
Abstract
Canine obesity is a major health concern that predisposes dogs to various disorders. The fecal microbiome has been attracting attention because of their impact on energy efficiency and metabolic disorders of host. However, little is known about specific microbial interactions, and how these may be affected by obesity in dogs. The objective of this study was to investigate the differences in fecal microbiome and specific microbial networks between obese and normal dogs. A total of 20 beagle dogs (males = 12, body weight [BW]: 10.5 ± 1.08 kg; females = 8, BW: 11.3 ± 1.71 kg; all 2-year-old) were fed to meet the maintenance energy requirements for 18 weeks. Then, 12 beagle dogs were selected based on body condition score (BCS) and divided into two groups: high BCS group (HBCS; BCS range: 7–9, males = 4, females = 2) and normal BCS group (NBCS; BCS range: 4–6, males = 4, females = 2). In the final week of the experiment, fecal samples were collected directly from the rectum, before breakfast, for analyzing the fecal microbiome using 16S rRNA gene amplicon sequencing. The HBCS group had a significantly higher final BW than the NBCS group (P < 0.01). The relative abundances of Faecalibacterium, Phascolarctobacterium, Megamonas, Bacteroides, Mucispirillum, and an unclassified genus within Ruminococcaceae were significantly higher in the HBCS group than those in the NBCS group (P < 0.05). Furthermore, some Kyoto Encyclopedia of Genes and Genomes (KEGG) modules related to amino acid biosynthesis and B vitamins biosynthesis were enriched in the HBCS group (P < 0.10), whereas those related to carbohydrate metabolism were enriched in the NBCS group (P < 0.10). Microbial network analysis revealed distinct co-occurrence and mutually exclusive interactions between the HBCS and NBCS groups. In conclusion, several genera related to short-chain fatty acid production were enriched in the HBCS group. The enriched KEGG modules in the HBCS group enhanced energy efficiency through cross-feeding between auxotrophs and prototrophs. However, further studies are needed to investigate how specific networks can be interpreted in the context of fermentation characteristics in the lower gut and obesity in dogs.
Similar content being viewed by others

Introduction
Owing to the growing interest in dog welfare, more people have started to take an interest in the health problems of dogs1. Among them, canine obesity is considered a major metabolic disease because it can predispose dogs to a variety of disorders, such as cardiovascular disease2, expiratory airway dysfunction3, and diabetes mellitus4, and has detrimental effects on longevity5. The main cause of obesity in dogs is an imbalance between energy intake and expenditure6. Furthermore, some studies suggest that breed, feed quality, and the owner’s interest in maintaining pet’s health are also possible causes7,8,9. Therefore, the veterinary community is paying increasing attention to the prevention of obesity in dogs.
Previously, companion dogs have been evaluated for being overweight and obese based on their body weight (BW), body condition score (BCS), and body composition (fat mass and lean body mass)10,11,12. These methods have been recognized to be efficient in terms of cost and convenience. However, given the limited data on the ideal weight for each breed and the subjective opinions of various veterinarians, these methods need to be improved for better reliability6. Accordingly, recent advances in omics technology are expected to replace traditional methods and provide new methods for investigating obesity indicators.
The gastrointestinal tract of dogs, especially the colon, is habituated by a complex microbiota that contributes to energy source absorption, metabolic functions, and immunogenic responses13. Several studies have investigated changes in the fecal microbiota in response to the modulation of dietary components, including carbohydrates14, proteins15, and fats16. In addition, some studies have reported differences in the fecal microbiota of obese dogs after a weight-loss program17,18,19,20. Taken together, these studies highlight the importance of the fecal microbiota in sustaining canine health and controlling obesity.
Previous studies have also reported differences in the gut microbiota among obese and lean individuals in humans and mice21,22,23,24,25. These changes are characterized by a reduced diversity of gut microbiota21,22, increased Firmicutes/Bacteroidota ratio23,24, and better energy harvesting25 in the obese group.
However, only a few studies have investigated the differences in the fecal microbiota between obese and lean dogs17,20,26. The composition of the gut microbiota in dogs can differ based on several factors, including diet, age, breed, and disorders27,28,29. Previous studies had several limitations in interpreting the relationship between obesity and gut microbiota, including the lack of unification in terms of breeds or ages17,20,26. Therefore, the present study investigated the effects of obesity on the gut microbiota and microbial functional features of dogs of a similar age and breed. We hypothesized that fecal microbiota, microbial functional profiles, and networks among microbiota would differ between obese and normal dogs.
Therefore, this study aimed to investigate differences in fecal microbiota composition, microbial functional profiles, and specific networks between obese and normal BCS groups after maintenance energy requirements were met.
Methods
Animals and experimental design
This study was conducted using neutered beagles owned by the National Institute of Animal Science in Korea. This experiment was conducted in accordance with the methods approved by the Animal Care and Use Committee National Institute of Animal (NIAS-2019-370). All methods for animal care are performed in accordance with the ARRIVE guidelines. Twenty beagle dogs (males = 12, initial body weight [BW]: 10.5 ± 1.08 kg; females = 8, initial BW: 11.3 ± 1.71 kg; all 2-year-old) were enrolled in this study. The beagles lived individually in an indoor space (1.8 m × 2.6 m) maintained at a temperature of 22–24 °C and relative humidity from 60 to 80% using automatic temperature control and continuous ventilation. All dogs were fed twice a day (at 10:00 and 16:00), had access to drinking water ad libitum, and engaged in approximately 3 h of outdoor activity every day. During the outdoor activity, all dogs were in the outdoor playground (1.8 m × 2.6 m), individually. In the outdoor playground, the floor was tiled and the side walls were bricked. One side was made of a stainless fence that allows the dog to see outside. All dogs did not have access to any grass, soil, or plant, and freely come and go indoor and outdoor environments during the outdoor activity. The dogs were fed an extruded pellet-type commercial dry food (Iskhan All-life33®; Wooriwa Ltd., Korea) that met the maintenance energy requirements estimated based on the formula suggested by the Association of American Feed Control Officials (AAFCO; metabolizable energy requirement = 132 kcal × kg BW0.75/day)30 (Table 1). The experiment was conducted for 18 weeks. In the first and final day of the experiment, the BCS was measured on a nine-point scale according to the criteria of Laflamme D12, and 12 neutered beagles (eight males and four females, all 2-years-old) were selected based on the final BCS 24 h before collecting feces and divided into two groups, each consisting of four males and two females. The normal BCS group (NBCS) was composed of six healthy dogs with a normal BCS (4–6), and the high BCS group (HBCS) was composed of six dogs with a high BCS (7–9) (Table 2).
