
Abstract
The invasion success of a species in an agrosystem is greatly influenced by environmental factors such as the use of insecticides, by the intrinsic evolutionary capabilities of the species, and also by interactions with resident species. On the island of La Réunion, the successive invasions of MEAM1 and MED whitefly species over the last 20 years have not only led an increased use of insecticides, but have also challenged the resident IO species. To trace the evolution of the 3 species, and the distribution of the kdr mutation (resistance to pyrethroid) in the para-type voltage-gated sodium channel, we genotyped 41 populations (using neutral nuclear markers) and look at the prevalence of the kdr allele. MEAM1 was predominantly present in agrosystems showing quasi fixation of the resistant kdr allele whereas IO was mainly in natural environments and did not have any resistant allele. Hybridization between the two former species was detected in low frequency but has not led to introgression of resistant alleles in the resident species so far. MED showed a limited distribution in agrosystems but all individuals displayed a resistant allele. These highly contrasting patterns of distribution and resistant mutations between invasive and resident whitefly species are further discussed.
Similar content being viewed by others

Introduction
Biological invasions, which could be defined as the successful establishment and spread of species outside their native range1, are one of the world’s costliest ecological concerns2. They are partly responsible for the loss of biodiversity, by altering ecosystems and natural habitats3, disrupting agriculture and posing multiple threats to native species (through competition, predation, hybridization or even introduction of parasites and diseases4,5,6,7). Not all introduced species settle permanently in a new environment, they must be able to pass through a few critical steps: transport or emigration, introduction, establishment, expansion and proliferation8,9. In each of these different steps, evolutionary mechanisms may play an important role10.
During biological invasion, previously isolated species or populations can be brought into contact and hybridize. Hybridization can generate new evolutionary solutions, through the pooling of differentiated genetic backgrounds and the increase of genetic variation. Due to the creation of novel genotypes, hybrids may exhibit more extreme transgressive phenotypes (compared to their parents), and may display an enhanced fitness5,11. Admixture offers many benefits to invaders, suggesting that hybridization could play an important role in biological invasions12. Hybridization can also alter the effectiveness of control strategies toward agricultural pests.
As they arrive in a new environment, invasive species have to face novel selection pressures, biotic as well as abiotic. Invasion success in agroecosystems may be constrained by insecticide treatments. In fact, insecticide use is a major selective force driving the evolution of insect pests and it may also increase the rate of mutations via epigenetic processes13. For example, one possible reason for the spread of thrips Frankliniella occidentalis is that intensive insecticide use in horticulture have selected an insecticide resistant strain, allowing its establishment in glasshouses across North America, and later to Europe, Asia, Africa and Australia14. Piiroinen, et al.15 have demonstrated that the phenotypic traits of insect pests that allow them to thrive under insecticide exposure, may also facilitate global invasions. For instance, sublethal pyrethroid insecticide exposure were found to have transgenerational positive effects on fitness-related traits in the Colorado potato beetle Leptinotarsa decemlineata16. Although many insect pest populations have shown resistance to pyrethroids, it remains one of the most commonly used insecticide class because it shows low risk to mammals17,18. A well-known and major pyrethroid resistance mechanism in insects, commonly referred to as knockdown resistance or ‘kdr’, involved a mutation on the pyrethroids’ target, the voltage-gate sodium channel gene19,20,21,22. Thus, monitoring the spread of kdr mutant alleles among pest populations in the field is considered to be an accurate method to assess the extent of pyrethroid resistance problems.
Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) is a highly adaptable insect pest, distributed throughout tropical and subtropical regions worldwide, and responsible for heavy crop losses23,24. Because of its ability to transmit more than 200 phytoviruses to an impressive range of host plants, it is threatening food security around the world25. B. tabaci is considered to be a cryptic species complex with at least 40 morphologically indistinguishable species23,26,27,28. Two of these cryptic species are invasive worldwide: the Middle East Asia Minor 1 species (MEAM1) and the Mediterranean species (MED), formerly known as B and Q biotypes, respectively23,29.
The MEAM1 whitefly species (originating from the Middle East-Asia Minor region) was responsible of a first worldwide invasion, observed in the late 1980s24,30. It has been followed in early 2000s by the MED species, which has spread globally from the countries bordering the Mediterranean basin23,31. Both species have evolved resistance to insecticides from most chemical classes32, but MED was reported to have a larger spectrum of resistance than MEAM1 to insecticides such as pyriproxyfen and neonicotinoids33. This difference between resistance statuses may have played a role in the displacement of the MEAM1 species by the MED species in some countries34.
In the island of La Réunion, three species of this whitefly complex have been described. The indigenous species Indian Ocean (IO), coexisting with two invasive species: MEAM1 and MED. The arrival of the MEAM1 species was dated to the late 90’s, together with the first description of the Tomato yellow leaf curl virus (TYLCV, genus: Begomovirus, family: Geminiviridae)35,36, that implied high yield losses on tomato crops, and the use of phytosanitary control measures for this pest37. Two studies based on the analysis of the genetic diversity of the whitefly populations have described the occurrence of interspecific hybridization between both MEAM1 and IO species38,39. Then in 2010, the MED species was first detected on the island40. Therefore, to further explore the interactions between those resident and invasive species in agrosystems, subjected to high insecticide pressure, we studied (i) the evolution of the distribution of the three species since their first description on the island, (ii) their genetic diversity and structure, and (iii) the evolution of the interspecific hybridization, in relation to the distribution of the kdr alleles conferring resistance to pyrethroids.
