
Abstract
The neotropical region ranks third in the number of termites and includes five different families. Of these, Termitidae is the most diverse and includes the species Nasutitermes ephratae, which is widespread in the neotropics. To date, only one study has been published about phylogeography in neotropical termites (N. corniger). Here, we explored the population genetic patterns of N. ephratae and also evaluated the phylogeographical processes involved in the evolutionary history of the species. We used the mitochondrial genes 16S rRNA and COII as molecular markers: these were sequenced for 128 samples of N. ephratae. We estimated the genetic diversity and divergence time as well as the demography and genetic structure. We also performed an ancestral area reconstruction and a haplotype network. The results showed high genetic variability, recent demographic expansion, and strong genetic structure. A dispersal route for the species, that occurred in both directions between South and Central America, was inferred. The results emphasize a temporary separation between the South and Central America populations that affected the origin of the current Central America populations. These populations were formed from different phylogeographic histories.
Similar content being viewed by others

Introduction
The neotropics host a large diversity of species and habitats that arose from the complex geological history occurring along environmental and climatic changes. Evolutionary phenomena such as vicariance, dispersion, and extinction shaped the geographical distribution patterns of the species found in this region1.
The neotropical termite fauna ranks third in the number of species. Of the five neotropical termite families, Termitidae is the most diverse and includes the subfamily Nasutitermitinae with 171 species described in the neotropical region, including 67 in the genus Nasutitermes—corresponding to almost 40% of the number of species of the subfamily2.
Like the other Nasutitermitinae, the soldiers of this genus are characterized by a conic-shaped frontal projection with the opening of an exocrine gland located at the top. This gland produces substances used for defense against predators3.
Nasutitermes ephratae (Fig. 1) was described by Holmgren4 using alates and workers collected in Ephrata, Suriname. Banks5 described the soldiers using specimens of N. creolina collected in Panama. Later Snyder6 synonymized N. creolina with N. ephratae. The nests of this species are arboreal and show a light to dark brown coloration and a leathery surface as if they were enveloped. Internally, the nests are reinforced around the royal camara that harbors the queen and the king in the center of the nest (closer to the trunk or branch of the tree)7.

Soldier of N. ephratae.
Nasutitermes ephratae is reconstructed close to N. corniger in various phylogenetic studies8,9,10,11. Both species are very common in the neotropics including most of Central and South America, e.g., Brazil, Bolivia, Colombia, Ecuador, French Guiana, Guyana, Peru, Suriname, and Venezuela2,6,12,13,14,15.
To date, only one phylogeographic study addressed neotropical termites, focusing on N. corniger15. The results showed high variability and strong genetic structure for the populations sampled. These were divided in haplogroups along its occurrence area associated with South American biomes. The authors also proposed a dispersal route for N. corniger, which would have left Central America towards South America, where the populations dispersed toward the eastern regions.
Phylogeographic and biogeographic studies help to explain how the species responded to geological and climatic changes over time. This fact highlights the importance of phylogeographic studies that present a variety of taxonomic groups, including insects, that have population and evolutionary dynamics different from other taxa16. Thus, the association of various phylogeographic data can help us to understand the evolutionary history of neotropical biota.
The main aim of this study was to explain the phylogeographical and historical processes that gave rise to population patterns of N. ephratae in the neotropics. For this purpose, we leveraged the mitochondrial molecular markers 16S rRNA and the subunit II of the code gene to cytochrome oxidase (COII). It is important to highlight that the mitochondrial DNA (mtDNA) have been extensively used for the investigation of phylogeographic issues as well as other evolutionary questions of a large variety of species, including termites15,17,18,19,20,21,22,23.
We performed demographic analyses, estimates of variability and genetic structure, analyses of divergence time, and ancestral area reconstruction. We also performed a haplotype network and proposed a dispersal route for the species.
Methods
Biogeographic units
The biogeographic units defined for the analyses are according to the neotropical regionalization in dominions proposed by Morrone24, as follows (considering the areas that were sampled): Antillean subregion (ANT) and Mesoamerican dominion (MES) in Central America; Pacific dominion (PAC) in Central and South America; Boreal Brazilian dominion (BOR), Chacoan dominion (CHA), Parana dominion (PAR), and south Brazilian dominion (SOU) in South America.
Sampling
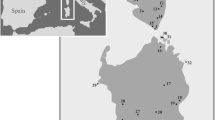
The samples of N. ephratae (Table 1; Fig. 2a) analyzed here have been obtained from the Isoptera collections of the Museu de Zoologia da Universidade de São Paulo (MZUSP) and the University of Florida (UF). The samples were collected in different campaigns and stored in 96% ethanol (MZUSP) and in 85% ethanol (UF) to better preservation of the DNA. Extracted DNA are stored in the Molecular Collection of the Laboratório de Biologia Evolutiva (LaBE) of the Faculdade de Ciências Agrárias e Veterinárias, UNESP (Jaboticabal, SP, Brazil). In addition to these samples, 12 nucleotide sequences of N. ephratae obtained in GenBank (public access) were included in the analyses, totaling 128 samples analyzed.

(a) Collection locations of the N. ephratae samples. The colors of the points are corresponding to the haplogroups; (b) Haplotype network generated using the concatenated sequences (16S + COII) of the N. ephratae samples. The white dots on the branches are indicating the mutational steps between the related haplotypes. The map (a) was generated by AFS using the software QGis v. 3.6.3 (http://qgis.org)40.
