
Abstract
The widespread leafhopper genus Scaphoideus Uhler is the most diverse genus in Scaphoideini and includes some species that are serious pests and vectors of plant pathogens. Here the first Scaphoideus mitogenome sequences are provided for three species, S. maai, S. nigrivalveus and S. varius, representing three main species groups in the Oriental region based on color pattern. The lengths of these three mitogenomes were 15,188, 15,235 and 15,207 bp, respectively. Gene order of three mitogenomes is highly conserved and identical to that of the putative ancestral insect. All three mitogenomes exhibited similar AT nucleotide bias, AT-, GC-skews and codon usage. One large 101 bp intergenic spacer between trnY and cox1 was in S. varius. All 22 tRNA genes had typical cloverleaf secondary structures, except for trnS1 (AGN) which appears to lack the dihydrouridine arm. Genes atp8, nad6 and nad2 were highly variable while cox1 showed the lowest nucleotide diversity. Phylogenetic analyses of three concatenated nucleotide datasets using maximum likelihood and Bayesian methods, comprising all 13 mitogenomes currently available for Membracoidea plus mitogenomes for eight outgroup species representing other cicadomorphan superfamilies, yielded the same topology in which Scaphoideus species formed a monophyletic group within a larger clade comprising three other included Deltocephalinae.
Similar content being viewed by others


Introduction
Insect mitochondrial genomes are typically small double-stranded circular molecules containing 37 genes including 13 protein-coding genes (PCGs), two ribosomal RNAs (rRNAs), 22 transfer RNAs (tRNAs) genes, and one non-coding A + T-rich region (or control region, CR)1,2. Owing to its high genome copy numbers, multiple genome-level characteristics, relatively high evolutionary rate and greater phylogenetic informativeness than single mitochondrial genes, mitogenome sequences have been widely used in the comparative and evolutionary genomics, molecular evolution, phylogenetics and population genetics although such data remain sparse or unavailable for many insect groups2,3,4,5,6.
One such group, Deltocephalinae, the largest subfamily of leafhoppers, is distributed worldwide and contains the majority of leafhopper vectors of economically important plant diseases. The tribe Scaphoideini, comprising over 630 species in 61 genera, is one of the more diverse, widespread and economically important groups within the subfamily Deltocephalinae. Although the circumscription and morphological characterization of Scaphoideini Oman were substantially revised by Zahniser & Dietrich7, the phylogenetic relationships among genera and species of this tribe have not been explored.
The type genus Scaphoideus Uhler is the largest and most diverse genus in this tribe, comprising over 200 described species in all major biogeographic regions and differing widely in color and male genitalia. Although some authors attempted to define the genus, its classification remains unsatisfactory. Ball divided the species of the genus into three groups based on face color8. DeLong divided it into three distinct subgenera according to male genitalia9. However, these two classifications applied only to North American species and have not been widely adopted. Recently, the Oriental species of the genus were also divided into three groups according to coloration10. Until now, phylogenetic studies, utilizing morphological and molecular data, have focused more broadly on Deltocephalinae and included few representatives of Scaphoideini7,11,12. The small amounts of DNA sequence data currently available for this tribe in GenBank mainly include DNA barcodes of partial cox1 sequences. No species of Scaphoideini has had its mitochondrial genome sequenced so far. To further elucidate the phylogenetic status and relationships of Scaphoideini, much more data are needed.
Here, we analyze the first three complete mitogenomes for Scaphodeini, based on three Oriental species of the genus Scaphoideus representative of the three main color forms found among the Asian fauna: S. maai, a species representing the common group with a median longitudinal yellowish or whitish stripe; S. nigrivalveus, representing the group with transverse bands on the head, pronotum and scutellum; and S. varius, representing the group with dark brown spots or bands on vertex and longitudinal bands on pronotum and scutellum. General genome features including base composition and codon usage of PCGs were compared to explore the sequence variability among these three groups, and tRNA secondary structures were predicated. To examine the phylogenetic utility of complete mtDNA sequences, we reconstructed the phylogenetic relationships among the three newly sequenced species and other leafhoppers for which mtDNA genome data are available, using the concatenated nucleotide sequences of 13 protein-coding genes (PCGs) and two ribosomal RNA genes.
