
Abstract
Here, we present new molecular and morphological evidence that contributes towards clarifying the phylogenetic relations within the family Hesperiidae, and overcomes taxonomic problems regarding this family. First, nine new complete mitogenomes, comprising seven newly sequenced species and two samples of previously sequenced species collected from different localities, were obtained and assembled to analyze characteristics. The length of the mitogenomes ranges from 15,284 to 15,853 bp and encodes 13 protein-coding genes, two ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes, and a control region. Two model-based methods (maximum likelihood and Bayesian inference) were used to infer the phylogenetic relationships. Based on the mitogenomic phylogenetic analyses and morphological evidence, we claim that the lineage that comprises two Asian genera, Apostictopterus Leech and Barca de Nicéville, should be a tribe Barcini stat. nov. of the subfamily Trapezitinae, Pseudocoladenia dea (Leech, 1894), P. festa (Evans, 1949), and Abraximorpha esta Evans, 1949 are considered distinct species. Finally, we suggest that Lotongus saralus chinensis Evans, 1932 should belong to the genus Acerbas de Nicéville, 1895, namely Acerbas saralus chinensis (Evans, 1932) comb. nov..
Similar content being viewed by others


Introduction
Advances in molecular techniques accelerate our understanding of biological diversity and phylogenetic relationships of taxa. Taxonomic research now largely relies on molecular phylogeny. However, there is still opportunity for taxonomic decisions to determine the taxonomic rank that should be assigned to a particular taxon.
Hesperiidae is the third-largest butterfly family, containing approximately 600 genera and 4300 species1,2,3. Recent higher-level molecular phylogenetic studies of the family have unveiled taxonomic inconsistencies that have attracted research attention1,2,4,5,6,7,8,9. The taxonomic interpretation is under more debate than the differences in phylogeny. Warren et al.10 proposed seven subfamilies, which is still followed by current reserach4,6. However, Zhang et al. divided the family into additional subfamilies including Katreinae, Chamundinae, and Barcinae8. The treatment of Pyrrhopyginae as a subfamily was followed in other literature10,11. However, these taxonomic conflicts remain unsettled.
The subfamily level, Pyrginae sensu lato and Barcinae are the focus of current study. Morphologically, the subfamily Pyrginae had been speculated as a polyphyletic group12,13,14, However, taxonomists have not attempted to divide the subfamily into appropriate groups until this is supported by molecular phylogenetic studies using several gene markers1,4,15. Based on three genes and 49 morphological characteristics, Warren et al.16 treated Pyrginae as a monophyletic group and the finding was extensively supported by current research4,6,7,17. Meanwhile, Li et al.7 divided the subfamily Pyrginae sensu lato (i.e., sensu Warren et al., 2009) into three subfamilies, namely Tagiadinae, Pyrrhopyginae, and Pyrginae based on their relative divergence time compared to other subfamilies7.
Two Asian genera, Apostictopterus and Barca, have been traditionally classified in the Heteropterus genus group of the subfamily Hesperiinae9,18,19 or in a distinct subfamily Heteropterinae1,20 based on morphology. Analysis of mitogenomic data, however, suggested that both Barca and Apostictopterus are not within the Heteropterin genera and should tentatively be assigned to Hesperiinae, owing to the absence of the subfamily Trapezitinae17 in the phylogeny. Phylogenetic analyses based on whole-genome data have demonstrated showed that the clade of these two genera is a sister to that of the Trapezitinae8. Based on the result, Zhang et al. proposed that Barca and Apostictopterus should be classified as members of a new subfamily Barcinae8. In our study, we aimed to challenge this taxonomic assumption.