Fecal sample collection and DNA extraction
Twenty-four hours before collecting feces, feeding was restricted. To avoid possible contamination and minimize external exposure, fecal samples were collected directly from the rectum using cotton swabs before breakfast (9:00 to 10:00), immersed in liquid nitrogen, and stored in a deep freezer at − 80 °C until analysis. DNA was extracted from the fecal samples using the NucleoSpin DNA Stool kit (Macherey–Nagel, Germany) according to the manufacturer’s instructions. Briefly, fecal samples (180–220 mg) were lysed using the MN Bead Tube Holder, and the genomic DNA was harvested through NucleoSpin DNA stool column centrifugation at 13,000 × g for 1 min. In the final step, genomic DNA was eluted into the elution buffer after centrifugation at 13,000 × g for 1 min and stored at − 20 °C.
16S rRNA gene amplicon sequencing and data processing
For PCR amplification targeting the V3 and V4 variable regions of 16S rRNA, the following primer sets were used31: forward primer 5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG-3′ and reverse primer 5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C-3′. Illumina adapter overhang nucleotide sequences were added to the gene-specific sequences. The locus-specific sequences were as follows: forward overhang 5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG-3′ and reverse overhang 5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G-3′. To purify the PCR products, KAPA HiFi HotStart ReadyMix (KAPA Biosystems, USA) and Agencourt AMPure XP system (Beckman Coulter Genomics, USA) were used. The libraries were sequenced using the Illumina MiSeq instrument (2 × 300 paired-end sequencing; Illumina, CA, USA).
The barcode sequences were trimmed using Cutadapt (version 1.11)32. Then, the raw amplicon sequences were analyzed using built-in plugins within Quantitative Insights into Microbial Ecology 2 (QIIME2) (version 2021.02)33. First, the DADA2 plugin was used to remove primer sequences, denoise low-quality reads (Q < 25), merge forward and reverse reads, and remove chimeric sequences. The raw 16S rRNA gene sequences are available in the National Center for Biotechnology Information sequence read archive (BioProject accession: PRJNA818097). The amplicon sequence variants (ASVs) were taxonomically classified using the Silva 16S rRNA gene database (version SSU138)34. Taxonomically unassigned ASVs were excluded. The alpha diversity of each sample was examined with respect to ACE (abundance-based coverage estimator) and Chao1 estimates, observed ASVs, Pielou's evenness index, Shannon diversity index, and Simpson diversity index based on rarefied ASV tables using randomly selected 67,151 ASVs per sample. Principal coordinate analysis (PCoA) was conducted based on unweighted and weighted UniFrac distance matrices to compare the overall dissimilarity of microbiota between the HBCS and NBCS groups and visualized using Emperor implemented in QIIME2. The number of common and exclusively identified bacterial taxa between the HBCS and NBCS groups at the phylum, family, and genus levels were visualized using Venn diagrams. Functional metagenomic features from the 16S rRNA gene sequences were predicted based on ASVs and the corresponding biological observation matrix (BIOM) table using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved State (PICRUSt2) (version 2.3.0-b) with default options35. Principal component analysis (PCA) was performed to compare the overall differences in predicted functional profiles based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologs between the HBCS and NBCS groups. The PCA plot was visualized using the ggfortify R package36. FastSpar was used to examine the correlations among the major bacterial genera in the HBCS and NBCS groups37, and the correlations were computed based on the sparse correlations for compositional data38. Only significant microbial interactions (P < 0.05) were further analyzed using co-expression differential network analysis (CoDiNA)39 to identify specific microbial networks in the HBCS and NBCS groups.
Statistical analysis
Before statistical analysis, all data were tested for normal distribution using the Shapiro–Wilk test in SAS 9.4 (SAS Institute Inc., NC, USA). Then, normally distributed data, including those on animal performance (BW, BCS, and BW gain) and alpha diversity indices (ACE, Chao1, observed ASVs, Pielou's evenness index, Shannon index, and Simpson diversity index) were analyzed using a t-test in SAS 9.4. Data that were not normally distributed, including major microbial taxa and predicted microbial functional features from KEGG categories and modules, were analyzed using a non-parametric Wilcoxon rank-sum test. Permutational multivariate analysis of variance (PERMANOVA) was used to further analyze the PCoA and PCA results to assess whether the overall microbial community and predicted functional profiles (KEGG orthologs) differed significantly between the HBCS and NBCS groups. Statistical significance was declared at P < 0.05, and a statistical trend was speculated at 0.05 ≤ P ≤ 0.10.
Ethical approval and consent to participate
The animal study was reviewed and approved by the National Institute of Animal Science in Korea. This experiment was conducted in accordance with the methods approved by the Animal Care and Use Committee National Institute of Animal (NIAS-2019-370).
Results
Relation between BCS and animal performance
Both the initial BW and BCS were higher in the HBCS group than those in the NBCS group (Table 2). Even both HBCS and NBCS groups fed maintenance energy requirements to maintain stable weight, the HBCS group had a significantly higher final BW and BCS than the NBCS group (P < 0.05), resulting in a higher BW gain in the HBCS group (Table 2).
Quality assessment and sample statistics
A total of 1,826,502 sequences were obtained from 16S rRNA amplicon sequencing analysis, with an average of 152,209 ± 30,573 sequences per fecal sample (Table S1). After quality filtering using QIIME 2 (Q score > 25), 1,193,079 sequences (65% of the raw reads) were generated, with an average of 99,423 ± 19,814 sequences per sample. Good’s coverage was greater than 99% for all fecal samples.
Relation between BCS and the alpha and beta diversity of fecal microbiota
In fecal microbiota, the difference in BCS did not result in significant changes in any of the alpha diversity measurements (Table 3). The PCoA results based on both unweighted and weighted UniFrac distances also did not present any significant difference in the fecal microbiota of the HBCS and NBCS groups (Fig. 1).

