
Abstract
Steroid-resistant asthma is a troublesome clinical problem in public health. The pathogenesis of steroid-resistant asthma is complex and remains to be explored. In our work, the online Gene Expression Omnibus microarray dataset GSE7368 was used to explore differentially expressed genes (DEGs) between steroid-resistant asthma patients and steroid-sensitive asthma patients. Tissue-specific gene expression of DEGs was analyzed using BioGPS. The enrichment analyses were performed using GO, KEGG, and GSEA analysis. The protein–protein interaction network and key gene cluster were constructed using STRING, Cytoscape, MCODE, and Cytohubba. A steroid-resistant neutrophilic asthma mouse model was established using lipopolysaccharide (LPS) and ovalbumin (OVA). An LPS-stimulated J744A.1 macrophage model was prepared to validate the underlying mechanism of the interesting DEG gene using the quantitative reverse transcription-polymerase chain reaction (qRT-PCR). A total of 66 DEGs were identified, most of which were present in the hematologic/immune system. Enrichment analysis displayed that the enriched pathways were the IL-17 signaling pathway, MAPK signal pathway, Toll-like receptor signaling pathway, and so on. DUSP2, as one of the top upregulated DEGs, has not been clearly demonstrated in steroid-resistant asthma. In our study, we observed that the salubrinal administration (DUSP2 inhibitor) reversed neutrophilic airway inflammation and cytokine responses (IL-17A, TNF-α) in a steroid-resistant asthma mouse model. We also found that salubrinal treatment reduced inflammatory cytokines (CXCL10 and IL-1β) in LPS-stimulated J744A.1 macrophages. DUSP2 may be a candidate target for the therapy of steroid-resistant asthma.
Similar content being viewed by others


Introduction
Asthma is commonly recognized as a chronic inflammatory respiratory disease, accompanied by airway reversible obstruction and respiratory symptoms, including cough, shortness of breath, and chest tightness1. Steroid inhalation is widely used for alleviating airway inflammation in asthma and reducing asthma exacerbations2. Despite high doses of steroid inhalation or systemic steroid usage, about 10% of asthma patients develop steroid resistance and lose control of symptoms3,4, whose phenotype is usually neutrophilic dependent5. The steroid is the main therapy for asthma but has an unsatisfactory response in neutrophilic asthma patients6. It was reported that IL-17A induced Th17 cells to recruit neutrophils in the airway of steroid-resistant asthma and Th17 cells played an important part in steroid-resistant asthma with neutrophilic airway inflammation7. A previous study suggested that steroid inhibited Th1/Th2‐related inflammatory cytokine production8, but failed to affect the production of Th17-related cytokines in steroid-resistant asthma9,10. The mechanisms of steroid resistance related to the IL-17A pathway remained to be elusive. The multiple mechanisms underlying the etiology of steroid-resistant asthma are complex11,12.
Given that steroid-resistant asthma is a complex clinical problem in public health, it is urgent to find an effective therapeutic strategy for steroid-resistant asthma. Therefore, well-established computational data-mining strategies were developed to explore the responsible genes for steroid-resistant asthma. Microarray dataset GSE7368 of bronchoalveolar lavage fluid (BALF) cells was used to identify the differentially expressed genes (DEGs) between steroid-resistant (SR) asthma patients and steroid-sensitive (SS) asthma patients, which aimed to provide a better understanding of the genetic etiology of steroid-resistant asthma.
Materials and methods
All the experimental protocols were approved by the Ethics Committee of Xiangya Hospital, Central South University (committee Reference Number: 201803691). Methods used in experimental animals in the study were approved by the Central South University Experimental Animal Ethics Committee of Xiangya Hospital and complied with ARRIVE guidelines and American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (2020). The datasets generated from human participants can be found in the GEO dataset: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE7368, which was performed following the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Microarray data
The microarray dataset GSE7368, constructed by Goleva et al., was retrieved from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/), which allows researchers to search and download expression data for analysis based on the GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array platform. The experiment contained gene array studies of BALF cells, consisting of 3 steroid-resistant (SR) asthma patients and 3 steroid-sensitive (SS) asthma patients. The data were normalized using the gcrma package (version 2.0.0)13. All analyses were performed in the R environment (version 3.6.2). The probes that have no expression in most of the samples were removed. The probes were annotated as gene symbols based on the annotation information of the GPL570 platform. An expression value for every gene was acquired based on mean expression estimates. As a result, we obtained the gene expression matrix.
Differential expression analysis
Differential expression analysis was performed to identify the DEGs using a limma package (version 3.42.2)14. P values were calculated using the t-tests. The DEGs were determined according to the following criteria: (1) a |log2 (fold-change)|> 1 and (2) a P value < 0.05. The volcano plot and the heatmap for the DEGs were constructed using ggplot2 (version 3.3.1) and pheatmap (version 1.0.12) packages15,16. We selected the top 6 upregulated DEGs to create a violin plot.
Tissue-specific expressed gene analysis
Online database BioGPS (http://biogps.org) (version 94eefe6) was used to analyze the tissue-specific expressed DEG genes. The genes identified as highly tissue-specific genes were similar to the previous study described, with more than > 30 multiples of the median (MoM) and less than 1/3 of the highest expressions in the second highest level17.
Functional enrichment analysis of DEGs
The functional enrichment analysis of DEGs was performed using the clusterProfiler package (version 3.14.3)18. The enrichment analyses were performed for predicting protein functions. The biological process (BP), cellular component (CC), and molecular function (MF) of DEGs were identified using the enrichGO function in the clusterProfiler package. The pathway enrichment was identified using the enrichKEGG function in the clusterProfiler package.