Results
Species identification and distribution
1562 adult whiteflies sampled in 41 different sites around the island were genotyped (Fig. 1). Bayesian clustering analyses performed with STRUCTURE and the DAPC allowed the clear determination of three main genetic groups in our dataset (Fig. 2). The sequencing of the COI barcoding region of 286 individuals from each group identified these groups to the three species already present in La Réunion: MEAM1, IO and MED. All sequences had 100% identity with accessions already published on La Réunion: no. AJ550175 for MEAM1, no. AJ877264 for IO, no. JN090173 for MED (Q1).

Global repartition of B. tabaci collection sites in and around market gardening areas of La Réunion. Each site is named as referred to in Table 1.

DAPC based on n = 1562 individuals using 11 microsatellite loci. Dots of different colours indicate individuals from different genetic clusters. PCs eigenvalues and discriminant factors retained are indicated.
The whole dataset comprises 61.1% of MEAM1, 33.5% of IO and 3.5% of MED, respectively (Table 1). In the sampled sites, MEAM1 was mainly found in greenhouses, open fields and field surroundings. The IO species was found mostly in non-cultivated areas and in field surroundings. Finally, the MED species was a minority present in 4 sites, only in the cultivated areas of the south-west of La Réunion.
Genetic diversity
No linkage disequilibrium was observed between all pairs of loci tested within each species, and between each pair of populations of those species. The average allelic richness was very similar between the three species, with in average 2.4 ± 0.03 (SE) for MEAM1, 2.5 ± 0.03 for IO and 2.3 ± 0.06 for MED, respectively. A higher level of heterozygosity was observed for IO (in average over all populations: 40.3% ± 0.7%) than for the two other species (29.7% ± 0.6% for MEAM1 and 30.3% ± 3.3% for MED). The FIS values ranged from -0.05 to 0.25 for MEAM1, from -0.76 to 0.096 for IO and 0.005 to 0.354 for MED, respectively. Significant departures from Hardy–Weinberg equilibrium were observed for 12 populations (out of 34) of the MEAM1, for 4 populations (out of 21) of the IO, and for 2 populations (out of 3) of the MED (Table 2).
The analysis of bottleneck performed with the SMM model did not reveal any significant signal on IO populations, whereas almost all MEAM1 populations had undergone a significant bottleneck (P < 0.05). On the contrary, the IAM model indicated no bottleneck for both species.
Population structure
Within species, the genetic differentiation between populations was low (overall FST for IO = 0.007, MEAM1 FST = 0.03, and MED FST = 0.07). In MEAM1, no significant genetic differentiation was found between different environments (greenhouse, open field or field surroundings, see Table 1) within sites; however, several pairwise genetic differentiations between distant sites were significant (see Supplementary Table S1 online). Nevertheless, no substructure was observed between groups of individuals of this species. No indication of isolation by distance for the MEAM1 species was found (Mantel tests p-value > 0.05).
Similar results were observed for IO populations, showing no substructure between groups of individuals or looking at FST genetic distances between different environments (see Supplementary Table S2 online). Indeed, only a single pairwise genetic distance was found to be significantly different (site 16 with site 41, two sites that were far apart: east and west coast of the island, respectively; Table 1, Fig. 1).
The MED species was the only species for which a substructure was observed with the Bayesian analysis (Fig. 3), with the best number of clusters of three. However, those clusters did not reflect differentiation between sampled sites or other obvious environmental constraints. It has to be noticed that this result involves a very small number of samples (n = 50), spread over four sites, close to each other (Fig. 1).

Genetic assignment (Bayesian assignment to K = 3 clusters, purple, pink and green bars) and kdr genotype at the loci L1 and L2 (green for SS, orange for RS and red for RR) for MED individuals sampled in greenhouse, open field and field surroundings. Missing data are indicated by white bars. Numbers on the X-axis (9, 11, 15 and 17) correspond to sampling sites as referred to in Table 1.
Hybrids
Individuals were considered hybrid when their Bayesian assignment (Fig. 4) to MEAM1 or IO clusters was between 10 and 90%, they were also well separated on the X-axis of the DAPC analysis (Fig. 2). Accordingly, there were 29 hybrids, which represents only 1.9% of the whole dataset. We found as many interspecific mating with female IO or with female MEAM1 (13 versus 16). The distribution of assignment probabilities indicate that most hybrids derived from backcrosses, mostly toward the MEAM1 species. Hybrids were found in all environments. However, assignment probabilities of hybrids were significantly linked with the sampled environment: individuals derived from backcrosses with MEAM1 (respectively IO) were preferentially found in agrosystems (respectively non-cultivated areas; χ2 = 8.28, p-value = 0.040) (see Supplementary Fig. S1 online).

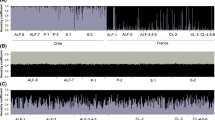
Genetic assignment to the MEAM1 (orange) and the IO (blue) genetic clusters, identification of hybrids (pink bars) and kdr genotype at the L1 locus (green for SS, orange for RS and red for RR) of individuals sampled in greenhouse, open field and field surroundings. Missing data are indicated by white bars. Individuals are ordered by their probability of assignment to the clusters. Numbers on the X-axis correspond to sampling sites as referred to in Table 1.
Kdr mutation
The region of the sodium channel gene carrying the kdr mutation was successfully genotyped for 98% (n = 1537) of the sampled individuals (Fig. 5). The L925I resistance mutation (here referred to as L1) was detected in different frequencies within species: 0.99 for MEAM1, 0.62 for MEAM1-IO hybrids, 0.60 for MED and 0 for IO.

Resistance genotypes of B. tabaci species collected in La Réunion, at a point mutation in the IIS4-5 linker of para-type voltage gated sodium channel gene: L925I. I925 and L925 are the resistant (R) and the susceptible (S) alleles, respectively.