Recent studies show that the mitochondrial genome can provide good phylogenetic signals for understanding taxonomic systematics8,17,21,22,23. Yoshizawa et al. used mitochondrial genomic data to construct phylogenetic trees and explore mitochondrial evolution problems in Psocodea, providing strong support for the Prionoglarididae family because its monophyletics were inconsistent in previous morphological and molecular studies23. To better understand the phylogenetic relationships among the subfamilies and genera of Hesperiidae, we sequenced and assembled the mitogenomes of 13 samples representing 13 species/subspecies, including four taxa (Pseudocoladenia dea, Pseudocoladenia festa, Abraximorpha esta, and Lotongus saralus chinensis) whose taxonomic statuses remain unsettled. In a previous study, Huang & Xue elevated three subspecies of Pseudocoladenia dan, Pesudocoladenia dan dea (type locality: Pu-tsu-fong, Sichuan), P. dan festa (type locality: Naga Hill, India), and P. dan fauta (type locality: Gangtok, Sikkim, India), to species level based on morphological characteristics24. Further, Lotongus saralus de Nicéville, 1889, a skipper from the Oriental region, was transferred to Acerbas based only on genome data2,9. Using publicly available mitogenomic sequences of 45 skipper species in GenBank (Supplementary Table S1) as well as our 13 mitogenomes, we reconstructed phylogenetic trees of the 58 skipper species using both maximum likelihood (ML) and Bayesian inference (BI) methods. Finally, based on the molecular results and morphological analysis, we aimed to clarify the monophyly of Pyrginae, the taxonomic positions of Apostictopterus and Barca, and the taxonomic status of the four taxa mentioned above.
Results and discussion
Mitogenome features
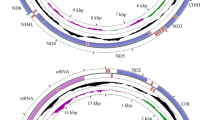
Nine new mitogenomes were assembled and annotated. Their circular maps were similar, thus only the map of Abraximorpha esta is shown (Fig. 1, Supplementary Fig. S1). The length of the mitogenomes ranged from the shortest, 15,284 bp (Coladenia maeniata) to the longest, 15,853 bp (Lotongus saralus chinensis and Trapezites iacchus). Each mitogenome was typically composed of 13 PCGs, 22 tRNA genes, 2 rRNA genes, and one major non-coding AT-rich region. Twelve of the 13 PCGs started with three typical start codon types (ATA, ATG, and ATT) in skippers, however one, COI, began with CGA, which is common across the order Lepidoptera. The PCGs terminated with stop codons TAA or TAG or an incomplete stop codon T–; incomplete stop codons always appear in lepidopteran mitogenomic PCGs. All start and stop codons are listed in Supplementary Table S2.

A circular map of the Abraximorpha esta mitochondrial genome.
The nucleotide proportion, GC content, AT-skew, and GC-skew of the nine sequences were comprehensively calculated and analyzed (Fig. 2). The fluctuation in adenine (A) content in butterflies is generally small25, and the variation range of the nine sequences base A was ± 0.01. Among the nine sequences, Lotongus saralus chinensis had the highest GC-skew (25.63%), whereas Pseudocoladenia dan fabia had the lowest GC-skew (19.25%). The mean GC-skew was 20.88%, indicating that cytosine was present more frequently in genes than guanine. We analyzed the characteristics of the PCG nucleotide sequences (Table 1). Among the 13 PCGs, the COI gene had the highest number of invariant sites and the lowest percentage of variable sites. According to the percentage of variable sites, ND6 has the highest mutation rate, and COI has the smallest mutation rate.

Analysis of base composition and skewness of nine mitochondrial genomes.
Most tRNA genes were folded into a cloverleaf secondary structure using MITOS, except for trnS, which lacked the DHU arm in nine species of skipper. Owing to the lack of the DHU stem, trnS was consistently the shortest among the tRNAs of the skippers (Supplementary Fig. S2). The two rRNA genes (rrnL and rrnS) encoding the small and large ribosomal subunits were located between trnL and trnV, as well as trnV and the AT-rich region, respectively.
The AT-rich region, also called the control region, exerts an important function in initiating replication in invertebrates. However, the control region does not encode any known functional genes26,27. In our study, the length of the AT-rich region in the nine skipper species ranged from 351 bp (Darpa inopinata) to 852 bp (Lotongus saralus chinensis). In addition to AT-rich regions, non-coding regions commonly exist between two genes, called spacer regions. Generally, butterflies have a 60–90 bp interval between the mitochondrial genes trnQ and ND2, which may be caused by gene rearrangement22. In terms of mitochondrial genome rearrangement, most mitochondrial genomes of Lepidoptera are reported to have trnM rearrangements17,22,28; here, the original trnI-trnQ-trnM was rearranged to trnM-trnI-trnQ (Fig. 3).

Characters of the nine sequenced hesperiid species’ mitochondrial genomes. Genetic names are replaced by abbreviations.
Phylogenetic relationships
In this study, 58 species were analyzed based on 37 mitochondrial genes. The total concatenated alignment length after removing ambiguous regions was 15,066 bp.
