
Abstract
α-Fe2O3/Fe3O4/g-C3N4 nanocomposites were prepared in-situ by solution combustion as magnetically separable photocatalysts using ferric nitrate as oxidant, glycine as organic fuel, and g-C3N4. The effects of various amounts of iron oxides, on the magnetic, optical, and photocatalytic properties were explored by different characterization methods. The magnetite (Fe3O4) phase as ferrimagnetic material disappeared with the increase in ferric nitrate contents, leading to the decrease of magnetic properties. The bandgap energy decreased from 2.8 to 1.6 eV with the increase of the hematite (α-Fe2O3) phase.The photocatalytic results showed that the type and amount of iron oxides had a significant effect on the decolorization of methylene blue, rhodamine B and methyl orange dyes under visible-light irradiation. The activity of the nanocomposite sample containing 37 wt. % iron oxides was more effective than that of the pristine g-C3N4 sample to photodegrade the methylene blue, rhodamine B and methyl orange, respectively. Moreover, the nanocomposites exhibited a higher photocurrent density than that of the pristine g-C3N4, mainly due to their lower charge recombination rate.
Similar content being viewed by others


Introduction
Semiconductor photocatalysts have been the subject of significant attention as a simple time and energy-efficient technology to convert organic pollutants into eco-friendly mineralized byproducts.under solar irradiation1. Several versatile semiconductor photocatalysts, including transition metal oxides, hydroxides, sulfides, phosphates and transition metal dichalcogenides of various particle sizes and shapes and also two dimensional (2D) materials were widely applied for wastewater treatment by degrading organic dyes under sunlight2,3,4,5.But the application of photocatalysts suffers from poor photodegradation efficiency, lower chemical stability, and poor exploitation of the solar spectra. Therefore, many efforts have been made to find a photocatalyst system without these shortcomings6,7.
Graphitic carbon nitride (g-C3N4) has been considered a metal-free semiconductor for water splitting, photodegradation of toxic organic pollutants, CO2 reduction and antibacterial agents to disinfect the antibiotic-resistant microorganism strains8,9. The non-toxicity, low cost, narrow bandgap (Eg≈2.7 eV), and high chemical stability are unique features of 2D polymeric g-C3N4 material8,10. Furthermore, the g-C3N4 material can be easily synthesized by one-step polycondensation of organic materials such as urea, thiourea, melamine, cyanamid, and dicyandiamide containing nitrogen atoms11,12. However, the high recombination rate, low visible-light absorption coefficient and secondary pollution are three main limitations for efficient applications of g-C3N4 material on an industrial scale13. To address the disadvantages, the g-C3N4 material can be modified by other materials such as TiO2, ZnO, WO3 and α-Fe2O3 to extend the visible-light absorption capacity and separation of photogenerated electron–hole pairs that prevents the fast electron–hole recombination reaction14,15,16,17. Furthermore, magnetic materials such as Fe3O4, ZnFe2O4, CoFe2O4, and BiFeO3 can be loaded on the pristine g-C3N4 material to form a magnetically separable photocatalyst18,19,20,21.
Among various materials, iron oxides such as α-Fe2O3, γ-Fe2O3, and Fe3O4 with chemical and thermal stability, moderate magnetic properties, and optical properties have been widely used in several magnetic, catalyst, and photocatalyst applications because of their ease of synthesis, abundance, low cost, and environmentally benign22,23,24. Therefore, the iron oxides can be suitable candidates to combine with g-C3N4 material to improve the photocatalytic performance via enhancing visible-light absorbance, separation of the charge carriers, and magnetic recyclability25,26,27. For example,α-Fe2O3 has high absorption (~ 43%) in the red region of visible light, making it a proper candidate for coupling with g-C3N4 for photodegradation28,29. Ghane et al.30 reported the in-situ preparation of g-C3N4/α-Fe2O3 nanocomposite by solution combustion synthesis method for decolorization of methylene blue (MB) dye under visible light irradiation. They optimized the amount of α-Fe2O3 phase to obtain high photodegradation and photocurrent density. However, the α-Fe2O3 phase has antiferromagnetic behavior with negligible magnetic properties, which are insufficient for recycling the g-C3N4/α-Fe2O3.
It was observed that high conductivity and suitable energy band structure of Fe3O4 can enhance the photocatalytic performance of composite photocatalysts due to the separation of electron − hole pairs31,32,33. Mousavi and Habibi-Yangjeh also reported the effective role of bismuth oxyiodide (BiOI) species on the photodegradation of rhodamine B (RhB) by g-C3N4/Fe3O4 nanocomposites34. The BiOIphase was used for electron trapping, while the magnetic recycling was obtained by the Fe3O4 phase34. The Fe2O3/Fe3O4/g-C3N4 nanocomposite was previously synthesized by the hydrothermal method31. Up to our knowledge, it was not prepared by the simple and cost-effective solution combustion route in which the amounts of various iron oxides can be easily changed by tuning the synthesis conditions.
In this work, the solution combustion method was applied for the in-situ preparation of α-Fe2O3/Fe3O4/g-C3N4 nanocomposites with the various amounts of α-Fe2O3 and Fe3O4 phases. The nanocomposites' structural, microstructural, and photoelectrochemical properties were characterized by different techniques including X-ray diffractometry, thermogravimetry, Raman spectroscopy, electron microscopy, N2 adsorption–desorption isotherms, vibrating sample magnetometry, diffuse reflectance spectroscopy, photoluminescence spectroscopy, dye photodegradation, electrochemical impedance spectroscopy, and photoelectrochemical tests.
