
Abstract
A selective synthesis of unsymmetrically functionalized disiloxanes via the subsequent hydrosilylation of internal alkynes in the first step, and alkynes (terminal or internal) or 1,3-diynes in the second, with 1,1,3,3-tetramethyldisiloxane (1) is presented for the first time. Using developed approaches performed in a stepwise or one-pot manner a new family of disubstituted disiloxanes was obtained which had previously been inaccessible by other synthetic methods. Moreover, symmetrically functionalized disiloxanes were obtained by direct hydrosilylation of 2 equivalents of terminal or internal alkynes with 1, showing the unique versatility of the hydrosilylation process. Three examples of symmetric disiloxanes were characterized by single crystal X-ray diffraction for the first time. As a result, a wide group of new compounds which can find potential applications as building blocks or coupling agents was obtained and characterized.
Similar content being viewed by others
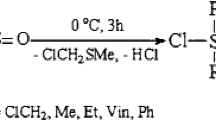

Introduction
The organosilicon compounds are one of the most important classes of molecules used in modern organic and materials chemistry in academia and industry1,2,3,4. Due to the almost unlimited possibilities of designing their structure, which influences their properties, such compounds are widely used as e.g., hybrid materials5, anti-corrosive coatings6, silicon rubbers7, and active pharmaceutical indigents (API)8. Among several methods for the preparation of organosilicon compounds, transition metal-catalyzed (TM) hydrosilylation of unsaturated bonds is one of the most important because of its 100% atom economy, high selectivity, and good functional group tolerance9.
The bifunctional organosilicon compounds, such as 1,1,3,3-tetramethyldisiloxane (1), are attractive building blocks that can be applied as polymeric electrolytes10, coupling agents11, or conjugated polymers12, depending on the groups attached to the silicon atom. The simplest dihydride siloxane 1 is produced as a by-product of the silicon industry. Because of its low-boiling point, relatively low price, and good stability in air and moisture, it is used as a versatile scaffold for the synthesis of advanced molecules13.
The mono- or bifunctionalization of 1 via the hydrosilylation of alkenes or allyl compounds with various substituents (e.g., epoxy14, aminopropyl15, γ-methacryloxypropyl16, boryl/germyl17, silyl18) is well-established. The synthesis of the symmetrically functionalized disiloxanes by hydrosilylation of alkynes19,20,21,22,23,24,25 or other methods26,27,28 was described in a few works in which disiloxanes mainly with monosubstituted alkenyl groups20,21,22,23,24,25,26,28,29,30,31 were obtained that were subsequently used in Hiyama coupling19,20,21,22,24,25,30,31. Reports on the synthesis of disiloxanes with disubstituted alkenyl groups are almost neglected and are limited to four compounds with no functional groups attached19,29,32. On the other hand, there are no examples of unsymmetrical functionalized derivatives bearing two different alkenyl groups.
Bearing in mind the above-described information, based on our experience in the utilization of the carbon-carbon triple (C≡C) bonds in hydrosilylation reactions33,34,35,36,37,38,39,40,41,42,43, we decided to develop a simple and straightforward synthetic protocol for obtaining new unsymmetrically functionalized disiloxanes via subsequent hydrosilylation of alkynes in the first step, and alkynes or 1,3-diynes in the second, using stepwise or one-pot approaches. The stepwise method gives an opportunity for the detailed characterization of both hydrosilylation processes and products of monofunctionalization (RSiMe2OSiMe2H). Isolation of monofunctionalization products allows the type of catalyst to be changed after the first hydrosilylation which opens new possibilities for the modification of the Si-H group. On the other hand, the one-pot method allows for avoiding the isolation of products (RSiMe2OSiMe2H) and the addition of a second portion of catalyst if the same systems can be used for both steps. Moreover, we planned to study the synthesis of the symmetrically functionalized disiloxanes obtained by the direct hydrosilylation of two equivalents of alkynes (terminal or internal) with disiloxane 1, in the presence of different catalysts. This will show the unique utility of the hydrosilylation process to obtain all possible products using one method. It cannot be done by the use of any other reported method.