Individuals of the MEAM1 species were overwhelmingly (99%) homozygous resistant (RR), whereas all individuals of the IO species were homozygous susceptible (SS) at this locus (Figs. 3, 5). Those results were confirmed by sequencing the kdr region for a fraction of individuals (L1, Table 3).
MED individuals showed a combination of RR (~ 36%), RS (~ 49%) and SS (~ 15%) at this L1 locus (Figs. 3, 5), and displayed a mutation conferring resistance to pyrethroids at a second locus (T929V; frequency = 0.41), here referred to as L241 (Table 3). All MED individuals sequenced carried at least one resistance allele (Fig. 3). This mutation at the L2 locus was also observed in one MEAM1 individual (frequency = 0.01; n = 58), but was totally absent in IO as in MEAM1-IO hybrids (Table 3).
Another mutation was detected in the intron region right after the 5’ end of all sequences of IO and MED species (locus L3, Table 3). In MEAM1, only 9 over the 58 sequenced individuals carried this mutation among which 5 were heterozygous both at this locus and at the L1 locus and 4 were homozygous at this locus and SS at the L1 locus.
For the hybrids, the kdr genotype was linked with assignment probability to parental species (χ2 = 18.616, p value = 9.069e−05). Hybrid individuals backcrossed to IO were all homozygous susceptible (SS), whereas two individuals backcrossed towards MEAM1 were heterozygotes (RS). The mutation at the L3 locus was also detected in 11 out of the 29 hybrids (Table 3), most of which (9/11) were backcrossed towards IO.
To summarize, if we consider both resistance mutations (L1 and L2) when analyzing the genotyping results presented in Fig. 5 and Table 3: IO appeared 100% susceptible, as we did not detect any resistance allele at any locus; MED appeared 100% resistant, as each individual carried at least one resistance allele at one locus (L1, L2 or both). For MEAM1, we detected 99% of resistant individuals, as they carried at least one resistance mutation at one locus (L1, L2 or both). Concerning MEAM1-IO hybrids, 65% of individuals seemed to be resistant as they carried at least one resistance mutation (at L1).
Discussion
The B. tabaci complex species in La Réunion is still composed of three species: MEAM1, IO and MED. The three species, which coexist on this relatively small island, display contrasting patterns in terms of ecological niches occupancy, genetic structure and diversity, and resistant genotypes in response to anthropic pressures.
Our study clearly indicates that the resident indigenous species IO was mainly associated with non-cultivated environments or field surroundings, whereas the two invasive species were almost restricted to agrosystems. Those ecological preferences were expected according to previous studies conducted in La Réunion, where MEAM1 was dominant on vegetable crops whereas IO was found on weeds38,40,42. In addition to host plant preferences, non-agricultural environments or wild environments are more likely to be non-pesticide environments.
MEAM1 species, an invader of this insular ecosystem, was first reported in 1997 in the southern part of the island, and in a very short period of time (3 years) it has colonized the entire tomato growing area of the west coast of the island. In 2003, MEAM1 was found in the whole fringe around the island (0–600 m above sea level), including the eastern part. Nevertheless, despite its resident status of over 20 years on this island, it seems that this species has not yet reached an equilibrium state, with lower heterozygosity compared to the indigenous species, genetic differentiation between distant sites (but without any IBD), and still detection of signals of bottlenecks. However, the absence of nuclear substructure within MEAM1 species, moderate genetic diversity and low COI haplotype diversity (no new haplotype found), are not in favor of a new recent invasion or multiple invasions of this species on the island, but might be the signal of a founder effect on the initial invasive population. On the contrary, IO was found to have a higher level of heterozygosity and higher diversity compared to both invasive species, with most of its sampled populations being at the equilibrium of Hardy–Weinberg. In addition, no bottleneck was found associated to any populations of this species, and low levels of genetic differentiation even between distant populations are in agreement with an indigenous, well-established species.
Opposite to the evolution of MEAM1, there is no demographic or geographic expansion of the worldwide invasive MED species. MED was first detected in 2010 in La Réunion40, on eggplant in Saint-Pierre (south part of the island). Since then, this species has only been found in four very close localities of the southern part of the island (mostly on crops), less than 30 km from the epicenter of the invasion. MED is expected to have higher competitive abilities compared to MEAM1, as indicated by the frequent replacement of MEAM1 by MED observed in several countries: in China, Japan, South Korea or Spain31,43,44,45,46,47. This replacement can even be fairly rapid as in China or in Florida, MED replaced MEAM1 in less than 5 years48,49. It was hypothesized that the higher competitive ability of MED compared to MEAM1 was linked to a higher insecticide resistance in the former species33,46,50. It appears that MED has not expanded its range during the last 8 years in La Réunion, and did not displaced MEAM1 in agricultural areas. Genotyping and sequencing of the kdr mutation indicates that the MED species in La Réunion is as well armed against pyrethroids as MEAM1, but this species may lack other resistance mechanisms in those particular populations. Unfortunately, because of the low occurrence of the MED species in La Réunion, insecticide resistance could not be assessed using bioassays so far51.
Finally, we found a genetic structure within the MED species. This substructure, which does not match with any factors explored here, might also reflect a potential signal of multiple invasion events for this species, as suggested in early studies using either microsatellites markers40, or SNP data over the whole genome52. However, the limited number of MED individuals sampled prevents from further analyses on this aspect.