Four datasets yielded mostly congruent results, differing only in the position of Eudaminae and Pyrginae; we used the results based on the PRT datasets for further analyses. PartitionFinder suggested 53 partitions as optimal for the PRT datasets (Supplementary file S1).
The phylogenetic trees reconstructed using the ML and BI methods result in an identical topology with strong support (Fig. 4); indicating that Hesperiidae is a strongly supported monophyletic group. Within this family, seven major clades correspond to seven subfamilies: Coeliadinae, Euschemoninae, Eudaminae, Pyrginae, Heteropterinae, Trapezitinae, and Hesperiinae. Similarly to previous studies4,7,9,16,17,29, (1) Coeliadinae was branched out at the base of the family; (2) Euschemoninae was sister to the other Hesperiidae excluding Coeliadinae; and (3) Pyrginae sensu lato was a monophyletic group. Within the subfamily Pyrginae, tribes Tagiadini and Celaenorrhinini were sister groups. Li et al. divided Pyrginae sensu lato into three subfamilies: Tagiadinae, Pyrrhopyginae, and Pyrginae7. In the current study we did not follow this taxonomy for two reasons. First, we lacked sufficient mitogenomic data for more material. Second, such fragmentation of subfamily opposes the purpose of taxonomically robust and stable classification. Notably, phylogeny of mitogenomes is not equal to the phylogeny of organisms; which are better represented by their nuclear genomes30,31. Morphological features, such as venation and genitalia, are encoded in the nuclear genome. Therefore, we also illustrated morphological characters, instead of the phylogenetic tree, and the information of nuclear genome was implicitly used in this study and projected onto the mitochondrial genome phylogeny. Herein, we discuss the following taxonomic problems in subfamily and species level.

Phylogenetic tree based on PRT dataset. Numbers at node indicates posterior probabilities (PP) and bootstrap value (UFBoot) based on ML analyses were also given. * represents the newly sequenced species.
Apostictopterus and Barca. Our molecular phylogeny indicates that the Trapezitinae and the clade composed of two Asian genera, Apostictopterus and Barca, are strongly supported monophyletic groups (PP = 1, UFBoot = 90), which is consistent with the finding of Zhang et al.8 These results suggest that the lineage of Apostictopterus and Barca belong to the subfamily Trapezitinae or represent a separate subfamily. Zhang et al.8 claimed that no apparent morphological synapomorphies unified the group of the two genera with the Trapezitinae and treated the lineage as a separate subfamily, Barcinae. Nevertheless, we did not follow this classification. Instead, we classified them as a tribe, Barcini stat. nov., within the subfamily Trapezitinae. The Trapezitinae in conventional sense is a monophyletic group which has been characterized by single synapomorphy: the discocellular vein on the hindwing is directed toward the wing apex14. Braby noted that the apex of the hindwing cell, in the Trapezitinae, is truncated, with the discocellular vein between M2 and M3 angled or inclined toward the dorsum, and that vein M2 is always present but usually weakly developed in the Trapezitinae32. Our morphological study also demonstrates that vein 1A + 2A on the forewing is a bow-like in shape that changes gradually: faintly curved in Trapezites (Fig. 5C), moderately curved in Atkinsia33, and prominently arched in Signeta (Fig. 5D). Similarly, Zhang et al.8 proposed the subfamily Barcinae, characterized by a vein 1A + 2A bow-like shape on the forewing. Apostictopterus and Barca share similar characteristics of the discocellular vein and weakly developed vein M2 on the hindwing and bow-like shaped vein 1A + 2A on the forewing with genera of the Trapezitinae (Fig. 5). During immature stages, eggs of Apostictopterus and Barca, like most eggs of the Trapezitinae, are strongly ribbed33. Biogeographically distributions such as this one in southern China and Australia are also found in Miletinae. Both Apostictopterus and Barca are distributed throughout Asia, while all the Trapezitinae members are distributed in Australia and adjacent locations. These disjunct distributions (e.g., southern China and Australia) were also identified in Miletinae, Lycaenidae34,35. Alternative taxonomic treatment includes Herteropterinae + Trapezitinae (including Apostictopterus and Barca) + Hesperiinae as a single subfamily Hesperiinae (PP = 1, UFBoot = 100) or Trapezitinae (including Apostictopterus and Barca) + Hesperiinae, excluding Heteropterinae, as a subfamily Hesperiinae (PP = 1, UFBoot = 88). Further extensive examination of morphology, life history and other biological data will increase our ability to determine which taxonomic treatment is the most suitable taxonomic framework.