Results and discussion
In the first stage of the research, the monofunctionalization of 1,1,3,3-tetramethyldisiloxane (1) was optimized. A series of terminal and internal alkynes (2) were tested in the hydrosilylation with 1 in the presence of various catalysts and under different conditions. It was found that for diphenylethyne 2a, a high yield of monofunctionalization product 3a was observed if the reaction was performed at room temperature. The requirement for selective activation of only one Si-H bond was the use of a 10-fold excess of disiloxane 1 in the presence of Karstedt’s catalyst (Pt2(dvs)3) (Fig. 1). If a lower amount of 1 was added, both Si-H groups reacted and symmetrical disiloxanes with two of the same alkenyl groups were formed. After the full conversion of 2a, the excess of 1 was evaporated from the post-reaction mixture, and the targeted product was isolated by filtration through silica pad to remove the catalyst and some by-products obtained from the decomposition of disiloxane 1.

Hydrosilylation of internal alkynes 2a‒c with 1 towards the monofunctionalized disiloxane derivatives 3a‒c.
With the optimized reaction conditions in hand, we tested two other internal alkynes. The hydrosilylation of 1,2-bis(4-bromophenyl)ethyne (2b) and aliphatic 4-octyne (2c) gave hydrosilylation products 3b and 3c with excellent selectivity and high isolation yields. For both, the formation of side hydrosilylation products was not observed. Moreover, the synthesis of 3a and 3c was performed on a gram-scale, giving monoalkenyl-functionalized disiloxane derivatives 3a‒c with very high isolation yields. These compounds have been reported for the first time and are perfect reagents for further transformations typical of the Si-H group. The attempts to obtain analogue products with terminal alkynes were unsuccessful despite using different catalysts, the stoichiometry of reagents, and reaction conditions.
In the next stage of the study, we used 3a‒c in the subsequent hydrosilylation of internal and terminal alkynes, as well as 1,3-diynes, to obtain unsymmetrically functionalized disiloxanes 4a‒n (Fig. 2). The hydrosilylation of 4-octyne (2c) with 3a yielded 4a with a good isolation yield in 18 h at 100 °C. The same product was obtained through the hydrosilylation of diphenylethyne (2a) with 3c, with a similar yield and selectivity. The application of bis(4-bromophenyl)ethyne (2b) and 3a gave 4b in a good isolation yield. Similarly, 4b could also be obtained via the hydrosilylation of 2a with 3b, without the loss of selectivity. In contrast to internal alkynes, hydrosilylation of terminal alkynes with monofunctionalized disiloxanes 3 was performed in the presence of the PtO2/Xphos catalytic system under inert gas, to ensure the highest possible selectivity of the process.

The synthesis of unsymmetrical disiloxane derivatives 4a‒n via the hydrosilylation of alkynes and 1,3-diynes with 3a‒c.
The hydrosilylation of phenylacetylene (2d) with 3a provided 4c with excellent selectivity (trans/gem = 99/1) and a very good isolation yield. Very similar results were observed when 4-bromophenylacetylene (2e) was used as the reagent. The applied catalytic system was also efficient for the hydrosilylation of sterically hindered 3,3-dimethylbut-1-yne (2f) and trimethyl((2-methylbut-3-yn-2-yl)oxy)silane (2g) to give β-(E)-isomers 4e and 4f exclusively. Compound 4f was further easily converted through hydrolysis to 4g with a good isolation yield (68%), without any changes in selectivity. It is worth underlining that the possibility of the use of a different catalyst in the second step of modification is undoubtedly an advantage of the stepwise method since Karstedt’s catalyst is not selective in the reaction with terminal alkynes and thus the above-described products cannot be obtained.