Results
General features of three Scaphoideus mitogenomes
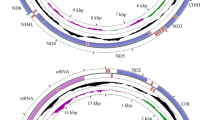
The sizes of the three Scaphoideus mitogenomes were 15,188 bp in S. maai (GenBank: KY817243), 15,235 bp in S. nigrivalveus (GenBank: KY817244) and 15,207 bp in S. varius (GenBank: KY817245), respectively. Circular maps of three mitogenomes are shown in the Fig. 1. Each newly sequenced mitogenome contained a typical set of 37 mitochondrial genes (13 PCGs, 22 tRNAs and two rRNAs) and one control region (Supplementary Tables S1–S3). Gene order was invariant and identical to that of Drosophila yakuba and to most other previously sequenced Membracoidea1,2,13,14,15.

Mitochondrial genomes of three sequenced Scaphoideus leafhoppers. Protein coding and ribosomal genes are shown with standard abbreviations. Transfer RNA (tRNA) genes are indicated using the IUPAC-IUB single letter amino acid codes (L1:CUN; L2:UUR; S1:AGN; S2:UCN).
Base composition
All three Scaphoideus mitogenomes exhibited heavy AT nucleotide bias, with 77.2%, 76.5% and 75.9% in S. maai, S. nigrivalveus and S. varius respectively. The A + T content of the CR (mean value = 82.8%) was always significantly higher (P = 0.000) than in other regions, while PCGs showed the lowest A + T content values (mean value = 75.6%) (Supplementary Fig. S1). All three species had higher A + T content in rrnL than rrnS, with significantly different mean values (80.2% and 77.9%) (Table 1; Supplementary Fig. S1). All three mitogenomes showed positive AT-skews (0.075 to 0.093) and negative GC-skews (−0.154 to −0.178). CR sequences always were slightly T-skewed (−0.014 to −0.026) and C-skewed (−0.006 to −0.100), while the two rRNA genes were moderately T-skewed (−0.102 to −0.130) and markedly G-skewed (0.178 to 0.300) (Table 1).
Protein-coding genes and codon usage
Of the 13 PCGs, nine were located on the majority strand (J-strand) while the other four PCGs were encoded by the minority strand (N-strand). The third codon position had a significantly higher (P = 0.000) A + T content than that of the first and second positions (87.5% versus 71.2% and 67.9%) (Supplementary Fig. S1). Pairwise comparisons among the three Scaphoideus species (Table 2) indicate that, except for cox2, PCGs had fewer variable sites between S. maai and S. nigrivalveus than species pairs.
Except for nad5, which began with TTG, all other PCGs started with the standard ATN codons, as in other leafhopper mitogenomes (Tambocerus sp., Idioscopus nitidulus) (Table 3). Most of the PCGs terminated with a TAA stop codon, while atp6 in S. maai, cox1 and cob in S. nigrivalveus end with a TAG codon. Two PCGs (cox2 and nad4) terminated with truncated T stop codons in all three species; nad5 in S. maai and nad1 in S. varius also end with the incomplete codon T (Table 3).
After excluding the stop codons, the relative synonymous codon usage (RSCU) was calculated and summarized in Fig. 2. The total numbers of non-stop codons were 3632, 3633 and 3635 in S. maai, S. nigrivalveus and S. varius respectively. Leucine (Leu), Serine (Ser), Isoleucine (Ile) and Methionine (Met) were the most frequently used amino acids. Within each amino acid codon, third positions ending with A/T were more frequent than those terminated with G/C, causing the highest A + T content to occur in third positions. The codons Arg (CGC) and Ser1 (AGG) were missing in S. maai, and Pro (CCG) and Thr (ACG) were missing in S. nigrivalveus, while S. varius had the full 62 available codons (Fig. 2).

Relative synonymous codon usage (RSCU) of three sequenced Scaphoideus mitochondrial genomes. The stop codon is not given. Codons absent in mitogenomes are shown at the top of columns.