Gene set enrichment analysis
We performed GSEA analysis using GSEA software (version 4.0.3)19. KEGG enrichment pathways were identified with the following criteria: a P value < 5% and a false discovery rate (FDR) < 25% for every analysis.
Protein–protein interaction (PPI) network analysis
Search Tool for the Retrieval of Interacting Genes (STRING; http://string-db.org) (version 10.0)20 was used to predict and construct the protein–protein interactions (PPI) network of DEGs. DEGs in the PPI network arrived at a minimum required interaction score > 0.4. The nodes indicate genes, and the edges indicate the associations between genes. The PPI network was visualized using Cytoscape (version 3.7.1). Besides, Molecular Complex Detection (MCODE) (version 1.6) in Cytoscape was used to assess the key clustered module with a degree cutoff of 2, a node score cutoff of 0.2, and a K-Core of 221. The clustered module of the top 10 genes was identified using CytoHubba (version 0.1) in Cytoscape with the MCC method22.
Animal experiments
Male C57BL/6 mice (6–8 weeks of age) were used to develop a steroid-resistant neutrophilic asthma (NA) model as described by previous studies with minor modification23,24,25,26. All mice were divided into five groups (n = 4 per group) randomly: Control group, NA group, NA + salubrinal group, NA + Dex group, and Control + salubrinal group. Briefly, ovalbumin (OVA, 100 μg) and lipopolysaccharides (LPS, 10 μg) in 50 μl PBS were intratracheally delivered to the mice under anesthesia with subcutaneous pentobarbital on day 0 and day 6. Then the mice were challenged with 5% aerosolized OVA for 40 min on day 13. The mice were sacrificed on day 14, and the lungs were extracted for histology analysis using hematoxylin and eosin (H&E) and Masson staining27,28. In some groups, the LPS/OVA-sensitized mice were injected intraperitoneally with dexamethasone (Dex, 1 mg/kg) or salubrinal (2 mg/kg) before the OVA challenge on day 13. The control mice were administered intratracheally with 50 μL PBS, then challenged with PBS for 40 min on day 13. In another group, the control mice were injected intraperitoneally with salubrinal (2 mg/kg) before the PBS challenge on day 13.
LPS stimulation and salubrinal treatment in J744A.1 macrophages
J774A.1 macrophages were retrieved from the cell repository of Advanced Research Center, Central South University. Cells were cultured in a completed medium at 5% CO2 and 37 °C. There are four groups in vitro experiment: the control group (control), unstimulated cells; the LPS group (LPS), cells stimulated with LPS (10 ng/ml) for 24 h; the LPS + salubrinal groups, cells costimulated with salubrinal (10uM) and LPS (10 ng/ml) for 24 h; the salubrinal group, cells stimulated with salubrinal (10uM) for 24 h.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from cells with Trizol (Invitrogen). The cDNA was obtained using the reverse transcription kit (Vazyme). qRT-PCR was conducted according to the instructions of the manufacturer (Vazyme). The primers are displayed in Table 1.
Flow cytometric analysis
The bronchoalveolar lavage (BAL) fluids were obtained by injecting 1 ml PBS into the murine trachea, repeating ten times. The cells of BAL fluid were stained with anti-CD45 (APC/Cy7; BioLegend), anti-CD11c (PE; BioLegend), anti-Siglec-F (APC; BioLegend), anti-MHCII (Percp/Cy5.5; BioLegend), anti-CD11b (BV605; BioLegend), anti-Ly6G (BV421; BioLegend), and anti-CD3 (AF488; BioLegend) from light for 30 min. The cell gating strategy in BAL fluids was conducted according to the methods previously reported29. The signals were obtained from a Cytek Dxp Athena flow cytometer. And the data were calculated using FlowJo software (version 10).
Statistical analysis
All data were expressed as mean ± standard error (SE) and analyzed with SPSS 19.0 statistical software. The student’s t-test was performed to calculate the difference between the two groups. The one-way ANOVA followed by Tukey’s post hoc analysis was used to calculate the differences in more than two groups. The difference was significant when a P value was < 0.05.
Results
Differentially expressed genes
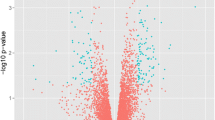
We analyzed the microarray dataset GSE7368 from the GEO database and selected DEGs between BALF cells from steroid-resistant asthma (SR) and steroid-sensitive (SS) asthma using R packages. Principal-component analysis displayed that the center of the SR group lay far apart from the center of the SS group in space, indicating the different expressions of genes between the two groups (Fig. 1A). A total of 57 upregulated and 9 downregulated genes were determined as DEG genes from SR patients and SS patients, as displayed in Table 2. The volcano plots showed significantly upregulated or downregulated DEGs in SR patients compared with SS patients (Fig. 1B). The red dots indicate the significantly upregulated genes, while the blue dots indicate the significantly downregulated genes. Hierarchical clustering heatmaps manifested the distinguishable DEGs expression pattern between SR patients and SS patients (Fig. 1C). These results above showed that the top 6 DEGs between SR patients and SS patients were IL6, CXCL8, TNF, DUSP2, ADM, and CXCL1.

Profiles of expressed genes in patients with steroid-resistant asthma (SR) and steroid-sensitive (SS) asthma. (A) Principal-component analysis (PCA) among samples. (B) The volcano plot of gene expressions in SR patients and SS patients. The red dots and the blue dots indicate upregulated DEGs and downregulated DEGs, respectively. (C) A heatmap for the DEGs was constructed using pheatmap (version 1.0.12) packages in the R environment (version 3.6.2).