Similar to the internal and terminal alkynes, the reactivity of more challenging reagents 1,3-diynes (symmetrical and unsymmetrical) was tested. The presence of two C≡C bonds makes this class of compounds attractive building blocks and at the same time, it is a great challenge to modify them in a selective manner. The hydrosilylation of 1,4-bis(trimethylsilyl)buta-1,3-diyne (2h) with 3a in the presence of Karstedt’s catalyst yielded product 4h with alkenyl and 1,3-enynyl moieties attached to the disiloxane core. The silicon atom was bonded with the internal carbon adjacent to the second C≡C bond. This was confirmed by 1D nOe NMR (see ESI, Fig. S38, page S35). The same regio- and stereoselectivity was observed for aliphatic 1,3-diynes substituted with methyl (2i) and cyclohexyl groups (2j). The hydrosilylation of symmetrical 1,4-substituted 1,3-diynes with thienyl (2k) and phenyl (2l) groups led to the stereoselective formation of expected addition products 4k and 4l with high isolation yields. We also tested the hydrosilylation of two unsymmetrical 1,3-diynes (2m and 2n) with 3a. The functionalization of 2m substituted with phenyl and triisopropyl groups occurred in a stereo- and regioselective manner, giving a pure product 4m. The silicon atom was attached to the internal carbon of the 1,3-diyne scaffold, whereas the alkenyl proton was bonded to the Cα connected to the phenyl ring. The hydrosilylation of 2n diyne with aliphatic t-butyl and n-hexyl substituents led to the product of monoaddition 4n with a 72% yield. The structures of products 4m and 4n and the regioselectivity of the processes were confirmed by 1D nOe NMR (see ESI, Figs. S57 and S59, pages S45 and S46).
The facts that in the stepwise, subsequent hydrosilylations of internal alkynes 2a‒c with disiloxane 1 in the first step, and internal alkynes (2a‒c) or 1,3-diynes (2h‒n) with monofunctionalized siloxanes 3a‒c in the second, Karstedt’s catalysts were used, encouraged us to perform the synthesis of 4a and 4m in a one-pot manner (Fig. 3). Moreover, we could check if one portion of the catalyst was going to be efficient for both transformations. Moreover, the work-up concerning the isolation of monofunctionalization products 3a‒c would be unnecessary. Therefore, we reacted alkyne 4-octyne (2c) with the 10-fold excess of disiloxane 1 in the presence of Karstedt’s catalyst (10−4 mol) at room temperature. The excess of 1 was evaporated after 3 hours when the complete conversion of alkyne 2c was confirmed. Subsequently, 1,4-diphenyletyne (2a) and toluene were added, and the reaction mixture was heated up to 100 °C. After 18 hours, the reaction had finished and product 4a was obtained. The selectivity of the one-pot process and the isolated yield of 4a were found to be similar to those obtained using the stepwise method. By the same method product 4m was synthesized. However, the synthesis of 3a was performed at room temperature for 18 hours as it was optimized for the stepwise approach.

Synthesis of unsymmetrical disiloxanes 4a and 4 m via one-pot, subsequent hydrosilylation reactions.
All synthesized unsymmetrically substituted disiloxanes 4a‒n were obtained and characterized for the first time. They constitute a novel class of organosilicon compounds possessing the Si-O-Si core suited to further functionalization due to the presence of reactive alkenyl, 1,3-enynyl moieties and other functional groups like Br or OH. It is worth emphasizing that, although studies on the synthesis of molecules based on a disiloxane core have been broadly carried out, there is no other method which allows the synthesis of analogues to 4a‒n systems possessing both di- or di- and monosubstituted alkenyl groups in the structure. Subsequent hydrosilylations of the C≡C of internal and terminal alkynes and/or 1,3-diynes gives such a unique opportunity and has herein been described for the first time.
In the last stage of our study, we performed the synthesis of symmetrically monoalkenyl-substituted disiloxanes (Fig. 4). This class of compounds can easily be prepared through the reaction of monofunctionalized siloxane 3a‒c with the corresponding alkyne 2a‒c in the presence of Karstedt’s catalyst or directly from the modification of both Si-H bonds with the application of 2 equiv. of alkyne 2 over disiloxane 1. The hydrosilylation of internal alkynes 2a‒c with disiloxane 1 selectively gave the cis-addition products 5a‒c with isolation yields from good to excellent. Products 5a and 5c were obtained selectively, isolated, and characterized for the first time. Product 5b was also synthesized with a high yield using the heterogenous system PtO/PtO2-Fe3O431.

The synthesis of symmetrical disiloxanes 5a‒k via the hydrosilylation of internal and terminal alkynes with 1.
Moreover, for compound 5b, the crystal structure was confirmed by single-crystal X-ray diffraction (Fig. 5a). To the best of our knowledge, this is the first example of the crystal structure of disiloxane 5 with two disubstituted alkenyl moieties.

The molecular structure of compounds 5b (a), 5e (b), and 5g (c). Displacement ellipsoids are shown at the 50% probability level. Selected geometrical parameters are summarized in Table S2 in ESI.