We found around 2% of hybrids between MEAM1 and IO, the two major species found in La Réunion, in our dataset. This confirms that the reproductive isolation between the two species is incomplete38,53. A few individuals were found to be first generation hybrids, but most were found to result from multiple generations of backcrossing between the two species. Hybrids from backcrosses were frequently assigned more strongly to the MEAM1 species, which may be explained by the predominance of this species in the sampled sites. Hybrids between those two species have been described in La Réunion since 2006. In the early invasion process of MEAM1 (2001–2002) on the island, higher rate of hybridization was found, reaching up to 38%38, then it decreased over years reaching around 11% in 200653. This decrease in the frequency of hybrids may either be due to the sampled environments: a larger diversity of environments was studied in 2001–2006 in both studies. Another hypothesis could be that the MEAM1 species is progressively genetically overwhelming the IO species in the agricultural niche, because of its greater fitness in this environment. In La Réunion, these two species have been shown recently to radically differ in terms of insecticide resistance, the MEAM1 species being highly resistant to two heavily used insecticides, whereas the IO species seems to remain susceptible to both insecticides51.
All the MED individuals in La Réunion displayed either one or the two mutations conferring the kdr resistance to pyrethroids, with 26 individuals carrying both mutations. Such a high level of allele frequencies has never been reported, at least never to that extent54. This result might be explained by (i) a low number of samples; (ii) the fact that MED populations were found in agrosystems and might have been selected due to insecticide pressure. In contrast, the second mutation (L2) was never found in our sampled MEAM1 individuals as expected55, except from one individual. Further investigation should be conducted to assess the occurrence of this mutation (hybridization between both species, high selection pressure on those populations leading to this mutation…).
Our study also reveals a contrasted pattern in the frequency of the first kdr mutation (L1) between the invasive MEAM1 and the resident IO species. The L1 mutation has been found to be nearly fixed in La Réunion MEAM1 populations, and fully absent from the IO populations. In Cyprus, several MEAM1 populations displayed high frequency of this allele and also showed an association between the frequency of the resistant allele and bifenthrin (a pyrethroid insecticide) resistance56. We have not formally tested the association between genotypic and phenotypic resistance to pyrethroids. However, we estimated the LC50 associated with deltamethrin for several MEAM1 populations and found that it was very high, which is in agreement with the high frequency of resistant allele (> 150 mg.L−1 of Decis Protech®). Recently, Gnankiné, et al.57 showed that in less than 10 years this mutation (initially present in 2% of the population) reached complete fixation for all MED (MED Q and MED ASL) populations in Burkina Faso. Such a surge in a resistance allele frequency would not take place without a high selection pressure linked to a heavy use of pyrethroid insecticides. We do not know if the same phenomenon occurred for MEAM1 in La Réunion where pyrethroid are commonly used, but it can be hypothesized. Another explanation would be that the mutation was already fixed in the MEAM1 individuals which invaded La Réunion in the late 90’s. Indeed, resistance to pyrethroids was described in whiteflies as early as 199558.
In contrast to MEAM1, the kdr mutation was never found in the IO populations, which implies either that it was not subjected to the same insecticide pressure as MEAM1 (i.e., IO is mostly found out of agrosystems38,42), or that it was not able to evolve resistance due to molecular constraints59. We hypothesized that the hybridization between MEAM1 and IO could have allowed the introgression of resistant alleles from the invasive species to the indigenous one. However, we found no indication of introgression of any kdr allele from MEAM1 to IO, but rather a potential introgression of the susceptible allele from IO to MEAM1. Furthermore, the mutation (L3) found in the exon, always associated with IO genotypes and in a few hybrid genotypes, can also be considered as an additional marker of introgression from IO to MEAM1. The susceptible allele is present at low frequency in the MEAM1 species. This further suggests that the initial invasive MEAM1 populations came with the resistant allele in the homozygous state, whereas this susceptible allele might originate from the IO species. Thus, the hybridization between MEAM1 and IO could explain the maintenance of a low frequency of susceptible alleles in MEAM1, despite a probable strong counter-selection of these alleles in the homozygous state. The level of dominance of kdr alleles is not known in B. tabaci, but a codominance between resistant and susceptible alleles has been described in other system, for example in a Lepidoptera species60. Heterozygotes would therefore have a better fitness than susceptible homozygotes when treated with insecticide, and would thus be less counter-selected.
The lack of introgression of kdr alleles in the other direction from MEAM1 to IO may seem surprising. One hypothesis would be that this resistant allele has a higher fitness cost for IO than for MEAM1. This could be explained if MEAM1 has a reduced cost of resistance according to the selection of a modifier gene elsewhere in the genome. In this case, IO needs to acquire both the resistant allele and the modifier gene to avoid counter-selection in absence of insecticide treatment. This would decrease the probability of introgression of the resistant allele in IO genome. The low number of hybrids leads to caution regarding the conclusions that can be drawn, however, we did not find any indication of a role of hybridization in the evolution of resistance in whitefly in La Réunion.
Following the two invasion events, the whitefly species distribution has not drastically evolved in the past years, and each species seems to be contained in its initial ecological niche. The initial hypothesis of indigenous species not being equally armed regarding to insecticide resistance, facing both invasive MEAM1 and MED species, is validated. The potential insecticide pressure of MED and MEAM1 can be analyzed and discussed in conjunction with more detailed local pesticide practices. Very high levels of resistance for both invasive species were found together with generally high gene flow between whitefly populations of the broadly spread MEAM1 species. As a conclusion, the non-spread of the invasive MED in the insular environment cannot only be explained by differential levels of pyrethroid resistance, but other insecticide resistance or factors must be in play, in particular ecological or environmental factors.
Methods
Bemisia tabaci collection and DNA extraction
La Réunion is a French subtropical island, with a high-altitudinal gradient (from 0 to 3071 m asl), located in the southwest Indian Ocean at 700 km east of Madagascar. Adult whiteflies were sampled in 41 different sites of this island, from April 2016 to December 2017 (Table 1 and Fig. 1). Sampling was mostly done in agricultural areas, all over the island, along the coastal strip from 0 to 500 m asl, where market gardening production is more important. In some sites, different environments were sampled: greenhouse, open field, field surroundings or non-cultivated area.
