
Abstract
The intensified quest for efficient materials drives us to study the alkali (Na)-based niobate (NaNbO3) and tantalate (NaTaO3) perovskites while exploiting the first-principles approach based on density functional theory, coded within WIEN2K. While using the Birch Murnaghan fit, we find these materials to be stable structurally. Similarly, the ab-initio molecular dynamics simulations (AIMD) at room temperature reveals that the compounds exhibit no structural distortion and are stable at room temperature. By using the recommended modified Becke–Johnson potential, we determine the electronic characteristics of the present materials providing insight into their nature: they are revealed to be indirect semiconductors with the calculated bandgaps of 2.5 and 3.8 eV for NaNbO3 and NaTaO3, respectively. We also determine the total and partial density of states for both materials and the results obtained for the bandgap energies of these materials are consistent with those determined by the band structure. We find that both compounds exhibit transparency to the striking photon at low energy and demonstrate absorption and optical conduction in the UV region. The elastic study shows that these compounds are mechanically stable, whereas NaNbO3 exhibits stronger ability to withstand compressive as well as shear stresses and resists change in shape while NaTaO3 demonstrates weaker ability to resist change in volume. We also find that none of the compound is perfectly isotropic and NaNbO3 and NaTaO3 are ductile and brittle in nature, respectively. By studying the optical properties of these materials, we infer that they are promising candidates for applications in optoelectronic devices. We believe that this report will invoke the experimental studies for further investigation.
Similar content being viewed by others

Introduction
Due to potential applications in photovoltaic cells1, lasers2, X-ray detectors3, light-emitting nanoantenna4, tunable and efficient light-emitting diodes5, 3-D nano-printing6 and ultra-high resolution color displays and multilevel anticounterfeiting7, superconductors, multiferroics, batteries, fuel cells, photovoltaic electrodes, catalysts, resistive switches, and sensing materials8,9,10, perovskites materials, have garnered considerable attention. They have the structure of CaTiO3, which was reported by Dick Megaw with X-ray diffractions (XRD)11.
A formula of ABX3 represents the perovskite where A and B are cations with A typically of a larger radius than the B and X denoting an anion. Exhibiting wonderful optoelectronic characteristics, the halide perovskites, an important section of the perovskites, have been under investigation for thermoelectric, memory, and artificial synapse applications12. They have shown large absorption coefficients, charge carriers with high mobility13, and carrier diffusing relatively more14. While demonstrating these properties and given the fact that they possess more-than-25-% power conversion efficiency15, they are potential photovoltaic candidates1,16,17. Since halide perovskites are highly versatile, their bandgaps are engineerable while altering the composition of the inorganic framework, by the selectivity of cation (both organic and inorganic), stoichiometric changes, structure with different layers18,19,20,21, and nanoparticles22. Since halide perovskites are interesting optically, electrically, and magnetically, they are good candidates for optoelectronic applications. Similarly, the oxide perovskites have drawn great attention thanks to their seamless availability and distinctive properties which are considerably tailorable, thus making them suitable for numerous applications23. They are represented by ABO3, A and B indicate cations. Generally, the radius of the cation residing on site A is larger than that of the cation located on site B. These cations correspond to a divalent (or monovalent) metal bonding with 12 O anions and a tetra (or pentavalent) atom, making bonds with 6 O anions. Their properties have widely been investigated. Muhammad Saeed et al. studied the alkali niobates, i.e., ANbO3, structurally and opto-electronically, and found that these niobates are semiconductors exhibiting indirect bandgaps24. SrTMO3 (TM = Mn, Fe, Co, Tc, Ru, Rh, Re, Os, Ir) were studied electronically, elastically and magnetically by Uzma Qazi et al. where they were revealed to be anisotropic, ductile, and mechanically stable25. Computationally investigated by Somia et al., AMoO3 (A = Ca, Sr, and Ba) oxide perovskites have been found to exhibit metallic nature, para-magnetism, mechanical stability, ductility, and anisotropy26. Shahid Mehmood et al. studied the SrFeO3 perovskites oxides electronically and magnetically where they (SrFeO3) demonstrated ferromagnetism and metallic nature27, although their previous investigation has revealed them to be helicoidal anti-ferromagnet28. Akbar Ali et al. investigated the BiFeO3 and BaTiO3 perovskites oxides42. L.G. Tejuca and his coworker investigated optical, electric, and magnetic characteristics of perovskite-type oxides29,30. Smyth et al. have thoroughly studied the optoelectronic characteristics of the Ca2Fe2O5 perovskites where they inferred that the studied perovskites can be efficient for photocatalysis and photosynthesis29. Nancy et al. have studied the brownmillerite perovskites where they investigated the influence of vacancies on the mechanical properties of these perovskites43. While studying the La2Ni2O5 metallic perovskites, Synagues et al. found that these perovskites are metallic and stable which is predominantly due to the substitution of cations on cites A and B partially, leading to the remarkable optical characteristics31. J. Sfeir et al. studied the CaCr2O4 and CaZrO3 perovskites where they looked for fuel oxidation by the anode contribution31. Pen˜a and Fierro et al. investigated the utility of oxide perovskites for applications in surface chemistry and heterogenous catalysis by determining the physical and optoelectronic characteristics for potential optoelectronic applications32. Since the experimental investigations require resources (materials and experimental setups) as well as are time consuming, computational study of the materials is a promising approach to explore the new materials for wide-ranging applications. This can not only validate the already-carried-out experimental studies, but also persuade the experimentalists to investigate the simulated new materials.
