. & Ryde, U. Comparison of methods for deriving atomic charges from the electrostatic potential and moments. J. Comput. Chem. 19, 377–395 (1998).
Ullmann, G. M. & Knapp, E.-W. Electrostatic models for computing protonation and redox equilibria in proteins. Eur. Biophys. J. 28, 533–551 (1999).
Warshel, A. & Dryga, A. Simulating electrostatic energies in proteins: Perspectives and some recent studies of pK as, redox, and other crucial functional properties. Proteins Struct. Funct. Bioinform. 79, 3469–3484 (2011).
Kent, T. A., Huynh, B. H. & Münck, E. Iron-sulfur proteins: Spin-coupling model for three-iron clusters. Proc. Natl. Acad. Sci. U. S. A. 77, 6574–6576 (1980).
Papaefthymiou, V., Girerd, J. J., Moura, I., Moura, J. J. G. & Muenck, E. Moessbauer study of D. gigas ferredoxin II and spin-coupling model for Fe3S4 cluster with valence delocalization. J. Am. Chem. Soc. 109, 4703–4710 (1987).
Greco, C., Fantucci, P., Ryde, U. & de Gioia, L. Fast generation of broken-symmetry states in a large system including multiple iron–sulfur assemblies: Investigation of QM/MM energies, clusters charges, and spin populations. Int. J. Quantum Chem. 111, 3949–3960 (2011).
Ryde, U. The coordination of the catalytic zinc in alcohol dehydrogenase studied by combined quantum-chemical and molecular mechanics calculations. J. Comput. Aided. Mol. Des. 10, 153–164 (1996).
Ryde, U. & Olsson, M. H. M. Structure, strain, and reorganization energy of blue copper models in the protein. Int. J. Quantum Chem. 81, 335–347 (2001).
Reuter, N., Dejaegere, A., Maigret, B. & Karplus, M. Frontier bonds in QM/MM methods: A comparison of different approaches. J. Phys. Chem. A 104, 1720–1735 (2000).
Hu, L., Söderhjelm, P. & Ryde, U. On the convergence of QM/MM energies. J. Chem. Theory Comput. 7, 761–777 (2011).
Perdew, J. P., Ernzerhof, M. & Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 105, 9982–9985 (1996).
Adamo, C. & Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999).
Acknowledgements
This investigation has been supported by grants from the Swedish research council (project 2018-5003) and the Vice President for Research and Technology, University of Kurdistan (project 01/9/3778). The computations were performed on computer resources provided by the Swedish National Infrastructure for Computing (SNIC) at Lunarc at Lund University and HPC2N at Umeå University, partially funded by the Swedish Research Council (Grant 2018-05973).
Author information
Authors and Affiliations
Contributions
S.J. Conceptualization; computations; data curation, formal analysis; validation; visualization; review & editing. U.R. Conceptualization; funding acquisition; investigation; project administration; supervision; resources; software; methodology; review & editing. M.I. Conceptualization; funding acquisition; investigation; project administration; supervision; writing; review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jafari, S., Ryde, U. & Irani, M. Two local minima for structures of [4Fe–4S] clusters obtained with density functional theory methods. Sci Rep 13, 10832 (2023). https://doi.org/10.1038/s41598-023-37755-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-37755-0
- Springer Nature Limited