At each collection site, GPS coordinates were recorded and about 50 individuals were sampled at random on crops or weeds, by vacuuming plant foliage with a mouth aspirator. According to the research method, it was not possible to determine whether there is mixed population on a single plant. Cultivated host plants included tomato Solanum lycopersicum L. (Solanaceae), eggplant Solanum melongena L. (Solanaceae), cucumber Cucumis sativus L. (Cucurbitaceae), and melon Cucumis melo L. (Cucurbitaceae); weeds included Mexican fireplant Euphorbia heterophylla L. (Euphorbiaceae), bean Vigna sp. L. (Fabaceae), lantana Lantana camara L. (Verbenaceae), pricklyburr Datura innoxia Mill. (Solanaceae), and turpeth Operculina turpethum L. (Convolvulaceae). The study complies with local and national guidelines on field studies on cultivated and wild plants.
Adult whiteflies were conserved in tubes containing 95% ethanol, and held at −20 °C in the laboratory until DNA extraction. Collected adult whiteflies were sexed under a Leica MZ6 stereomicroscope61. Indeed, only adult females were used for the population genetic analyses due to the haplo-diploid status of the species. A non-destructive DNA extraction method was used as described in Tocko-Marabena, et al.62. Then, extractions were conserved at −30 °C until further use.
Mitochondrial DNA amplification and sequencing
Taxonomic identification of the cryptic species of the B. tabaci complex is based on the sequencing of the partial 3′ mitochondrial cytochrome oxidase I gene (COI)23. Polymerase chain reactions (PCR) were performed as described in Ally, et al.63, using the primer pair designed by Mugerwa, et al.26 (see Supplementary Table S3 online). DNA Amplicons were then sent to Macrogen Europe laboratory for sequencing. Sequences were manually edited and aligned using Geneious™ software R10.2.664, and then compared with reference sequences from GenBank using the BLAST algorithm.
Nuclear microsatellite PCR amplification and genotyping
Nuclear microsatellite PCR amplification was done using 11 loci, combined in three multiplex primer reactions (see Supplementary Table S3 online). PCR amplification, dilution of PCR products, formamide denaturation of DNA and genotyping was according to Ally et al.63. Nuclear microsatellite genotyping was carried out with an Applied Biosystem© 3130XL DNA sequencer. Peaks were scored manually using GeneMapper™ software v4.0 (Applied Biosystems, Foster City, CA).
Population genetic analysis
Micro-checker software was used to correct genotyping errors in our microsatellite data, identifying non-amplified alleles, short allele dominance and the scoring of stuttering peaks65. For each of our three cryptic species, mean number of allele per population, allelic richness, observed heterozygosity, expected heterozygosity, and fixation indices66 were calculated using Genepop v4.767. Hardy–Weinberg equilibrium was tested and p-value adjusted with Bonferroni correction. Linkage disequilibrium was tested with Genepop between pairwise populations and markers. The software Bottelneck v1.2.0268 was used to test the temporary excess of heterozygosity that results from a decrease of the effective population size. Deviations from expected heterozygosity were computed through 1,000 permutations, with the stepwise mutation model (SMM) and the two-phased model of mutation (TPM).
Population structure analysis
Bayesian clustering analyses were performed to assess genetic population structure between our populations. The first analysis was done with Structure software (2.3.4 version)69. We first analyzed the whole dataset, including MEAM1, IO and MED species; setting Structure with a 100,000 burn in iteration (10%) and followed by 1,000,000 Markov Chain Monte-Carlo (MCMC) iterations, using an admixture model allowing correlation between allele frequencies between populations. The number of assumed clusters (K) was set to a range going from 1 to 20, each step being repeated 5 times. The best K was estimated by means of ∆k as described by Evanno, et al.70, using the online program Structure Harvester71. Then, a Discriminant Analysis of Principal Components (DAPC) was performed in R72, between those genetic clusters, using the ‘adegenet’ package v2.1.373. The two (Structure and DAPC) analyses were congruent and analyses were further carried out.
Because of the existence of hybrids between both MEAM1 and IO species38, we analyzed a first subset excluding MED species, in order to better identify these individuals. We further split the whole dataset into three other subsets, each species being taken alone to decipher their substructure. For these four analyses of population structure (MEAM1 & IO, MEAM1, IO and MED), Structure was also set with 100,000 burn-in length with run length of 1,000,000 MCMC. The number of assumed clusters was set to a range going from 1 to 20, but each step was repeated 5 times. The best K was estimated with Structure Harvester71, then Structure software was run again for the best K, 50 times. Clumpak on the web74 was used to summarize the best K posterior probabilities and to graphically represent the bar plot output average over the 50 repetitions.
Matrix of the pairwise FST genetic distances between populations of each species, together with Bonferroni corrected p value, were achieved using Genepop v4.767. Correlation between genetic differentiation and geographic distances between sampling locations were tested (either FST vs the combined distances along the minimum spanning tree between locations or FST/(1 − FST) vs log-transformed Euclidean distances between locations), in order to detect isolation by distance (IBD)75, using the ISOLDE program in Genepop67.