In this work, we study alkali (Na)-based niobate (NaNbO3) and tantalate (NaTaO3) perovskites while exploiting the density functional theory (DFT) and the full potential linearized augmented plane wave (FP-LAPW), coded in WIEN2K. We believe the optical and mechanical properties of these materials haven’t been studied theoretically. We determine their structural, optoelectronic, and elastic properties. The structural study suggests that these compounds are stable. While using the mBJ method, the present perovskites exhibit an indirect bandgap semiconducting nature where NaNbO3 and tantalate NaTaO3 have 2.5 eV and 3.8 eV bandgap energy. Both compounds are mechanically stable with NaNbO3 exhibiting stronger ability of withstanding compressive and shear stresses and opposing change in shape and NaTaO3 demonstrating weaker ability of resisting change in volume. While none of the compound is perfectly isotropic, NaNbO3 and NaTaO3 exhibit ductility and brittleness, respectively. The computed optical properties demonstrate that with distinctive properties they are potentially useful for UV-devices applications.
Computational details
Using the full-potential linearized augmented-plane wave (FP-LAPW) method33,34, coded in the WIEN2K35, was exploited for the solution of Kohn Sham equations. This is a well-considered method, developed while using the DFT, for the determination of materials’ electronic structure36,37. While exploiting the exchange–correlation potential in the application of generalized gradient approximation (GGA) structural properties of the materials were determined35. Following the Tran and Blaha modified Becke–Johnson potential 38, which doesn’t underestimate the bandgaps of the materials unlike the generalized gradient approximation (GGA), the optoelectronic properties of the present alkali (Na)-based niobate (NaNbO3) and tantalate (NaTaO3) perovskites were determined. Functions based on FP-LAPW having a minimum radius of the muffin-tin sphere (RMT) x Kmax (which was 8) were used to get the required convergence. The energy loss function was calculated by employing the Fermi’s golden rule and dipole matrix elements39,40,41. For alkali (Na)-based niobate, the RMT values were taken as 2.50, 1.87 and 1.69 a. u. for Na, Nb and O, respectively, while 2.50, 1.92 and 1.65 a. u. were the values respectively for Na, Ta, and O in case of alkali (Na)-based tantalate. An extension in the spherical harmonics within the muffin-tin spheres was made up to the Imax = 10, while Fourier expanded charge density was taken to be Gmax = 12 (a. u.)−1. The energy volume curve was fit with Birch–Murnaghan equation providing the structural parameters42.
Results and discussion
Structural properties
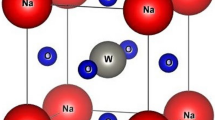
Figure 1 illustrates a simple cubic structure of one of the two alkali-based perovskite oxides, i.e., NaNbO3 with Na residing on-site A while Nb on B where A and B are cations in the general formula of perovskite oxides (ABO3). The structure of the other compound, i.e., alkali-based tantalate perovskite oxide (NaTaO3) can be estimated like this one where Na is replaced with Ta. As can be seen from Fig. 1, the atom residing on site A, i.e., Na, is larger than that placed on B (Nb), which is compatible with the general structure of the perovskite oxides. With a space group of pm-3 m (221), in the simple cubic structure of the compound, the sodium cation coordinates with 12 oxygen anions while niobium with 6. The coordinates of Na and Nb are respectively (0.0, 0.0, 0.0) and (0.5, 0.5, 0.5) while those of oxygen are (0.0, 0.5, 0.5), (0.5, 0.0, 0.5) and (0.5, 0.5, 0.0).