Tissue-specific expression of genes
We used BioGPS to identify 20 DEG genes as tissue-specific or organ system-specific expressed genes. Most of the tissue-expressed genes (65%, 13/20) were present in the hematologic/immune system. The respiratory and neurologic systems showed similar levels of tissue-expressed genes (10%, 2/20; 10%, 2/20), while the digestive, circulatory systems, and skin/skeletal muscle showed relatively low levels of tissue-expressed genes (5%, 1/20; 5%, 1/20; 5%, 1/20) (Table 3). Collectively, our results displayed that most tissue specific expressed DEG genes were distributed in the hematologic/immune system.
Functional and pathway enrichment analyses of DEGs
To explore the function of DEGs between SR patients and SS patients, we performed GO and KEGG enrichment analyses. The biological processes (BPs) of DEGs were mainly enriched in cytokine activity, chemokine activity, and cytokine receptor binding. The cellular components (CCs) of DEGs were mainly involved in the membrane region, membrane microdomain, and membrane draft. The molecular functions (MFs) of DEGs were mainly enriched in the cellular response to lipopolysaccharide, response to the molecule of bacterial region, and response to lipopolysaccharide (Fig. 2A). The DEGs enriched in GO function enrichment were visualized using the ClusterProfiler package. The yellow dots indicated the GO categories, the color of the line indicated the association between dots and the category, and the size of a dot showed the gene numbers (Fig. 2B). As for the KEGG pathway enrichment analysis, DEGs were enriched in the IL-17 signaling pathway, NF-kappa B signaling pathway, TNF signaling pathway, NOD-like receptor signaling pathway, and cytokine-cytokine receptor interaction (Fig. 2C). Our results showed that the functional and pathway enrichment of DEGs was mainly related to cytokine responses.

The results of GO and KEGG enrichment analysis of DEGs between SR patients and SS patients. (A) Bar plot of the functional GO terms, including biological process (BP), cellular component (CC), and molecular function (MF); (B) Circle graph showing the relationship between GO enrichment and DEGs. (C) Bar plot of KEGG enriched terms colored by a P value. The terms with more genes indicate a more significant P value.
Gene set enrichment analysis
The GSEA analysis was performed to explore the enriched gene-related pathways. The pathways related to cytokine cytokine receptor interaction, MAPK signal pathway, Toll-like receptor signaling pathway, T cell receptor signaling pathway, natural killer cell mediated cytotoxicity, and hematopoietic cell lineage pathway were mainly enriched in the SR group (Fig. 3A–F). Our results suggested that the enriched gene-related pathways were mainly associated with cytokine or immune responses.

Gene set enrichment analysis (GSEA) of the enriched genes in the SR and SS group. (A) cytokine cytokine receptor interaction; (B) MAPK signal pathway; (C) Toll-like receptor signaling pathway; (D) T cell receptor signaling pathway; (E) Natural killer cell mediated cytotoxicity; (F) Hematopoietic cell lineage pathway.
PPI networks of DEGs between SR patients and SS patients
The protein association networks of DEGs between SR patients and SS patients were constructed by STRING. A PPI network with 60 nodes and 191 edges had an interaction score > 0.4 visualized by Cytoscape software (Fig. 4A). The nodes indicate genes, and the edges indicate the associations between genes. We used the MCODE plugin in Cytoscape to identify the key PPI network module with the highest score, consisting of 14 genes (Fig. 4B). Furthermore, the top 10 hub genes were identified by the Cytohubba in Cytoscape (Fig. 4C). The functional enrichment analysis showed that these top 10 hub genes were primarily related to the IL-17 signaling pathway, TNF signaling pathway, and NF-kappa B signaling pathways (Fig. 4D). These results revealed that DEGs between SR patients and SS patients were more likely to be related to inflammatory cytokine response.

PPI Networks of DEGs. (A) The PPI network of DEGs was constructed using the STRING online database. (B) The key module with the highest score was identified by the MCODE in Cytoscape. (C) The top 10 hub gene cluster constructed by the Cytohubba in Cytoscape. (D) Functional enrichment analysis for the top 10 hub genes.
Validation of DUSP2 in an asthma mouse model
Our analysis indicated that DUSP2 was potentially associated with steroid-resistant asthma. To our knowledge, there are few studies relating to the roles of DUSP2 in steroid-resistant asthma. Therefore, we hypothesized that inhibition of DUSP2 showed a protective effect on the airway inflammation of steroid-resistant asthma. We generated a steroid-resistant NA mouse model and administrated salubrinal according to the experimental procedure shown in Fig. 5A. It was demonstrated that more neutrophil infiltrations in BAL fluids were observed in the steroid-resistant murine asthma model30. Our study suggested that neutrophil numbers were not decreased by Dex treatment in the NA group (LPS/OVA + Dex) but were reduced after salubrinal administration in the NA group (LPS/OVA + salubrinal) (Fig. 5B). No significant changes in eosinophil, macrophage, and lymphocyte numbers of BAL fluids were observed in the NA group with or without salubrinal treatment (Fig. 5B). Besides, our study revealed that peribronchial inflammation infiltration and subepithelial collagen deposition were more severe in the NA group compared to the control group, and Dex failed to reverse lung morphological changes based on the inflammation score and Ashcroft score of fibrosis (Fig. 5C,D). As mentioned in the bioinformatic analysis above, DEGs were mainly related to IL-17 and TNF pathways. As a result, our study displayed that the NA group had the highest mRNA levels of IL-17A, TNF-α, and DUSP2 (Fig. 5E–G). The mRNA levels of IL-17A, TNF-α, and DUSP2 were not reduced by Dex administration in the NA group but were significantly reduced after salubrinal treatment in the NA group (Fig. 5E–G). These results indicated that the DUSP2 inhibitor ameliorated neutrophilic airway inflammation and cytokine responses (IL-17A and TNF-α)in an asthma mouse model with steroid-resistant.