Laboratory procedures
The total DNA was extracted from the head of an individual per colony following Liu and Beckenbach25 protocol, which includes phenol–chloroform for the extraction and 100% and 70% ethanol for the DNA washing. The amplification of the 16S rRNA and COII (gene regions of mtDNA) was performed by PCR following the conditions described in Supplementary Table S1. The PCR reaction was composed by 0.6 pmol/μL of each primer (forward and reverse)26,27,28, PCR Master Mix (Promega) 1x, nuclease free water, and 2.4 ng/μL of target DNA. The PCR product was purified using the Wizard® SV Gel and PCR Clean-Up System (Promega) according to the manufacturer’s instructions, followed by Sanger sequencing in ABI 3730 XL DNA Analyzer (Applied Biosystems) automatic sequencer.
Data analysis
Genetic diversity and neutrality tests
The nucleotide sequences were read in Chromas Lite v. 2.6.5 (Technelysium Ltd., 2005). The sequences of 16S were aligned using Mafft v. 729,30 and the sequences of COII were aligned using Geneious v. 7.1.9 (https://www.geneious.com) by ClustalW, both followed by inspection by eye. Geneious also was used to concatenate the 16S and COII sequences of the samples in which both regions could by sequenced.
To quantify the genetic diversity, the following parameters were estimated using DnaSP v. 631: number of polymorphic sites (S), nucleotide diversity (π), average number of nucleotide differences (k), and haplotype diversity (Hd). In addition to these parameters, the value of θ-W per sequence was estimated using Arlequin v. 3.5.1.232. This software also was used to perform the Fu’s Fs33 and Tajima’s D34 neutrality tests—these tests were performed for the entire sample set and, separately, for each neotropical dominion sampled. The other neutrality tests Fu and Li’s F* and D* and Achaz Y*35 were performed using DnaSP v. 631.
The nucleotide composition of the sequences and the pairwise genetic distances (among individuals, among dominions and within dominions) were estimated using MEGA-X36 (the model Kimura 2-parameter37 was used to estimate the genetic distances). The genetic distances and the neutrality tests were estimated for both the concatenated sequences (16S + COII) and the single genes (16S or COII). Only the single genes were considered to estimate the genetic diversity indexes.
Haplotype network
The haplotype network was generated with the concatenated sequences (16S + COII) using TCS v. 1.2138, that uses parsimony method to establish the relationship among the haplotypes, with 95% of connection limit. The haplogroups were defined based on the shape of the network, considering the distance among the haplotypes and among the tips and central haplotypes of the group. The network was colored using tcsBU39 considering the frequency of the haplotype in each neotropical dominion sampled.
The map showing the distribution of the samples, colored according to the observed haplogroups, was performed using QGIS v. 3.6.340 based on the datum WGS 1984 and on the shapefile of the neotropical region developed by Löwenberg-Neto41, considering the regionalization proposed by Morrone24.
Genetic structure and Mantel test
The Analysis of Molecular Variance (AMOVA) was performed using Arlequin v. 3.5.1.232 to assess the possibility of genetic structure among the sampled populations. Using the concatenated sequences, tree constructions were defined for this analysis: (i) an AMOVA was performed considering the haplogroups of the haplotype network; (ii) the second AMOVA was performed separating the samples in seven groups according to their neotropical dominion (ANT, BOR, CHA, MES, PAC, PAC, and SOU); (iii) the third AMOVA was also performed considering the dominions as groups, but regrouping them into two major groups: Central America (ANT, MES, and northern PAC) and South America (BOR, CHA, southern PAC, PAR, and SOU). The F indexes, calculated by AMOVA, range from 0 to 1 and indicate high differentiation above 0.25 and moderately high differentiation between 0.15 and 0.2542.
Even to evaluate the inference of genetic structure, we performed an analysis of DNA clustering using the package rhierBAPS43,44 implemented in R v. 4.0.145. The results were obtained for both the concatenated sequences and the single genes. The graphs showing the geographical distribution of each genetic cluster (considering the concatenated sequences results) were obtained using Microsoft Excel (2008).
The Mantel test was performed using the package vegan46, implemented in R v. 4.0.1, considering the Pearson’s correlation as method with a number of permutation equal to 10,000. We inputted the concatenated sequences and the geographical coordinates of each sample to perform this analysis.
Analysis of divergence time and ancestral area reconstruction
We estimated the divergence time among the samples of N. ephratae relying on Bayesian inference. The tree was generated in BEAST v. 2.6.347 using strict clock and chain length equal to 90 million. Two partitions were included in the analysis, the first containing the 16S sequences and the second containing the COII sequences. The tree models (Fossilized Birth Death Model)48 and the clock models for both partitions were linked, and the substitution model TrN + I + G were applied for 16S and COII49. The selection of the best-fit model of nucleotide substitution was done based on the results of jModelTest50 considering the lower values of BIC.