Principal coordinates analysis based on (A) unweighted UniFrac and (B) weighted UniFrac distance matrices of the fecal microbiota. Permutational multivariate analysis of variance was used to compare differences in fecal microbiota composition between high and normal body condition score (BCS) groups. HBCS, high BCS group (BCS range: 7–9, 4 males and 2 females, all 2-year-old); NBCS, normal BCS group (BCS range: 4–6, 4 males and 2 females, all 2-year-old).
Relation between BCS and the fecal microbiota
In this study, only bacterial taxa have been presented because archaeal reads could not be detected. The bacterial taxa that were detected in over 50% of the samples and had a relative abundance of over 0.05% in at least one of the BCS groups were defined as major microbial taxa.
The Venn diagram shows common and exclusively identified prokaryotic taxa in the HBCS and NBCS groups (Fig. 2). Eight of the nine phyla were shared between the HBCS and NBCS groups, and only Desulfobacterota was exclusively detected in the NBCS group (Fig. 2A); moreover, Desulfobacterota was detected in only one of the six samples. At the family level, 50 families (approximately 75.8%) were shared between the HBCS and NBCS groups and no major families were found exclusively in the BCS group (Fig. 2B). At the genus level, more than 100 genera (approximately 72.7%) were shared between the HBCS and NBCS groups, and only 14 and 24 genera were exclusively detected in the HBCS and NBCS groups, respectively (Fig. 2C). Succinivibrio and an unclassified genus within Atopobiaceae were the only major genera (present in at least 50% of the samples in each group) found exclusively in the HBCS and NBCS groups, respectively; however, they only accounted for a minor proportion (relative abundance < 0.01%).

Venn diagram of common and exclusively identified bacterial taxa in collapsed (A) phyla, (B) families, and (C) genera based on biological observation matrix (BIOM) tables between high and normal body condition score (BCS) groups. HBCS, high BCS group (BCS range: 7–9, 4 males and 2 females, all 2-year-old); NBCS, normal BCS group (BCS range: 4–6, 4 males and 2 females, all 2-year-old).
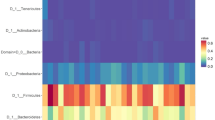
The major phyla, families, and genera with relative abundances of over 0.5% in at least one group are shown in Figure S1. Among them, the major taxa that showed statistical tendency (P < 0.10) are shown in Fig. 3 (relative abundance ≥ 0.05% in at least one group). Firmicutes was the most predominant phylum (accounting for at least 77% of the total microbiota), followed by Fusobacteriota (Figure S1A). Of the five predominant phyla, the relative abundance of Bacteroidota tended to be higher in the HBCS group than that in the NBCS group (HBCS, 7.61% vs. NBCS, 2.32%; P = 0.0656), whereas Actinobacteria showed an opposite trend (HBCS, 2.62% vs. NBCS, 4.32%; P = 0.0927) (Fig. 3A). The relative abundance of Deferribacterota, accounting for a minor proportion, was significantly higher in the HBCS group than that in the NBCS group (HBCS, 0.051% vs. NBCS, 0.008%; P = 0.0278) (Fig. 3A). At the family level, Lachnospiraceae, Peptostreptococcaceae, and Lactobacillaceae were the three most abundant families in the HBCS group, whereas Peptostreptococcaceae, Erysipelotrichaceae, and Lactobacillaceae were the most abundant in the NBCS group (Figure S1B). Among the major classified families, the relative abundances of Ruminococcaceae, Bacteroidaceae, Acidaminococcaceae, Selenomonadaceae, and Deferribacteraceae were higher in the HBCS group than those in the NBCS group (Fig. 3B). At the genus level, Peptoclostridium was the most predominant genus in both BCS groups (HBCS, 14.90% vs. NBCS, 23.79%), and more than 84% of the entire fecal microbiota was assigned to 54 major classified genera (representing > 0.05% of relative abundance in at least one BCS group). At the genus level, the relative abundances of Bacteroides, Mucispirillum, and the four genera within Firmicutes (Faecalibacterium, Phascolarctobacterium, Megamonas, and UG_Ruminococcaceae) were significantly higher in the HBCS group than those in the NBCS group (Fig. 3C, P < 0.05). Meanwhile, the relative abundances of Sutterella, Prevotellaceae Ga6A1 group, and four genera within Firmicutes (UG_Erysipelotrichaceae, Lachnospiraceae NK4A136 group, Catenibacterium, and Fournierella) tended to be higher in the HBCS group than those in the NBCS group (Fig. 3C, P < 0.10).

Differences in fecal microbiota at (A) phylum, (B) family, and (C) genus levels (each representing ≥ 0.05% in at least one of the treatments). Taxa representing at least statistical tendency (P < 0.10) are presented. HBCS, high BCS group (BCS range: 7–9, 4 males and 2 females, all 2-year-old); NBCS, normal BCS group (BCS range: 4–6, 4 males and 2 females, all 2-year-old).
Relation between BCS and the predicted functional profiles from fecal microbiota
The overall distribution of the microbial functional profiles predicted based on KEGG orthologs was visualized using PCA plots (Fig. 4A). There was no significant difference in the overall distribution of KEGG orthologs between the HBCS and NBCS groups based on the PERMANOVA analysis. Among the top 10 KEGG categories (Level 2), only carbohydrate metabolism was significantly higher in the NBCS group than that in the HBCS group (Fig. 4B, P < 0.05). Among the major KEGG modules, three modules related to amino acid metabolism (lysine biosynthesis [M00030], serine biosynthesis [M00020], and tryptophan biosynthesis [M00023]) and five modules related to the metabolism of cofactors and vitamins (cobalamin biosynthesis [M00122], pyridoxal-P biosynthesis [M00124], phylloquinone biosynthesis [M00932], and lipoic acid biosynthesis [M00881, M00884]) were markedly enriched in the HBCS group (Fig. 4B, P < 0.10). In contrast, three modules related to carbohydrate metabolism (galactose degradation [M00632], nucleotide sugar biosynthesis [M00549], and D-galactonate degradation [M00552]), two modules related to energy metabolism (reductive pentose phosphate cycle [M00165] and methanogenesis [M00357]), and one related to cysteine biosynthesis [M00609] tended to be higher in the NBCS group (Fig. 4C, P < 0.10).