Subsequently, we investigated the functionalization of terminal alkynes with 1. Similar to the hydrosilylation of terminal alkynes with 3a, the reactions were performed in the presence of the in situ generated PtO2/Xphos catalytic system, which selectively led to the formation of trans-isomers in the cis-addition of the SiH bond to the C≡C bond. The addition of Si–H bonds to 2 equiv. of phenylacetylene 2d gave product 5d, as expected. The same regio- and stereoselectivity was observed for the hydrosilylation of phenylacetylene derivatives substituted with electron-withdrawing (–Br (2d), –F (2o), -CF3 (2p)) or electron-donating (–OMe (2r)) groups. Products 5e‒h were easily isolated with high yields, and the process occurred with excellent selectivity (97% or greater) of β-(E)-isomers. The synthesis of symmetrically alkenyl-substituted disiloxanes was also applicable for sterically hindered terminal alkynes such as 3,3-dimethylbut-1-yne (2f), trimethyl((2-methylbut-3-yn-2-yl)oxy)silane (2g), and ethynyltriisopropylsilane (2s). Products 5j and 5k could be obtained in almost quantitative yields. Disiloxanes 5a, c, g, j, and k were obtained and fully characterized for the first time. On the other hand, compounds 5b, d‒f, h‒i, and analogue symmetrical systems were synthesized using various approaches19,21,22,26,27,28,31,44. It is worth emphasizing that compounds 5e and 5g, were characterized by single crystal X-ray diffraction (Fig. 5b,c) and their crystal structures are the only representatives of such a family of compounds.
Finally, to prove that alkenyl-functionalized disiloxanes 4 and 5 constitute potentially useful building blocks in the formation of new carbon-carbon bonds, a Pd-catalyzed Hiyama coupling of 5a with 4-iodotoluene 6a was carried out. The cross-coupling product 7a was obtained with 92% of the isolated yield. This result encouraged us to improve the 2-step, sequential method and perform hydrosilylation and Hiyama reactions one after the other, in the one-pot manner (Fig. 6). Such an approach led directly to product 7a with excellent selectivity (> 99%) and the same high isolation yield (92%) as was observed for the sequential processes.

Synthesis of 7a by the one-pot hydrosilylation and Hiyama reactions, starting from disiloxane 1.
Conclusion
In summary, we synthesized novel bifunctional disiloxanes via the subsequent hydrosilylation of internal alkynes in the first step and alkynes (terminal or internal) or 1,3-diynes in the second. As a result, unsymmetrical bisfunctionalized disiloxanes with two disubstituted (symmetrical (4a–b) and unsymmetrical (4h–n)) or one di- and one monosubstituted (4c–g) alkenyl groups were obtained which became a completely new family of compounds unavailable through other reported methods.
At the same time, hydrosilylation of two equivalents of the same alkyne (internal or terminal) led to a known type of symmetrical siloxanes with two mono- (5d–k) or disubstituted (5a–c) alkenyl functionalities. Even though analogues systems were obtained by other methods, hydrosilylation was proved to be the most versatile, straightforward, and tolerant for many functional groups, based on commercially available reagents and catalysts and an easily accessible method. Only with the use of hydrosilylation process all types of disiloxanes (unsymmetrical and symmetrical) with mono- and disubstituted alkenyl groups could be achieved.
Therefore, by the developed method 27 compounds were obtained, 21 of which were described for the first time (3a‒c, 4a‒n, 5a, c, g, j, k). Crystal structures for compounds 5b, 5e, and 5g were determined for the first time and represent the only examples of such systems.
Finally, disiloxane 5a was reacted with 4-iodotoluene in the presence of Pd2(dba)3 and TBAF, and gave the product of cross-coupling 7a in high yield. The high efficiency of both processes (hydrosilylation and Hiyama coupling) allowed the development of the one-pot method of 7a synthesis. This example proved the concept that obtained compounds can find potential applications as building blocks or coupling agents, or reagents in many powerful transformations such as Heck, Suzuki, and Hiyama couplings.