Validation of DUSP2 in an asthma mouse model. (A) Flow chart for a neutrophilic asthma mouse model establishment. (B) Inflammatory cells in BAL fluids. (C) Representative histological analysis of lung sections with H&E staining and a bar graph displaying the inflammation scores. Scale bar = 50 μm. (D) Representative histological analysis of lung sections with Masson staining and a bar graph displaying the Ashcroft scores. Scale bar = 50 μm. (E) Real-time PCR of IL-17A mRNA levels normalized to GAPDH. (F) Real-time PCR of TNF-α mRNA levels normalized to GAPDH. (G) Real-time PCR of DUSP2 mRNA levels normalized to GAPDH. n = 4, data were expressed as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. NA neutrophilic asthma.
Inhibition of inflammatory cytokines by salubrinal in LPS-stimulated J744A.1 macrophages
Incubation of J774A.1 macrophages with salubrinal significantly inhibited DUSP2 mRNA (Fig. 6A). Furthermore, the elevation of inflammatory cytokines (CXCL10, IL-1β) in LPS-stimulated J774A.1 macrophages was suppressed by salubrinal (Fig. 6B,C). Taken together, our results revealed that the correlation between DUSP2 and inflammatory cytokines (CXCL10, IL-1β) may be involved in the inflammatory response of LPS-stimulated J744A.1 macrophages.

Inhibition of inflammatory cytokines by salubrinal in LPS-stimulated J774A.1 macrophages. (A–C) Effects of salubrinal on DUSP2, IL1β, and CXCL10 mRNAs in J774A.1 macrophage, respectively. n = 3. Data were expressed as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, vs LPS group. #P < 0.05, ##P < 0.01, ###P < 0.001, vs control group.
Discussion
Increased morbidity is observed in steroid-resistant asthma patients, and steroid-resistant asthma is more likely to be related to neutrophilic inflammation5,31,32. In this respect, it is urgent to explore alternative treatment options for steroid-resistant asthma. Our computational data-mining analysis explored several genes associated with steroid-resistant asthma, aiming to provide new insight into the pathogenesis of steroid-resistant asthma. Different from the original source of the data set, our study pays more attention to the evaluation and correction of data. We use the limma package to search for differential genes, thereby improving the power of the test.
In the current study, we conducted a discovery-driven analysis to identify DEGs in cells of BAL fluids from SR asthma patients and SS asthma patients. 66 DEGs were identified, consisting of 57 upregulated genes and 9 downregulated genes, which were consistent with activated macrophage-related genes. The top six upregulated DEG genes (IL6, CXCL8, TNF, DUSP2, ADM, and CXCL1) in SR asthma patients compared with SS asthma patients were identified. Recent studies have indicated that reduced expression of IL6 suppressed the development of steroid asthma33,34. CXCL8, also named IL8, is a CXC chemokine that could recruit neutrophils in steroid-resistant airway inflammation35,36. Dex could not reduce the levels of IL6, and IL8 in the macrophages of severe asthma patients37. It was found that patients with steroid-resistant showed a higher level of TNF-α than patients with steroid-sensitive in C. pneumoniae seropositive asthmatics38. Dex could not suppress the levels of TNF-α in steroid-resistant asthma mouse models32. LIGHT and LTαβ, which belong to the TNF family, were reported to induce a steroid-resistant inflammatory response in airway epithelial cells39. DUSP2, also named phosphatase of activated cells 1 (PAC-1), was reported to function as dephosphorylating threonine and tyrosine residues of targeting substrates40,41. DUSP2, predominantly expressed in hematopoietic cells, was demonstrated to upregulate the production of proinflammatory cytokines in immune cells42,43. However, few studies have investigated the role of DUSP2 in steroid-resistant asthma. It is necessary to further delineate the pathogenesis role of DUSP2 in steroid-resistant asthma. One study has revealed that plasma ADM was increased in the acute attack period of asthma compared to the remission period of asthma44, suggesting ADM may be involved in the process of asthma. CXCL1 was observed to be a kind of neutrophil-related chemokine45, which was elevated in neutrophilic asthma patients46. Immune cells were reported to play important roles in the pathogenic development of asthma, and asthma is characterized by granulocytic inflammation infiltration in the airways44,47. In our study, the tissue-specific expression analysis indicated that the hematologic/immune system was the most highly specific system of the DEGs, suggesting the common occurrence of immune response in steroid-resistant asthma.