Transfer RNA and ribosomal RNA genes
For the 22 typical animal tRNA genes in each Scaphoideus mitogenome, 14 tRNAs were encoded by the J-strand and the remaining eight were located on the N-strand, ranging from 61 to 71 bp in length. TrnM, trnK and trnI showed the highest identical sites percentage when aligned using MAFFT (92.5%, 90.1% and 89.6%, respectively), while trnY presented the lowest similarity (62.5%) (Table 4). All tRNAs could be folded into the canonical cloverleaf secondary structure except for trnS1 (AGN), which lacks the dihydrouridine (DHU) arm and instead forms a loop in all three Scaphoideus leafhoppers (Figs S2–S4). Besides the classic A-U and C-G pairs in the secondary structure, there were 27, 25 and 19 G-U base pairings in S. maai, S. nigrivalveus and S. varius respectively. Some other mismatched base pairs (A-A, U-U, A-G and A-C) were also found in the acceptor arm and anticodon arm (Figs S2–S4).
The lengths of the two rRNA genes (rrnL and rrnS) in the Scaphoideus mitogenomes were about 1200 and 740 bp, with the mean A + T contents of 80.2% and 77.9% respectively (Table 1), and both rrn genes were encoded on the N-strand. The large rRNA subunit was located at a conserved position between trnL1 (CUN) and trnV, while the small rRNA subunits was between trnV and the control region (Fig. 1). The percentage of pairwise identity between the three Scaphoideus species based on the MAFFT alignment is summarized in Table 5. As for the PCGs S. maai was more similar to S. nigrivalveus than to S. varius in both rrnL or rrnS (80.4% versus 78.3%, 84.3 versus 79.4%, respectively) (Table 5).
Gene overlaps and non-coding regions
All three Scaphoideus mitogenomes had gene overlaps and each single overlap ranged from 1 to 8 bp. S. maai had a total of 31 bp in overlaps between 10 gene junctions, while S. nigrivalveus and S. varius had 34 bp overlaps between 11 gene junctions and 30 bp overlaps between nine gene junctions, respectively (Supplementary Tables S1–S3). Except for the two existing common pairs of gene overlaps: atp8-atp6 (7 bp) and nad4-nad4l (7 bp) which are found in other leafhoppers (D. nuchalis, E. vitis, H. vitripennis, I. nitidulus, N. cincticeps), all three Scaphoideus species also share the same five other pairs of gene overlaps: trnR-trnN (1 bp), trnN-trnS1 (AGN) (1 bp), nad6-cob (1 bp), trnI-trnQ (3 bp) and trnW-trnC (8 bp) (Supplementary Tables S1–S3).
Excluding the control region, there were 10, nine and 13 intergenic spacers totaling 70, 46 and 152 bp non-coding bases in S. maai, S. nigrivalveus and S. varius respectively. All three mitogenomes have the same two intergenic spacer patterns, between nad4l-trnT (2 bp) and trnP-nad6 (2 bp) respectively. The longest intergenic spacers in Scaphoideus mitogenomes were present between trnY and cox1 (with 26, 21 and 101 bp respectively). A much larger, 101 bp, intergenic spacer in S. varius made it distinctly different from other two species (Supplementary Tables S1–S3). We confirmed the presence of this spacer by Sanger sequencing using primers (Forward 5′-3′: CGTTTAGCTTTTACTTC and Reverse 5′-3′: GTTCCAGGGTGTGCTAAT) located in flanking regions of nad2 and cox1.
The putative control region, or A + T rich region, located between rrnS and trnI, was the most variable region in the whole mitogenome, with the pairwise identity between Scaphoideus species were relatively low (all < 55%) (Table 5). The full lengths of CR in three Scaphoideus mitogenomes were 847, 902 and 762 bp respectively, comparable to other sequenced leafhoppers (from 399 bp in N. cincticeps to 1581 bp in Tambocerus sp.). No tandem repeat units were found in Scaphoideus species.
Nucleotide diversity among three Scaphoideus mitogenomes
Nucleotide diversity of the 13 PCGs and two rRNA genes among three Scaphoideus mitogenomes were shown in Fig. 3. The most variable region (729–978) was in nad2 (Pi = 0.30) while the most conserved fragment (11475–11730) was in rrnL (Pi = 0.11). Within each gene, the shortest PCG atp8 (153 bp, Pi = 0.27) present the highest variability, nad6 (Pi = 0.24) and nad2 (Pi = 0.23) were also highly variable. The most conserved PCGs were cox1 (Pi = 0.15) and nad4l (Pi = 0.16). Two rRNA genes were highly conserved, with the value 0.20 in rrnL and 0.17 in rrnS respectively.