Methods
Synthesis of monofunctionalized disiloxanes 3a‒c by the hydrosilylation of internal alkynes 2a‒c with 1,1,3,3-tetramethyldisiloxane (1)
Alkyne (2a‒c) (0.5 mmol) and 1 (5 mmol) were placed in a 100 mL round-bottom flask equipped with a stirring bar containing 2 mL of toluene (0.25M). Reaction for 3a was performed without solvent. Subsequently, the reaction mixture was stirred at room temperature and Karstedt’s catalyst (5 × 10−5 mmol of Pt) was added. All volatiles were removed under vacuum from the post-reaction mixture after 18 h for 3a‒b and 3h for 3c. The residue was dissolved in n-hexane and filtered through a pad of silica using TLC to control the separation process. Compounds were characterized by GC-MS, FT-IR and 1H, 13C and 29Si NMR and elemental analyses. The gram-scale synthesis of 3a and 3c was performed in the same manner with the application of 50 mmol of 1 and 5 mmol of alkynes 2a and 2c, respectively.
Synthesis of unsymmetric disiloxanes (4a‒b, 4h‒n) by the hydrosilylation of internal alkynes (2a‒c) or 1,3-diynes (2h‒n) with monofunctionalized disiloxanes (3a‒c)
Alkyne (2a‒c) or 1,3-diyne (2h‒n) (0.5 mmol) and monofunctionalized disiloxane (3a‒c) (0.5 mmol) were placed in a 50 mL round-bottom flask equipped with stirring bar containing 2 mL of dry toluene. Subsequently, the reaction mixture was heated up to 100 °C and Karstedt’s catalyst (5 × 10−4 mmol of Pt2(dvs)3) was added. After 18 h post-reaction mixture was cooled down and all volatiles were removed under a vacuum. Products were purified by flash chromatography and characterized by GC-MS, FT-IR and 1H, 13C, 29Si NMR and elemental analyzes.
Synthesis of unsymmetric disiloxanes (4c‒f) by the hydrosilylation of terminal alkynes (2c‒f) with monofunctionalized disiloxane (3a)
Platinum(IV) oxide (0.005 mmol) and Xphos (0.01 mmol) were placed in a 25 mL Schlenk vessel equipped with a stirring bar under an argon atmosphere. 2 mL of dry tetrahydrofuran was added and the reaction mixture was heated up to 60 °C and stirred for 1 hour. Subsequently, terminal alkyne 2 (0.5 mmol) and 3 (0.5 mmol) were added and heated up to 100 °C. After 18 h post-reaction mixture was cooled down, and filtered through a syringe filter and all volatiles were removed under a vacuum. flash chromatography was performed and products were characterized by GC-MS, FT-IR and 1H, 13C, 29Si NMR and elemental analyzes.
Synthesis of unsymmetric disiloxanes (4a, 4m) by the subsequent, one-pot bishydrosilylation of alkyne 2c and alkyne 2a with 1,1,3,3-tetramethyldisiloxane (1) as well as alkyne 2a and 1,3-diyne (2m) with 1,1,3,3-tetramethyldisiloxane (1)
Alkyne (2c for 4a and 2a for 4m) (0.5 mmol) and 1 (5 mmol) were placed in a 100 mL round-bottom flask equipped with a stirring bar containing 2 mL of toluene (0.25M). Reaction with 2a was performed without solvent Subsequently, the reaction mixture was stirred at room temperature and Karstedt’s catalyst (5 × 10−4 mmol of Pt2(dvs)3) was added. All volatiles were removed under vacuum from the post-reaction mixture after 3 h or 18 h for 2c and 2a, respectively. Afterwards, 2 mL of toluene and appropriate alkyne or 1,3-diyne (0.5 mmol) (2a for 4a and 2m for 4m) were added and heated over 18 h at 100 °C. Obtained via one-pot protocol products were purified and characterized analogously like for the stepwise procedure.
Synthesis of symmetric disiloxanes (5a‒c) by the hydrosilylation of internal alkynes (2a‒c) with 1,1,3,3-tetramethyldisiloxane (1)
To the 100 mL, round-bottom flask equipped with a stirring bar and 2 mL of toluene, the internal alkynes (2a‒c) (1 mmol) and 1,1,3,3-tetramethyldisiloxane (1) (0.5 mmol) were added and heated up to 60 °C (2c) or 100 °C (2a-b).
Subsequently, Karstedt’s catalyst (5 × 10−4 mmol of Pt2(dvs)3) was added. After 18 h, the reaction mixture was cooled down and all volatiles were removed under a vacuum. Products were purified by flash chromatography and characterized by GC–MS, FT–IR and 1H, 13C, 29Si NMR and elemental analyzes.