Kdr mutation identification
PCR–RFLP visual reading
A polymerase chain reaction–restriction fragment length polymorphism (PCR‐RFLP) diagnostic assay developed by Tsagkarakou, et al.41 was used in this study to detect the mutation known to confer resistance to pyrethroids (i.e., the resistant kdr). Two mutations on a portion of the voltage-gated sodium channel gene are responsible for this resistance for the MED species (linked to two described non-synonymous mutations, either of them conferring an amino acid change: L925I and T929V), and one for the MEAM1 (involving the first locus and amino acid change: L925I). This diagnostic assay was performed on the whole dataset. Briefly, a 184 bp fragment was amplified (see Supplementary Table S3 online) following the PCR amplification protocol described by Tsagkarakou, et al.41—with slight modifications—and using GoTaq® DNA Polymerase provided by Promega™. The PCR amplified fragment was then fully digested with restriction enzyme DdeI (Promega™), yielding fragments of different size depending on the kdr genotype. As susceptible allele contains one site for restriction enzyme DdeI, digestion of the PCR product results in a restriction pattern of two fragments (124 and 60 pb). The restriction site is non-functional when the resistant allele is present, leading to an intact fragment of 184 pb after enzyme digestion. This method allows to visualize homozygous such as heterozygous individuals at this locus. The QIAxcel® Advanced Instrument, an automated capillary electrophoresis device supplied by Qiagen™, was used to perform gel electrophoresis and to visualize restriction patterns.
Kdr mutation sequencing
The sodium channel kdr region was sequenced for 12% of the whole dataset (including: 98% of all MED, 7% of all MEAM1, 7% of all IO, and 100% of hybrid genotypes) using the above-described primers and methodology. Then, PCR products were sequenced in both directions by Macrogen Europe laboratory (see Supplementary Table S3 online). Sequences were manually edited and aligned using Geneious™ software R10.2.664. The resulting consensus sequences were compared with reference sequences from GenBank using the BLAST algorithm.
Interspecific hybridization
Interspecific hybridization between the different species was assessed using the Bayesian clustering analysis performed by structure software, and further confirmed by the DAPC analysis. Individuals were considered hybrid when their assignment to the MEAM1 or IO cluster was comprised between 10 and 90%39,76. We used Kruskal–Wallis one-way analysis of variance (i) to assess the effect of the environment (greenhouse, open field, field surroundings or non-cultivated area) on assignment probabilities of hybrids (i.e., direction of backcrosses), and (ii) to test if there was a link between the kdr genotype and assignment probability to parental species.
Data availability
The datasets generated during and/or analysed during the current study are available in the zenodo repository at https://doi.org/10.5281/zenodo.5565437 and at https://dataverse.cirad.fr/.
References
Pimentel, D. et al. Economic and environmental threats of alien plant, animal, and microbe invasions. Agric. Ecosyst. Environ. 84, 1–20 (2001).
Wilcove, D. S. & Chen, L. Y. Management costs for endangered species. Conserv. Biol. 12, 1405–1407 (1998).
Singer, M. C., Wee, B., Hawkins, S. & Butcher, M. Rapid natural and anthropogenic diet evolution: three examples from checkerspot butterflies in The Evolutionary Biology of Herbivorous Insects: Speciation, Specialization and Radiation (ed. Tilmon, K. J.). 311–324. (University of California Press, 2008).
Ruesink, J. L., Parker, I. M., Groom, M. J. & Kareiva, P. M. Reducing the risks of nonindigenous species introductions. Bioscience 45, 465–477 (1995).
Rhymer, J. M. & Simberloff, D. Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. 27, 83–109 (1996).
Vitousek, P. M., D’Antonio, C. M., Loope, L. L. & Westbrooks, R. Biological invasions as global environmental change. Am. Sci. 84, 468–478 (1996).
Daszak, P., Cunningham, A. A. & Hyatt, A. D. Emerging infectious diseases of wildlife-threats to biodiversity and human health. Science 287, 443–449 (2000).
Lockwood, J. L., Cassey, P. & Blackburn, T. The role of propagule pressure in explaining species invasions. Trends Ecol. Evol. 20, 223–228 (2005).
Blackburn, T. M. & Jeschke, J. M. Invasion success and threat status: two sides of a different coin?. Ecography 32, 83–88 (2009).
Facon, B. et al. A general eco-evolutionary framework for understanding bioinvasions. Trends Ecol. Evol. 21, 130–135 (2006).
Ellstrand, N. C. & Schierenbeck, K. A. Hybridization as a stimulus for the evolution of invasiveness in plants?. Proc. Natl. Acad. Sci. USA 97, 7043–7050 (2000).
Verhoeven, K. J. F., Macel, M., Wolfe, L. M. & Biere, A. Population admixture, biological invasions and the balance between local adaptation and inbreeding depression. Proc. R. Soc. B-Biol. Sci. 278, 2–8 (2011).
Brevik, K., Lindström, L., McKay, S. D. & Chen, Y. H. Transgenerational effects of insecticides-implications for rapid pest evolution in agroecosystems. Curr. Opin. Insect Sci. 26, 34–40 (2018).
Kirk, W. D. J. & Terry, L. I. The spread of the western flower thrips Frankliniella occidentalis (Pergande). Agr. Forest. Entomol. 5, 301–310 (2003).
Piiroinen, S., Lyytinen, A. & Lindström, L. Stress for invasion success? Temperature stress of preceding generations modifies the response to insecticide stress in an invasive pest insect. Evol. Appl. 6, 313–323 (2013).
Margus, A. et al. Sublethal pyrethroid insecticide exposure carries positive fitness effects over generations in a pest insect. Sci. Rep. 9, 1–10 (2019).
Vais, H., Williamson, M. S., Devonshire, A. L. & Usherwood, P. N. R. The molecular interactions of pyrethroid insecticides with insect and mammalian sodium channels. Pest Manag. Sci. 57, 877–888 (2001).
Smith, L. B., Kasai, S. & Scott, J. G. Voltage-sensitive sodium channel mutations S989P+ V1016G in Aedes aegypti confer variable resistance to pyrethroids, DDT and oxadiazines. Pest Manag. Sci. 74, 737–745 (2018).