(Color online) A simple cubic structure of alkali (Na)-based niobate (NaNbO3): the blue-, red- and green-colored spheres represent sodium, niobium, and oxygen, respectively. The structure of alkali (Na)-based tantalate (NaTaO3) is like this one where Na is replaced by Ta.
To carry out all characterizations of material, volume optimization of its unit cell is imperative as a different unit cell results in changes in the properties of the material. To get the volume optimization, we use the Birch–Murnaghan equation of state for fitting. Illustrating energy depending on volume, the Birch Murnaghan fit is employed to interpolate our determined points analytically for finding the ground-state parameters like lattice constant a0, and the bulk modulus B.
Figure 2 depicts the volume-optimization curve for simple cubic of alkali (Na)-based niobate (NaNbO3) and tantalate (NaTaO3) perovskite oxides, which have been achieved by reducing the total energy of a unit cell by changing the volume. The total energy for which the volume is minimum is known as ground state energy (E0), while the volume is ground state volume (V0). A material with optimized energy is expected to have a stable structure.

Volume-optimization curve of alkali (Na)-based (a) niobate (NaNbO3) and (b) tantalate (NaTaO3), fitted with Birch–Murnaghan equation.
Table 1 lists all structural parameters such as optimized lattice constants (a0), ground state energy(E0), volume (V0), and the bulk modulus (B0), obtained while optimizing the volume of the materials. Since NaTaO3 has more minimum energy than NaNbO3 and the energy-volume curve for NaTaO3 is sharper than that for NaNbO3, the former is predicted to have more structural stability than the latter (NaNbO3). These optimized parameters, particularly the lattice constants (a0), were used to carry out further calculations.
To determine the thermal stability of NaNbO3 and NaTaO3, we have carried out their ab-initio molecular dynamics simulations (AIMD)43 at room temperature for 6 ps with a time interval of 1 fs, as shown in Fig. 3. Figure 3 exhibits that there is very small fluctuation in energy and both perovskites retain their geometries without any structural distortion, hence confirming that NaNbO3 and NaTaO3 are stable at room temperature.

Thermal stability of alkali (Na)-based (a) niobate [NaNbO3] and (b) tantalate [NaTaO3] perovskites.
Electronic properties
Band structure describes the behavior of electrons and their state in a given material44 by providing information regarding their energy and momentum. Both electrical and optical properties, i.e., how a material responds when subjected to electromagnetic radiations, of material are functions of electronic state and behavior. The optical properties include dielectric functions, electrical conductivity, absorbance, refractive index, and reflectivity. For the determination of electronic characteristics of the studied alkali (Na)-based niobate (NaNbO3) and tantalate (NaTaO3) perovskite oxides, we exploited modified Becke–Johnson (mBJ) potential to find their band-structure profile. Figure 4 illustrates the band profile of the simple cubic niobate and tantalate oxides perovskites: the first Brillion zone with high symmetry is shown as well. The valence and conduction bands overlap with each other at zero energy, known as the Fermi energy. mBJ was employed for the calculation of results, which demonstrate that these results are in accord with those obtained by FP-LAPW41.

Band-structure diagrams for alkali (Na)-based (a) niobate (NaNbO3) and (b) tantalate (NaTaO3).
Both parts of Fig. 4, i.e., (a) and (b), show that the valence band minima lie at M point in the Brillion zone while at Γ point, the conduction band maxima occur: both niobate and tantalate perovskite oxides exhibit the indirect semiconducting nature. The bandgap of NaNbO3 and NaTaO3 turned out to be 2.5 eV and 3.8 eV, respectively, that is the latter is a more wide-bandgap semiconductor than the former. The electronic characteristics also are determined by investigating the density of states (DOS) whose results are to be compatible with those of the band structure. The DOS indicates the number of states in which the electrons are allowed to reside at a particular energy. In other words, it tells us how many numbers of electronic levels per unit volume per unit of energy are there.