Synthesis of symmetric disiloxanes (5d‒k) by the hydrosilylation of terminal alkynes (2c‒f, 2o‒s) with 1,1,3,3-tetramethyldisiloxane (1)
Platinum(IV) oxide (0.005 mmol) and Xphos (0.01 mmol) were placed in a 25 mL Schlenk vessel equipped with a stirring bar under an argon atmosphere. 2 mL of dry tetrahydrofuran was added and the reaction mixture was heated up to 60 °C and stirred for 1 hour. Subsequently, terminal alkyne 2 (0.5 mmol) and 1 (0.25 mmol) were added and heated up to 100 °C. After 18 h, the post-reaction mixture was cooled down, filtered through a syringe filter and all volatiles were removed under a vacuum. flash chromatography was performed and products were characterized by GC–MS, FT-IR and 1H, 13C, 29Si NMR and elemental analyzes.
One-pot synthesis of 7a via hydrosilylation and Hiyama reactions
To a Schlenk flask with a Rotaflo® stopcock equipped with a magnetic stirring bar the 1,1,3,3-tetramethyldisiloxane (1, 1 equiv.) and 4-octyne (2c, 2 equiv.) were added and the mixture was purged with argon for 5 min after which THF (1M) and Karstedt’s catalyst (10−4 mol of Pt per mol of 1) were added. The mixture was stirred at 60 °C for 18 h. Subsequently, the 4-iodotoluene (6a, 2 equiv.) was added, followed by the addition of TBAF (4 equiv. 1M in THF) and Pd2(dba)3 (3.5 × 10−2 mol) dissolved in THF (0.11M). The obtained dark brown solution was stirred at 60 °C for 18 h. The mixture was diluted with dichloromethane, passed through a pad of Celite and concentrated. The residue was purified by flash column chromatography to afford the product 7a with 92% of yield.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information file.
References
Marciniec, B., Maciejewski, H., Pietraszuk, C. & Pawluć, P. in Applied Homogeneous Catalysis with Organometallic Compounds, 569–620 (2017).
Duke, B. J. et al. Nano-dispersed platinum(0) in organically modified silicate matrices as sustainable catalysts for a regioselective hydrosilylation of alkenes and alkynes. N. J. Chem. 42, 11782–11795. https://doi.org/10.1039/c8nj01889h (2018).
Li, Z., Chevalier, P. M. & Niu, Z. Investigation of β-alkynol inhibition mechanism and Ru/Pt dual catalysis in Karstedt catalyzed hydrosilylation cure systems. J. Organometal. Chem. 928, 121541. https://doi.org/10.1016/j.jorganchem.2020.121541 (2020).
Guan, J. et al. Conjugated copolymers that shouldn’t be. Angew. Chem. Int. Edn 60, 11115–11119. https://doi.org/10.1002/anie.202014932 (2021).
Yasuhara, K., Miki, S., Nakazono, H., Ohta, A. & Kikuchi, J.-I. Synthesis of organic–inorganic hybrid bicelles–lipid bilayer nanodiscs encompassed by siloxane surfaces. Chem. Commun. 47, 4691–4693. https://doi.org/10.1039/c1cc10254k (2011).
Bakhshandeh, E., Jannesari, A., Ranjbar, Z., Sobhani, S. & Saeb, M. R. Anti-corrosion hybrid coatings based on epoxy–silica nano-composites: Toward relationship between the morphology and EIS data. Prog. Org. Coat. 77, 1169–1183 (2014).
Troegel, D. & Stohrer, J. Recent advances and actual challenges in late transition metal catalyzed hydrosilylation of olefins from an industrial point of view. Coord. Chem. Rev. 255, 1440–1459. https://doi.org/10.1016/j.ccr.2010.12.025 (2011).
Franz, A. K. & Wilson, S. O. Organosilicon molecules with medicinal applications. J. Med. Chem. 56, 388–405. https://doi.org/10.1021/jm3010114 (2013).
Marciniec, B. in Hydrosilylation 3–51 (Springer, 2009).
Lee, I. J., Song, G. S., Lee, W. S. & Suh, D. H. A new class of solid polymer electrolyte: Synthesis and ionic conductivity of novel polysiloxane containing allyl cyanide groups. J. Power Sources 114, 320–329 (2003).