Experimental procedures
Analytical grade of ferric nitrate (Fe(NO3)3.9H2O, ≥ 98%), glycine (C2H5NO2, ≥ 99%), and melamine (C3H6N6, ≥ 99%) were purchased from Merck company.
Synthesis of α-Fe2O3/Fe3O4/g-C3N4nanocomposites
The g-C3N4powder was prepared by heating 10 g melamine powder from room temperature up to 550 °C at a rate of 10 °C/min in a muffle furnace. After holding at 550 °C for 3 h, the products were cooled in the furnace8.
The α-Fe2O3/Fe3O4/g-C3N4 nanocomposites were prepared as follows: 2 g graphitic carbon nitride powder was dispersed into 30 mL distilled water. The ferric nitrate as oxidant and glycine as fuel were added to the suspension. The amounts of Fe(NO3)3.9H2O were adjusted to form 0.05, 0.10, and 0.20 g iron oxides in the final products which were related to 2.5 wt. %, 5 wt. %, and 10 wt. %, respectively. The molar ratio of glycine fuel to Fe(NO3)3.9H2O was set to 1.6735. The precursor solution was stirred and evaporated at 80 °C until completely dried. The combustion reaction was started by heating the dried gel to 250 °C on a hotplate. For easy presentation, the nanocomposites were coded on the base of intended iron oxide contents; for example, the symbol “X5” shows that the desired mass ratio of iron oxide was 5 wt. %.
Characterization methods
The structure and phase of the combusted products were characterized by the X-ray diffractometry (XRD) technique. The diffraction patterns were collected on a D8 ADVANCE (Bruker, Kanagawa, Japan) using Cu Kα radiation (λ = 1.5418 A°). The combusted powders were thermally analyzed on an STA 503 instrument (Bahr, Germany) with a heating rate of 10 °C/min in the air atmosphere. Raman spectra were recorded on TEKSAN N1-541 spectrophotometer with Nd:YAG laser source. The saturation magnetization of the combusted powders was obtained from hysteresis loops measured on a vibrating sample magnetometer (MeghnatisKavir Kashan Co., Iran). The particle size and elemental distribution were obtained on MIRA3(TESCAN, Czech Republic) scanning and CM200 (Philips, UK) transmission electron microscopy. The specific surface areas, pore size distribution, and pore volume were calculated from the N2 adsorption/desorption isotherms measured on PHSCHINA (PHS-1020, China) instrument. The bandgap energy was obtained from diffuse reflectance spectra recorded on a 52550 UV–Vis (Shimadzu, Japan) spectrophotometer. The charge separation was studied by photoluminescence (PL) spectra which were obtained on a G9800A (Agilent, USA) fluorescence spectrophotometer (λEx. = 320 nm).
Photocatalytic test
In order to evaluate the photocatalytic performance of the nanocomposite for positively and negatively-charged dyes, 5 ppm solution of methylene blue (MB), rhodamine B (RhB) and methyl orange (MO) dyes were photodegraded by 0.1 g samples under irradiation of two 100 W Xenon lamps with the intensity of 1000 \(\frac{mW}{{cm}^{2}}\) as visible light source. The UV light was filtered by a cutoff filter (λ = 420 nm). Before turning on the light, the solution was stirred in dark for 1 h to establish the adsorption/desorption equilibrium of the dye. A 2600 UV–Vis spectrophotometer (Shimadzu, Japan) was used to monitor the relative concentrations of the dyes versus time.
Photoelectrochemical (PEC) test
The working electrode was prepared by dispersing 0.01 g nanocomposites in 0.5 mL NMP solution containing PVDF by sonication. Then the slurry was coated on 2 × 2 cm2 fluorine-doped tin oxide (FTO) glass and dried in an oven overnight. The working electrode was used in a three-electrode system in which Ag/AgCl and Pt were referenceand counter electrodes, respectively. The electrolyte was an aqueous solution of Na2SO4 (0.1 M). All of the electrochemical tests including electrochemical impedance spectroscopy, linear scanning voltammetry, and chronoamperometry were carried out on an OrigaFlexOGF01SOLARERON electrochemical workstation. The 500 W Xe lamp with an IR filter was used as a light source.
Results and discussion
Materials characterization
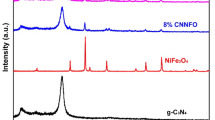
Figures 1a–c show the XRD patterns, Raman spectra, and TGA curves of the pristine g-C3N4 powder and nanocomposites. The two reflections at 2θ = 13.1° and 27.3° for the pristine g-C3N4 powders are related to the (100) plane of the in-planar motif of tri-s-triazine units and (002) plane of interlayer stacking of conjugated carbonaceous rings, respectively27. In addition to the reflections of the g-C3N4, the X2.5 sample shows mainly the diffraction peaks of the Fe3O4 phase (PDF2#00-033-0664) accompanied by a few amounts of α-Fe2O3 phase (PDF2#00-033-0664). The diffraction peaks of the α-Fe2O3 phase are intensified, while those of the Fe3O4 phase are weakened in the X5 and X10 samples. The X10 sample has a higher amount of α-Fe2O3 phase and some of the Fe3O4 phase. However, the (100) and (002) reflections of the g-C3N4 phase are absent in the X5 and X10 samples. The solution combustion synthesis (SCS) involves an exothermic reaction between ferric nitrate as oxidant and glycine as fuel. The combustion reaction begins by endothermal decomposition of ferric nitrate to α-Fe2O3 as solid products and O2 and N2 as gaseous products as follows36,37:

(a) XRD patterns, (b) TGA curves, and (c) Raman spectra of the pristine gC3N4 powders and the X2.5, X5, and X10 samples.