Enrichment analyses were constructed to explore the roles of DEGs. Results from the GO analysis revealed that the DEGs were mainly enriched in cytokine activity, chemokine activity, cytokine receptor binding, membrane region, membrane microdomain, membrane draft, cellular response to lipopolysaccharide, response to molecule of the bacterial region, and response to lipopolysaccharide. Cytokine-cytokine receptor interaction was reported to be more enriched in neutrophilic asthma than in eosinophil asthma48. Lipopolysaccharide (LPS), a component of gram-negative bacteria, was reported to chemoattract neutrophils49. These biological pathways are mainly associated with an inflammatory response. KEGG analysis demonstrated that the DEGs were mainly enriched in the IL-17 signaling pathway, NF-kappa B signaling pathway, TNF signaling pathway, NOD-like receptor signaling pathway, and cytokine-cytokine receptor interaction. After analyzing the PPI network of DEGs, we found that the top 10 hub genes identified by the Cytohubba in Cytoscape were primarily enriched in the IL-17 signaling pathway, TNF signaling pathway, and NF-kappa B signaling pathways, which were consistent with the KEGG enrichment analysis. IL-17 signaling pathway has been suggested to mediate the pathogenesis of steroid-resistant asthma, which has the function of neutrophil chemotaxis50,51. Nuclear factor-κB (NF-κB) was reported to lead to epithelial cell inflammation of asthma and may be a potential marker for asthma severity52. Tumor necrosis factor-alpha (TNF-α) was reported to be associated with steroid-resistant asthma and asthma exacerbation frequency53,54. These functional enrichment analyses may exhibit the enrichment of DEGs in a neutrophil-related inflammatory response in steroid-resistant asthma. Different from the previous study55, our study has added the enrichment analyses of DEG genes, which suggested that neutrophil-related inflammatory responses were implicated in steroid-resistant asthma.
The GSEA data revealed that cytokine cytokine receptor interaction, MAPK signal pathway, Toll-like receptor signaling pathway, T cell receptor signaling pathway, natural killer cell mediated cytotoxicity, and hematopoietic cell lineage pathway were mainly enriched in SR patients. The p38 MAPK pathway activation was reported to be involved in steroid resistance of asthma56. Dex failed to inhibit NTHi-induced steroid-resistant allergic airway inflammation, by activating the p38 MAPK pathway57. Immune cells were involved in the pathogenic process of asthma44. More than half of the pulmonary CD4+CD3+ cells in moderate-to-severe asthma patients were natural killer T cells, with elevated invariant T-cell receptor expression58. Toll-like receptors (TLRs) were reported to be involved in macrophage-related steroid-resistant AHR59,60. The modulation of the T-cell receptor (TCR) in CD4+T cells recruited inflammatory cells and elevated cytokine production in the airway of asthma61. Our findings support the association between immune cell activation, inflammatory response, and steroid-resistant asthma.
The DUSP2 gene was reported to be involved in the immune activation process, which could enhance inflammatory responses43. However, few studies have reported the role of DUSP2 in steroid-resistant asthma. Since our study suggested that DUSP2 was upregulated in the BAL cells of SR asthma patients as compared with SS asthma patients, we further investigate the role of DUSP2 on steroid-resistant asthma. Previous studies have suggested that salubrinal was a candidate drug for inhibiting the expression of DUSP262,63. Salubrinal is a 480-Da agent that inhibits a serine phosphatase, protein phosphatase 1 (PP1) and elevates the phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α)64. It was suggested that LPS significantly increased DUSP2 expression but caused a decrease in the p-eIF2a/eIF2a ratio, which was reversed by salubrinal in the LPS-induced intraneural hemi-Parkinson disease (PD) model62. Through eIF2α, salubrinal could mediate transcriptional and translational regulation63,65. Therefore, we can assume that salubrinal may inhibit the transcriptional and translational process of DUSP2 by acting on p-eIF2a/eIF2a ratio. It was found that the administration of salubrinal in mouse models of arthritis significantly reduced inflammatory responses63. Interestingly, our findings revealed that administration of a candidate synthetic DUSP2 inhibitor (salubrinal) before the challenge reduced the neutrophilic airway inflammation and cytokine responses (IL-17A, TNF-α) of the steroid-resistant asthma mouse model, suggesting a protective effect of salubrinal in steroid-resistant asthma. Macrophages are considered to be the main immune cells in asthma66. Results from a previous study indicated that salubrinal reduced the DUSP2 expressions in both RAW264.7 macrophages and Jurkat cells63. In line with the previous studies, our study supports that salubrinal is an inhibitor of DUSP2. It was founded that salubrinal inhibited inflammatory cytokines (IL-1β, IL-6, TNF-α), and DUSP2 expressions stimulated by LPS62,63. CXCL10 is a kind of inflammatory cytokine, which enhanced type 1 inflammation resulting in severe pathology67. IL-1β was reported to induce neutrophilic inflammation68. Of note, CXCL10 and IL-1β were reported to be elevated in steroid-resistant asthma30,69,70. Consistent with the previous studies, our study revealed that salubrinal inhibited LPS-driven DUSP2 and IL-1β expressions in J774A.1 macrophage. Besides, we first demonstrated that salubrinal reduced LPS-driven CXCL10 expression in J774A.1 macrophage, indicating the interaction between DUSP2 and CXCL10 may be the potential mechanism for steroid-resistant asthma.
Limitations
That said, there are some limitations of the present study. First, the sample size of asthma patients in the public datasets was small. More BAL cell expression profiles of asthmatics will be needed in future analysis. Second, more potential in vitro mechanisms should be determined in further study.
Conclusion
In conclusion, several DEGs and enriched pathways in the SR patients compared to the SS patients were identified. Our study not only provides an insight into the role of DUSP2 and its inhibitor (salubrinal) in steroid-resistant neutrophilic airway inflammation but also lay a foundation for the development of alternative therapy options in steroid-resistant asthma.
Data availability
The datasets generated for this study can be found in the GEO dataset: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE7368. The permission is granted to (Springer Nature Limited) to publish the images under a CC BY open access license and in all formats i.e. print and digital.