Differences in predicted functional profiles of fecal microbiota. (A) Principal component analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologs89. (B) Distribution of top 10 KEGG categories. (C) Distribution of major KEGG modules (of which relative abundances were over 0.05% in at least one group) showing at least statistical tendency (P < 0.10). All microbial functional profiles were predicted using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States version 2.0. HBCS, high BCS group (BCS range: 7–9, 4 males and 2 females, all 2-year-old); NBCS, normal BCS group (BCS range: 4–6, 4 males and 2 females, all 2-year-old).
Differential network analysis based on BCS
The statistics for the differential networks of the major genera in the HBCS and NBCS groups are listed in Table 4. The total number of nodes for the HBCS and NBCS groups was 31 and 39, respectively, and the total number of edges was 74 and 71, respectively. In the differential network of the HBCS group, positive edges were detected more frequently than negative edges (Table 4; 51 positive edges vs. 23 negative edges), whereas the number of positive edges was similar to that of negative edges in the differential network of the NBCS group (Table 4; 36 positive edges vs. 35 negative edges). In the differential networks of each treatment, the γ link, which was specific to one of the networks, was assigned to 29% of the HBCS group and 23.1% of the NBCS group (Table 4). The specific genera (γ link) in the differential network of the HBCS group were Allobaculum, Lachnospiraceae NK4A136 group, UG_Ruminococcaceae, Collinsella, UG_Erysipelotrichaceae, Phascolarctobacterium, Muribaculaceae, Megamonas, and Fusobacterium (Fig. 5A), and those of NBCS group were Veillonella, Prevotella, Negativibacillus, Bifidobacterium, Bacillus, Oribacterium, Turicibacter, Faecalibacterium, and Helicobacter (Fig. 5B). Based on the two centrality measurements (degree and eigenvector centrality) and hub score within each differential network, UG_Erysipelotrichaceae and Phascolarctobacterium were the keystone genera in HBCS, while Negativibacillus and Bifidobacterium were those in the NBCS group (Fig. 5 and Figure S2).

Exclusive co-occurrence and mutual exclusion networks in (A) high body condition score group (HBCS; BCS range: 7–9, 4 males and 2 females, all 2-year-old) and (B) normal BCS group (NBCS; BCS range: 4–6, 4 males and 2 females, all 2-year-old). Only genera accounting for ≥ 0.05% abundance in at least one of the treatments were used. Node color represents exclusive microbial nodes (green) and keystone genera (magenta). Keystone genera were detected based on the hub score, degree centrality, and eigenvector centrality measurements. Edge color represents co-occurrence (blue) or mutually exclusive (red) interactions. Edge thickness was adjusted based on the absolute value of the correlation coefficients. UG, unclassified genus.
Discussion
Several human obesity studies have reported that obesity is generally linked to altered gut microbiota, presenting lower diversity in obese humans than that in lean humans21,22. In addition, a recent metagenomic study revealed that the dog microbiota was similar to human microbiota in comparison with pig or mouse microbiota, and the alteration of microbiota in response to dietary changes was also in agreement with that observed in human studies40. Therefore, we expected the HBCS group to have a lower microbiota diversity than the NBCS group. However, the present study revealed no significant differences in alpha diversity indices explaining richness (observed ASVs, Chao1, ACE), evenness, and overall diversity represented by Shannon's index and Simpson’s index between the HBCS and NBCS groups. In addition, there was no significant difference in beta diversity measurements between the HBCS and NBCS groups. Similar to the results of microbial diversity in this study, Handl et al.17 reported no significant differences in alpha diversity (Shannon’s index, Pielou's evenness index, and the number of operational taxonomic units, i.e. OTUs) and beta diversity between lean and obese companion dogs. In addition, a recent study that evaluated differences in the fecal microbiota among obese, lean, and weight loss dogs showed that there were no significant differences in alpha diversity (Faith’s phylogenetic diversity, Shannon’s index, and the number of OTUs) and beta diversity, whereas the Pielou's evenness index was significantly higher in the obese group than that in the lean and weight loss groups20.
Previous studies have reported that the five predominant phyla in the fecal microbiota of dogs are Firmicutes, Bacteroidota, Proteobacteria, Actinobacteriota, and Fusobacterium27,40 as shown in the present study. The Firmicutes/Bacteroidota ratio is generally considered an indicator of obesity because several studies in humans and mice have shown that the obese group has a higher Firmicutes/Bacteroidota ratio than the lean group23,24. In addition, a previous study proposed that Firmicutes was more effective at harvesting energy from a diet than Bacteroidetes; therefore, a higher Firmicutes/Bacteroidota ratio may promote weight gain41. However, in the present study, the relative abundance of Bacteroidota tended to be higher in the HBCS group, whereas there was no significant difference in the relative abundance of Firmicutes, thereby reducing the Firmicutes/Bacteroidota ratio in the HBCS group (data not shown). Meanwhile, some studies on gut microbiota composition in obese humans or mice have also reported conflicting results, such as a lowerFirmicutes/Bacteroidota ratio42 or no difference in the Firmicutes/Bacteroidota ratio43. In addition, several studies have observed no significant differences in both Firmicutes and Bacteroidota between obese and lean dogs15,17,44. Park et al.26 reported that the relative abundance of Firmicutes was significantly lower in the obese group than that in the lean group, but no difference was observed in Bacteroidota. In a recent study, Macedo et al.20 showed a significantly higher proportion of Bacteroidota and a lower proportion of Firmicutes in obese dogs than that in lean dogs, which is similar to our findings. Therefore, a high relative abundance of Firmicutes, low relative abundance of Bacteroidota, and a high Firmicutes/Bacteroidota ratio might not be reliable biomarkers to identify obese dogs.