We determined the total and partial density of states, i.e., (TDOS) and (PDOS), respectively, for both materials. The TDOS shows the total DOS of material and the contribution each of its constituents makes to the TDOS while the contribution of subshells, such as p, s, and d, to the density of states of an individual atom, is shown by PDOS. Both TDOS and PDOS for NaNbO3 and NaTaO3 are respectively shown in Figs. 5 and 6. As estimated from the band-structure profile in Fig. 4, DOS compatibly reveals both materials to be semiconductors in nature, although their nature, i.e., direct, or indirect, cannot be estimated by DOS as can be done by the band-profile structure in Fig. 4. Figure 5 shows the DOS for NaNbO3 perovskite oxides: parts (a) and (b), (c) and (d) illustrate TDOS and PDOS for Na, Nb, and O, respectively. Figure 5a exhibits that TDOS in the valence band of the NaNbO3 is predominantly contributed by the oxygen atoms followed by a contribution by Na and Nb, minorly. The absence of DOS across the Fermi energy state shows the semiconducting nature of the compound with a bandgap energy of 2.5 eV, consistent with the value revealed by its band structure in Fig. 4a. In the valence band of Fig. 5a, the contribution of Nb to the DOS between 2.6 and 6.5 eV exceeds those of the other two atoms, i.e., Na and O. The relative contribution of these atoms to the TDOS lies in their electronic configuration: the Na, Nb, and O have electronic configurations as [Ne]3s1, [Kr] 4d45s1 and [He] 2s2 2p4, respectively. Due to a greater number of unpaired valence electrons (three) than in the Na and Nb each with one valence electron, oxygen causes a greater contribution than Na and Nb. Similarly, the relative contribution of s, p, and d subshells to the partial density of states of Na, Nb, and O can be explained based on the number of unpaired valence electrons: Due to one electron in s subshell in the [Ne]3s1 electronic configuration of Na, the s-subshell contributes more to the PDOS of individual oxygen than the other subshells. The TDOS of Na in Fig. 5c is.

(Color online) DOS for NaNbO3 oxide perovskites (a) TDOS and PDOS for (b) Na, (c) Nb and (d) O.

DOS for NaTaO3 oxide perovskites (a) TDOS and PDOS for (b) Na, (c) Nb and (d) O.
predominantly caused by its d-subshell PDOS followed by a contribution from the p-subshell while the contribution of its s-subshell is negligible. The TDOS for NaTaO3 is exhibited in Fig. 6a, while (b), (c), and (d) parts of Fig. 6 illustrate the PDOS of individual Na, Ta, and O atoms, respectively. Figure 6a shows that the majority of the TDOS occurs in valence bands ranging from -5.8 to 0 eV and which are mainly caused by the PDOS of the oxygen atom. The zero density of states from 0 to 3.8 eV in the conduction band indicates the bandgap of this material to be 3.8 eV, consistent with the result of the band structure in Fig. 4b. Figure 6b shows the PDOS of Na in NaTaO3, where the TDOS of Na is contributed more by s- and p-subshells of Na in the valence band, while in the conduction band there is less population of states. The PDOS of Ta is caused by its d-subshell while there is a smaller contribution of its other subshells, i.e., s and p, and the states in the valence band are denser than in the conduction band (Fig. 6c). Figure 6d exhibits that lies mostly in the valence band, the TDOS is mainly caused by the p-subshell of O while the contribution from other subshells is negligible.
Elastic properties
To explore the elastic properties of a material, the determination of elastic constants is important as they establish a material’s response to external forces and provide insight into the mechanical characteristics of the material. Thus, such constants reveal how material is mechanically stable and tough. At zero pressure, the desired constants were computed where the components of the stress tensor for small strains were calculated, and the energy was applied according to the lattice strain which kept the volume constant44. Interfaced with WIEN2K and developed particularly for cubic systems, the IRelast package was used to determine the elastic constants. In the case of cubic structure, there is only three total independent elastic constants Cij, i.e., C11, C12, and C14. Equations (1)–(7)45,46 were used to find out various elastic properties of the materials.
whereas, \({G}_{R}\) and \({G}_{v}\) are Reuss and Voigt shear moduli respectively47, \({C}_{11}\), \({C}_{12}\) and \({C}_{44}\) are elastic stiffness coefficients46, \(V\) is the Poisson’s ratio, B is bulk modulus, \(A\) is Zener’s constant that predicts the anisotropy, \(\Delta V\) is change in volume while \(p\) is pressure.
Table 2 lists the measured elastic parameters for both materials of sodium niobate (NaNbO3) and sodium tantalate (NaTaO3).
For mechanical stability of cubic crystals, the elastic constants are to be related with each other such as C11–C12 > 0 and C11 > 0, C44 > 0, C11 + 2C12 > 0 and B > 045. Since the studied materials obey these conditions, they are suggested to be mechanically stable. From Table 2, it is evident that NaNbO3 has larger elastic stiffness coefficients suggesting strong ability of the material to withstand compressive as well as shear stresses. On the other hand, NaTaO3 has the lower bulk modulus thus suggesting weaker ability to resist change in volume. It can also be seen that NaNbO3 has a larger modulus of rigidity G than that of NaTaO3 suggesting stronger ability to resist change in the shape. The values of A show that none of the compound is perfectly isotropic and both are half isotropic. Interestingly, the materials’ B/G ratio of the materials is larger than 1.75 for NaNbO3 while smaller for NaTaO3. The B/G larger than 1.75 suggests ductile nature of the NaNbO3 while the smaller suggests brittleness of the NaTaO348.