The glycine molecules can be exothermally oxidized to CO2, N2, and H2O gases by the liberated O2 gas as follows38:
Reaction (2) releases higher thermal energy than that of the required heat to proceed with the reaction (1), leading to the self-propagating nature of the SCS39. Moreover, the liberated gases can reduce, oxidize, and sulfidize the solid products40. Therefore, the appearance of the Fe3O4 phase instead of the α-Fe2O3 phase in the X2.5 sample can be attributed to the reduction of α-Fe2O3 phase by reductive H2 and CO gases. However, the higher combustion temperature in the X5 and X10 samples prevents the effective reduction of α-Fe2O3 phase to Fe3O4 phase. Furthermore, the absence of g-C3N4 peaks in the X5 and X10 samples can be attributed to the stacking disorders by introducing a mixture of α-Fe2O3/Fe3O4 phases41,42. Figure 1b shows TGA curves of the X0, X5, and X100 powders to calculate the amount of loaded α-Fe2O3/Fe3O4 nanoparticles on g-C3N4. The g-C3N4 starts to decompose at 600 °C, which can be completed at higher temperatures (> 700 °C) by burning g-C3N443,44. The α-Fe2O3/Fe3O4 powder shows a low weight loss of 7%, while the X5 composite has a higher weight loss of 73% due to the burning of g-C3N4. Therefore, the residual fraction of the X5 sample is 37 wt. %. Furthermore, the g-C3N4/α-Fe2O3/Fe3O4 nanocomposite begins to lose its weight at the lower temperature of 400 °C, which can be attributed to the higher induced crystal defects in g-C3N4 species by combustion reaction. The higher amount of solid product of the as-combusted X5 sample can be attributed to the burning of g-C3N4 during combustion temperature. Figure 1c shows Raman spectra of the g-C3N4 and X5 samples. The characteristic Raman bands of the g-C3N4 phase can be identified at 220, 262, 311, 350, 480, and 708 cm−145. The X5 sample shows the bands at 221, ~ 294, ~ 410, ~ 498, and ~ 607 cm−1 for the α-Fe2O3 phase and 370, 520, and 670 cm−1 for the Fe3O4 phase46, in addition to the bands related to the g-C3N4 phase. The most intense peak at 708 cm−1 corresponding to the s-triazine ring confirms the existence of g-C3N4 despite its absence in XRD patterns (Fig. 1a).
The magnetization curves of the X2.5 and X5 samples and the separation of the composite from solution by a magnet are presented in Fig. S1 (supplementary information). The ferromagnetic behavior of the composite powders is due to the presence of the ferrimagnetic Fe3O4 phase. By the appearance of the antiferromagnetic α-Fe2O3 phase, the saturation magnetization (Ms) decreases from 22 to 18 emu/g. Furthermore, the lower Ms value of the X2.5 sample than that of bulk Fe3O4 can be attributed to the nonmagnetic g-C3N4. The magnetic properties of the composite are enough for recycling the photocatalyst following the decontamination processes. Wang et al.31 synthesized a similar composite by hydrothermal method. The Ms value of the α-Fe2O3/Fe3O4/g-C3N4 nanocomposite is in the same range as this work (17.98 emu/g). Furthermore, the high magnetic properties confirm the existence of Fe3O4 as ferrimagnetic phase, because the α-Fe2O3 phase is antiferromagnetic material with very low saturation magnetization.
Figure 2 shows the FESEM images of the pristine g-C3N4 powder and X2.5 and X5 nanocomposites. The pure g-C3N4 powders are composed of secondary particles (1–3 μm) (Fig. 2a) with small primary particles of multiple wrinkled-layer-stack of g-C3N4 (Fig. 2b). After impregnating g-C3N4 powders with the precursor solution of ferric nitrate and rendering the combustion reaction, the nanoparticles of iron oxides are distributed on the edge and surface of g-C3N4 powders (Fig. 2c,d). With the increase of the amount of nanoparticles of iron oxides, the nanoparticles are aggregated into larger particles (Fig. 2e,f). The non-uniform distribution of the nanoparticles of iron oxides can be attributed to the hydrophobic behavior of the surface of g-C3N447. TEM images of the X5 composite powders are given in Fig. 3a–b. The particles of iron oxides are aggregated on the edge and surface of g-C3N4 powders. Furthermore, there are some pores between iron oxide nanoparticles.

SEM and images of (a and b) g-C3N4, (c and d) X2.5, and (e and f) X5 powders.

TEM images of the X5 composite powders.