Abbreviations
- GEO:
-
Gene Expression Omnibus
- DEGs:
-
Differentially expressed genes
- SR:
-
Steroid-resistant
- SS:
-
Steroid-sensitive
- PPI:
-
Protein–protein interaction
- LPS:
-
Lipopolysaccharide
- OVA:
-
Ovalbumin
- qRT-PCR:
-
Quantitative reverse transcription-polymerase chain reaction
- GCs:
-
Glucocorticoids
- BALF:
-
Bronchoalveolar lavage fluid
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
- FDR:
-
False discovery rate
- MCODE:
-
Molecular Complex Detection
- NA:
-
Neutrophilic asthma
- H&E:
-
Hematoxylin and eosin
- Dex:
-
Dexamethasone
- SD:
-
Standard deviation
- NF-κB:
-
Nuclear factor-κB
- TNF-α:
-
Tumor necrosis factor-alpha
- NLRP3:
-
Nod-like receptor family pyrin domain containing 3
- TLRs:
-
Toll-like receptors
References
Holgate, S. T. et al. Asthma. Nat. Rev. Dis. Primers 1, 15025. https://doi.org/10.1038/nrdp.2015.25 (2015).
Hudson, W. H. et al. Cryptic glucocorticoid receptor-binding sites pervade genomic NF-κB response elements. Nat. Commun. 9, 1337. https://doi.org/10.1038/s41467-018-03780-1 (2018).
Chung, K. F. et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur. Respir. J. 43, 343–373. https://doi.org/10.1183/09031936.00202013 (2014).
Sullivan, S. D., Rasouliyan, L., Russo, P. A., Kamath, T. & Chipps, B. E. Extent, patterns, and burden of uncontrolled disease in severe or difficult-to-treat asthma. Allergy 62, 126–133. https://doi.org/10.1111/j.1398-9995.2006.01254.x (2007).
Zhang, F. et al. Adenosine triphosphate promotes allergen-induced airway inflammation and Th17 cell polarization in neutrophilic asthma. J. Immunol. Res. 2017, 5358647. https://doi.org/10.1155/2017/5358647 (2017).
Hansbro, P. M. et al. Mechanisms and treatments for severe, steroid-resistant allergic airway disease and asthma. Immunol. Rev. 278, 41–62. https://doi.org/10.1111/imr.12543 (2017).
Liang, S. C. et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J. Immunol. 179, 7791–7799. https://doi.org/10.4049/jimmunol.179.11.7791 (2007).
Ano, S. et al. Transcription factors GATA-3 and RORγt are important for determining the phenotype of allergic airway inflammation in a murine model of asthma. J. Immunol. 190, 1056–1065. https://doi.org/10.4049/jimmunol.1202386 (2013).
Schewitz-Bowers, L. P. et al. Glucocorticoid-resistant Th17 cells are selectively attenuated by cyclosporine A. Proc. Natl. Acad. Sci. USA 112, 4080–4085. https://doi.org/10.1073/pnas.1418316112 (2015).
Ramesh, R. et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J. Exp. Med. 211, 89–104. https://doi.org/10.1084/jem.20130301 (2014).
Mohamed, N. A. & Abdel-Rehim, A. S. Influence of vitamin D receptor gene FokI and ApaI polymorphisms on glucocorticoid response in patients with asthma. Int. Forum Allergy Rhinol. 10, 556–563. https://doi.org/10.1002/alr.22511 (2020).
Goleva, E., Hauk, P. J., Boguniewicz, J., Martin, R. J. & Leung, D. Y. Airway remodeling and lack of bronchodilator response in steroid-resistant asthma. J. Allergy Clin. Immunol. 120, 1065–1072. https://doi.org/10.1016/j.jaci.2007.07.042 (2007).
Gharaibeh, R. Z., Fodor, A. A. & Gibas, C. J. Background correction using dinucleotide affinities improves the performance of GCRMA. BMC Bioinform. 9, 452. https://doi.org/10.1186/1471-2105-9-452 (2008).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Villanueva, R. A. M. & Chen, Z. J. (Taylor & Francis, 2019).
Kolde, R. J. R. p. v. Pheatmap: Pretty heatmaps. 1, 726 (2012).
Massingham, L. J. et al. Amniotic fluid RNA gene expression profiling provides insights into the phenotype of Turner syndrome. Hum. Genet. 133, 1075–1082. https://doi.org/10.1007/s00439-014-1448-y (2014).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Szklarczyk, D. et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605-d612. https://doi.org/10.1093/nar/gkaa1074 (2021).
Bader, G. D. & Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 4, 2. https://doi.org/10.1186/1471-2105-4-2 (2003).
Zheng, P. F. et al. Identifying patterns of immune related cells and genes in the peripheral blood of acute myocardial infarction patients using a small cohort. J. Transl. Med. 20, 321. https://doi.org/10.1186/s12967-022-03517-1 (2022).
Wang, M. et al. Impaired anti-inflammatory action of glucocorticoid in neutrophil from patients with steroid-resistant asthma. Respir Res. 17, 153. https://doi.org/10.1186/s12931-016-0462-0 (2016).
Wilson, R. H. et al. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 180, 720–730. https://doi.org/10.1164/rccm.200904-0573OC (2009).
Fang, S. B. et al. Human iPSC-MSCs prevent steroid-resistant neutrophilic airway inflammation via modulating Th17 phenotypes. Stem Cell Res. Ther. 9, 147. https://doi.org/10.1186/s13287-018-0897-y (2018).
Whitehead, G. S., Thomas, S. Y. & Cook, D. N. Modulation of distinct asthmatic phenotypes in mice by dose-dependent inhalation of microbial products. Environ. Health Perspect. 122, 34–42. https://doi.org/10.1289/ehp.1307280 (2014).