Wing venation of four genera of Trapezitinae. (A) Apostictopterus fuliginosus (B) Barca bicolor (C) Trapezites symmomus (D) Signeta flammeata.
Pseudocoladenia dea and P. festa. In our study, three members of the genus Pseudocoladenia, P. dea (from Yingjing, Sichuan), P. festa (from Moxi, Sichuan), and P. dan fabia (from Yingde, Guangdong), were clustered into a highly supported clade, and P. dea was a sister to P. festa + P. dan fabia. Male genitalia differ among these taxa. In P. dea, the valva had a ventrodistal process vertically elongated (long and pointed distally in P. festa and short and small in P. dan fabia) and the aedeagus possesses a dorsal spiny process (no such process in the other two).
The genetic distance was calculated for the COI barcoding region (658 bp), and the results indicated that the distance between P. dea and P. dan fabia was 4.9%, 3.7% between P. festa and P. dan fabia, and 4.7% between P. dea and P. festa. Although we could not sample the nominate subspecies P. dan dan (type locality: Tranquebar, India), P. dan fabia and the nominate subspecies in our study shared similar wing patterns and male genitalia. Thus, it appears appropriate to discuss the status of P. dea and P. festa with P. dan fabia.
Recently, Huang restored the subspecies of P. dan, fatih (type locality: Mussoorie, India), to species level and classified P. festa as a subspecies of P. fatih because of its allopatric distribution to P. festa and a lack of differences in their male genitalia36. Further, based on the distributions of P. fatih and P. festa in India (https://www.ifoundbutterflies.org/pseudocoladenia-fatih or festa, Anonymous 2022) as well as our specimens from Zhang Mu, Tibet, the two taxa appear sympatric. In addition, P. fatih can be morphologically distinguished from P. festa based on differences in the white sub-hyaline spots on the forewing and the arc-shaped outward edge of the ventrodistal process of valva (Huang, 2021, Fig. 85: 22-1, 23-1). Lastly, we obtained a 658 bp COI sequence for P. fatih and estimated the genetic distance between P. fatih and P. festa to be 2.3%. Based on the differences in wing pattern and male genitalia, with supporting molecular data, we suggest that all P. dea, P. fatih, and P. festa should be regarded as distinct species.
Abraximorpha esta. Evans described A. esta as a subspecies of A. davidii12. Devyatkin & Monastyrskii37, however, regarded A. esta as a separate species based on morphological examination of the type specimens stored in the Natural History Museum, London. In this study, A. esta and A. davidii were recovered as sister taxa. The COI-based sequence divergence between these two taxa was 0.9%. Genetic divergence values between closed sister taxa are often lower than 1%, which is very common in Lepidoptera because hybridization, introgression or incomplete lineage sorting can cause low interspecific divergence or produce young species in which divergence is too recent for lineage sorting to complete38,39,40. Our morphological study showed that A. esta can be distinguished from A. davidii based on the following characteristics: In A. esta, the wing pattern on the dorsal side is the same as that on the ventral side, the spots at the apex are radial, and the spots in spaces CuA1 and CuA2 are long-rectangular shaped. In contrast, in A. davidii, white spots on the ventral side are more developed than those of the dorsal side, the spots at the apex are small and rectangular, and only the spot in the space CuA1 is long-rectangular shaped. The male genitalia show significant differences between these two taxa (Fig. 6): in A. esta, the dorsal process in the middle of valva is long, thin, and pointed, reaching dorsal swelling and its ventrodistal process with the inner edge is S-shaped, whereas in A. davidii, the dorsodistal process is short with a blunt tip, not reaching the swelling in left valva; the ventrodistal process of the inner edge is straight, although there are individual variations in A. davidii. In summary, based on the difference in wing pattern and male genitalia described above combined with the work of Devyatkin and Monastyrskii37 and Osada et al.41 suggesting their sympatric distribution, we agree with Devyatkin & Monastyrskii37 that A. esta is a distinct species.

Valvae of two Abraximorpha skippers. (A,B) A.esta (A: Baoshan, Yunnan; B: Vietnam);. (C, D) A.davidii (C:Baoxin, Sichuan; D: Chongqing).