The Bayesian inference was calibrated with the ages of four fossil records, following Heath, Huelsenbeck, and Stadler instructions48: Valditermes brenanae51, 136.4 to 130 million of years ago (My); Nanotermes isaace52, 56 to 47.8 My; Nasutitermes electrinus53, 23.03 to 15.97 My; and Atlantitermes caribea54, 20.44 to 13.82 My. Besides the fossil records, the origin dates estimated by Bourguignon55 for Termitidae (54 My), Nasutitermitinae (26.2–19.4 My), and Nasutitermes (22.6–16.4 My) were used as an additional calibration to improve de analysis estimation (Supplementary Figure S2; Supplementary Table S2). For the MRCA (most recent common ancestor) priors we used exponential distribution. As outgroups, we included the species Mastotermes darwiniensis (Mastotermitidae), Amitermes dentatus (Termitidae: Termitinae), Atlantitermes snyderi (Termitidae: Nasutitermitinae), and Nasutitermes longinasus (Termitidae: Nasutitermitinae).
The MCMC trace files generated by Bayesian inference were viewed and analyzed using Tracer v. 1.7.156 to check the values of Effective Sample Size (ESS > 200). The trees were resampled using the BEAUti v. 2.6.3 application “Full To Extant Tree Combiner”47 to remove the fossil taxa of the topology, keeping only the extant species. The best tree was annotated by TreeAnnotator v. 2.6.357 with 10% burn-in, and it was viewed and draw using FigTree v. 1.4.458.
The tree topology, as well as the divergence times estimated by this analysis, were used for the ancestral area reconstruction analysis, which was performed using the package BioGeoBEARS59 in R v. 4.0.145. We defined seven occurrence areas for this analysis, according to the neotropical dominions which were sampled (ANT, BOR, CHA, MES, PAC, PAR and SOU). We tested the models DEC, DIVALIKE and BAYAREALIKE with and without j (i.e., six models tested). The best-fit model was selected based on the lower values and higher weights of AIC and AICc.
Results
Genetic diversity and demographic inferences
We obtained 123 nucleotide sequences for 16S [396 base pairs (bp)] and 108 for COII (742 bp) within the samples, totaling 103 specimens with both mtDNA genes sequenced. The nucleotide composition of the concatenated sequences corresponds to 40.27% of adenine (A), 25.35% of thymine (T), 21.20% of cytosine (C), and 13.19% of guanine (G). The mean genetic distance among the samples was 0.011 for the concatenated sequences, 0.007 for the 16S sequences, and 0.015 for COII sequences (the pairwise genetic distances are presented in Supplementary Tables S3, S4, and S5). After estimating the genetic distances among and within the dominions, higher values were obtained for the BOR-MES and BOR-PAC pairwise comparisons (Table 2). The mean genetic distance within dominions was 0.008.
We found 66 haplotypes considering the concatenated sequences: 41 haplotypes (Hd = 0.890) for 16S and 53 haplotypes (Hd = 0.932) for COII (Table 3a), indicating high genetic variability for the populations sampled. High values also were found for the number of polymorphic sites (S) of both mtDNA genes (24 for 16S and 87 for COII). The COII synonym sites showed the higher value of nucleotide diversity (π), which was 200 times higher than the value observed for the non-synonym sites (Table 3a). The 16S π value was lower than the COII π value, which tends to present more polymorphism.
Regarding to the neutrality tests performed with the whole sample set (Table 3b), the Tajima’s D value was not significant. This means that there is not enough evidence to discriminate between demographic expansion or population bottlenecks from these indices. The negative and significant value of Fu’s Fs, i.e., the most sensitive among the neutrality tests performed60, suggest demographic expansion for the populations, which is also suggested from the negative value of Achaz Y*. Regarding the neutrality tests performed per dominion (Table 3c), the negative and significant values of Fu’s Fs also indicate demographic expansion for all the dominions Only the value of Tajima’s D for CHA was significantly negative. The results of neutrality tests performed for each gene are presented in Supplementary Table S6.
Haplotype network
We generated a haplotype network to reconstruct the relations among the 66 haplotypes found for N. ephratae (Fig. 2b). We observed four haplogroups (Hg) composed, in general, by haplotypes close each other and originated from a more frequent central haplotype. Haplotypes that were very distant from the central haplotype of the haplogroup (i.e., that present many mutational steps) were not grouped. The star shape of the haplogroups suggests recent demographic expansion events for the populations analyzed.
The network also showed a clear geographic differentiation among South American populations composed of haplogroup 1 and Central American populations composed mostly of haplogroups 2 and 4 (Fig. 2b; Supplementary Table S7). The haplogroup 3 is formed mainly by populations located in northern South America (BOR) and in the Antillean islands. Haplogroup 3 also includes haplotypes from eastern Brazil (PAR – Espírito Santo, Brazil), which are intermediaries between this haplogroup and the haplogroup 1. The geographical distribution of the haplogroups is also showed in Fig. 2a.
Population structure and isolation by distance
The inference of population structure in N. ephratae was verified from the results of the AMOVA (Table 4) and rhierBAPS (Fig. 3). Regarding the AMOVA among haplogroups (Table 4a), the FST value was significantly very high indicating strong differentiation among the haplogroups established in the network (Fig. 2b). A high FST was also observed in the AMOVA among dominions (Table 4b), showing strong genetic differentiation among the dominions sampled. In the AMOVA among continents (Table 4c), the high FCT also indicates strong differentiation among the populations located in different continents while a moderate differentiation can be observed among the dominions within continents, as showed by FSC.