Li, R. et al. The role of macrophage migration inhibitory factor (MIF) in asthmatic airway remodeling. Allergy Asthma Immunol. Res. 13, 88–105. https://doi.org/10.4168/aair.2021.13.1.88 (2021).
Fuhrmann, D. C., Mondorf, A., Beifuß, J., Jung, M. & Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 36, 101670. https://doi.org/10.1016/j.redox.2020.101670 (2020).
Van Hoecke, L., Job, E. R., Saelens, X. & Roose, K. Bronchoalveolar lavage of murine lungs to analyze inflammatory cell infiltration. J. Vis. Exp https://doi.org/10.3791/55398 (2017).
Kim, R. Y. et al. Role for NLRP3 inflammasome-mediated, IL-1β-dependent responses in severe, steroid-resistant asthma. Am. J. Respir. Crit. Care Med. 196, 283–297. https://doi.org/10.1164/rccm.201609-1830OC (2017).
Hew, M. et al. Relative corticosteroid insensitivity of peripheral blood mononuclear cells in severe asthma. Am. J. Respir. Crit. Care Med. 174, 134–141. https://doi.org/10.1164/rccm.200512-1930OC (2006).
Ito, K. et al. Steroid-resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. Am. J. Respir. Cell Mol. Biol. 39, 543–550. https://doi.org/10.1165/rcmb.2008-0028OC (2008).
Esty, B. et al. Treatment of severe persistent asthma with IL-6 receptor blockade. J. Allergy Clin. Immunol. Pract. 7, 1639-1642.e1634. https://doi.org/10.1016/j.jaip.2019.02.043 (2019).
Chen, S. et al. Prevention of IL-6 signaling ameliorates toluene diisocyanate-induced steroid-resistant asthma. Allergol. Int. Off. J Jpn. Soc. Allergol. 71, 73–82. https://doi.org/10.1016/j.alit.2021.07.004 (2022).
Clifford, R. L. et al. CXCL8 histone H3 acetylation is dysfunctional in airway smooth muscle in asthma: Regulation by BET. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, 962–972. https://doi.org/10.1152/ajplung.00021.2015 (2015).
Liu, R. et al. Multi-allergen challenge stimulates steriod-resistant airway inflammation via NF-κB-mediated IL-8 expression. Inflammation 36, 845–854. https://doi.org/10.1007/s10753-013-9611-5 (2013).
Bhavsar, P. et al. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax 63, 784–790. https://doi.org/10.1136/thx.2007.090027 (2008).
Paróczai, D. et al. Chlamydia pneumoniae Influence on cytokine production in steroid-resistant and steroid-sensitive asthmatics. Pathogens https://doi.org/10.3390/pathogens9020112 (2020).
da Silva-Antunes, R., Madge, L., Soroosh, P., Tocker, J. & Croft, M. The TNF family molecules LIGHT and Lymphotoxin αβ induce a distinct steroid-resistant inflammatory phenotype in human lung epithelial cells. J. Immunol. 195, 2429–2441. https://doi.org/10.4049/jimmunol.1500356 (2015).
Jeffrey, K. L., Camps, M., Rommel, C. & Mackay, C. R. Targeting dual-specificity phosphatases: Manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug Discov. 6, 391–403. https://doi.org/10.1038/nrd2289 (2007).
Wei, W. et al. Dual-specificity phosphatases 2: Surprising positive effect at the molecular level and a potential biomarker of diseases. Genes Immun. 14, 1–6. https://doi.org/10.1038/gene.2012.54 (2013).
Lu, D. et al. The phosphatase DUSP2 controls the activity of the transcription activator STAT3 and regulates TH17 differentiation. Nat. Immunol. 16, 1263–1273. https://doi.org/10.1038/ni.3278 (2015).
Jeffrey, K. L. et al. Positive regulation of immune cell function and inflammatory responses by phosphatase PAC-1. Nat. Immunol. 7, 274–283. https://doi.org/10.1038/ni1310 (2006).
Kucukosmanoglu, E., Keskin, O., Karcin, M., Cekmen, M. & Balat, A. Plasma adrenomedullin levels in children with asthma: Any relation with atopic dermatitis? Allergol. Immunopathol. 40, 215–219. https://doi.org/10.1016/j.aller.2011.05.014 (2012).
Rutledge, H. et al. Genetic regulation of Zfp30, CXCL1, and neutrophilic inflammation in murine lung. Genetics 198, 735–745. https://doi.org/10.1534/genetics.114.168138 (2014).
Kivihall, A. et al. Reduced expression of miR-146a in human bronchial epithelial cells alters neutrophil migration. Clin. Transl Allergy 9, 62. https://doi.org/10.1186/s13601-019-0301-8 (2019).
Lambrecht, B. N., Hammad, H. & Fahy, J. V. The cytokines of asthma. Immunity 50, 975–991. https://doi.org/10.1016/j.immuni.2019.03.018 (2019).
Chen, G. et al. Identification of key signaling pathways and genes in eosinophilic asthma and neutrophilic asthma by weighted gene co-expression network analysis. Front. Mol. Biosci. 9, 805570. https://doi.org/10.3389/fmolb.2022.805570 (2022).
Nakagome, K. & Nagata, M. Involvement and possible role of eosinophils in asthma exacerbation. Front. Immunol. 9, 2220. https://doi.org/10.3389/fimmu.2018.02220 (2018).
Zeng, Z., Lin, X., Zheng, R., Zhang, H. & Zhang, W. Celastrol alleviates airway hyperresponsiveness and inhibits Th17 responses in obese asthmatic mice. Front. Pharmacol. 9, 49. https://doi.org/10.3389/fphar.2018.00049 (2018).