Acerbas saralus chinensis comb. nov.. Cong et al.2 and Zhang et al.9 reclassified Lotongus saralus into the genus Acerbas based on genomic phylogenetic analyses of the nominate subspecies. Our phylogenetic analyses also indicated that the genus Lotongus is not monophyletic. Further, in our phylogeny, Lotongus saralus chinensis was a sister to Erionota, whereas Lotongus taprobanus42 was closely related to Matapa. The male genitalia of L. calathus differed from those of L. saralus, including a pair of lateral cone-shaped process of dorsum and apically broad uncus, with small and pointed dorso-lateral processes (Fig. 7), implying that Lotongus is not a monophyletic group. Although we failed to sample the type species of Acerbas and the nominate subspecies of L. saralus in this study, L. saralus chinensis exhibited a similarity in male genitalia implying its close relationship with Acerbas: the socius is absent and the uncus is shallowly indented apically (Fig. 7A,C), resembling its nominate subspecies in terms of wing pattern and male genitalia. Therefore, based on our morphological and molecular evidence, as well as the results of Cong et al.2 and Zhang et al.9, we agree that Lotongus saralus was misclassified in Lotongus and should be reclassified to the genus Acerbas along with the subspecies chinensis.

Male genitalia of Acerbas and Lotongus. (A) A. anthea; (B) L.calathus calathus; (C) L. saralus chinensis. I: Left valva, inner view; II: Genitalia ring, lateral view; III: Tegument, dorsal view.
Conclusion
Stable phylogenetic relationships are key to understanding the role of speciation in the formation of biodiversity. In this study, a phylogenetic tree was constructed based on mitogenome data and morphological analysis. We suggest that (1) Barca and Apostictopterus should be reclassified together as a tribe Barcini in the subfamily Trapezitinae; (2) Pseudocoladenia festa, P. dea, and Abraximorpha esta are distinct species and; (3) Lotongus saralus chinensis should be transferred to the genus Acerbas. Although this study shares similarities with previously published research3,8,17,24, our results were based on a molecular phylogenetic analysis, integrated morphological characters and Bayesian statistics which allowed us to better evaluate the state of these groups. Thus, this study confirms the results of previous studies, thereby improving confidence in the respective suggestions for taxonomic rank. Additionally, mtDNA data alone cannot identify issues on different taxonomical levels. In the study of the Hesperiidae, information from morphology is vital for classification at different levels. Finally, in some branches of the phylogenetic tree, there were no high node support values. These issues will be addressed in future studies, adding nuclear genomic data and additional materials to reveal the phylogenetic relationships within the family Hesperiidae.
Materials and methods
Taxon sampling and morphological study
Thirteen species belonging to eight genera of the family Hesperiidae were sampled (Supplementary Table S3). Among these, whole-genome data of four species were available at NCBI (Tagiades japentus, T. symmonus, Signeta flammeata, and Lotongus calathus taprobanus). Seven species were sequenced for the first time, including Pseudocoladena festa, P. dea, Darpa inopinata, Abraximorpha esta, Coladenia maeniata, Trapezites iacchus, and L. saralus chinensis. Additionally, P. dan fabia and Matapa aria have been sequenced in previous studies9,29; however, the collection locality differed from that in this study. The specimens used in this study were stored at South China Agricultural University (SCAU), Guangzhou, China. Methods of morphological studies of adult specimens and male genitalia described Fan et al. were followed43, and the wing venation treatment was performed according to the method outlined by Hou et al.44.
Laboratory protocols
Total DNA was extracted from the leg muscle tissue of a single adult butterfly for each sample using the HiPure Insect DNA Kit (Magen, China), following the manufacturer’s instructions. For amplification, complete mitogenomes were divided into 27 overlapping fragments. The primers and amplification fragment steps were performed as described by Kim et al.25 and Han et al.17. Owing to the AT-rich region instability, we cloned this fragment, using methods outlined by Fan et al.45, after amplification.
Mitogenome annotation and data analyses
The sequences obtained were proofread and then assembled using the software Geneious v7.1.446. Protein-coding genes (PCGs) were identified by finding the respective ORFs on the NCBI website (https://www.ncbi.nlm.nih.gov/oreeinder) with the invertebrate mitochondrial genetic codes. Next, tRNAs and rRNAs were identified using the MITOS Web Server (http://mitos.bioinf.uni-leipzig.de/index.py). Each new sequence was aligned against the complete mitogenomes of other skippers using MAFFT v7.31347 to determine gene boundaries and relative positions. The AT-rich region was identified by recognizing the boundaries between rrnS and trnM. Nucleotide composition bias was calculated using the formula: AT-skew = (A − T)/(A + T); GC-skew = (G − C)/(G + C)48.