In SCS, for the synthesis of g-C3N4/α-Fe2O3/Fe3O4, the g-C3N4 particles were dispersed in a precursor solution containing the ferric nitrate and glycine. The precursor solution was dried by heating up at 80 °C, leading to the formation of gel. Therefore, if the surface of g-C3N4 had hydrophilic nature, a thin and continuous film of gel can be easily formed on it which transformed into well-dispersed nanoparticles during the combustion reaction. The non-uniform distribution of the nanoparticles of iron oxides on the surface of g-C3N4 may be caused by the hydrophobic behavior of precursor solution. It is worth noting that the iron oxide particles have a quasi-spherical shape. Elemental distributions of the C, O, and Fe and the superposition of elemental distribution of Fe and C in the region shown in Fig. S2a are given in Figs. S2b–e, respectively. There is C element over all regions, while the Fe element is concentrated on the brighter aggregate.
N2 adsorption–desorption isotherms and pore size distribution plots of the X2.5 and X5 composite powders are presented in Fig. S3. The isotherms are IV type with H3 hysteresis loop related to the mesoporous network according to the IUPAC classification48. The X5 composite has a higher specific surface area (32 m2/g) and pore volume (0.2 cm3/g) than those of the X2.5 sample (19 m2/g and 0.1 cm3/g). The textural properties of the combusted products are a compromise of the amount of liberated gases and heat in which the lower released heat and higher exhausted gases lead to higher specific surface areas, higher pore volume, and larger pores due to the suppression of particles’ sintering and disintegration of large particles49. Therefore, the higher specific surface of the X5 composite sample can be s ascribed to the release of higher liberated gaseous products. The mesoporous nature of the composite samples provides further reaction sites, which improves the photocatalytic efficiency via charge separation.
It is well-known that photodegradation performance strictly depends on the light absorption coefficient. The UV–Vis diffuse reflectance spectroscopy was applied to show the photoabsorption ability of the composite samples (Fig. S4a). The pristine g-C3N4 powders have an absorption edge of about 470 nm, leading to a negligible absorption in the visible range. However, the ability of the composite samples is considerably higher for light absorption in the visible range. Furthermore, the light absorption in the visible range increases with the increase of α-Fe2O3 content, appropriating the application of α-Fe2O3/Fe3O4/g-C3N4 nanocomposites as visible-light-driven photocatalysts. According to Taucʼs equation, the bandgap energy (Eg) can be estimated as follows50:
where α is the light absorption coefficient, and the n value is related to the transition type51. Therefore, the bandgap energy (Eg) can be obtained by extrapolating the linear part of the (αhν)2 curves versus hν to 0 (Fig. S4b). The Eg values of g-C3N4, X2.5, X5, and X10 samples are 2.8, 2.5, 1.6, and 2 eV, respectively. The bandgap of the composite samples lies between the pristine g-C3N4 and pristine α-Fe2O3 powders, indicating a good interaction between both components, resulting in the bandgap alignment52. The very narrow bandgap of the X5 sample can be attributed to the more heterogeneous distribution of the α-Fe2O3 phase.
Photocatalytic performance
Figure 4a,b show the UV–Vis spectra of the MB and RhB solutions versus light illumination time in the presence of the X5 sample as photocatalyst. The weakening of the characteristic peaks of MB and RhB dyes at 665 and 550 nm, respectively, shows the degradation of dye structure with time. The dependence of the relative concentration of dyes versus time in the presence of g-C3N4, X2.5, X5, and X10 samples are given in Fig. 4c,d. The photodegradation rate of MB and RhB dyes can be fitted by the pseudo-first kinetics as follows:

UV–Vis spectra of (a) MB and (b) RhB dye in the presence of the X5 composite sample, the relative concentration of (c) MB and (d) RhB dyes versus illumination time, Ln(C0/C) versus time for (e) MB and (f) RhB dyes, and removal efficiency of (g) MB and (h) RhB dyes by various catalysts.
The values of k (min−1) as rate constant calculated by plotting Ln \(\left(\frac{C}{C0}\right)\) versus irradiation time, as shown in Fig. 4e,f. The constant rate of photodegradation increases 2.5 and 4 times for MB and RhB dyes, respectively, by the combination of g-C3N4 with a proper amount of iron oxides. The removal efficiencies, including adsorption and photodegradation processes of MB and RhB dyes in the presence of various catalysts, are summarized in Fig. 4g,h. About 40% of MB is photodegraded by the g-C3N4 catalyst, which increases to 62 and 75 for the X2.5 and X5 powders, respectively. However, the photodegradation decreases to 54% for the X10 catalysts after 60 min of illumention. For RhB dye and following illumination for three h, the pristine g-C3N4 powders photodegrade about 35% while the amount of photodegradation firstly increases up to 58% with the addition of α-Fe2O3/Fe3O4 nanoparticles and then decreases to 8% for the X10 sample, possibly due to the disappearance of Fe3O4 as the charge acceptor. The separation of photogenerated charges, higher light absorbance, and higher specific surface areas are responsible factors for the higher photocatalytic activity of the nanocomposites30. Furthermore, the adsorption capacity of the MB dye is higher than that of the RhB dye because of the strong interaction between the surface ligands of the catalyst and MB dye molecules34.