LaRonde, F. J., Ragheb, A. M. & Brook, M. A. Controlling silica surfaces using responsive coupling agents. Colloid Polym. Sci. 281, 391–400 (2003).
Ding, Y. et al. Linear hybrid siloxane-based side chains for highly soluble isoindigo-based conjugated polymers. Chem. Commun. 56, 11867–11870. https://doi.org/10.1039/d0cc01497d (2020).
Deng, J. 1,1,3,3-Tetramethyldisiloxane. Synlett 22, 2102–2103 (2011).
Zhang, D. et al. Environment-friendly synthesis and performance of a novel hyperbranched epoxy resin with a silicone skeleton. RSC Adv. 3, 3095–3102. https://doi.org/10.1039/c2ra22969b (2013).
Niu, Y.-Z., Zhang, L., Liang, S.-J., Wang, D.-X. & Feng, S.-Y. Synthesis of ester-capped carbosilane dendrimers via a hybrid divergent–convergent method. Chin. Chem. Lett. 25, 1419–1422 (2014).
Guzman, G. et al. High strength bimodal amphiphilic conetworks for immunoisolation membranes: Synthesis, characterization, and properties. Macromolecules 48, 6251–6262 (2015).
Januszewski, R., Grzelak, M., Orwat, B., Dutkiewicz, M. & Kownacki, I. Simple catalytic approach to highly regioselective synthesis of monofunctionalized disiloxanes decorated with metalloids. J. Catal. 390, 103–108 (2020).
De Vekki, D. & Skvortsov, N. Hydrosilylation of vinylsiloxanes with hydrosiloxanes in the presence of thermo-and photoactivated platinum (II) phosphine complexes. Russ. J. Gen. Chem. 74, 197–206 (2004).
Perry, R. J., Karageorgis, M. & Hensler, J. Hydrosilylation reactions of 1, 3-diynes and bis (silyl hydrides): Model studies and polymerizations. Macromolecules 40, 3929–3938 (2007).
Sore, H. F. et al. Fluoride-free cross coupling using vinyldisiloxanes. Org. Biomol. Chem. 7, 1068–1072 (2009).
Sore, H. F. et al. Vinyldisiloxanes: Their synthesis, cross coupling and applications. Org. Biomol. Chem. 9, 504–515 (2011).
Frye, E. C. et al. Palladium-catalysed cross-coupling of vinyldisiloxanes with benzylic and allylic halides and sulfonates. Chem. Eur. J. 18, 8774–8779 (2012).
Rivero-Crespo, M. A., Leyva-Pérez, A. & Corma, A. A ligand-free Pt3 cluster catalyzes the markovnikov hydrosilylation of alkynes with up to 106 turnover frequencies. Chem. Eur. J. 23, 1702–1708 (2017).
Zhang, X., Ji, X., Xie, X. & Ding, S. Construction of highly sterically hindered 1, 1-disilylated terminal alkenes. Chem. Commun. 54, 12958–12961 (2018).
Wang, D. et al. Markovnikov hydrosilylation of alkynes with tertiary silanes catalyzed by dinuclear cobalt carbonyl complexes with NHC ligation. J. Am. Chem. Soc. 143, 12847–12856 (2021).
Marciniec, B. et al. Trans-silylation vs cross-metathesis of styrene with 2, 2, 4, 4, 6, 6, 8, 8-octamethyl-1, 5-dioxo-2, 4, 6, 8-tetrasila-3, 7-exo-dimethylenecyclooctane catalyzed by ruthenium complexes. Organometallics 18, 3968–3975 (1999).
Zhou, H., Zhang, Q.-Y. & Lu, X.-B. Synthesis and catalytic application of N-heterocyclic carbene copper complex functionalized conjugated microporous polymer. RSC Adv. 6, 44995–45000 (2016).
Zhou, H. & Wang, Y. B. Copper (I)-catalyzed highly regio-and stereoselective hydrosilylation of terminal alkynes with boryldisiloxane. ChemCatChem 6, 2512–2516 (2014).
Tanaka, M., Uchimaru, Y. & Lautenschlager, H. J. Platinum-complex-catalyzed dehydrogenative double silylation of acetylenes, dienes, and olefins with bis(hydrosilanes). Organometallics 10, 16–18. https://doi.org/10.1021/om00047a010 (1991).