Phylogenetic analysis
The ingroup for phylogenetic analysis included 13 sequences from this study and 45 publicly available mitogenomes, collectively representing 58 hesperiid species (Supplementary Table S1). The mitogenomes of six species were obtained from GenBank as outgroup (Supplementary Table S1).
Each of the 13 PCGs was aligned individually using the software MAFFT V7.3.1349. Specifically, tRNAs and rRNAs were aligned separately using the Q-INS-i strategy through the MAFFT V7.313 online alignment server (https://maf.cbrc.jp/alignment/server/)50. Gaps and ambiguous sites from the 13 PCGs were removed using the Gblocks V0.91 online server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html).
We reconstructed ML and BI tree of four datasets (PRT, PCGD, PCGC, and PCGR)14, in which we used the partition scheme produced by PartitionFinder v2.1.1 under the Bayesian information criterion (BIC)47. ML analyses were performed on the IQ-TREE web online server (http://iqtree.cibiv.univie.ac.at/, accessed in May 2022)51 with 1000 ultrafast bootstraps (UFBoot) to estimate branch support. We set bootstrap analysis to ultrafast and the number of bootstrap alignments to 1000, UFBoot indicates the confidence level of each branch, generally considered to be high when UFBoot ≥ 9552, and the rest were use as default parameters. BI analyses were performed using MrBayes V3.2.6 on the CIPRES Science Gateway 3.3 (http://www.phylo.org/)53. The phylogenetic tree construction model setting adopts reversible jump Markov chain Monte Carl (MCMC), the specific parameter settings: set 4 MCMC chains (1 cold chain and 3 hot chains) each chain ran 5 × 108 generations, sampling once every 1 × 103 generations, running independently twice, discarding the 25% aging tree that just runs. The file obtained after running uses Tracer V1.654 to verify whether the ESS value of each valid sample size was greater than 200, that is, whether Bayesian converges. The phylogenetic tree is shown with FigTree v1.4.3. Overall, this study is almost identical to methods used by Han et al.17, with the only difference being our larger sample size with additional species. In this current study, the genetic distances for the COI barcodes were calculated using Kamar 2–parameter model in MEGA X55.
Data availability
The data that support the findings of this study are available from [National Center for Biotechnology Information]. We have provided GenBank accession numbers: OP723917-OP723926, OQ784637-OQ784639.
References
Warren, A. D., Ogawa, J. R. & Brower, A. V. Phylogenetic relationships of subfamilies and circumscription of tribes in the family Hesperiidae (Lepidoptera: Hesperioidea). Cladistics 24, 642–676 (2008).
Cong, Q., Zhang, J., Shen, J. H. & Gtishin, N. V. Fifty new genera of Hesperiidae (Lepidoptera). Insecta Mundi 731, 1–56 (2019).
Toussaint, E. A. et al. Historical biogeography of Heteropterinae skippers via Beringian and post-Tethyan corridors. Zoolog. Scr. 00, 1–12 (2020).
Sahoo, R. K. et al. genes and two topologies: An exploration of higher relationships in skipper butterflies (Hesperiidae). PeerJ 4, e2653 (2016).
Sahoo, R. K., Warren, A. D., Collins, S. C. & Kodandaramaiah, U. Hostplant change and pale oclimatic events explain diversification shifts in skipper butterflies (Family: Hesperiidae). BMC Evol. Biol. 17, 1–9 (2017).
Toussaint, E. F. et al. Anchored phylogenomics illuminates the skipper butterfly tree of life. BMC Evol. Biol. 18, 101 (2018).
Li, W. et al. Genomes of skipper butterflies reveal extensive convergence of wing patterns. Proc. Natl. Acad. Sci. 116, 6232–6237 (2019).
Zhang, J., Cong, Q., Shen, J. H., Brockmann, E. & Grishin, N. Three new subfamilies of skipper butterflies (Lepidoptera, Hesperiidae). ZooKeys 861, 91–105 (2019).
Zhang, J., Cong, Q., Shen, J. H. & Grishin, N. Taxonomic changes suggested by the genomic analysis of Hesperiidae (Lepidoptera). Insecta mundi 2022, 1409 (2022).