Sliding window analyses of protein coding genes and ribosomal RNA genes among three Scaphoideus mitogenomes. The blue line was drawn using each mid-point value with its nucleotide diversity (Pi) (a sliding window of 250 bp with the step size of 25 bp). Pi value of each PCG was shown under the gene name.
Phylogenetic relationships
In this study, no saturation was detected for the three candidate nucleotide sequence datasets (P123, P123DEGEN and P123R) prepared for ML and BI analyses (all Iss < Iss.cSym or Iss.cAsym, P < 0.05) (Table 6), suggesting that the concatenated data were suitable for phylogenetic analysis. Thus, we analyzed these three different datasets, in addition to the corresponding amino acid sequence data, to evaluate the tree topology and nodal support. The phylogenetic tree topologies yielded by all six analyses of nucleotide sequence data were identical to each other (Fig. 4). A separate analysis of amino acid sequence data yielded a topology that was identical except for two deep internal nodes within Membracoidea that received only low to moderate ML bootstrap support in all analyses (Fig. S5). The lower overall branch support for the tree resulting from analysis of amino acid sequences indicates that the nucleotide sequence data contain phylogenetic signal (e.g., in third codon positions) that is lost when the data are translated to amino acids.

Phylogenetic relationships for Scaphoideus based on the P123/P123DEGEN/P123R datasets inferred from RaxML and MrBayes. Numbers on branches are Bootstrap values (BS) and Bayesian posterior probabilities (PP) (ML-P123; ML-P123DEGEN; ML-P123R; BI-P123; BI-P123DEGEN; BI-P123R, respectively). An asterisk indicates BS = 100 and PP = 1.0 in all six inferences.
Monophyly of the three cicadomorphan superfamilies Cicadoidea, Cercopoidea and Membracoidea was strongly supported in all analyses (Bootstrap support values (BS) = 100, Bayesian posterior Probability (PP) = 1.00). All nodes received high support (PP > 0.96) in Bayesian analyses of nucleotide sequence data but a few nodes received only moderate or low support ML analyses of some datasets (BS < 75). Within Cicadellidae, the six Deltocephalinae species constituted one clade and formed a sister group to other leafhoppers. Within Deltocephalinae, Scaphoideus species (tribe Scaphoideini) formed a monophyletic sister group to Tambocerus sp. (tribe Athysanini) and Drabescus nuchalis (tribe Drabescini) was sister to Nephotettix cincticeps (tribe Chiasmini). Of the three Scaphoideus species, S. maai was sister clade to S. nigrivalveus, consistent with the relatively high pairwise sequence similarity of these two species.
Discussion
Consistent with previous observations of Membracoidea, mitogenome sequences of Scaphoideus were highly conserved in gene content, gene size, gene order, base composition, codon usage of PCGs and tRNA secondary structures. Variation in the length of mt genomes is mostly due to length variation in the control region, which ranges from 70 bp (Orthoptera: Ruspoliadubia)16 to 4.6 kb (Diptera: Drosophila melanogaster) in insects17. The sizes of the A + T-rich region in S. maai, S. nigrivalveus and S. varius are consistent with those of the A + T-rich regions of other leafhoppers, which range from 399 bp in N. cincticeps to 1581 bp in Tambocerus sp., but are relatively short compared to most other insect mitogenomes. Moreover, the three newly sequenced species share 32.3% sequence identity in this region.