Denmark, S. E. & Wang, Z. Highly stereoselective hydrocarbation of terminal alkynes via Pt-catalyzed hydrosilylation/Pd-catalyzed cross-coupling reactions. Org. Lett. 3, 1073–1076 (2001).
Martin, S. E. & Watson, D. A. Preparation of vinyl silyl ethers and disiloxanes via the silyl-Heck reaction of silyl ditriflates. J. Am. Chem. Soc. 135, 13330–13333 (2013).
Cano, R., Yus, M. & Ramón, D. J. Impregnated platinum on magnetite as an efficient, fast, and recyclable catalyst for the hydrosilylation of alkynes. ACS Catal. 2, 1070–1078. https://doi.org/10.1021/cs300056e (2012).
Franczyk, A., Stefanowska, K., Dutkiewicz, M., Frąckowiak, D. & Marciniec, B. A highly selective synthesis of new alkenylsilsesquioxanes by hydrosilylation of alkynes. Dalton Trans. 46, 158–164 (2017).
Stefanowska, K. et al. An effective hydrosilylation of alkynes in supercritical CO2–A green approach to alkenyl silanes. J. Catal. 356, 206–213 (2017).
Walczak, M. et al. Hydrosilylation of alkenes and alkynes with silsesquioxane (HSiMe2O)(i-Bu) 7Si8O12 catalyzed by Pt supported on a styrene-divinylbenzene copolymer. J. Catal. 367, 1–6 (2018).
Stefanowska, K. et al. Selective hydrosilylation of alkynes with octaspherosilicate (HSiMe2O) 8Si8O12. Chem. Asian J. 13, 2101–2108 (2018).
Walkowiak, J. D. et al. Pt-catalyzed hydrosilylation of 1, 3-diynes with triorganosilanes: Regio-and stereoselective synthesis of mono-or bis-silylated adducts. J. Organ. Chem. 84, 2358–2365 (2019).
Stefanowska, K., Franczyk, A., Szyling, J. & Walkowiak, J. Synthesis of functional 3-buten-1-ynes and 1, 3-butadienes with silsesquioxane moiety via hydrosilylation of 1, 3-diynes. ChemCatChem 11, 4848–4853 (2019).
Szyling, J. et al. Synthesis of bifunctional disiloxanes via subsequent hydrosilylation of alkenes and alkynes. Chem. Commun. 57, 4504–4507 (2021).
Stefanowska, K., Szyling, J., Walkowiak, J. & Franczyk, A. Alkenyl-functionalized open-cage silsesquioxanes (RSiMe2O) 3R′ 7Si7O9: A novel class of building nanoblocks. Inorg. Chem. 60, 11006–11013 (2021).
Sokolnicki, T., Franczyk, A., Janowski, B. & Walkowiak, J. Synthesis of bio-based silane coupling agents by the modification of eugenol. Adv. Synth. Catal. 363, 5493–5500 (2021).
Stefanowska, K., Sokolnicki, T., Walkowiak, J., Czapik, A. & Franczyk, A. Directed cis-hydrosilylation of borylalkynes to borylsilylalkenes. Chem. Commun. 58, 12046–12049 (2022).
Walkowiak, J., Szyling, J., Franczyk, A. & Melen, R. L. Hydroelementation of diynes. Chem. Soc. Rev. (2022).
Prukała, W. A novel approach to stilbenoid dendrimer core synthesis. Synlett 2008, 3026–3030 (2008).
Acknowledgements
This work was supported by the National Centre for Research and Development in Poland, Lider Programme No. LIDER/6/0017/L-9/17/NCBR/2018 and the National Science Centre in Poland No. UMO-2019/32/C/ST4/00235, UMO-2018/31/G/ST4/04012.
Author information
Authors and Affiliations
Contributions
J.S.: investigation and writing original draft; J.W.: review and editing; A.C.: determination of crystal structures; A.F.: conceptualization, writing original draft, review and editing, work supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Szyling, J., Walkowiak, J., Czapik, A. et al. Synthesis of unsymmetrically and symmetrically functionalized disiloxanes via subsequent hydrosilylation of C≡C bonds. Sci Rep 13, 10244 (2023). https://doi.org/10.1038/s41598-023-37375-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-37375-8
- Springer Nature Limited