
Abstract
The genus Hoya is highly diverse and many of its species are popular ornamental plants. However, the relationships between Hoya and related genera (the Hoya group) are not fully resolved. In this study, we report 20 newly sequenced plastomes of species in the Hoya group. The complete plastomes vary in length from 175,405 to 178,525 bp while the LSCs vary from 90,248 to 92,364 bp and the complete SSCs vary from 2,285 to 2,304 bp, making the SSC in the Hoya group one of the shortest known in the angiosperms. The plastome structure in the Hoya group is characterised by a massive increase in the size of the inverted repeats as compared to the outgroups. In all ingroup species, the IR/SSC boundary moved from ycf1 to ndhF while this was not observed in outgroup taxa, making it a synapomorphy for the Hoya group. We have also assembled the mitogenome of Hoya lithophytica, which, at 718,734 bp, is the longest reported in the family. The phylogenetic analysis using exons from 42 taxa in the Hoya group and three outgoups confirms that the earliest divergent genus in the Hoya group is Papuahoya, followed by Dischidia. The relationship between Dischidia and the clade which includes all Hoya and Oreosparte taxa, is not fully supported. Oreosparte is nested in Hoya making it paraphyletic unless Clemensiella is recognised as a separate genus.
Similar content being viewed by others


Introduction
Hoya R.Br., with 350–450 species1 is the largest genus in Apocynaceae-Asclepiadoideae-Marsdenieae, and the second largest genus in Apocynaceae after Ceropegia L.2. It includes epiphytic or more rarely terrestrial and hemi-epiphytic vines and shrubs with leaves ranging from coriaceous to very thick and succulent. The distribution area spans from the Himalayan foothills to the northwest, Okinawa (Japan) to the northeast, Australia to the south and the Fiji Islands to the southeast. Hoya and the similar genera Absolmsia Kuntze (1 sp.), Anatropanthus Schltr. (1 sp.), Clemensiella Schltr. (2 spp.), Dischidia R.Br. (ca. 80 spp.), Heynella Backer (1 sp.), Madangia P.I.Forst., Liddle & I.M.Liddle (1 sp.), Micholitzia N.E.Br. (1 sp.), and Oreosparte Schltr. (3 spp.), have been generally called the “Hoya group”3,4,5,6. Absolmsia, Anatropanthus, Clemensiella, Micholitzia, as well as Eriostemma (Schltr.) Kloppenb. & Gilding and Hiepia V.T.Pham & Aver., have been subsumed under Hoya based on molecular data7,8,9,10 and therefore at present the Hoya group includes only Dischidia, Heynella (for which no molecular data is available), Hoya, and the recently published Papuahoya Rodda & Simonsson (3 species)7.
The best sampled analysis of the morphological and taxonomic diversity of the Hoya group conducted to date is based on three chloroplast loci (trnT-UGU–trnL-UAA–trnF-GAA, psbA–trnH, matK) and two nuclear loci (ITS and ETS)7. The Hoya group clade, including Hoya s.l., Dischidia, Oreosparte and Papuahoya, is nested within Marsdenieae in a clade with other Asian and Australasian species. Hoya is paraphyletic unless Dischidia and Oreosparte are synonymised. However, the relationships between the two main Hoya clades, Oreosparte and Dischidia are not supported and there is no sufficient evidence to synonymise Oreosparte and Dischidia with Hoya. Papuahoya, from New Guinea, originally suspected to be part of Oreosparte based on morphological similarities, is sister to the rest of the Hoya group but only with 79% bootstrap support (BS).
Several studies have focused on evolution of plastomes in Apocynaceae, spanning much of the diversity of the family. The first complete plastomes in the family were of Asclepias syriaca L.11 and Catharanthus roseus (L.) G.Don12. These plastomes were compared to the available plastomes in Gentianales: Straub et al.11 reported the loss of accD, clpP, and ycf1 in A. syriaca, and Ku et al.12 found that the plastome of C. roseus is highly similar to that of Coffea arabica L. (Rubiaceae), with no gene losses. Ku et al.12 noted another missing gene in A. syriaca compared to C. roseus (ycf15), as well as an expansion in some intergenic regions in A. syriaca and a difference in the position of inverted repeat boundaries. The plastome changes in Asclepias L., and the dynamics between plastomes and mitogenomes were reported in detail by Straub et al.13. They found evidence of gene movement from the mitogenome to the plastome, which is unusual in angiosperms14. Straub et al.15 expanded the taxonomic sampling to 12 plastomes within Apocynaceae, and concluded that plastome structure is highly conserved in the family, with the exception of the genus Asclepias. Fishbein et al.16 published 73 complete plastomes from the family, and their plastome phylogeny was used to assess taxonomic relationships within the family. Several other plastomes have also been published17,18,19,20,21,22,23,24. Only three plastomes of Hoya have been published so far. Hoya pottsii Traill (a synonym of Hoya verticillata (Vahl) G.Don) and Hoya liangii Tsiang (a synonym of Hoya diversifolia Blume)25 were reported to have a plastome architecture similar to that of other Apocynaceae, and Hoya carnosa (L.f.) R.Br.26 was reported to have a near complete loss of the small single copy regions (SSC) of the plastome due to a boundary shift leading to a large expansion of the two inverted repeats (IRs).
Mitogenomes of Apocynaceae have been less thoroughly studied. In Asclepias (subfamily Asclepiadoideae), Straub et al.13 reported substantial import of plastome regions to the mitogenomes, and major restructuring of the genomes when compared to closest relatives; both are common features in plant mitogenomes. Park et al.27 similarly reported in Rhazya Decne. (informal group Rauvolfioids) repeat regions and movement of genetic material from the nucleus and plastome to the mitogenome, but no movement from the mitogenome to plastome. In Cynanchum L. (subfamily Asclepiadoideae) the mitogenome is reported to be multipartite, consisting of two chromosomes28.
In this paper, we sequenced and assembled the complete or near complete plastomes of 20 species in the Hoya group. Our aim was to investigate the evolutionary position of the structural changes reported by Wei et al.26 with a broader sampling of taxa. In addition, we assembled the plastome exons of a larger number of species (39) to provide maximum support for a phylogenetic reconstruction of the evolution of plastomes in this group. This complete plastome phylogeny (omitting only poorly aligning intergenic areas and very short exons) will be an invaluable resource when interpreting nuclear phylogenies in the group, and will provide a backbone against which reticulation events and poorly resolved trees can be compared.
Results
Plastome structure in the Hoya group
We acquired complete plastomes for ten species in the ingroup and two in the outgroup. For a further ten species in the ingroup, we acquired near complete plastomes, with 1–6 gaps in mononucleotide regions with low coverage (Table 1). For the remaining 19 species (18 ingroup, one outgroup), all targeted exons were acquired.
We observed that species in the Hoya group have a very large copy number of plastomes per cell: 3.55–21.70% of all sequencing reads mapped to the plastome (Table 1).
The total length of the complete plastomes varies from 175,405 bp (Dischidia milnei Hemsl.) to 178,525 bp (Hoya omlorii (Livsh. & Meve) L.Wanntorp & Meve) (161,660–161,700 bp in the outgroups) (Table 1). The length of complete IRs varies from 41,272 (D. milnei) to 42,069 bp (Hoya exilis Schltr.) (26,117–28,287 bp in the outgroups). The complete LSCs of the ingroups varies in length from 90,248 bp (Hoya ignorata T.B.Tran, Rodda, Simonsson & Joongku Lee) to 92,364 bp (Hoya megalaster Warb. ex K.Schum. & Lauterb.) and the complete SSCs varies from 2,285 bp (H. diversifolia) to 2,304 bp (H. omlorii), making the SSC in Hoya group one of the shortest known in angiosperms29.
The plastome structure in all species in the Hoya group (Fig. 1) is characterised by a massive increase in the size of inverted repeats as compared to the outgroups; the IR/SSC boundary moved from ycf1 to ndhF (Fig. 2). The outgroup species that is most closely related to the Hoya group, Marsdenia flavescens A.Cunn. ex Hook., lacks this boundary change, but it is characterised by a smaller change in the IR/LSC boundary (loss of rps19 from IR to LSC).