Phylogenetic analyses suggest that the S. maai group is more closely related to the S. nigrivalveus group than to the S. varius group, consistent with the pairwise differences in both PCGs and rRNA genes. While Scaphoideini was found to be relatively close to Drabescini in the phylogeny of Deltocephalinae based on combined morphological, 28 S and Histone H3 data7, our analysis placed a species of the poorly defined and polyphyletic tribe Athysanini (Tambocerus, not included in Zahniser and Dietrich’s dataset) as sister to Scaphoideus. In recent phylogenetic studies of the entire subfamily Deltocephalinae based on partial gene sequences and morphological characters, Scaphoideini was resolved as paraphyletic with respect to Drabescini with low branch support. Further phylogenetic studies are needed to elucidate the status and relationships within this group. Although our taxon sample includes only a small fraction of the diversity of leafhoppers, a group comprising >21,000 known species, our phylogenetic analyses of mitogenome sequences indicate that such data are able to resolve relationships at various levels in the taxonomic hierarchy of this insect group. Thus, addition of taxa to our leafhopper mitogenome dataset may help improve resolution of the still poorly understood relationships among major leafhopper lineages. Interestingly, as in our analysis of mitogenome data, a recent larger-scale phylogenomic study of Membracoidea that included nucleotide sequence data from 388 gene regions was also unable to resolve some deep internal branches of the phylogeny of this group18. This suggests that some ancient divergences among leafhoppers may be difficult to resolve, even after the addition of much more data.
Mitochondrial genes have been widely used as genetic markers, especially with cox1 partial sequences gaining widespread popularity as convenient DNA barcodes for species identification19. Our sliding window analysis indicated that cox1 is one of the most conserved protein-coding genes of the mitogenome and the two rRNA genes are also highly conserved when compared to other regions. Similar results were found in recent studies of Psylloidea and Psychodidae species20,21. On the other hand, mitogenome sequence data are sufficiently variable to also have potential use in single nucleotide polymorphism (SNP) studies.
Partial mitogenome sequence data were recently used in a population genetic study of Scaphoideus. Papura et al. used 10 polymorphic microsatellite loci and a variable 623 bp region of the mitogenome (trnL2(UUR) - cox2) in an attempt to elucidate the colonization scenario of the phytoplasma vector species S. titanus, which is native to northeastern North America but is now well established and spreading in Europe22. Consistent with microsatellite data, the mitochondrial data indicated much higher haplotype diversity among the native North American populations than observed in European samples, which displayed low levels of genetic diversity and no isolation by distance22. The three most variable coding regions in the mitogenomes of Scaphoideus species sequenced for our study were atp8 (Pi = 0.27), nad6 (Pi = 0.24) and nad2 (Pi = 0.23), while cox2 (Pi = 0.17) was relatively conserved and slightly below the mean Pi value for PCGs (0.20). This suggests that the first three mentioned markers may be more useful than cox2 for future population genetic studies. Although atp8 (153 bp) may be too short to be very informative, nad6 (477 bp) and nad2 (975 bp) should be considered for use in future Scaphoideus population genetic studies.
Materials and Methods
Sample collection and DNA extraction
All species used in this study were collected in China between 2011 and 2015 (Table S4). Fresh specimens were captured and preserved in 100% ethanol, and stored at −20 °C in the laboratory. After morphological identification, voucher specimens with male genitalia prepared were deposited in the Entomological Museum of Northwest A&F University, and total genomic DNA was extracted from muscle tissues of the thorax using the DNeasy DNA Extraction kit (Qiagen).
Next generation sequence assembly
Most of the mitochondrial genome sequences of the three species were generated using Illumina HiSeq™2500 with paired reads of 2 × 150 bp. A total of 23,047,208, 16,162,636 and 26,342,108 raw paired reads were retrieved and quality-trimmed using CLC Genomics Workbench v7.0.4 (CLC Bio, Aarhus, Denmark) with default parameters for S. maai, S. nigrivalveus and S. varius respectively. Subsequently, with the mitochondrial genome of Drabescus nuchalis (KR349344)14 employed as a bait sequence, the resultant 23,047,153, 16,162,588 and 26,342,065 clean paired reads were used for mitogenome reconstruction using MITObim v1.7 software23 with default parameters. A total of 18,361, 15,925 and 18,166 individual mitochondrial reads yielded an average coverage of 120.9 × , 81.9 × and 252.0 × for S. maai, S. nigrivalveus and S. varius respectively.