Optical properties
Dielectric function
For numerous optical properties of NaNbO3 and NaTaO3, the determination of dielectric function is instructive. How the electromagnetic waves propagate when the medium for them is changed and the interaction between phonons and electrons are described by the complex dielectric function. Given by Eq. (8),
where the \({\varepsilon }_{1}\left(\omega \right)\) and \({\varepsilon }_{2}(\omega )\) are respectively real and imaginary parts, the dielectric function’s determination is imperative for the optical properties of NaNbO3 and NaTaO3. Figure 7 exhibits the dielectric function for both NaNbO3 and NaTaO3 where part (a) of Fig. 7 illustrates the real part [ε1(ω)], while the imaginary part, ε2(ω), is shown in its part (b): the real part elucidates how the photons are dispersed in the material and the degree of polarization. The ε1(ω) initially increases with increasing energy while reaching its maximum values at 3.40 and 3.27 eV for NaNbO3 and NaTaO3, respectively, followed by a dramatic drop to the negative values at 5.88- and 6.34-eV energy, respectively, while fluctuating afterward for both materials. The static dielectric function, ε1(0), for both materials is found to be the same, i.e., 3.87. Figure 7b illustrates that the ε2(ω) starts increasing from zero at around 3.0 eV, which is an absorption edge, and reaches its maximum value of 6.41 at 5.69 eV and 5.41 at 5.98 eV for NaNbO3 and NaTaO3, respectively, indicating that the absorption is maximum in the ultra-violet (UV)-visible (Vis) region. This drops abruptly to 6.17 and 7.26 eV respectively and afterward starts fluctuating. The absorption in the UV–Vis region makes these materials promising for applications in the optoelectronic devices such as light emitting diodes (LEDs).

(Color Online) (a) real and (b) imaginary parts of dielectric function for NaNbO3 (red) and NaTaO3 oxide perovskites.
Optical conductivity
Figure 8 compares the optical conductivity of NaNbO3 and NaTaO3: the real and imaginary parts of the conductivity are illustrated in parts (a) and (b), respectively.

(Color Online) (a) real and (b) imaginary parts of optical conductivity for NaNbO3 (red) and NaTaO3 oxide perovskites.
The real part of optical conductivity (Fig. 8a) shows that the conductivity is maximum at 5.65 and 6.02 eV for NaNbO3 and NaTaO3, respectively. The conductivity of NaNbO3 at lower energy than that of NaTaO3 is attributed to the lower bandgap energy of the former than the latter as revealed by Fig. 4, i.e., due to the lower bandgap energy, the electrons are moved from the valence band to the conduction band more easily (at lower energy), thus contributing to the conductivity. Figure 8b shows the imaginary part of the optical conductivity, describing the screening of the applied field. The imaginary part of the optical conductivity decreases from 0 to − 2.91 in the 0–3.25-energy range for NaTaO3 while for the NaNbO3, it decreases from -3.63 in the energy ranging from 0 to 3.55. They respectively reach their maximum value at 6.26 and 5.77 followed by a dramatic drop in both. After around 7.7 eV, however, its starts fluctuations with no clear trend for both materials.
Refractive index and reflectivity
Figure 9A shows the determined refractive index for NaNbO3 (red) and NaTaO3 (blue) in the energy range of 0–14 eV, where the static refractive index is 1.97 for both materials. The curves for both materials while coinciding with each other start increasing with increasing energy and get to the maximum values of 2.67 at 3.30 eV and 2.9 at 3.53 eV for NaTaO3 and NaNbO3, respectively.

(Color Online) (a) refractive index and (b) reflectivity as a function of energy for NaNbO3 (red) and NaTaO3 (blue) oxide perovskites.