Butyrate is a major short-chain fatty acid in the gut environment because it is the main fuel for epithelial cells in the colon and exerts anti-inflammatory and anti-carcinogenic effects45; therefore, butyrate-producing bacteria are considered important microbiota46. In this study, Faecalibacterium and Lachnospiraceae NK4A136 group were enriched in the HBCS group. Faecalibacterium is the predominant genus belonging to Clostridial cluster IV and can produce butyrate and D-lactate by degrading a wide range of carbohydrate substrates47. Previous studies have reported conflicting results regarding the relative abundance of Faecalibacterium between obese and lean humans, such as a lower48,49 or higher50,51,52 abundance in obese humans. In the fecal microbiome of dogs, there are few reports on the relationship between members of Faecalibacterium and obesity. Recently, Macedo et al.20 reported that the relative abundance of Faecalibacterium was significantly higher in obese dogs than that in lean dogs, and it decreased significantly after the obese group underwent a weight loss program. Thus, further studies are needed to confirm the role of Faecalibacterium in dogs with obesity. Lachnospiraceae NK4A136 group is a potential butyrate producer53, and Companys et al.52 previously reported that the relative abundance of the Lachnospiraceae NK4A136 group was enriched in obese humans, which is supported by our results. Hence, the presence of butyrate-producing bacteria in the fecal microbiome of dogs may be related to obesity. The abundance of the family Ruminococcaceae was higher in the HBCS group. Many genera under Ruminococcaceae belong to cluster IV and possess enzymes that degrade recalcitrant carbohydrate sources and produce acetate as a major fermentation product that can be used as an energy source by the host47. In addition, four major genera related to acetate production as the major fermentation product, specifically Fournierella54, Catenibacterium55, Megamonas56, and unclassified genera within Ruminococcaceae47, were enriched in the HBCS group. Faecalibacterium prausnitzii, which is a major species belonging to Faecalibacterium, requires acetate for growth57. Therefore, the increase in Faecalibacterium may be linked to the high abundance of acetate-producing bacteria. Furthermore, several studies have suggested a relationship between Sutterella spp. and obesity in humans58,59. A previous study reported a higher relative abundance of Sutterella in obese dogs20, which is supported by our results. Thus, we speculate that the increase in Sutterella may be related to obesity in dogs. Serena et al.60 revealed that obesity in humans is associated with elevated levels of circulating succinate concomitant with specific changes in succinate-related microbiota, especially higher succinate producers (Prevotellaceae and Veillonellaceae) and lower succinate consumers (Odoribacteraceae and Clostridiaceae). In the present study, there were no significant differences in these families between the HBCS and NBCS groups. Instead, we found that the relative abundances of Bacteroides61,62 and Mucispirillum63, which are considered to be succinate producers63, and that of Phascolarctobacterium, which is a propionate producer through the succinate pathway, were higher in the HBCS group at the genus level. We thus suggest that obese dogs may have a higher relative abundance of succinate producers and succinate utilizer than lean dogs.
The alteration in fecal microbiota composition is linked to changes in microbial functional features. In this study, the difference in BCS did not affect the overall predicted functional features, as expected from the lack of differences in the overall bacterial diversity (Table 3). An excessive energy supply to the host by increasing carbohydrate metabolism is an important factor in obese animals64. In addition, several studies have reported an increase in the functional profile of carbohydrate metabolism in obese humans65,66. Therefore, we expected an increased relative abundance of functional profiles of carbohydrate metabolism in the HBCS group. However, carbohydrate metabolism among the top 10 KEGG categories showed a significant increase only in the NBCS group. Some KEGG modules involved in carbohydrate metabolism (galactose degradation [M00632], nucleotide sugar biosynthesis [M00549], and D-galactonate degradation [M00552]) were significantly enriched in the NBCS group. The unexpected increase in carbohydrate metabolism can be explained by two possible reasons: (1) the functional profile was predicted based on DNA; therefore, the RNA-based results (i.e.. RNA sequencing) in carbohydrate metabolism may be different from this result, and (2) the lack of a dog microbiome-specific PICRUSt2 database. In this study, some KEGG modules related to B vitamins biosynthesis (cobalamin biosynthesis [M00122] and pyridoxal-P biosynthesis [M00124]) and amino acid biosynthesis (lysine biosynthesis [M00030], serine biosynthesis [M00020], and tryptophan biosynthesis [M00023]) were enriched in the HBCS group. The microbiota produces many types of micronutrients that play an important role in host homeostasis67. B vitamins acts as an enzymatic cofactor or a precursor of cofactors68 and can be exchanged and shared between microbiota, thereby stabilizing the gut microbiota by cross-feeding69. A recent study that investigated the effects of co-culturing butyrate producers revealed that Faecalibacterium prausnitzii, a major species belonging to Faecalibacterium, requires several B vitamins and tryptophan for growth (auxotrophs)70. They also demonstrated that co-culturing Faecalibacterium prausnitzii with species belonging to Lachnospiraceae, which are prototrophic (capable of de novo synthesis) for all amino acids and several vitamins, stimulates the growth of Faecalibacterium prausnitzii through cross-feeding70. In this study, the HBCS group showed a higher relative abundance of butyrate producers (Faecalibacterium and Lachnospiraceae NK4A136 group) and obesity-related genera (Lachnospiraceae NK4A136 group and Sutterella). Turnbaugh et al.25 previously suggested that the microbiota of obese mice may be more efficient in energy harvesting than that of lean mice. Therefore, we speculate that obesity in dogs may be influenced by the enhancement of energy-harvesting efficiency through cross-feeding among fecal microbiota, especially among auxotrophs and prototrophs.