(a) Results of the analysis of clustering (rhierBAPS) performed with the concatenated sequences; (b) Pie charts generated from the frequency of the dominions in each cluster recovered by rhierBAPS;
The rhierBAPS results (Fig. 3) showed five genetic clusters (represented by different shades of gray in Fig. 3a) for the N. ephratae samples. The samples of the cluster 1 are distributed in South America; the samples of cluster 2 are distributed in northern South America (Venezuela and Colombia in southern portion of PAC) and Central America (MES dominion). Cluster 3 includes only samples from Central America (MES and northern portion of PAC), and cluster 4 includes samples from BOR, ANT, and mainly from PAC (northern and southern portions) and MES. Cluster 5 includes samples from BOR and southern portion of PAC (Fig. 3b). These results also suggest strong genetic differentiation among the populations located in South and Central America.
Mantel’s test was performed to verify the existence of isolation by distance and resulted in a significantly positive r value (0.1197; p = 0.0005), thus indicating that there is a positive correlation between the genetic distance and the geographic distance, i.e., the genetic distances can increase as geographic distance between populations increase.
Divergence time and ancestral area reconstruction
The divergence times of the N. ephratae populations were estimated from a Bayesian inference (BI) analysis performed using the haplotypes of the 16S and COII sequences (Fig. 4; Supplementary Fig. S2). According to the tree obtained, three important cladogenetic events seem to have given rise to the N. ephratae populations analyzed. The first event (6.42 My) separated the Hg 4 + H25-H59 clade (that includes samples from Central America and northern South America) from the other clades – the ancestral node of this clade was dated to 2.19 My by the analysis and the Hg 4 clade was dated to 1.74 Ma. The second event (2.98 My) separated the Hg 1 clades + Hg 3 clade from the Hg 2 clade, which ancestral node was dated to 2.57 My. The last one includes only specimens distributed in Central America (except for one sample of H19 found in BOR). The third event (2.52 My) caused the divergence among the Hg 1 main clade (formed by South American populations) from the Hg 1 + Hg 3 clade (formed by samples from northern South America and Antillean islands). The ancestral nodes of the Hg 3 and the Hg 1 main clade were dated to 1.59 My and 1.73 My, respectively.

Bayesian inference tree generated using the haplotypes of N. ephratae, associated to the results of the ancestral area reconstruction. The numbers near to the nodes correspond to the estimated divergence times in million of years (My). The “N” inside the PAC circles (close to the taxa names) are indicating that the respective haplotype was observed in the northern portion of PAC dominion, located in Central America; PAC circles without the “N” are indicating the haplotypes from southern portion of PAC dominion, located in South America. The posterior probabilities and the 95% HPD intervals for this BI are showed in Supplementary Fig. S2. The map was generated by AFS using the software QGis v. 3.6.3 (http://qgis.org)40.
For the ancestral area reconstruction, the best-fit model selected, DEC + j, presented AIC and AICc equal to 211.7 and 212.4 with weights equal to 0.85 and 0.84, respectively. The results showed that the common ancestor of all the clades was distributed in MES and PAC dominions and were dispersed posteriorly to the other areas (Fig. 4; Supplementary Fig. S3).
Discussion
Data on the genetic diversity obtained for the N. ephratae populations showed high genetic variability for the species, as evidenced by the large number of haplotypes in relation to the total number of samples as well as by the high haplotype diversity. As expected, the nucleotide diversity observed for the COII sequences was greater than the nucleotide diversity found for the 16S. Furthermore, the π value obtained for the COII synonyms sites was higher than the π value for the non-synonym sites (Table 3a).
The high genetic variability observed for N. ephratae seems not to be widely shared among the populations, as highlighted by the AMOVA results (Table 4), which showed strong genetic structure for the species. That is, there are a lot of genetic variants that are exclusive of one or few regions suggesting low or moderated gene flow among distant populations. This limitation in gene flow may have occurred due to isolation by distance as confirmed by Mantel’s test. The clustering analysis results also corroborated the population structure inference because five clusters with a distribution limited to adjacent dominions were recovered. Cluster 1 shows a larger geographic distribution and occurs in all South American dominions. Great genetic variability with haplotypes little shared among distant geographic regions was also observed very similar to N. corniger15.
Regarding to the demographic history of the N. ephratae populations, the neutrality tests (Table 3b,c), specifically Fu’s Fs, indicated demographic expansion for the species (considering the whole sample set) and particularly for each neotropical dominion sampled. This also can be inferred from the star shape of the haplogroups in the haplotype network, thus suggesting that a lot of descendent haplotypes had recently risen from the ancestor haplotypes located in the center of the haplogroups.
The haplotype network (Fig. 2b) also showed four haplogroups presenting clear differences about the geographical distribution of the haplotypes. Haplogroup 1 is entirely composed of haplotypes from the South America dominions while haplogroups 2 and 4 are mostly formed by Central American haplotypes. Some haplotypes included in haplogroup 4 can also be found in northern South America (Venezuela and Trinidad and Tobago) but were not often observed at latitudes below this. Haplogroup 3 is composed of haplotypes from northern South America (Trinidad and Tobago and French Guiana) and from the Antillean islands (Dominica and Guadeloupe).
These groups were also recovered by the clustering analysis (Fig. 3a) except for a few differences among them. In general, the results of the analyses are linked as following: haplogroup 1 corresponds to cluster 1; haplogroup 2 corresponds to cluster 3; haplogroup 3 corresponds to cluster 5; and haplogroup 4 corresponds to cluster 4.