The higher refractive index for NaNbO3 than that for NaTaO3 suggests that the photons are more retarded, resulting from the electronic polarization while traveling through the medium of the former. The degree of electronic polarization depends upon the size of the constituent atoms of material: the Ta atom with a larger size produces a higher polarization, leading to the higher retardation (slower velocity) of the photon inside the material, and, hence, lower refractive index. It reduces to 0.82 at 6.14 and 0.92 at 6.67 eV for NaNbO3 and NaTaO3, respectively. This is followed up with increasing energy before dropping down at higher energies (7.87 – 14 eV). Figure 9b exhibits reflectivity of the NaNbO3 (red) and NaTaO3 (blue) in the energy range of 0-to-14 eV. For both materials, the zero-frequency reflectivity is found to be matching with the value of 0.10, which starts increasing with increasing energy of photons and get the maximum value of 0.4 at 5.73 eV and 0.34 at 6.15 eV for NaNbO3 and NaTaO3, respectively. The lower value for reflectivity at low energy (in the range of bandgap of the material) is attributed to the transparency of the materials to the incidence of photons. This makes these materials promising for applications in lens fabrication.
Extinction coefficient
Demonstrating the degree of light absorption by a material, the extinction coefficient, \(k\left(\omega \right)\), was calculated by Eq. (9):
Figure 10 exhibits the extinction coefficient for both NaNbO3 and NaTaO3, represented by red- and blue-colored data, respectively, where it is zero for the photon energy ranging from 0 to 3.0 eV and starts increasing with further increasing energy. The extinction coefficient gets to its maximum value at 5.87 and 6.17 eV for NaNbO3 and NaTaO3, respectively. This decreases at higher energy followed by fluctuations with increasing energy beyond 7.5 eV.

(Color Online) Extinction coefficient as a function of energy for NaNbO3 (red) and NaTaO3 (blue) oxide perovskites.
Energy-loss function
Describing the intra-band, inter-band, and plasmon interdependencies, the energy-loss function for both NaNbO3 and NaTaO3 is shown in Fig. 11, represented by red and blue data points, respectively. The energy-loss function also describes the energy lost by an electron while moving fast through a material. Figure 11 shows that the energy-loss function for both materials is zero for the energy of the photon ranging from 0 to 3.18 eV and starts increasing with increasing photon energy while peaking at 6.24 and 6.86 eV for NaNbO3 and NaTaO3, respectively. At higher energies, i.e., beyond 7.75 eV, energy loss hikes up for NaNbO3 while there is no significant variation in it for NaTaO3.

(Color Online) Energy loss function as a function of energy for NaNbO3 (red) and NaTaO3 (blue) oxide perovskites.
Conclusion
In conclusion, we studied the structural and optoelectronic properties of alkali (Na)-based niobate (NaNbO3) and tantalate (NaTaO3) perovskites while using the DFT, embedded in WIEN2K. Fitted with the Birch–Murnaghan equation of state, the energy-volume curve demonstrated the structural stability of both materials. The ab-initio molecular dynamics simulations (AIMD) showed that compound both perovskites retain their geometries without any structural distortion, hence confirming that they are stable at room temperature. By exploiting the mBJ method, the calculated electronic properties revealed the semiconductor nature of the materials with indirect bandgap energy of 2.5 and 3.8 eV for NaNbO3 and NaTaO3, respectively. The computed total and partial density of states also exhibited the same values of bandgap energy. The optical properties such as dielectric function, optical conductivity, reflectivity, refraction, energy loss function, and extinction coefficient exhibited that these materials have potential application in UV devices. It was also found that these materials are mechanically stable. NaNbO3 exhibited stronger ability to bear the compressive and shear stresses and resist change in shape while NaTaO3 showed poor ability to oppose the change in volume. None of the compound was found to be perfectly isotropic and NaNbO3 and NaTaO3 showed ductility and brittleness, respectively. Both materials exhibited transparency to the incident photon at low energy and absorption and optical conduction in the UV–Vis region, making them promise for optoelectronic applications. We hope that for further investigations, this study will draw the attention of experimental studies.
Data availability
Data can be provided on request made to the first author, Shaukat Ali Khattak.
References
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Deschler, F. et al. High photoluminescence efficiency and optically pumped lasing in solution-processed mixed halide perovskite semiconductors. J. Phys. Chem. Lett. 5, 1421–1426 (2014).
Glushkova, A. et al. Ultrasensitive 3D aerosol-jet-printed perovskite X-ray photodetector. ACS Nano 15, 4077–4084 (2021).
Tiguntseva, E. Y. et al. Light-emitting halide perovskite nanoantennas. Nano Lett. 18, 1185–1190 (2018).
Sichert, J. A. et al. Quantum size effect in organometal halide perovskite nanoplatelets. Nano Lett. 15, 6521–6527 (2015).
Chen, M. et al. 3D nanoprinting of perovskites. Adv. Mater. 31, 1904073 (2019).
Chen, M. et al. Three-dimensional perovskite nanopixels for ultrahigh-resolution color displays and multilevel anticounterfeiting. Nano Lett. 21, 5186–5194 (2021).