Gap closing-PCR amplification and sequencing
According to the flanking sequences assembled from the NGS data, we designed three pairs of primers to amplify the control region (CR) (S. maai: Forward 5′-3′: TAGGGTATCTAATCCTAGTTTA and Reverse 5′-3′: TGTTGATGCTACTCTTTG; S. nigrivalveus: Forward 5′-3′: CGCCAAATTCTTTGAGCT and Reverse 5′-3′: ATTGTGAAATGGTGCTGA; S. varius: Forward 5′-3′: TTAACCGCGAATGCTGGCAC and Reverse 5′-3′: GAATGGAATAAACGACAG). PCR reactions were performed with TaKaRa LA-Taq Kits (TaKaRa Co., Dalian, China) under the following cycling conditions: 5 min at 94 °C, 38 cycles of 30 s at 94 °C, 1 min at 45–50 °C, 2–3 min at 68 °C, and a final elongation step at 68 °C for 10 min. PCR products were eletrophoresed on 1% agarosegels, purified and then sequenced in both directions on an ABI 3730 XL automated sequencer (Applied Biosystems).
Genome annotation and bioinformatic analyses
All the three Scaphoideus mitochondrial genomes were annotated with GENEIOUS R8 (Biomatters Ltd., Auckland, New Zealand). All 13 protein-coding genes and two rRNA genes were identified by comparison with the homologous sequences of other leafhoppers from GenBank. The 22 tRNA genes were determined using both of the tRNAScan-SE server v 1.2124 and MITOS WebSever25, the clover-leaf secondary structures were also predicted by the MITOS WebSever.
The base composition and relative synonymous codon usage (RSCU) values of each protein coding gene (PCG) were calculated with MEGA 6.0626. Strand asymmetry was calculated using the formulas AT skew = [A−T]/[A + T] and GC skew = [G−C]/[G + C]27. Polymorphic sites and nucleotide diversity (Pi) of each PCG among three Scaphoideus species were calculated with DnaSP 5.028. A sliding window of 250 bp (in 25 bp overlapping steps) was used to estimate Pi among PCGs and rRNA genes across the alignment of three Scaphoideus mitogenomes.
Phylogenetic analyses
Taxa selection
A dataset consisting of the three newly sequenced taxa and 10 previously available Membracoidea mitogenomes (3 treehoppers and 10 leafhoppers), plus outgroups consisting of three species of Cicadoidea and five species of Cercopoidea was compiled for phylogenetic analysis (Table 7).
Sequence alignment and substitution saturation test
Each of the 13 PCGs and two rRNA genes was aligned separately based on the invertebrate mitochondrial genetic code using the MAFFT algorithm in the TranslatorX online server (http://translatorx.co.uk/)29, with poorly aligned sites removed from the protein alignment before back-translating to nucleotides using GBlocks under default settings. Each of the two rRNAs was aligned using the Q-INS-i method through MAFFT version 7 alignment server (http://mafft.cbrc.jp/alignment/server/)30. Potential substitution saturation of each dataset was assessed using the index of substitution saturation (Iss) of Xia et al.31 implemented in the DAMBE 532.
Dataset concatenation, partitioning and substitution model selection
Alignments of individual genes were concatenated using SequenceMatrix 1.7.833. To compensate for nucleotide compositional heterogeneity between taxa, the “Degen” approach was selected to make synonymous changes largely invisible but leaving the inference of non-synonymous change largely intact, as implemented in the online server (http://www.phylotools.com/)34,35. To assess the stability of phylogenetic results under strategies for reducing noise in the data, fourdatasets were generated: 1) P123: 13 PCGs with 9912 nucleotides; 2) P123DEGEN: 13 PCGs with all coding positions with 9912 nucleotides; 3) P123R: 13 PCGs and two rRNAs with 12047 nucleotides; 4) AA: amino acid sequences of 13 PCGs with 3304 amino acids. PartitionFinder v1.1.136 was employed to infer the optimal nucleotide substitution models and partition strategies. Data blocks for PCGs were defined by codon positions. The Bayesian information criterion (BIC) was chosen as the metric for the partitioning scheme under the “greedy” search algorithm. Details of the best-fit schemes calculated for each dataset are shown in Supplementary Table S5.
Phylogenetic inference
ML analyses were implemented using raxmlGUI 1.537 under the GTRGAMMAI model, and support for nodes was assessed by performing 1000 rapid bootstrap replicates (BS). Bayesian analyses were conducted using MrBayes 3.2.638. Following the partition schemes suggested by PartitionFinder, all model parameters were set as unlinked across partitions. Two simultaneous runs with four independent Markov chains were run for seven million generations and trees were sampled every 1000th generation. After the average standard deviation of split frequencies fell below 0.01, the first 25% of samples were discarded as burn-in and the remaining trees were used to generate a consensus tree and calculate the posterior probabilities (PP).