In order to evaluate the photocatalytic performance of the nanocomposites to the negatively charged dyes, MO dye photodegradation by the composites was also tested. Fig. S4a shows the dependence of the relative concentration of methy orange (MO) in the presence of samples g-C3N4, X2.5, X5 versus time. MO degradation was not occurred in presence of sample X10. The photodegradation rate of MO was fitted by the pseudo-first kinetics (Fig. S4b). The constant rate of photodegradation increases 3 times for MO by the combination of g-C3N4 with a proper amount of iron oxides (sample X5). The removal efficiencies, including adsorption and photodegradation processes of MO dyes in the presence of various catalysts, are summarized in Fig. S5c. About 30% of MO is photodegraded by the g-C3N4 catalyst, which decreases to 14 and then increases to 53 for the X2.5 and X5 powders, respectively. This reveals that the the adsorption capacity of the MO dye is lower than the other two dyes since the electrostatic interactions of the MO (which is an anionic dye) is different with the surface of the catalyst53.
The photocatalytic process involves three main steps: (i) generation of electron (e−) and hole (h+) pairs by absorbing the light photons, which have higher energy than that of the bandgap energy of semiconductor; (ii) migration of charge carries without their recombination to the surface of catalyst; (iii) redox reactions between the photogenerated charges with solvated species such as O2 and H2O to produce highly oxidant agents like.O2 and OH on the surfaceof catalyst54. Therefore, the bandgap energy, absorption coefficient, and charge recombination as optical characteristics of catalyst have an effective role on the photocatalytic activity6. Because the g-C3N4 and α-Fe2O3 phases in the composites show different ranges of photoabsorption, their combination broadens the visible-light photoresponse and narrows the bandgap (Fig. S4), leading to the generation of a great number of charge carriers under visible light illumination.
The charge separation can be described on the base of the band structure between g-C3N4 and α-Fe2O3 phases. The position of conduction and valence levels of g-C3N4 and α-Fe2O3 can be calculated as follows25:
which X is the absolute electronegativity (6.90 and 5.83 eV for g-C3N4 and α-Fe2O3, respectively), Ee is the energy of free electrons on the hydrogen scale (4.5 eV), and Eg is the bandgap energy of the semiconductor, respectively55. The positions of EVB and ECB for g-C3N4 with the bandgap Eg of 2.80 eV are + 1.57 and − 1.12 eV, whereas those are + 2.48 and + 0.28 eV for α-Fe2O3 with the Eg of 1.9 eV, respectively. The photogenerated charges can be transferred between the various bands of the combined semiconductors in which the electrons go to the less negative CB. In contrast, the holes are reversely transferred to the higher energy level of VB, as schematically shown in Fig. 5a. In fact, the g-C3N4/α-Fe2O3/Fe3O4 nanocomposite follows Z-scheme for charge transfer, because the g-C3N4 is reduction photocatalyst, while the α-Fe2O3 is oxidation photocatalyst. In this scheme, the photogenerated electrons with strong reduction abilities in CB of g-C3N4 and holes with strong oxidation abilities in VB of α-Fe2O3 are preserved, while the photogenerated electrons in CB of α-Fe2O3 and holes in VB of g-C3N4 with inferior redox power recombine56. Therefore, the efficient charge separation decreases the recombination rate of electron–hole pairs in the nanocomposites, as can be revealed by PL spectra (Fig. 5b). The X2.5 and X5 composite samples show a weaker PL intensity than pristine g-C3N4 because of the formation of heterojunction structure between g-C3N4 and α-Fe2O3, preventing the electron–hole recombination57.

(a) Schematic of the band structure and charge transfer mechanism and (b) PL spectra of the g-C3N4, X2.5, and X5 samples.
Because of the low bandgap energy, the electron–hole pairs are produced on both g-C3N4 and α-Fe2O3 under visible-light irradiation. The CB and VB positions of g-C3N4 are higher than those of the α-Fe2O3. Some photogenerated electrons on the CB of g-C3N4 can quickly react with O2 to produce •O2−, because of the more negative CB potential of g-C3N4 than the potential of the O2/•O2− (-0.33 eV)58. However, the more positive CB potential of α-Fe2O3 than that of the O2/•O2- shows that the electrons at CB of α-Fe2O3 cannot reduce O2 to •O2-. Moreover, the more negative CB potential of α-Fe2O3 than the potential of O2/H2O2 (+ 0.682 eV) results in the production of H2O2 by transferring the accumulated electrons in the CB of α-Fe2O3 to adsorbed oxygen59. The produced H2O2 molecules can react with the electrons to produce hydroxyl radicals (•OH). Furthermore, the photogenerated holes can react with adsorbed H2O to produce the hydroxyl radicals (•OH) on account of the more positive VB potential than that of •OH/ − OH (+ 2.38 eV)60. The photogenerated holes can directly react with the adsorbed dye molecules to produce harmless CO2 and H2O products. The main reactive species can be realized by using a series of scavengers. Figure 6 shows the photodegradation of MB dye in the presence of 1 mmol disodium ethylenediaminetetracetate (Na2-EDTA) and 1 mmol isopropyl alcohol (IPA) for quenching the role of h+and •OH radicals, respectively34. The decrease of photodegradation in the presence of Na2-EDTA is greater than that of IPA. Hence, the effect of photogenerated holes is higher than that of the hydroxyl radicals.

Photodegradation of the MB dye by the X5 composite powders in the presence of a series of scavengers.
Electrochemistry analysis
The electrochemical impedance spectra (EIS) of the pristine g-C3N4 and the X5 composite are shown in Fig. 7. The excitation and transfer process of photogenerated charges can be determined from the Nyquist plots. The X5 composite powders have a smaller arc size compared to the pristineg-C3N4 powders, implying a lower resistance in charge transfer and higher efficiency in charge separation between electron–hole pairs55.