The BI analysis (Fig. 4; Supplementary Fig. S2) shows that some nodes of the tree presented posterior probabilities below 0.50 possibly due to the difficulties of the algorithm, implemented in BEAST, in solving datasets containing very similar sequences, which occurs in many intraspecific analyses61. Despite this, large clades of this tree include haplotypes from the same haplogroup (Fig. 4), helping to support the phylogenetic inferences raised by BI. Observing the topology of the tree, the haplotype network, and the ancestral ranges reconstructed, we inferred dispersal events and then proposed a dispersal route for the N. ephratae populations (Fig. 5).

Dispersal route inferred for N. ephratae based on the results obtained in the ancestral area reconstruction. The arrows are indicating the direction of the dispersion. *Reconnection between the N. ephratae populations of South and Central America. The map was generated by AFS using the software QGis v. 3.6.3 (http://qgis.org)40.
The ancestral populations distributed in MES and PAC (“1”; Fig. 5a) suffered a temporary separation that split the populations of South America (southern PAC) from the Central American (northern PAC + MES) populations (“2”; Fig. 5b). The ancestor of the haplogroups 1, 2, and 3 occurred in MES during this separation (“3”; Fig. 5c). This leads to the origin of haplogroup 2 that was restricted to Central America (northern PAC + MES). After the reconnection of the N. ephratae populations (indicated with an asterisk in Fig. 5), the Mesoamerican ancestor dispersed to the South American portion of PAC (“5”; Fig. 5e) from where there was a new dispersion to SOU (“6”, Fig. 5f) and to ANT-BOR (“7”, Fig. 5f). This last dispersion gave rise to haplogroup 3 composed mainly of French Guiana and Antillean populations. The ancestral populations of the haplogroup 1 that had arisen in SOU were widely dispersed to CHA and PAR (Atlantic Forest lato sensu) reaching to BOR and southern PAC (“8”, Fig. 5f), but remaining limited to South America.
Still during the temporary separation between the South and Central America N. ephratae populations, the South American ancestor of the haplogroup 4 arose in southern PAC (“4”; Fig. 5d). After the populations’ reconnection, there was a dispersal from southern PAC to northern PAC and to MES originating as the late ancestor of haplogroup 4 and clade H25-A59, which were restricted to these dominions (“5”; Fig. 5e).
Based on the dispersal route proposed, we inferred that the N. ephratae populations currently distributed in Central America arose from distinct dispersal events. This becomes clearer when we also observe the geographic distribution of haplogroups 2 and 4 (Fig. 2) whose occurrence areas overlap, but who have different genetic groups. That is, even though these haplogroups are distributed in the same area, they are results of different evolutionary events. The ancestors of these haplogroups possibly diverged during a temporary split among the ancestral N. ephratae populations from South and Central American between the late Pliocene and early Pleistocene (around 5.06 and 2.78 My; Figs. 4 and 5). Haplogroup 2 originated from populations that were restricted to Central America during this split while the haplogroup 4 originated after the reconnection perhaps arising from populations that were dispersed from the northern South America (southern PAC) to Central America (northern PAC + MES). The same direction of dispersion (South America to Central America) was also identified for the ant species Neoponera villosa16 dated from 0.46 to 0.28 My; earlier dispersions have been observed for N. ephratae.
Haplogroup 1 is exclusively South American and arose from a Mesoamerican ancestor that dispersed to the northern South America and then to the SOU dominion. Therefore, the dispersal events of the N. ephratae populations among Central and South America occurred in both senses, thus shaping the genetic and phylogeographic patterns observed here.
Although the causes responsible for the temporary split between the South and Central American N. ephratae population are not clear, some hypotheses can be proposed. A population isolation caused by the geographic distance between the populations may have led to this split: The occurrence of isolation by distance was suggested by the Mantel’s test. The loss of distribution area could also have caused this effect in the populations. In this way, the reconnection of the populations could have occurred due to the demographic expansion that was detected by the neutrality tests and can be suggested from the star shape of the haplogroups in the haplotype network.
Geological and/or geographic factors could also help to explain this split. Following this approach, it is possible that the separation is related to the tertiary's tectonic and paleographical reorganization movements (in the late Pliocene), which led to the emergence of barriers and changes in dispersal routes in South America62. Specifically, this split between the N. ephratae populations may have been caused by the momentaneous effects of the elevation of the Panama isthmus. These effects would have lasted between 4.6 and 2.6 My62,63—a date close to the one estimated for the separations detected (5.06 to 2.78 My). Moreover, the early Quaternary climate changes (early Pleistocene) was characterized by temperature and dryness oscillations in the continents and was also impacted the adaptability and the migration of species and populations62.
Although dispersion by water (inside flotsam carried by ocean or river currents or ferried by vessels) helps to explain dispersion patterns for termite species55,64,65, it is more likely that the paths taken by the populations of N. ephratae were overland, which makes the PAC dominion an obligatory passage for the dispersal of populations between South and Central America. This dominion harbor peculiarly haplotypes from all haplogroups and present a higher value of intra-dominion genetic distance (Table 2b). These features help to infer the PAC intermediary position for the N. ephratae dispersions.
The dispersal route traced for the populations analyzed here are very similar to the dispersal route observed for N. corniger species15 including the dispersion from Central to South America, the eastward dispersion on South America lands, and the late occupation of Atlantic Forest. Nevertheless, the dispersal from South to Central America and the temporary split among the population have not yet been detected for N. corniger unlike N. ephratae. However, it is important to note that only the 16S mitochondrial marker was used for the N. corniger analyses15. The addition of other markers can lead to a more robust comparison among the population patterns of the two species.