Kubicek, M., Bork, A. H. & Rupp, J. L. Perovskite oxides–a review on a versatile material class for solar-to-fuel conversion processes. J. Mater. Chem. A 5, 11983–12000 (2017).
Pandey, R. et al. Mutual insight on ferroelectrics and hybrid halide perovskites: a platform for future multifunctional energy conversion. Adv. Mater. 31, 1807376 (2019).
Yin, W. et al. Oxide perovskites, double perovskites and derivatives for electrocatalysis, photocatalysis, and photovoltaics. Energy Environ. Sci. 12, 442 (2019).
Megaw, H. D. Crystal structure of barium titanate. Nature 155, 484–485 (1945).
Seok, S. I. & Guo, T.-F. Halide perovskite materials and devices. MRS Bull. 45, 427–430 (2020).
Dong, Q. et al. Electron-hole diffusion lengths> 175 μm in solution-grown CH3NH3PbI3 single crystals. Science 347, 967–970 (2015).
Shi, D. et al. Low trap-state density and long carrier diffusion in organolead trihalide perovskite single crystals. Science 347, 519–522 (2015).
Best Research-Cell Efficiency Chart NREL from 1976 to the present, in N.g.t. energy (Ed.), 1976.
Burschka, J. et al. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 499, 316 (2013).
Lee, M. M., Teuscher, J., Miyasaka, T., Murakami, T. N. & Snaith, H. J. Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites. Science 338, 643 (2012).
Akkerman, Q. A. et al. Solution synthesis approach to colloidal cesium lead halide perovskite nanoplatelets with monolayer-level thickness control. J. Am. Chem. Soc. 138, 1010–1016 (2016).
Shamsi, J. et al. Colloidal synthesis of quantum confined single crystal CsPbBr 3 nanosheets with lateral size control up to the micrometer range. J. Am. Chem. Soc. 138, 7240–7243 (2016).
Song, J. et al. Monolayer and few-layer all-inorganic perovskites as a new family of two-dimensional semiconductors for printable optoelectronic devices. Adv. Mater. 28, 4861–4869 (2016).
Dou, L. et al. Atomically thin two-dimensional organic-inorganic hybrid perovskites. Science 349, 1518–1521 (2015).
Schmidt, L. C. et al. Nontemplate synthesis of CH3NH3PbBr 3 perovskite nanoparticles. J. Am. Chem. Soc. 136, 850–853 (2014).
Zhu, J. et al. Perovskite oxides: preparation, characterizations, and applications in heterogeneous catalysis. ACS Catal. 4, 2917–2940 (2014).
Saeed, M. et al. First-principles study of the structural and optoelectronic properties of ANbO3 (A= Na, K and Rb) in four crystal phases. Mater. Sci. Semicond. Process. 139, 106364 (2022).
Qazi, U., Mehmood, S., Ali, Z., Khan, I. & Ahmad, I. Electronic structure and magnetic properties of the perovskites SrTMO3 (TM= Mn, Fe Co, Tc, Ru, Rh, Re, Os and Ir). Physica B 624, 413361 (2022).
Mehmood, S., Ali, Z., Khan, I., Khan, F. & Ahmad, I. First-principles study of perovskite molybdates AMoO3 (A= Ca, Sr, Ba). J. Electron. Mater. 48, 1730–1739 (2019).
Mehmood, S., Ali, Z., Khan, I. & Ahmad, I. Effects of Ni substitution on the electronic structure and magnetic properties of perovskite SrFeO3. J. Electron. Mater. 49, 3780–3790 (2020).
Takeda, T., Yamaguchi, Y. & Watanabe, H. Magnetic structure of SrFeO3. J. Phys. Soc. Jpn. 33, 15 (1972).
Tejuca, L. G. & Fierro, J. L. Properties and Applications of Perovskite-Type Oxides (CRC Press, 2000).
Tejuca, J. L. G. Properties and Applications of Perovskite-Type Oxides (Marcel Dekker Inc, 1993).
Sayagués, M. J., Vallet-Regí, M., Caneiro, A. & Gonzalez-Calbet, J. M. Microstructural characterization of the LaNiO3-y system. J. Solid State Chem. 110, 295 (1994).
Pena, M. & Fierro, J. Chemical structures and performance of perovskite oxides. Chem. Rev. 101, 1981–2018 (2001).
Madsen, G. K., Blaha, P., Schwarz, K., Sjöstedt, E. & Nordström, L. Efficient linearization of the augmented plane-wave method. Phys. Rev. B 64, 195134 (2001).