EIS of the pristine g-C3N4 powders and the X5 composite powders.
Figure 8a compares the photocurrents produced from the g-C3N4, X2.5, and X5 samples versus time at a constant applied bias of + 0.5 V. The photocurrent intensity of the X2.5 and X5 composite samples is approximately 3 and 9 times as high as that of the pristine g-C3N4 powders. Therefore, the photogenerated electrons and holes are effectively separated and easily transferred at the interface of the various semiconductors. Furthermore, the chopping cycles are repeated in each interval time without any changes, showing the photostability of the materials for use in photoelectrochemical cells. Figure 8b illustrates the linear sweep voltammetry (LSV) of the X5 composite sample. A high anodic current is obtained in the presence of visible light irradiation, indicating a facilitated charge transfer to the electrode's surface without significant charge recombination30.

(a) Photocurrent versus time under a chopped condition at a constant applied bias of + 0.5 V and (b) LSV plot of the X5 composite sample in dark and under visiblelight.
Conclusion
The g-C3N4/α-Fe2O3/Fe3O4 nanocomposites with various types and amounts of iron oxides were successfully synthesized by the solution combustion method. The magnetic properties of the nanocomposites were related to the amount of Fe3O4 phase, while the optical properties such as absorption coefficient and bandgap energy were dependent on α-Fe2O3 content. With the combination of 37 wt.% iron oxides to g-C3N4 (sample X5), the photodegradation of MB, RhB and MO dyes increases from 40 to 75%, from 35 to 58% and from 30 to 53%, respectively, under visible light irradiation. Furthermore, the photocurrent density increased from 0.46 μA/cm2 for g-C3N4 to 4.9 μA/cm2 for the α-Fe2O3/Fe3O4/g-C3N4 nanocomposites. The enhancement of photocatalytic performance was recognized by the higher specific surface area, more harvesting of the visible-light irradiation, and efficient separation of the electron–hole pairs.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Amanulla, M. et al. Fabrication and characterization of Th(MoO4)2/TiO2 nanocomposite for potential use in photocatalytic degradation of toxic pollutants. Appl. Phys. A 128, 397 (2022).
Maria Magdalane, C., Kanimozhi, K., Arularasu, M. V., Ramalingam, G. & Kaviyarasu, K. Self-cleaning mechanism of synthesized SnO2/TiO2 nanostructure for photocatalytic activity application for waste water treatment. Surf. Interfaces 17, 100346 (2019).
Abdul Rashid, N. M. et al. Structural- and optical-properties analysis of single crystalline hematite (α-Fe2O3) nanocubes prepared by one-pot hydrothermal approach. CrystEngComm 18, 4720–4732 (2016).
Karthigaimuthu, D. et al. Synthesis or MoS2/Mg(OH)2/BiVO4 hybrid photocatalyst by ultrasonic homogenization assisted hydrothermal methods and its application as sunlight active photocatalyst for water decontamination. Chemosphere 308, 136406 (2022).
Ramalingam, C. et al. Enhanced visible light-driven photocatalytic performance of CdSe nanorods. Environ. Res. 203, 111855 (2022).
Zhu, S. & Wang, D. Photocatalysis: Basic principles, diverse forms of implementations and emerging scientific opportunities. Adv. Energy Mater. 7, 1700841 (2017).
Zhao, D. et al. Boron-doped nitrogen-deficient carbon nitride-based Z-scheme heterostructures for photocatalytic overall water splitting. Nature Energy 6, 388 (2021).
Yuan, Y. et al. High-yield synthesis and optical properties of g-C3N4. Nanoscale 7, 12343–12350 (2015).
Naseri, A., Samadi, M., Pourjavadi, A., Moshfegh, A. Z. & Ramakrishna, S. Graphitic carbon nitride (g-C3N4)-based photocatalysts for solar hydrogen generation: Recent advances and future development directions. J. Mater. Chem. A 5, 23406–23433 (2017).
Barrio, J., Volokh, M. & Shalom, M. Polymeric carbon nitrides and related metal-free materials for energy and environmental applications. J. Mater. Chem. A 8, 11075–11116 (2020).
Hong, Y. et al. A direct one-step synthesis of ultrathin g-C3N4 nanosheets from thiourea for boosting solar photocatalytic H2 evolution. Int. J. Hydrogen Energy 44, 7194–7204 (2019).
Zhu, J., Xiao, P., Li, H. & Carabineiro, S. A. C. Graphitic carbon nitride: Synthesis, properties, and applications in catalysis. ACS Appl. Mater. Interface 6, 16449–16465 (2014).
Cao, S., Low, J., Yu, J. & Jaroniec, M. Polymeric photocatalysts based on graphitic carbon nitride. Adv. Mater. 27, 2150–2176 (2015).
Bai, X. et al. A novel Fe-free photo-electro-fenton-like system for enhanced ciprofloxacin degradation: Bifunctional Z-scheme WO3/g-C3N4, environmental science. NANO 6, 2850–2862 (2019).
Alcudia-Ramos, M. A. et al. Fabrication of g-C3N4/TiO2 heterojunction composite for enhanced photocatalytic hydrogen production. Ceram. Int. 46, 38–45 (2020).
Paul, D. R. et al. ZnO-Modified g-C3N4: A potential photocatalyst for environmental application. ACS Omega 5, 3828–3838 (2020).