Crews and Esposito66 also studied dispersal routes and identified that South America is most probably the origin of most of the Caribbean arthropod fauna including N. ephratae, N. corniger, and other species of the genus Nasutitermes. These data contradict the inferences raised by Santos et al.15 although there are some important methodological differences among the studies. Crews and Esposito66 analyzed 18 species of Nasutitermes using the same mtDNA genes used here as molecular markers (16S + COII). The dispersal route proposed herein is according to the inferences made by Crews and Esposito66 because the N. ephratae Antillean populations arose from a South America ancestor according to the ancestral area reconstruction. Specifically, for these island populations, it is possible that the dispersion from northern South America to the Antilles occurred via flotsam or floating wood carried to the islands by oceanic currents. This kind of dispersion has already been detected for Caribbean termite species65. Most of the islands’ termite species analyzed here are compose of haplogroup 3 together with samples from French Guiana (BOR dominion) and Trinidad and Tobago (located in the transition between BOR and PAC). This data suggests high genetic similarity among populations of these areas—although a larger sampling can better clarify the relations among the two dominions.
Regarding the BOR dominion, we found that the samples from this area have haplotypes typically found in other areas, which stayed distant from each other in the haplotype network (except for the haplotypes from French Guiana). This distance was also observed in BI. Some authors argue that the Amazonian biota (composed of parts of BOR and SOU dominions and the southeastern Amazon dominion—the latter was not sampled herein) have unnatural biogeographic origins67,68,69,70,71. As a speculation, this hypothesis could help clarify the existence of haplotypes that are genetically distant from each other in Amazonian localities that are geographically close to each other. However, we point that it is necessary to include a greater sampling of this region to clearly detect the genetic population patterns and the evolutionary events involving N. ephratae in this dominion.
In general, there are many questions to be explored about the phylogeographic processes of the South American and neotropical species especially for termites whose studies are still new. Better sampling of N. ephratae and/or including new molecular markers in future studies, as well as addressing phylogeographic issues of other species, can help solidify the inferences made in this work and can expand the understanding of the evolutionary history of the group and of the neotropics.
Conclusions
This study considered most area where N. ephratae occurs in the neotropics. It was possible to make important inferences about the general panorama of the evolutionary history of the species in this region although a broader sampling, especially from central-eastern South America, could better clarify some phylogeographic patterns. Our data also showed similarities on the population and dispersal patterns among N. ephratae and N. corniger. Here, it is possible to speculate that both species responded similarly to the biogeographic processes that have occurred in the neotropics although new comparative studies may better answer this question. In summary, this study offers important contributions to the understanding of biogeographic and phylogeographic issues in the neotropics especially evolutionary studies of termites and other insects.
Data availability
DNA sequences: Genbank accessions numbers OL830473–OL830583 (16S) and OL830584–OL830683 (COII).
References
Carvalho, C. J. B. & Couri, M. S. Biogeografia de Muscidae (Insecta: Isoptera) da América do Sul. In Biogeografia da América do Sul: padrões & processos (eds Carvalho, C. J. B. & Almeida, E. A. B.) 277–298 (Roca, 2010).
Constantino, R. On-line termite database http://164.41.140.9/catal/ (2020).
Eisner, T., Kriston, I. & Aneshansley, D. J. Defensive behavior of a termite (Nasutitermes exitiosus). Behav. Ecol. Sociobiol. 1, 83–125 (1976).
Holmgren, N. Versuch einer Monographie der amerikanische Eutermes-Arten. Jahrb. Hamburg. Wiss. Anst. 27, 235–243 (1910).
Banks, N. The termites of Panama and British Guiana. Bull. Am. Mus. Nat. Hist. 38, 659–667 (1918).
Snyder, T. E. Catalog of the termites (Isoptera) of the world. Smithsonian Misc. Collect. 2, 1–490 (1949).
Thorne, B. L. Differences in nest architecture between the neotropical arboreal termites Nasutitermes corniger and Nasutitermes ephratae (Isoptera: Termitidae). Psyche 87, 235–244 (1980).
Miura, T., Roisin, Y. & Matsumoto, T. Molecular phylogeny and biogeography of nasute termite genus Nasutitermes (Isoptera: Termitidae) in the pacific tropics. Mol. Phylogenet. Evol. 17, 1–10 (2000).
Scheffrahn, R. H., Krecek, J., Szalanski, A. L. & Austin, J. W. Synonymy of neotropical arboreal termites Nasutitermes corniger and N. costalis (Isoptera: Termitidae: Nasutitermitinae), with evidence from morphology, genetics, and biogeography. Ann. Entomol. Soc. Am. 98, 273–281 (2005).
Scheffrahn, R. H., Krecek, J., Szalanski, A. L., Austin, J. W. & Roisin, Y. Synonymy of two arboreal termites (Isoptera: Termitidae: Nasutitermitinae): Nasutitermes corniger from the neotropics and N. polygynus from New Guinea. Fla. Entomol. 88, 28–33 (2005).
Roy, V. et al. Species delimitation and phylogeny in the genus Nasutitermes (Termitidae: Nasutitermitinae) in French Guiana. Mol. Ecol. 23, 902–920 (2014).