References
Boore, J. L. Animal mitochondrial genomes. Nucleic. Acids. Res. 27, 1767–1780 (1999).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Simon, C., Buckley, T. R., Frati, F., Stewart, J. B. & Beckenbach, A. T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 37, 545–579 (2006).
Song, N., Liang, A. P. & Bu, C. P. A molecular phylogeny of Hemiptera inferred from mitochondrial genome sequences. PLoS One 7, e48778 (2012).
Li, H. et al. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 5, 8527 (2015).
Yuan, M. L. et al. High-level phylogeny of the Coleoptera inferred with mitochondrial genome sequences. Mol. Phylogenet. Evol. 104, 99–111 (2016).
Zahniser, J. N. & Dietrich, C. H. A review of the tribes of Deltocephalinae (Hemiptera: Auchenorrhyncha: Cicadellidae). Eur. J. Taxon. 45, 1–121 (2013).
Ball, E. D. New genera and species of leafhoppers related to Scaphoideus. J. Wash. Acad. Sci. 22, 9–19 (1932).
DeLong, D. M. A review of the genus Scaphoideus. P. Entomol. Soc. Wash. 41, 33–45 (1939).
Viraktamath, C. & Mohan, G. A revision of the deltocephaline leafhopper genus Scaphoideus (Hemiptera: Cicadellidae) from the Indian subcontinent. Zootaxa 578, 1–48 (2004).
Zahniser, J. N. & Dietrich, C. H. Phylogeny of the leafhopper subfamily Deltocephalinae (Insecta: Auchenorrhyncha: Cicadellidae) and related subfamilies based on morphology. Syst. Biodivers. 6, 1–24 (2008).
Zahniser, J. N. & Dietrich, C. H. Phylogeny of the leafhopper subfamily Deltocephalinae (Hemiptera: Cicadellidae) based on molecular and morphological data with a revised family-group classification. Syst. Entomol. 35, 489–511 (2010).
Zhao, X. & Liang, A. P. Complete DNA sequence of the mitochondrial genome of the treehopper Leptobelus gazella (Membracoidea: Hemiptera). Mitochondrial DNA A 27, 3318–3319 (2016).
Wu, Y. F., Dai, R. H., Zhan, H. P. & Qu, L. Complete mitochondrial genome of Drabescoides nuchalis (Hemiptera: Cicadellidae). Mitochondrial DNA A 27, 3626–3627 (2016).
Yu, P. F., Wang, M. X., Cui, L., Chen, X. X. & Han, B. Y. The complete mitochondrial genome of Tambocerus sp.(Hemiptera: Cicadellidae). Mitochondrial DNA A 28, 133–134 (2015).
Zhou, Z., Huang, Y. & Shi, F. The mitochondrial genome of Ruspolia dubia (Orthoptera: Conocephalidae) contains a short A + T-rich region of 70 bp in length. Genome 50, 855–866 (2007).
Lewis, O., Farr, C. & Kaguni, L. Drosophila melanogaster mitochondrial DNA: completion of the nucleotide sequence and evolutionary comparisons. Insect Mol. Biol. 4, 263–278 (1995).
Dietrich, C. H. et al. Anchored hybrid enrichment-based phylogenomics of leafhoppers and treehoppers (Hemiptera: Cicadomorpha: Membracoidea). Insect Syst. Divers. 1(1), 57–72 (2017).
Hebert, P. D., Cywinska, A. & Ball, S. L. Biological identifications through DNA barcodes. Proc. Roy. Soc. Lond. B Biol. Sci. 270, 313–321 (2003).
Ye, F., Liu, T., King, S. D. & You, P. Mitochondrial genomes of two phlebotomine sand flies, Phlebotomus chinensis and Phlebotomus papatasi (Diptera: Nematocera), the first representatives from the family Psychodidae. Parasite Vector 8, 472 (2015).
Wu, F. N. et al. The Complete Mitochondrial Genome Sequence of Bactericera cockerelli and Comparison with Three Other Psylloidea Species. PloS One 11, e0155318 (2016).