Rohan, P. J. et al. PAC-1: A mitogen-induced nuclear protein tyrosine phosphatase. Science 259, 1763–1766. https://doi.org/10.1126/science.7681221 (1993).
McKinley, L. et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. immunol. 181, 4089–4097. https://doi.org/10.4049/jimmunol.181.6.4089 (2008).
Nie, Z. et al. Role of TNF-α in virus-induced airway hyperresponsiveness and neuronal M2 muscarinic receptor dysfunction. Br. J. Pharmacol. 164, 444–452. https://doi.org/10.1111/j.1476-5381.2011.01393.x (2011).
Laza-Stanca, V. et al. Rhinovirus replication in human macrophages induces NF-kappaB-dependent tumor necrosis factor alpha production. J. Virol. 80, 8248–8258. https://doi.org/10.1128/jvi.00162-06 (2006).
Goleva, E. et al. Corticosteroid-resistant asthma is associated with classical antimicrobial activation of airway macrophages. J. Allergy Clin. Immunol. 122, 550-559.e553. https://doi.org/10.1016/j.jaci.2008.07.007 (2008).
Li, L. B., Leung, D. Y. & Goleva, E. Activated p38 MAPK in peripheral blood monocytes of steroid resistant asthmatics. PLoS ONE 10, e0141909. https://doi.org/10.1371/journal.pone.0141909 (2015).
Wang, G. et al. Combined treatment with SB203580 and dexamethasone suppresses non-typeable Haemophilus influenzae-induced Th17 inflammation response in murine allergic asthma. Eur. J. Pharmacol 862, 172623. https://doi.org/10.1016/j.ejphar.2019.172623 (2019).
Akbari, O. et al. CD4+ invariant T-cell-receptor+ natural killer T cells in bronchial asthma. N. Engl. J. Med. 354, 1117–1129. https://doi.org/10.1056/NEJMoa053614 (2006).
Yang, M., Kumar, R. K. & Foster, P. S. Pathogenesis of steroid-resistant airway hyperresponsiveness: Interaction between IFN-gamma and TLR4/MyD88 pathways. J. Immunol. 182, 5107–5115. https://doi.org/10.4049/jimmunol.0803468 (2009).
Kovach, M. A. & Standiford, T. J. Toll like receptors in diseases of the lung. Int. Immunopharmacol. 11, 1399–1406. https://doi.org/10.1016/j.intimp.2011.05.013 (2011).
Jang, H. Y. et al. SPA0355 suppresses T-cell responses and reduces airway inflammation in mice. Eur. J. Pharmacol. 745, 19–28. https://doi.org/10.1016/j.ejphar.2014.08.038 (2014).
Cankara, F. N. et al. The beneficial effect of salubrinal on neuroinflammation and neuronal loss in intranigral LPS-induced hemi-Parkinson disease model in rats. Immunopharmacol. Immunotoxicol. 44, 168–177. https://doi.org/10.1080/08923973.2021.2023174 (2022).
Hamamura, K. et al. Salubrinal acts as a Dusp2 inhibitor and suppresses inflammation in anti-collagen antibody-induced arthritis. Cell. Signal 27, 828–835. https://doi.org/10.1016/j.cellsig.2015.01.010 (2015).
Cnop, M. et al. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J. Biol. Chem. 282, 3989–3997. https://doi.org/10.1074/jbc.M607627200 (2007).
Wan, Q., Xu, W., Yan, J. L., Yokota, H. & Na, S. Distinctive subcellular inhibition of cytokine-induced SRC by salubrinal and fluid flow. PLoS ONE 9, e105699. https://doi.org/10.1371/journal.pone.0105699 (2014).
Fehervari, Z. J. N. i. Alveolar macrophages in asthma. 16, 64–64 (2015).
Antonelli, A. et al. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun. Rev. 13, 272–280. https://doi.org/10.1016/j.autrev.2013.10.010 (2014).
Mahmutovic Persson, I. et al. IL-1β mediates lung neutrophilia and IL-33 expression in a mouse model of viral-induced asthma exacerbation. Respir. Res. 19, 16. https://doi.org/10.1186/s12931-018-0725-z (2018).
Gauthier, M. et al. Severe asthma in humans and mouse model suggests a CXCL10 signature underlies corticosteroid-resistant Th1 bias. JCI Insight https://doi.org/10.1172/jci.insight.94580 (2017).
Clarke, D. L. et al. TNFα and IFNγ synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-κB, and the transcriptional coactivator CREB-binding protein. J. Biol. Chem. 285, 29101–29110. https://doi.org/10.1074/jbc.M109.0999952 (2010).
Funding
This work was supported by grants from the National Natural Science Foundation of China (81873406), Hainan Provincial Natural Science Foundation of China (No.823QN345), Key Laboratory of Emergency and Trauma of Ministry of Education (Hainan Medical University) (Grant. KLET-202213).
Author information
Authors and Affiliations
Contributions
C.H., C.W. and Y.W. performed study concept and design; Y.W. performed development of methodology and revision of the paper; C.W. and Y.W. provided acquisition, analysis and interpretation of data, and statistical analysis; C.W. and Y.W. provided technical and material support. C.W., Y.W. and C.H. prepared all figures and wrote the main manuscript text. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, C., Wang, Y. & Hu, C. Bioinformatic analysis and experimental validation of the potential gene in the airway inflammation of steroid-resistant asthma. Sci Rep 13, 8098 (2023). https://doi.org/10.1038/s41598-023-35214-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35214-4
- Springer Nature Limited