Snyder, T. E. Termites collected on the Mulford biological exploration to the Amazon Basin, 1921–1922. Proc. U. S. Natl. Mus. 68, 1–76 (1926).
Araujo, R. L. Catálogo dos Isoptera do novo mundo (ed. Araujo, R. L.) 1–92 (Academia Brasileira de Ciências, 1977).
Torales, G. J. & Armua, A. C. Contribución al conocimiento de las termites de Argentina (Provincia de Corrientes). Nasutitermes corniger (Isoptera: Termitidae). Primera Parte. Facena. 6, 203–222 (1986).
Santos, A. F., Carrijo, T. F., Cancello, E. M. & Morales, A. C. Phylogeography of Nasutitermes corniger (Isoptera: Termitidae) in the Neotropical Region. BMC Evol. Biol. 17, 1–12 (2017).
Mendoza-Ramírez, M., Gutiérrez-Rodríguez, J., Poteaux, C., Ornelas-García, P. & Zaldívar-Riverón, A. Late Pleistocene genetic diversification and demographic expansion in the widely distributed neotropical ant Neoponera villosa (Ponerinae). Mitochondrial DNA part A. 30, 296–306 (2019).
Li, H. F., Ye, W., Su, N. Y. & Kanzaki, N. Phylogeography of Coptotermes gestroi and Coptotermes formosanus (Isoptera: Rhinotermitidae) in Taiwan. Ann. Entomol. Soc. Am. 102, 684–693 (2009).
Jenkins, T. M. et al. Phylogeography illuminates maternal origins of exotic Coptotermes gestroi (Isoptera: Rhinotermitidae). Mol. Phylogenet. Evol. 42, 612–621 (2007).
Austin, J. W., Szalanski, A. L., McKern, J. A. & Gold, R. E. Molecular phylogeography of the subterranean termite Reticulitermes tibialis (Isoptera: Rhinotermitidae). J. Agric. Urban Entomol. 25, 63–79 (2008).
Kutnik, M., Uva, P., Brinkworth, L. & Bagnères, A. G. Phylogeography of two European Reticulitermes (Isoptera) species: The Iberian refugium. Mol. Ecol. 13, 3099–3113 (2004).
Tripodi, A. D. et al. Phylogeography of Reticulitermes Termites (Isoptera: Rhinotermitidae) in California inferred from mitochondrial DNA sequences. Ann. Entomol. Soc. Am. 99, 697–706 (2006).
Park, Y. C., Kitade, O., Schwarz, M., Kim, J. P. & Kim, W. Intraspecific molecular phylogeny, genetic variation and phylogeography of Reticulitermes speratus (Isoptera: Rhinotermitidae). Mol. Cells. 21, 89–103 (2006).
Bourguigon, T. et al. Towards a revision of the Neotropical soldierless termites (Isoptera:Termitidae): Redescription of the genus Anoplotermes and description of Longustitermes, gen. nov. Invertebr. Syst. 24, 357–370 (2010).
Morrone, J. J. Biogeographical regionalisation of the Neotropical region. Zootaxa 3782, 1–110 (2014).
Liu, H. & Beckenbach, A. T. Evolution of the mitochondrial cytochrome oxidase II gene among 10 orders of insects. Mol. Phylogenet. Evol. 1, 41–52 (1992).
Kambhampati, S. A phylogeny of cockroaches and related insects based on DNA sequence of mitochondrial ribosomal RNA genes. Proc. Natl. Acad. Sci. USA. 92, 2017–2020 (1995).
Xiong, B. & Kocher, T. D. Comparison of mitochondrial DNA sequences of seven morphospecies of black flies (Diptera: Simuliidae). Genome 34, 306–311 (1991).
Simon, C. et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Kuraku, S., Zmasek, C. M., Nishimura, O. & Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 41, 22–28. https://doi.org/10.1093/nar/gkt389 (2013).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166 (2017).
Rozas, J. et al. DnaSP v6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Excoffier, L. & Lischer, H. E. L. Arlequin suite v. 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Achaz, G. Testing for neutrality in samples with sequencing errors. Genetics 179, 1409–1424 (2008).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Clement, M., Posada, D. & Crandall, K. A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Santos, A. M., Cabezas, M. P., Tavares, A. I., Xavier, R. & Branco, M. tcsBU: A tool to extend TCS network layout and visualization. Bioinformatics 32, 627–628 (2016).
QGIS Development Team. QGIS Geographic Information System http://qgis.org (2009).
Löwenberg-Neto, P. Neotropical region: A shapefile of Morrone’s (2014) biogeographical regionalization. Zootaxa 3802, 300–300 (2014).
Wright, S. Variability within and among natural populations. In Evolution and the Genetics of Populations Vol. 4 (ed. Wright, S.) 1–590 (University of Chicago Press, 1978).
Cheng, L., Connor, T. R., Sirén, J., Aanensen, D. M. & Corander, J. Hierarchical and spatially explicit clustering of DNA sequences with BAPS software. Mol. Biol. Evol. 30, 1224–1228 (2013).
Tonkin-Hill, G., Lees, J. A., Bentley, S. D., Frost, S. D. & Corander, J. RhierBAPS: An R implementation of the population clustering algorithm hierBAPS. Wellcome Open Res. 3, 1–9 (2018).
R Core Team. R: A Language and Environment for Statistical Computing https://www.R-project.org/ (R Foundation for Statistical Computing, 2020).