Papura, D. et al. Microsatellite and mitochondrial data provide evidence for a single major introduction for the Neartic leafhopper Scaphoideus titanus in Europe. PLoS One 7, e36882 (2012).
Hahn, C., Bachmann, L. & Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic. Acids. Res. 41, e129 (2013).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic. Acids. Res. 25, 955–964 (1997).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Librado, P. & Rozas, J. DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Abascal, F., Zardoya, R. & Telford, J. M. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic. Acids. Res. 38, 7–13 (2010).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Xia, X., Xie, Z., Salemi, M., Chen, L. & Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 26, 1–7 (2003).
Xia, X. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30, 1720–1728 (2013).
Vaidya, G., Lohman, D. J. & Meier, R. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180 (2011).
Regier, J. C. et al. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 463, 1079–1083 (2010).
Zwick, A., Regier, J. C. & Zwickl, D. J. Resolving discrepancy between nucleotides and amino acids in deep-level arthropod phylogenomics: differentiating serine codons in 21-amino-acid models. PLoS One 7, e47450 (2012).
Lanfear, R., Calcott, B., Ho, S. Y. & Guindon, S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701 (2012).
Silvestro, D. & Michalak, I. RaxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12, 335–337 (2012).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Stewart, J. B. & Beckenbach, A. T. Insect mitochondrial genomics: the complete mitochondrial genome sequence of the meadow spittlebug Philaenus spumarius (Hemiptera: Auchenorrhyncha: Cercopoidae). Genome 48, 46–54 (2005).
Yang, H., Liu, J. & Liang, A. The complete mitochondrial genome of Cosmoscarata bispecularis (Hemiptera, Cicadomorpha, Cercopoidea, Cercopidae). Mitochondrial DNA A 27, 3957–3958 (2016).
Liu, J., Bu, C., Wipfler, B. & Liang, A. Comparative analysis of the mitochondrial genomes of callitettixini spittlebugs (Hemiptera: Cercopidae) confirms the overall high evolutionary speed of the at-rich region but reveals the presence of short conservative elements at the tribal level. PloS One 9, e109140 (2014).
Mao, M., Yang, X. & Bennett, G. The complete mitochondrial genome of Entylia carinata (Hemiptera: Membracidae). Mitochondrial DNA B 1, 662–663 (2016).
Liang, A. P., Gao, J. & Zhao, X. Characterization of the complete mitochondrial genome of the treehopper Darthula hardwickii (Hemiptera: Aetalionidae). Mitochondrial DNA A 27, 3291–3292 (2016).
Zhou, N. N., Wang, M. X., Cui, L., Chen, X. X. & Han, B. Y. Complete mitochondrial genome of Empoasca vitis (Hemiptera: Cicadellidae). Mitochondrial DNA A 27, 1052–1053 (2016).
Acknowledgements
We thank Dr. John Richard Schrock (Emporia State University, Emporia, KS, USA) for his comments on an earlier draft of this paper. We are also grateful to academic editor and two anonymous referees for constructive criticism that significantly improved the manuscript. The project was supported by National Natural Science Foundation of China (Nos. 31572306, 31272343, 31420103911).
Author information
Authors and Affiliations
Contributions
Wu Dai and Christopher H. Dietrich conceived and designed the experiments; Yimin Du performed the experiments; Yimin Du and Wu Dai analyzed the data; Yimin Du, Wu Dai and Christopher H. Dietrich wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, Y., Dai, W. & Dietrich, C.H. Mitochondrial genomic variation and phylogenetic relationships of three groups in the genus Scaphoideus (Hemiptera: Cicadellidae: Deltocephalinae). Sci Rep 7, 16908 (2017). https://doi.org/10.1038/s41598-017-17145-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17145-z
- Springer Nature Limited
This article is cited by
-
Comparative mitochondrial genome analysis of three leafhopper species of the genus Abrus Dai & Zhang (Hemiptera: Cicadellidae: Deltocephalinae) from China with phylogenetic implication
BMC Genomics (2023)
-
Structural and phylogenetic implications of the complete mitochondrial genome of Ledra auditura
Scientific Reports (2019)