Schwarz, K., Blaha, P. & Madsen, G. K. Electronic structure calculations of solids using the WIEN2k package for material sciences. Comput. Phys. Commun. 147, 71–76 (2002).
Blaha, P., Schwarz, K., Medsen, G. K. H., Kvasnicka, D. & Luitz, J. WIEN2k, An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties. (Vienna University Technology, Vienna, Austria, 2001).
Gao, S., Hahn, J. & Ho, W. Adsorption induced hydrogen bonding by CH group. J. Chem. Phys. 119, 6232–6236 (2003).
Schwarz, K. DFT calculations of solids with LAPW and WIEN2k. J. Solid State Chem. 176, 319–328 (2003).
Tran, F. & Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102, 226401 (2009).
Neckel, A., Eibler, R., Weinberger, P. & Schwarz, K. Results of self-consistent band-structure calculations for ScN, ScO, TiC, TiN, TiO, VC, VN and VO. J. Phys. C Solid State Phys. 9(4), 579 (1975).
Schwarz, J. S. H. Spontaneous breaking of supersymmetry through dimensional reduction. Phys. Lett. B 82, 60–64 (1979).
Schwarz, K. & Wimmer, E. Electronic structure and X-ray emission spectra of YS in comparison with NbC. J. Phys. F Metal Phys. 10, 1001 (1980).
Murnaghan, F. D. The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. 30, 244–247 (1944).
Rongfeng Yuan, J. A. N., Yan, C., Marsalek, O., Markland, T. E. & Fayer, M. D. Tracking aqueous proton transfer by two-dimensional infrared spectroscopy and ab initio molecular dynamics simulations. ACS Cent. Sci. 5, 1269–1277 (2019).
Ashcroft, N. W. & Mermin, N. D. Solid state physics (Holt, Rinehart and Winston, New York, 1976).
Khan, H. et al. Theoretical study of different aspects of Al-based Fluoroperovskite AlMF3 (M= Cu, Mn) compounds using TB-MBJ potential approximation method for generation of energy. Results Phys. 42, 105982 (2022).
Grimvall, G. Thermophysical Properties of Materials (Elsevier, 1999).
Luan, H. Q. X., Liu, F., Dai, Z., Yi, Y. & Li, Q. The mechanical properties and elastic anisotropies of cubic Ni3Al from first principles calculations. Crystals 8, 307 (2018).
Vaitheeswaran, G. et al. High-pressure structural, elastic, and electronic properties of the scintillator host material K Mg F 3. Phys. Rev. B 76, 014107 (2007).
Acknowledgements
The authors would like to thank the National Research Program for Universities (NRPU), Higher Education Commission Pakistan, No. 20–8862/NRPU/R&D/HEC/2017. The authors would like to thank Researchers Supporting Project Number (RSP2022R448), King Saud University, Riyadh, Saudi Arabia. The authors would like to thank the National Research Program for Universities (NRPU), Higher Education Commission Pakistan, No. 20-15131/NRPU/R&D/HEC/2021. The authors would like to thank National Research Program for Universities (NRPU), Higher Education Commission Pakistan, No. 20-15128/NRPU/R&D/HEC/2021.
Author information
Authors and Affiliations
Contributions
S.A.K. and S.M.W. designed the project, made the calculation and analysis, and wrote the manuscript. M.A.I., M.H., I.U., S.Z., G.R., N.R., M.S.K., G.K., and T.K. reviewed the manuscript. B.G. contributed to designing the project, analysis and manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khattak, S.A., Wabaidur, S.M., Islam, M.A. et al. First-principles structural, elastic and optoelectronics study of sodium niobate and tantalate perovskites. Sci Rep 12, 21700 (2022). https://doi.org/10.1038/s41598-022-26250-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26250-7
- Springer Nature Limited
This article is cited by
-
Exploring the Structural, Elastic and Optoelectronic Properties of Stable Sr2XSbO6 (X = Dy, La) Double Perovskites: Ab Initio Calculations
Journal of Inorganic and Organometallic Polymers and Materials (2024)
-
Stress-induced transformation on the cubic perovskite RbTaO3 for high-temperature applications: a DFT approach
Journal of Computational Electronics (2024)
-
Exploring the properties of quaternary X2NaTlF6 (X = Cs, Rb) halide double perovskite materials for energy conversion, harvesting, and storage using density functional theory
Optical and Quantum Electronics (2024)
-
Investigating the structural, elastic, and optoelectronic properties of LiXF3 (X = Cd, Hg) using the DFT approach for high-energy applications
Optical and Quantum Electronics (2024)