We also compared the overall interactions within the major fecal bacterial genera belonging to each BCS group. Unclassified genera within Erysipelotrichaceae and Phascolarctobacterium were the keystone genera within exclusive networks in the HBCS group. Previous studies have reported that several species belonging to the family Erysipelotrichaceae are associated with obesity in mouse models71 and humans72. Additionally, an association between Erysipelotrichaceae and host lipid metabolism has been suggested73. In the present study, UG_Erysipelotrichaceae had an exclusive positive interaction with five major genera, namely Allobaculum, Fusobacterium, Lachnospiraceae NK4A136 group, Megamonas, and Phascolarctobacterium. Among these genera, Lachnospiraceae NK4A136 group52 and Megamonas74, which were higher in abundance in the HBCS group, were also positively associated with obesity and weight gain, respectively. Phascolarctobacterium consumes succinate and produces propionate during carbohydrate fermentation75. Succinate is not only a metabolic end-product of some bacteria but also a primary cross-feeding metabolite in microbial propionate synthesis76. Phascolarctobacterium showed a strong positive correlation with 10 major genera, whereof three genera, namely Fusobacterium77, Bacteroides61,62, and Mucispirillum63 are considered to be succinate producers. Interestingly, Muribaculaceae, which showed no difference in the relative abundance between the HBCS and NBCS groups, exhibited exclusively negative correlations with four major genera, namely the Lachnospiraceae NK4A136 group, Megamonas, Phascolarctobacterium, and Prevotellaceae Ga6A1 group, which had significantly higher abundances in the HBCS group. Although little is known about the relationship between Muribaculaceae and obesity, a previous study revealed the resistance effect of Muribaculaceae in lean mice fed with a high-fat diet78. Therefore, we speculate that Muribaculaceae may play an anti-obesity role in dogs because of its negative correlation with obesity-related genera. In the exclusive network of the NBCS group, Bifidobacterium and Negativibacillus were the keystone genera. Bifidobacterium is well known as an important probiotic that exerts beneficial health effects, such as regulating microbial homeostasis, reducing intestinal lipopolysaccharide levels, and improving the mucosal barrier function79,80,81,82. In addition, several studies have reported that the relative abundance of Bifidobacterium is lower in mice fed a high-fat diet83,84. In this study, Bifidobacterium exhibited a strong negative correlation with Peptoclostridium and Blautia, which have previously been associated with obesity in dogs20,85, and a strong positive correlation with Lactobacillus, Sellimonas, and Negativibacillus. A previous study suggested that Lactobacillus may serve as a potential probiotic for dogs27 and Sellimonas plays an important role in gut recovery homeostasis86. Therefore, the co-occurrence interactions among potential probiotics and mutually exclusive interactions among obesity-related genera might have maintained the BW of the NBCS group.
The majority of previous studies investigating the fecal microbiome in obese dogs focused on the impact of dietary changes or physical training to reduce weight rather than on natural differences in the microbiome and microbial interactions. In this study, we investigated essential differences in the fecal microbiome and their interactions between normal and obese dogs. However, we found several results, especially regarding microbiota related to succinate metabolism and functional profiles of carbohydrate metabolism, that were inconsistent with the results of previous studies on obese mice and humans. Therefore, to better understand obesity in dogs, future studies should be conducted to further investigate the differences in microbial functional metabolism and metabolites between normal and obese dogs.
Conclusions
Differences in BCS did not affect the overall distribution of the microbiota and predicted functional features. However, several genera related to short-chain fatty acid production were enriched in the HBCS group. In the predicted microbial functional profiles, some KEGG modules related to B vitamins and amino acid biosyntheses were higher in the HBCS group, indicating enhanced energy efficiency through cross-feeding between auxotrophs and prototrophs. In addition, specific co-occurrence and mutually exclusive interactions among the different genera were detected among the HBCS and NBCS groups. Further studies are needed to investigate how the specific networks can be interpreted in the context of fecal fermentation characteristics and obesity in dogs.
Data availability
The raw 16S rRNA sequences have been deposited in the NCBI SRA ubder BioProject PRJNA818097. All analyzed microbioal datasets in the present study are available from the corresponding author on reasonable request.
Abbreviations
- BW:
-
Body weight
- BCS:
-
Body condition score
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- AAFCO:
-
Association of American Feed Control Officials
- QIIME2:
-
Quantitative insights into microbial ecology 2
- ASV:
-
Amplicon sequence variant
- ACE:
-
Abundance-based coverage estimator
- PCoA:
-
Principal coordinate analysis
- BIOM:
-
Biological observation matrix
- PICRUSt2:
-
Phylogenetic investigation of communities by reconstruction of unobserved state
- PCA:
-
Principal component analysis
- CoDiNA:
-
Co-expression differential network analysis
- PERMANOVA:
-
Permutational multivariate analysis of variance
- OUT:
-
Operational taxonomic unit
References
Houpt, K. A. et al. Proceedings of a workshop to identify dog welfare issues in the US, Japan, Czech Republic, Spain and the UK. Appl. Anim. Behav. Sci. 106, 221–233 (2007).
Mizelle, H. L., Edwards, T. C. & Montant, J.-P. Abnormal cardiovascular responses to exercise during the development of obesity in dogs. Am. J. Hypertens 7, 374–378 (1994).
Bach, J. F. et al. Association of expiratory airway dysfunction with marked obesity in healthy adult dogs. Am. J. Vet. Res. 68, 670–675 (2007).
Mattheeuws, D., Rottiers, R., Kaneko, J. & Vermeulen, A. Diabetes mellitus in dogs: Relationship of obesity to glucose tolerance and insulin response. Am. J. Vet. Res. 45, 98–103 (1984).
Kealy, R. D. et al. Effects of diet restriction on life span and age-related changes in dogs. J. Am. Vet. Med. Assoc. 220, 1315–1320 (2002).
German, A. J. The growing problem of obesity in dogs and cats. J. Nutr. 136, 1940S-1946S (2006).
Edney, A. & Smith, P. Study of obesity in dogs visiting veterinary practices in the United Kingdom. Vet. Rec. 118, 391–396 (1986).
Mason, E. Obesity to pet dogs. Vet. Rec. 86, 612–616 (1970).
Kienzle, E., Bergler, R. & Mandernach, A. A comparison of the feeding behavior and the human–animal relationship in owners of normal and obese dogs. J. Nutr. 128, 2779S-2782S (1998).
Burkholder, W. J. Precision and practicality of methods assessing body composition of dogs and cats. Compend. Contin. Educ. Pract. Vet. 23, 1–10 (2001).
Pendergrass, P. B., Bartley, C. M., Nagy, F., Ream, L. J. & Stuhlman, R. A rapid method for determining normal weights of medium-to-large mongrel dogs. J. Small Anim. Pract. 24, 269–276 (1983).
Laflamme, D. Development and validation of a body condition score system for dogs. Canine Pract (Santa Barbara, Calif: 1990)(USA) (1997).
Huang, Z., Pan, Z., Yang, R., Bi, Y. & Xiong, X. The canine gastrointestinal microbiota: early studies and research frontiers. Gut Microbes 11, 635–654 (2020).
Li, Q., Lauber, C. L., Czarnecki-Maulden, G., Pan, Y. & Hannah, S. S. Effects of the dietary protein and carbohydrate ratio on gut microbiomes in dogs of different body conditions. mBio 8, e01703-01716 (2017).