Guerrero, F. D., Jamroz, R. C., Kammlah, D. & Kunz, S. E. Toxicological and molecular characterization of pyrethroid-resistant horn flies, Haematobia irritans: Identification of kdr and super-kdr point mutations. Insect Biochem. Mol. 27, 745–755 (1997).
Morin, S. et al. Mutations in the Bemisia tabaci para sodium channel gene associated with resistance to a pyrethroid plus organophosphate mixture. Insect Biochem. Mol. 32, 1781–1791 (2002).
Kasai, S. et al. First detection of a putative knockdown resistance gene in major mosquito vector, Aedes albopictus. Jpn. J. Infect. Dis. 64, 217–221 (2011).
Brito, L. P. et al. Assessing the effects of Aedes aegypti kdr mutations on pyrethroid resistance and its fitness cost. PLoS ONE 8, e60678 (2013).
De Barro, P. J., Liu, S. S., Boykin, L. M. & Dinsdale, A. B. Bemisia tabaci: A statement of species status. Annu. Rev. Entomol. 56, 1–19 (2011).
Perring, T. M. The Bemisia tabaci species complex. Crop Prot. 20, 725–737 (2001).
Navas-Castillo, J., Fiallo-Olivé, E. & Sánchez-Campos, S. Emerging virus diseases transmitted by whiteflies. Annu. Rev. Phytopathol. 49, 219–248 (2011).
Mugerwa, H. et al. African ancestry of new world, Bemisia tabaci-whitefly species. Sci. Rep. 8, 2734 (2018).
Kanakala, S. & Ghanim, M. Global genetic diversity and geographical distribution of Bemisia tabaci and its bacterial endosymbionts. PLoS ONE 14, e0213946 (2019).
Hu, J. et al. New putative cryptic species detection and genetic network analysis of Bemisia tabaci (Hemiptera: Aleyrodidae) in China based on mitochondrial COI sequences. Mitochondr. DNA Part DNA Mapp. Seq. Anal. 29, 474–484 (2018).
Vyskocilova, S., Tay, W. T., van Brunschot, S., Seal, S. & Colvin, J. An integrative approach to discovering cryptic species within the Bemisia tabaci whitefly species complex. Sci. Rep. 8, 10886 (2018).
Cheek, S. & Macdonald, O. Statutory controls to prevent the establishment of Bemisia tabaci in the United Kingdom. Pestic. Sci. 42, 135–137 (1994).
Horowitz, A. R. et al. Biotype Q of Bemisia tabaci identified in Israel. Phytoparasitica 31, 94–98 (2003).
Basit, M. Status of insecticide resistance in Bemisia tabaci: Resistance, cross-resistance, stability of resistance, genetics and fitness costs. Phytoparasitica 47, 207–225 (2019).
Horowitz, A. R., Kontsedalov, S., Khasdan, V. & Ishaaya, I. Biotypes B and Q of Bemisia tabaci and their relevance to neonicotinoid and pyriproxyfen resistance. Arch. Insect Biochem. Physiol. 58, 216–225 (2005).
Horowitz, A. R., Ghanim, M., Roditakis, E., Nauen, R. & Ishaaya, I. Insecticide resistance and its management in Bemisia tabaci species. J. Pest. Sci. 93, 893–910 (2020).
Delatte, H. et al. A new silverleaf-inducing biotype Ms of Bemisia tabaci (Hemiptera: Aleyrodidae) indigenous to the islands of the south-west Indian Ocean. B. Entomol. Res. 95, 29–35 (2005).
Peterschmitt, M. et al. First report of tomato yellow leaf curl virus in Réunion Island. Plant Dis. 83, 303 (1999).
Delatte, H., Lett, J.-M., Lefeuvre, P., Reynaud, B. & Peterschmitt, M. An insular environment before and after TYLCV introduction in Tomato Yellow Leaf Curl Virus Disease: Management, Molecular Biology, Breeding for Resistance (ed. Czosnek, H.). 13–23. (Springer, 2007).
Delatte, H. et al. Microsatellites reveal extensive geographical, ecological and genetic contacts between invasive and indigenous whitefly biotypes in an insular environment. Genet. Res. 87, 109–124 (2006).
Delatte, H. et al. Genetic diversity, geographical range and origin of Bemisia tabaci (Hemiptera: Aleyrodidae) Indian Ocean Ms. B. Entomol. Res. 101, 487–497 (2011).
Thierry, M. et al. Mitochondrial, nuclear, and endosymbiotic diversity of two recently introduced populations of the invasive Bemisia tabaci MED species in La Réunion. Insect. Conserv. Divers. 8, 71–80 (2015).
Tsagkarakou, A. et al. Molecular diagnostics for detecting pyrethroid and organophosphate resistance mutations in the Q biotype of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae). Pestic. Biochem. Phys. 94, 49–54 (2009).
Delatte, H. et al. Differential invasion success among biotypes: case of Bemisia tabaci. Biol. Invasions 11, 1059–1070 (2009).
Chu, D., Tao, Y.-L., Zhang, Y.-J., Wan, F.-H. & Brown, J. K. Effects of host, temperature and relative humidity on competitive displacement of two invasive Bemisia tabaci biotypes [Q and B]. Insect Sci. 19, 595–603 (2012).
Chu, D., Wan, F. H., Zhang, Y. J. & Brown, J. K. Change in the biotype composition of Bemisia tabaci in Shandong Province of China from 2005 to 2008. Environ. Entomol. 39, 1028–1036 (2010).
Pascual, S. & Callejas, C. Intra- and interspecific competition between biotypes B and Q of Bemisia tabaci (Hemiptera: Aleyrodidae) from Spain. B. Entomol. Res. 94, 369–375 (2004).