Gopal, R. et al. Facile synthesis and defect optimization of 2D-layered MoS2 on TiO2 heterostructure for industrial effluent, wastewater treatments. Sci. Rep. 10, 21625. https://doi.org/10.1038/s41598-020-78268-4 (2020).
Hassannezhad, M., Hosseini, M., Ganjali, M. R. & Arvand, M. A graphitic carbon nitride (g-C3N4/Fe3O4) nanocomposite: An efficient electrode material for the electrochemical determination of tramadol in human biological fluids. Anal. Methods 11, 2064–2071 (2019).
Palanivel, B. et al. Rational design of ZnFe2O4/g-C3N4 nanocomposite for enhanced photo-Fenton reaction and supercapacitor performance. Appl. Surf. Sci. 498, 143807 (2019).
Yao, Y. et al. Enhanced photo-Fenton-like process over Z-scheme CoFe2O4/g-C3N4 Heterostructures under natural indoor light. Environ. Sci. Pollut. Res. 23, 21833–21845 (2016).
Deng, X.-Z. et al. Enhanced photocatalytic efficiency of C3N4/BiFeO3 heterojunctions: The synergistic effects of band alignment and ferroelectricity. Phys. Chem. Chem. Phys. 20, 3648–3657 (2018).
Liu, S. et al. Synthesis of Fe2O3 loaded porous g-C3N4 photocatalyst for photocatalytic reduction of dinitrogen to ammonia. Chem. Eng. J. 373, 572–579 (2019).
Duan, B. & Mei, L. A Z-scheme Fe2O3/g-C3N4 heterojunction for carbon dioxide to hydrocarbon fuel under visible illuminance. J. Colloid Interface Sci. 575, 265–273 (2020).
Xiao, D., Dai, K., Qu, Y., Yin, Y. & Chen, H. Hydrothermal synthesis of α-Fe2O3/g-C3N4 composite and its efficient photocatalytic reduction of Cr(VI) under visible light. Appl. Surf. Sci. 358, 181–187 (2015).
Bakr, A. E. A. et al. Synthesis and characterization of Z-scheme α-Fe2O3 NTs/ruptured tubular g-C3N4 for enhanced photoelectrochemical water oxidation. Sol. Energy 193, 403–412 (2019).
She, X. et al. High efficiency photocatalytic water splitting using 2D α-Fe2O3/g-C3N4 Z-scheme catalysts. Adv. Energy Mater. 7, 1700025 (2017).
Alduhaish, O. et al. Facile synthesis of mesoporous α-Fe2O3@g-C3N4-NCs for efficient bifunctional electro-catalytic activity (OER/ORR). Sci. Rep. 9, 14139 (2019).
Balu, S. et al. synthesis of α-Fe2O3 decorated g-C3N4/ZnO ternary Z-scheme photocatalyst for degradation of tartrazine dye in aqueous media. J. Taiwan Inst. Chem. Eng. 99, 258–267 (2019).
Balu, S., Chen, Y.-L., Juang, R. C., Yang, T. C. K. & Juan, J. C. Morphology-controlled synthesis of α–Fe2O3 nanocrystals impregnated on g-C3N4–SO3H with ultrafast charge separation for photoreduction of Cr (VI) under visible light. Environ. Pollut. 267, 115491 (2020).
Ghane, N., Sadrnezhaad, S. K. & Hosseini, S. M. Combustion synthesis of g-C3N4/Fe2O3 nanocomposite for superior photoelectrochemical catalytic performance. Appl. Surf. Sci. 534, 147563 (2020).
Wang, Z. et al. Novel magnetic g-C3N4/α-Fe2O3/Fe3O4 composite for the very effective visible-light-fenton degradation of orange II. RSC Adv. 8, 5180–5188 (2018).
Yang, J. et al. Synthesis of Fe3O4/g-C3N4 nanocomposites and their application in the photodegradation of 2,4,6-trichlorophenol under visible light. Mater. Lett. 164, 183–189 (2016).
Zhu, D., Liu, S., Chen, M., Zhang, J. & Wang, X. Flower-like-flake Fe3O4/g-C3N4 nanocomposite: Facile synthesis, characterization, and enhanced photocatalytic performance. Colloids Surf., A 537, 372–382 (2018).
Mousavi, M. & Habibi-Yangjeh, A. Magnetically separable ternary g-C3N4/Fe3O4/BiOI nanocomposites: Novel visible-light-driven photocatalysts based on graphitic carbon nitride. J. Colloid Interface Sci. 465, 83–92 (2016).
Fathi, H., Masoudpanah, S. M., Alamolhoda, S. & Parnianfar, H. Effect of fuel type on the microstructure and magnetic properties of solution combusted Fe3O4 powders. Ceram. Int. 43, 7448–7453 (2017).
Erri, P., Pranda, P. & Varma, A. Oxidizer−fuel interactions in aqueous combustion synthesis. 1. Iron(III) nitrate−model fuels. Ind. Eng. Chem. Res. 43, 3092–3096 (2004).
Ianoş, R., Tăculescu, A., Păcurariu, C. & Lazău, I. Solution combustion synthesis and characterization of magnetite, Fe3O4, nanopowders. J. Am. Ceram. Soc. 95, 2236–2240 (2012).