Xu, J. et al. The response of canine faecal microbiota to increased dietary protein is influenced by body condition. BMC Vet. Res. 13, 1–11 (2017).
Moinard, A. et al. Effects of high-fat diet at two energetic levels on fecal microbiota, colonic barrier, and metabolic parameters in dogs. Front. Vet. Sci. 7, 699 (2020).
Handl, S. et al. Faecal microbiota in lean and obese dogs. FEMS Microbiol. Ecol. 84, 332–343 (2013).
Salas-Mani, A. et al. Fecal microbiota composition changes after a BW loss diet in Beagle dogs. J. Anim. Sci. 96, 3102–3111 (2018).
Sanchez, S. B. et al. Fecal microbiota in client-owned obese dogs changes after weight loss with a high-fiber-high-protein diet. Peer J. 8, e9706 (2020).
Macedo, H. T. et al. Weight-loss in obese dogs promotes important shifts in fecal microbiota profile to the extent of resembling microbiota of lean dogs. Anim. Microbiome 4, 1–13 (2022).
Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009).
Le Chatelier, E. et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 (2013).
Ley, R. E. et al. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. 102, 11070–11075 (2005).
Ley, R. E., Turnbaugh, P. J., Klein, S. & Gordon, J. I. Human gut microbes associated with obesity. Nature 444, 1022–1023 (2006).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Park, H. J. et al. Association of obesity with serum leptin, adiponectin, and serotonin and gut microflora in beagle dogs. J. Vet. Intern. Med. 29, 43–50 (2015).
Grześkowiak, Ł, Endo, A., Beasley, S. & Salminen, S. Microbiota and probiotics in canine and feline welfare. Anaerobe 34, 14–23 (2015).
Lee, W.-J. & Hase, K. Gut microbiota–generated metabolites in animal health and disease. Nat. Chem. Biol. 10, 416–424 (2014).
Simpson, J., Martineau, B., Jones, W., Ballam, J. & Mackie, R. I. Characterization of fecal bacterial populations in canines: Effects of age, breed and dietary fiber. Microb. Ecol. 44, 186–197 (2002).
AAFCO: Association of American Feed Control Officials. The Official Publication of the Association of American Feed Control Officials 2019.
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1–e1 (2013).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688 (2020).
Tang, Y., Horikoshi, M. & Li, W. ggfortify: Unified interface to visualize statistical results of popular R packages. R J. 8, 474 (2016).
Watts, S. C., Ritchie, S. C., Inouye, M. & Holt, K. E. FastSpar: Rapid and scalable correlation estimation for compositional data. Bioinformatics 35, 1064–1066 (2019).
Friedman, J., & Alm, E. J. Inferring correlation networks from genomic survey data. (2012).
Morselli Gysi, D. et al. Whole transcriptomic network analysis using co-expression differential network analysis (CoDiNA). PLoS ONE 15, e0240523 (2020).
Coelho, L. P. et al. Similarity of the dog and human gut microbiomes in gene content and response to diet. Microbiome 6, 1–11 (2018).
Krajmalnik-Brown, R., Ilhan, Z. E., Kang, D. W. & DiBaise, J. K. Effects of gut microbes on nutrient absorption and energy regulation. Nutr. Clin. Pract. 27, 201–214 (2012).
Schwiertz, A. et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 18, 190–195 (2010).
Duncan, S. H. et al. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 32, 1720–1724 (2008).
Chun, J. L. et al. Difference of gut microbiota composition based on the body condition scores in dogs. J. Anim. Sci. Technol. 62, 239 (2020).
Hamer, H. M. et al. The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 27, 104–119 (2008).
Louis, P. & Flint, H. J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 294, 1–8 (2009).
Duncan, S. H., Louis, P. & Flint, H. J. Cultivable bacterial diversity from the human colon. Lett. Appl. Microbiol. 44, 343–350 (2007).
Yasir, M. et al. Comparison of the gut microbiota of people in France and Saudi Arabia. Nutr. Diabetes 5, e153–e153 (2015).
Andoh, A., Nishida, A., Takahashi, K., Inatomi, O., Imaeda, H., Bamba, S., Kito, K., Sugimoto, M., & Kobayashi, T. Comparison of the gut microbial community between obese and lean peoples using 16S gene sequencing in a Japanese population. J. Clin. Biochem. Nutr. 2016, advpub.
Chávez-Carbajal, A. et al. Gut microbiota and predicted metabolic pathways in a sample of Mexican women affected by obesity and obesity plus metabolic syndrome. Int. J. Mol. Sci. 20, 438 (2019).
Salah, M., Azab, M., Ramadan, A. & Hanora, A. New insights on obesity and diabetes from gut microbiome alterations in Egyptian adults. OMICS 23, 477–485 (2019).
Companys, J. et al. Gut microbiota profile and its association with clinical variables and dietary intake in overweight/obese and lean subjects: A cross-sectional study. Nutrients 13, 2032 (2021).
Stadlbauer, V. et al. Dysbiosis, gut barrier dysfunction and inflammation in dementia: a pilot study. BMC Geriatr. 20, 248 (2020).
Togo, A. H. et al. Fournierella massiliensis gen. nov., sp. nov., a new human-associated member of the family Ruminococcaceae. Int. J. Syst. Evol. Microbiol. 67, 1393–1399 (2017).
Kageyama, A. & Benno, Y. Catenibacterium mitsuokai gen. nov., sp. Nov., a gram-positive anaerobic bacterium isolated from human faeces. Int. J. Syst. Evol. Microbiol. 50, 1595–1599 (2000).
Chevrot, R., Carlotti, A., Sopena, V., Marchand, P. & Rosenfeld, E. Megamonas rupellensis sp. nov., an anaerobe isolated from the caecum of a duck. Int. J. Syst. Evol. Microbiol. 58, 2921–2924 (2008).
Duncan, S. H., Hold, G. L., Harmsen, H. J., Stewart, C. S. & Flint, H. J. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov.. Int. J. Syst. Evol. Microbiol. 52, 2141–2146 (2002).