Oksanen, F. J. et al. vegan: Community Ecology Package. R package version 2.5-7, https://CRAN.R-project.org/package=vegan (2020).
Bouckaert, R. et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650. https://doi.org/10.1371/journal.pcbi.1006650 (2019).
Heath, T. A., Huelsenbeck, J. P. & Stadler, T. The fossilized birth–death process for coherent calibration of divergence-time estimates. PNAS 111, E2957–E2966 (2014).
Tamura, K. & Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526 (1993).
Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008).
Jarzembowski, E. A. An early Cretaceous termite from southern England (Isoptera: Hodotermitidae). Syst. Entomol. 6, 91–96 (1981).
Engel, M. S. & Grimaldi, D. A. The termites of Early Eocene Cambay amber, with the earliest record of the Termitidae (Isoptera). ZooKeys. 148, 105–123 (2011).
Krishna, K. New fossil species of termites of the subfamily Nasutitermitinae from Dominican and Mexican amber (Isoptera, Termitidae). Am. Mus. Novit. 3176, 1–13 (1996).
Krishna, K. & Grimaldi, D. Diverse Rhinotermitidae and Termitidae (Isoptera) in Dominican amber. Am. Mus. Novit. 3640, 1–48 (2009).
Bourguigon, T. et al. Mitochondrial phylogenomics resolves the global spread of higher termites, ecosystem engineers of the tropics. Mol. Biol. Evol. 34, 589–597 (2016).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Rambaut, A. & Drummond, A. J. TreeAnnotator v. 2.6.3. http://beast.bio.ed.ac.uk (2020).
Rambaut, A. FigTree: Tree Figure Drawing Tool. v. 1.4.4. http://tree.bio.ed.ac.uk (2018).
Matzke, N. J. BioGeoBEARS: BioGeography with Bayesian (and Likelihood) Evolutionary Analysis in R Scripts. R package version 0.2.1, http://CRAN.R-project.org/package=BioGeoBEARS (2013).
Ramos-Onsins, S. E. & Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 19, 2092–2100 (2002).
Seal, J. N., Kellner, K., Trindl, A. & Heinze, J. Phylogeography of the parthenogenic ant Platythyrea punctata: Highly successful colonization of the West Indies by a poor disperser. J. Biogeogr. 38, 868–882 (2011).
Turchetto-Zolet, A. C., Pinheiro, F., Salgueiro, F. & Palma-Silva, C. Phylogeographical patterns shed light on evolutionary process in South America. Mol. Ecol. 22, 1193–1213 (2013).
Mann, P., Peterson, L. & Droxler, A. Tectonics, Circulation, and Climate in the Caribbean Gateway (eds Mann, P. et al.) 1–100 (Joint Oceanographic Institutions, Inc., 2006).
Scheffrahn, R. H., Křeček, J., Chase, J. A., Maharajh, B. & Mangold, J. R. Taxonomy, biogeography, and notes on termites (Isoptera: Kalotermitidae, Rhinotermitidae, Termitidae) of the Bahamas and Turks and Caicos Islands. Ann. Entomol. Soc. Am. 99, 463–486 (2006).
Janowiecki, M. A., Scheffrahn, R. H., Austin, J. W. & Szalanski, A. L. Population structure of the drywood termite Incisitermes schwarzi (Blattodea: Kalotermitidae) in the Caribbean. J. Agric. Urban Entomol. 36, 101–108 (2020).
Crews, S. C. & Esposito, L. A. Towards a synthesis of the Caribbean biogeography of terrestrial arthropods. BMC Evol. Biol. 20, 1–27 (2020).
Prum, R. O. Historical relationships among avian forest areas of endemism in the Neotropics. Acta Ornithol. 19, 2562–2572 (1988).
Dos Amorim, D. S. Amazonias. In Introducción a la biogeografia em Latino América: teorías, conceptos, métodos y aplicaciones (eds Llorente-Bousquets, J. & Morrone, J. J.) 245–255 (UNAM, 2001).
Cracraft, J. & Prum, R. O. Patterns and processes of diversification: Speciation and historical congruence in some Neotropical birds. Evolution 42, 603–620 (1988).
Bates, J. M. Avian diversification in Amazonia: evidence for historical complexity and a vicariance model for a basic diversification pattern. Diversidade biológica e cultural da Amazônia. 119–137 (2001).
Ribas, C. C., Aleixo, A., Nogueira, A. C., Miyaki, C. Y. & Cracraft, J. A palaeobiogeographic model for biotic diversification within Amazonia over the past three million years. Proc. R. Soc. Lond. B. 279, 681–689 (2012).
Acknowledgements
We thank Prof. Dr. Rudolf Scheffrahn (University of Florida) and the Isoptera Collection team of MZUSP for assistance in obtaining samples; the Laboratory of Genetic of Bacteria (FCAV/UNESP) for providing equipment; and CAPES for the scholarship granted to AFS.
Author information
Authors and Affiliations
Contributions
A.F.S. performed the laboratory procedures. A.F.S. and A.C.M. generated, analyzed, and interpreted the results. E.M.C. coordinated the obtention of the samples. All the authors contributed, read, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
de Faria Santos, A., Cancello, E.M. & Morales, A.C. Phylogeography of Nasutitermes ephratae (Termitidae: Nasutitermitinae) in neotropical region. Sci Rep 12, 11656 (2022). https://doi.org/10.1038/s41598-022-15407-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-15407-z
- Springer Nature Limited