Manukyan, K. V. et al. Solution combustion synthesis of nano-crystalline metallic materials: Mechanistic studies. J. Phys. Chem. C 117, 24417–24427 (2013).
Carlos, E., Martins, R., Fortunato, E.M.C., Branquinho, R. Solution combustion synthesis: Towards a sustainable approach for metal oxides, n/a (2020).
Thoda, O., Xanthopoulou, G., Vekinis, G. & Chroneos, A. Review of recent studies on solution combustion synthesis of nanostructured catalysts. Adv. Eng. Mater. 20, 1800047 (2018).
Lau, V.W.-H. et al. Low-molecular-weight carbon nitrides for solar hydrogen evolution. J. Am. Chem. Soc. 137, 1064–1072 (2015).
Hou, Y., Zuo, F., Dagg, A. P., Liu, J. & Feng, P. Branched WO3 nanosheet array with layered C3N4 heterojunctions and CoOx nanoparticles as a flexible photoanode for efficient photoelectrochemical water oxidation. Adv. Mater. 26, 5043–5049 (2014).
Elshafie, M., Younis, S. A., Serp, P. & Gad, E. A. M. Preparation characterization and non-isothermal decomposition kinetics of different carbon nitride sheets. Egypt. J. Pet. 29, 21–29 (2020).
Yi, X.-T., Zhao, T., Wang, F., Xu, J. & Xue, B. Palladium nanoparticles supported on exfoliated g-C3N4 as efficient catalysts for selective oxidation of benzyl alcohol by molecular oxygen. New J. Chem. 45, 13519–13528 (2021).
Rashid, J. et al. Facile synthesis of g-C3N4(0.94)/CeO2(0.05)/Fe3O4(0.01) nanosheets for DFT supported visible photocatalysis of 2-Chlorophenol. Sci. Rep. 9, 10202 (2019).
Namduri, H. & Nasrazadani, S. Quantitative analysis of iron oxides using Fourier transform infrared spectrophotometry. Corros. Sci. 50, 2493–2497 (2008).
Mao, Z. et al. Modification of surface properties and enhancement of photocatalytic performance for g-C3N4 via plasma treatment. Carbon 123, 651–659 (2017).
Sing, K.S.W., Everett, D.H., Haul, R.A.W., Moscou, L., Pierotti, R.A., Rouquerol, J., Siemieniewska, T., Reporting Physisorption Data for Gas/Solid Systems. in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA (2008).
Deganello, F. & Tyagi, A. K. Solution combustion synthesis, energy and environment: Best parameters for better materials. Prog. Cryst. Growth Charact. Mater. 64, 23–61 (2018).
Zuluaga, S. et al. Structural bandgap tuning in g-C3N4. Phys. Chem. Chem. Phys. 17, 957–962 (2015).
Dong, G., Zhang, Y., Pan, Q. & Qiu, J. A fantastic graphitic carbon nitride (g-C3N4) material: Electronic structure, photocatalytic and photoelectronic properties. J. Photochem. Photobiol., C 20, 33–50 (2014).
Jiang, T. et al. Surface modification of porous g-C3N4 materials using a waste product for enhanced photocatalytic performance under visible light. Green Chem. 21, 5934–5944 (2019).
Khalaji, A. D. Spherical α-Fe2O3 nanoparticles: Synthesis and characterization and its photocatalytic degradation of methyl orange and methylene blue. Phys. Chem. Res. 10, 473–483 (2022).
Ismail, A. A. & Bahnemann, D. W. Photochemical splitting of water for hydrogen production by photocatalysis: A review. Sol. Energy Mater. Sol. Cells 128, 85–101 (2014).
Kumar, S., Kumar, B., Baruah, A. & Shanker, V. Synthesis of magnetically separable and recyclable g-C3N4–Fe3O4 hybrid nanocomposites with enhanced photocatalytic performance under visible-light irradiation. J. Phys. Chem. C 117, 26135–26143 (2013).
Xu, Q. et al. Direct Z-scheme photocatalysts: Principles, synthesis, and applications. Mater. Today 21, 1042–1063 (2018).
Li, Y. et al. Synthesis of α-Fe2O3/g-C3N4 photocatalyst for high-efficiency water splitting under full light. Mater. Des. 196, 109191 (2020).
Wu, Y. et al. g-C3N4@α-Fe2O3/C Photocatalysts: Synergistically intensified charge generation and charge transfer for NADH regeneration. ACS Catal. 8, 5664–5674 (2018).
Li, G. et al. Degradation of acid orange 7 using magnetic AgBr under visible light: The roles of oxidizing species. Chemosphere 76, 1185–1191 (2009).
He, Z. et al. BiOCl/BiVO4 p–n heterojunction with enhanced photocatalytic activity under visible-light irradiation. J. Phys. Chem. C 118, 389–398 (2014).
Author information
Authors and Affiliations
Contributions
M.A.: Data curation, Formal analysis. S.M.M.: Supervision, Conceptualization, Writing—review & editing. M.H.: Supervision, Conceptualization, Writing—review & editing. S.A.: Supervision, Conceptualization, Writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Afkari, M., Masoudpanah, S.M., Hasheminiasari, M. et al. Effects of iron oxide contents on photocatalytic performance of nanocomposites based on g-C3N4. Sci Rep 13, 6203 (2023). https://doi.org/10.1038/s41598-023-33338-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33338-1
- Springer Nature Limited