Mielke, O. H. M., Brockmann, E & Mielke, C. G. C. Butterflies of the world part 49. Hesperiidae II (New World Pyrrhopiginae. Goecke & Evers, 120pp).
Brockmann, E., Mielke, C. G. C & Mielke, O. H. M. Butterflies of the world Part 50. Hesperiidae III (New world Pyrrhopiginae. Goecke & Evers. 40pp+76plates, 2022).
Evans, W. H. A catalogue of the Hesperiidae from Europe, Asia and Australia in the British Museum (Natural History). (Trust. Brit. Mus. London, 502 pp., 53 pis 1949).
Evans, W. H. A catalogue of the American Hesperiidae in the British Museum (Natural History). Part I. Introduction and Group A, Pyrrhopyginae. (British Museum Natural History 1951).
Ackery, P., de Jong, R. & Vane-Wright, R. The Butterflies: Hedyloidea, Hesperioidea and Papilionoidea 263–300 (Walter de Gruyter, New York, 1999).
Wahlberg, N. et al. Synergistic effects of combining morphological and molecular data in resolving the phylogeny of butterflies and skippers. Proc. R. Soc. B Biol. Sci. 272, 1577–1586 (2005).
Warren, A. D., Ogawa, J. R. & Brower, A. V. Revised classification of the family Hesperiidae (Lepidoptera: Hesperioidea) based on combined molecular and morphological data. Syst. Entomol. 34, 467–523 (2009).
Han, Y. K., Huang, Z. F., Tang, J., Chiba, H. & Fan, X. L. The complete mitochondrial genomes of two skipper genera (Lepidoptera: Hesperiidae) and their associated phylogenetic analysis. Sci. Rep. 8, 1–10 (2018).
Chou, I. (ed.) Monographia Rhopalocerorum Sinensium (Henan Scientifific and Technological Publishing House, Henan, 1994).
Chou, I. Classifcation and Identifcation of Chinese Butterfies (Henan Scientifc and Technological Press, Zhengzhou, 1998).
Yuan, X. Q., Gao, K., Yuan, F., Wang, P. & Zhang, Y. L. Phylogenetic relationships of subfamilies in the family Hesperiidae (Lepidoptera: Hesperioidea) from China. Sci. Rep. 5, 11140 (2015).
Cameron, S. L., Miller, K. B., D’Haese, C. A., Whiting, M. F. & Barker, S. Mitochondrial genome data alone are not enough to unambiguously resolve the relationships of Entognatha, Insecta and Crustacea sensu lato (Arthropoda). Cladistics 20, 534–557 (2005).
Kim, M. J., Wang, A. R., Park, J. S. & Kim, I. Complete mitochondrial genomes of five skippers (Lepidoptera: Hesperiidae) and phylogenetic reconstruction of Lepidoptera. Gene 549, 97–112 (2014).
Yoshizawa, K. et al. Mitochondrial phylogenomics and genome rearrangements in the barklice (Inscta: Psocodea). Mol. Phylogenet. Evol. 119, 118–127 (2018).
Huang, H. & Xue, Y. P. The Chinese Pseudocoladenia skippers. Neue Entomol. Nachr. 56, 161–170 (2004).
Kim, M. J., Wan, X. L., Kin, K. G., Hwang, J. S. & Kim, I. Complete nucleotide sequence and organization of the mitogenome of endangered Eumenis autonoe (Lepidoptera: Nymphalidae). Afr. J. Biotech. 9, 735–754 (2010).
Dai, L. S., Kausar, S., Abbas, M. N. & Wang, T. T. Complete sequence and characterization of the Ectropis oblique mitochondrial genome and its phylogenetic implications. Int. J. Biol. Macromol. 107, 1142–1150 (2018).
Zheng, N. et al. Characterization of the complete mitochondrial genome of Biston marginata (Lepidoptera: Geometridae) and phylogenetic analysis among lepidopteran insects. Int. J. Biol. Macromol. 113, 961–970 (2018).
Wang, X. et al. Phylogenetic relationships among Bombycidae s.l. (Lepidoptera) based on analyses of complete mitochondrial genomes. Syst. Entomol. 44, 490–498 (2018).
Xiao, J. T., Liu, J. Q., Ma, L. Y., Hao, X. Y. & Yuan, X. Q. Mitogenomes of nine asian skipper genera and their phylogenetic position (Lepidoptera: Hesperiidae: Pyrginae). Insects 13, 68 (2022).