Hou, Y. -P., He, Q. -Q., Ouyang, H. -M., Peng, H. -S., Wang, Q., Li, J., Lv, X. -F., Zheng, Y. -N., Li, S. -C., Liu, H.-L. Human gut microbiota associated with obesity in Chinese children and adolescents. BioMed. Res. Int. 2017, 2017.
Gao, X. et al. Body mass index differences in the gut microbiota are gender specific. Front. Microbiol. 9, 1250 (2018).
Serena, C. et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 12, 1642–1657 (2018).
Miller, T. L. The pathway of formation of acetate and succinate from pyruvate by Bacteroides succinogenes. Arch. Microbiol. 117, 145–152 (1978).
Johnson, J. L., Moore, W. & Moore, L. V. Bacteroides caccae sp. nov., Bacteroides merdae sp. nov., and Bacteroides stercoris sp. nov. isolated from human feces. Int. J. Syst. Evol. Microbiol. 36, 499–501 (1986).
Loy, A. et al. Lifestyle and horizontal gene transfer-mediated evolution of Mucispirillum schaedleri, a core member of the murine gut microbiota. mSystems 2, e00171-00116 (2017).
Wang, T.-Y. et al. A comparative study of microbial community and functions of type 2 diabetes mellitus patients with obesity and healthy people. Appl. Microbiol. Biotechnol. 104, 7143–7153 (2020).
Pedrogo, D. A. M., Jensen, M. D., Van Dyke, C. T., Murray, J. A., Woods, J. A., Chen, J., Kashyap, P. C., Nehra, V. Gut microbial carbohydrate metabolism hinders weight loss in overweight adults undergoing lifestyle intervention with a volumetric diet. In Mayo Clinic Proceedings. Elsevier; 2018: 1104–1110.
Del Chierico, F. et al. Gut microbiota markers in obese adolescent and adult patients: Age-dependent differential patterns. Front. Microbiol. 9, 1210 (2018).
Agus, A., Clément, K. & Sokol, H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 70, 1174–1182 (2021).
Madigan, M. T., Martinko, J. M. & Parker, J. Brock Biology of Microorganisms (Pearson Prentice Hall Upper Saddle River, 2006).
Sharma, V. et al. B-vitamin sharing promotes stability of gut microbial communities. Front. Microbiol. 10, 1485 (2019).
Soto-Martin, E. C. et al. Vitamin biosynthesis by human gut butyrate-producing bacteria and cross-feeding in synthetic microbial communities. mBio 11, e00886-00820 (2020).
Woting, A., Pfeiffer, N., Loh, G., Klaus, S. & Blaut, M. Clostridium ramosum promotes high-fat diet-induced obesity in gnotobiotic mouse models. MBio 5, e01530-e1514 (2014).
Zhang, H. et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. 106, 2365–2370 (2009).
Kaakoush, N. O. Insights into the role of Erysipelotrichaceae in the human host. Front. Cell Infect. Microbiol. 5, 84 (2015).
Kieler, I. N. et al. Gut microbiota composition may relate to weight loss rate in obese pet dogs. Vet. Med. Sci. 3, 252–262 (2017).
Del Dot, T., Osawa, R. & Stackebrandt, E. Phascolarctobacterium faecium gen. nov., spec. nov., a novel taxon of the Sporomusa group of bacteria. Syst. Appl. Microbiol. 16, 380–384 (1993).
Fernández-Veledo, S. & Vendrell, J. Gut microbiota-derived succinate: Friend or foe in human metabolic diseases?. Rev. Endocr. Metab. Disord. 20, 439–447 (2019).
McDonald, N. C. & White, R. L. Reduction of fumarate to succinate mediated by Fusobacterium varium. Appl. Biochem. Biotechnol. 187, 163–175 (2019).
Cao, W. et al. The role of gut microbiota in the resistance to obesity in mice fed a high fat diet. Int. J. Food Sci. Nutr. 71, 453–463 (2020).
Sela, D., Price, N. & Mills, D. Bifidobacteria: Genomics and Molecular Aspects 45–70 (Book Chapter, 2010).
Wang, Z. et al. The role of bifidobacteria in gut barrier function after thermal injury in rats. J. Trauma Acute Care Surg. 61, 650–657 (2006).
Griffiths, E. A. et al. In vivo effects of bifidobacteria and lactoferrin on gut endotoxin concentration and mucosal immunity in Balb/c mice. Dig. Dis. Sci 49, 579–589 (2004).
Wang, Z.-T., Yao, Y.-M., Xiao, G.-X. & Sheng, Z.-Y. Risk factors of development of gut-derived bacterial translocation in thermally injured rats. World J. Gastroenterol. 10, 1619 (2004).
Cani, P. D. et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772 (2007).
Cani, P. D. et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50, 2374–2383 (2007).
Thomson, P., Santibáñez, R., Rodríguez-Salas, C., Flores-Yañez, C. & Garrido, D. Differences in the composition and predicted functions of the intestinal microbiome of obese and normal weight adult dogs. Peer J. 10, e12695 (2022).
Muñoz, M., Guerrero-Araya, E., Cortés-Tapia, C., Plaza-Garrido, A., Lawley, T. D., & Paredes-Sabja, D. Comprehensive genome analyses of Sellimonas intestinalis, a potential biomarker of homeostasis gut recovery. Microb. Genom. 2020, 6.
Council, N. R. Nutrient Requirements of Dogs and Cats (National Academies Press, 2006).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 10008 (2008).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Funding
This work was carried out with the support of the Cooperative Research Program of Center for Companion Animal Research (Project No. PJ013984012022), Rural Development Administration, Korea. This study was supported by the 2022 RDA Fellowship Program of the National Institute of Animal Science, Rural Development Administration, Korea.
Author information
Authors and Affiliations
Contributions
The study design was performed by J.S. and K.H.K. K.S., H.C., and J.C. conducted the experiment and analyzed the animal data. H.K. and J.S. did the sequencing based analysis. All the visualization of data and statistical analyses were performed by H.K. H.K. and J.K. drafted the manuscript. K.H.K. reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, H., Seo, J., Park, T. et al. Obese dogs exhibit different fecal microbiome and specific microbial networks compared with normal weight dogs. Sci Rep 13, 723 (2023). https://doi.org/10.1038/s41598-023-27846-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-27846-3
- Springer Nature Limited
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.