Pan, H. et al. Insecticides promote viral outbreaks by altering herbivore competition. Ecol. Appl. 25, 1585–1595 (2015).
Shatters, R. G. et al. Population genetics of Bemisia tabaci biotypes B and Q from the Mediterranean and the U.S. inferred using microsatellite markers. in Fourth International Bemisia Workshop International Whitefly Genomics Workshop (3–8 December 2006). (Duck Key: USDA/ARS US Horticultural Research Laboratory, 2006).
McKenzie, C. L. & Osborne, L. S. Bemisia tabaci MED (Q biotype) (Hemiptera: Aleyrodidae) in Florida is on the move to residential landscapes and may impact open-field agriculture. Fla. Entomol. 100, 481–484 (2017).
Guo, X.-J. et al. Diversity and genetic differentiation of the whitefly Bemisia tabaci species complex in China based on mtCOI and cDNA-AFLP analysis. J. Integr. Agr. 11, 206–214 (2012).
Prabhaker, N., Castle, S., Henneberry, T. J. & Toscano, N. C. Assessment of cross-resistance potential to neonicotinoid insecticides in Bemisia tabaci (Hemiptera: Aleyrodidae). B. Entomol. Res. 95, 535–543 (2005).
Taquet, A. et al. Insecticide resistance and fitness cost in Bemisia tabaci (Hemiptera: Aleyrodidae) invasive and resident species in La Réunion Island. Pest Manag. Sci. 76, 1235–1244 (2020).
Elfekih, S. et al. Genome-wide analyses of the Bemisia tabaci species complex reveal contrasting patterns of admixture and complex demographic histories. PLoS ONE 13, e0190555 (2018).
Thierry, M. et al. Symbiont diversity and non-random hybridization among indigenous (Ms) and invasive (B) biotypes of Bemisia tabaci. Mol. Ecol. 20, 2172–2187 (2011).
Gauthier, N. et al. Genetic structure of Bemisia tabaci Med populations from home-range countries, inferred by nuclear and cytoplasmic markers: impact on the distribution of the insecticide resistance genes. Pest Manag. Sci. 70, 1477–1491 (2014).
Alon, M. et al. Multiple origins of pyrethroid resistance in sympatric biotypes of Bemisia tabaci (Hemiptera: Aleyrodidae). Insect Biochem. Mol. 36, 71–79 (2006).
Vassiliou, V. et al. Insecticide resistance in Bemisia tabaci from Cyprus. Insect Sci. 18, 30–39 (2011).
Gnankiné, O., Hema, O., Namountougou, M., Mouton, L. & Vavre, F. Impact of pest management practices on the frequency of insecticide resistance alleles in Bemisia tabaci (Hemiptera: Aleyrodidae) populations in three countries of West Africa. Crop Prot. 104, 86–91 (2018).
Cahill, M., Byrne, F. J., Gorman, K., Denholm, I. & Devonshire, A. L. Pyrethroid and organophosphate resistance in the tobacco whitefly Bemisia tabaci (Homoptera: Aleyrodidae). B. Entomol. Res. 85, 181–187 (1995).
Weill, M. et al. Insecticide resistance: A silent base prediction. Curr. Biol. 14, 552–553 (2004).
Bouvier, J.-C. et al. Deltamethrin resistance in the codling moth (Lepidoptera: Tortricidae): Inheritance and number of genes involved. Heredity (Edinb) 87, 456–462 (2001).
Calvert, L. A. et al. Morphological and mitochondrial DNA marker analyses of whiteflies (Homoptera: Aleyrodidae) colonizing cassava and beans in Colombia. Ann. Entomol. Soc. Am. 94, 512–519 (2001).
Tocko-Marabena, B. K. et al. Genetic diversity of Bemisia tabaci species colonizing cassava in Central African Republic characterized by analysis of cytochrome c oxidase subunit I. PLoS ONE 12, e0182749 (2017).
Ally, H. M. et al. What has changed in the outbreaking populations of the severe crop pest whitefly species in cassava in two decades?. Sci. Rep. 9, 1–13 (2019).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M. & Shipley, P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538 (2004).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984).
Raymond, M. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995).
Piry, S., Luikart, G. & Cornuet, J. M. BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 90, 502–503 (1999).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. & VonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
R Core Team. R: A Language and Environment for Statistical Computing. https://www.r-project.org/ (2020).
Jombart, T. & Ahmed, I. Adegenet 1.3–1: New tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011).
Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A. & Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191 (2015).
Slatkin, M. Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47, 264–279 (1993).
Vähä, J.-P. & Primmer, C. R. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol. Ecol. 15, 63–72 (2006).
Acknowledgements
The authors are grateful to all farmers from La Réunion whom welcomed us in their fields for sampling whitefly. This study was funded by the CIRAD, the ‘Conseil Régional de La Réunion’ and the European Agricultural Fund for Rural Development (EAFRD). A. Taquet is a recipient of a PhD fellowship from the CIRAD and Anses. The authors acknowledge the Plant Protection Platform (3P, IBISA) where all experiments were conducted.
Author information
Authors and Affiliations
Contributions
A.T, M.G. and H.J.-P. carried out the sampling. A.T. and C.S. performed all molecular work. A.T., H.J.-P., B.B. and H.D. analyzed the data, discussed the results and wrote the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taquet, A., Jourdan-Pineau, H., Simiand, C. et al. Distribution of invasive versus native whitefly species and their pyrethroid knock-down resistance allele in a context of interspecific hybridization. Sci Rep 12, 8448 (2022). https://doi.org/10.1038/s41598-022-12373-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-12373-4
- Springer Nature Limited