Edwards, S. V. & Bensch, S. Looking forwards or looking backwards in avian phylogeography? A comment on Zink and Barrowclough 2008. Mol. Ecol. 18, 2930–2933 (2009).
Toews, D. P. L. & Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 21, 3907–3930 (2012).
Braby, M. F. Butterflies of Australia: Their identification, Biology and Distribution Collingwood: CSIRO Publishing (2000).
Toussaint, E. F. Molecular phylogeny, systematics and generic classification of the butterfly subfamily Trapezitinae (Lepidoptera: Papilionoidea: Hesperiidae). Zool. J. Linn. Soc. 195, 1407–1421 (2022).
Wang, M. & Fan, X. L. Butterflies Fauna Sinica: Lycaenidae (Henan Science and Technology Press, Zheng Zhou, 2002).
D'Abrera, B. Butterflies of the Australian Region. (printed: Craftprint Ltd., Singapore 1990).
Huang, H. Taxonomy and morphology of Chinese butterflies 1 Hesperiidae: Pyrginae: Genera Coladenia Moore, [1881] and Pseudocoladenia Shirozu & SaiguSa, 1962. Atalanta 52, 569–620 (2021).
Devyatkin, M. Hesperiidae of Vietnam, 12.1 A further contribution to the Hesperiidae fauna of North and Central Vietnam. Atalanta 33, 1–2 (2002).
Nazari, V., Hagen, W. T. & Bozano, G. C. Molecular systematics and phylogeny of the “Marbled Whites” (Lepidoptera: Nymphalidae, Satyrinae, Melanargia Meigen). Syst. Entomol. 35, 132–147 (2010).
Habel, J. C. et al. Differentiation in the marbled white butterfly species complex driven by multiple evolutionary forces. J. Biogeogr. 44, 433–445 (2017).
Hausmann, A. et al. Now DNA-barcoded: the butterflies and larger moths of Germany (Lepidoptera: Rhopalocera, Macroheterocera). Spixiana 34, 47–58 (2011).
Osada, S., Uemura, Y. & J. Uehara: An illustrated checklist of the butterflies of Laos P.D.R. - Mokuyo-sha (Tokyo, 1999).
Zhang, J. et al. Taxonomic discoveries enabled by genomic analysis of butterflies. Taxon. Rep. 7, 2643–4776 (2022).
Fan, X. L., Chiba, H. & Wang, M. The genus Scobura Elwes & Edwards, 1897 from China, with descriptions of two new species (Lepidoptera: Hesperiidae). Zootaxa 2490, 1–15 (2010).
Hou, Y. X. et al. Molecular and morphological evidence reveals a new genus of the subfamily Heteropterinae (Lepidoptera, Hesperiidae) from China. ZooKeys 1055, 55–67 (2021).
Fan, X. L. et al. Clarification of the phylogenetic framework of the tribe Baorini (Lepidoptera: Hesperiidae: Hesperiinae) inferred from multiple gene sequences. PLoS ONE 11, 7 (2016).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773 (2016).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. Mol. Biol. Evol. 41, 353–358 (1995).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinf. 20, 1160–1166 (2017).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Minh, B. Q., Nguyen, M. A. T. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30(5), 1188–1195 (2013).
Miller, M. A., Pfeiffer, W. and Schwartz, T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In Gateway Computing Environments Workshop (GCE) 1–8 (2010).
Rambaut, A., Suchard, M. A., Xie, D., Drummond, A. J. Tracer v1.6. http://beast.bio.ed.ac.uk/Tracer (2014).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Acknowledgements
This work was supported by the National Nature Science Foundation of China of LX. Fan. (Grants No.31872264 and 31471984), LSh. Chen. (Grants No. 41661011) and ZF. Huang (Grants. No.32000326). We are grateful to the Cave Biology Laboratory of South China Agricultural University for taking the wing venation and genitalia photographs.
Author information
Authors and Affiliations
Contributions
L.Z. and Y.H. wrote the manuscript and did the experiments and data analyses, L.Z. prepared all the Figures and tables. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, L., Han, Y., Hou, Y. et al. Mitogenomic phylogenetic analyses provide novel insights into the taxonomic problems of several hesperiid taxa (Lepidoptera: Hesperiidae). Sci Rep 13, 7901 (2023). https://doi.org/10.1038/s41598-023-34608-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34608-8
- Springer Nature Limited