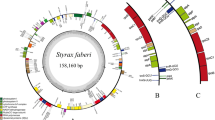
Chloroplast genome of Hoya lithophytica. The colour-coded bars indicate different functional groups. The darker grey area in the inner circle indicates GC content, while the lighter grey area indicates AT content. IR inverted repeat, SSC small single copy, LSC large single copy.

Mauve alignment of plastomes of Hoya group and selected other Apocynaceae species. The inverted repeat closest to psbA was removed, and the small single copy is displayed in a direction that best illustrates the shift in inverted repeat boundaries. The alignment colours refer to locally collinear blocks shared between plastomes. The extent of the inverted repeat is shown with a bar.
The nucleotide diversity (Pi) in the ingroup varies from 0 to 0.0433 (Fig. 3). The IRs were consistently less variable than the other parts of the plastomes, except for one highly variable gene (ycf1). One gene in the LSC (accD) and the region at IR-SSC boundaries (near ndhF) were similarly variable. ycf1 and accD were characterised by long aminoacid repeats.

Nucleotide diversity (Pi) values of 20 complete ingroup plastomes, showing genome parts, barcoding regions commonly used in the taxa in question (trnT-UGU–trnL-UAA–trnF-GAA, psbA–trnH, matK) and genes in highly variable regions (accD, ndhF and ycf1).
Mitogenome structure
The mitogenome structure of Hoya lithophytica Kidyoo (Fig. 4) shows massive restructuring in relation to the other complete mitogenomes available in Apocynaceae (Fig. 5). At 718,734 bp, it is the longest mitogenome reported in the family. Movement of plastome DNA to mitogenome explains at least 56,698 bp (7.889%) of the mitogenome.

Mitochondrial genome of Hoya lithophytica.

Mauve alignment of available mitogenomes in Apocynaceae, showing massive restructuring. (Hoya lithophytica on the top, Asclepias syriaca below). Corresponding blocks present in both mitogenomes are indicated by colour.
Phylogenetic analysis
The model choice in MrBayes had no effect on the tree topology, and only a minor effect on the node values. The two model options tested resulted in the same topology and highly similar BPP values (Bayesian Posterior Probability), differing at most by 0.05 for any node; both runs passed our quality control. The values indicated in the next paragraph and shown in the molecular phylogeny presented in Fig. 6 were acquired using GTR + Gamma.

Molecular phylogeny of representative species of the Hoya group based on exons longer than 90 bp (excluding accD, ycf1 and ycf2). Numbers at the nodes indicate bootstrap percentages followed by Bayesian Posterior Probability (only indicated when not fully supported).
To facilitate comparison, for the Hoya clades we refer to the clade names of Wanntorp et al.10 (their Figs. 3 and 4) and Rodda et al.7 (their Fig. 4) whenever possible. A new name is provided for one unidentified clade from previous studies, which includes Hoya imperialis Lindl. and H. obtusifolia Wight (Clade Y).
Within the Hoya group several well supported clades (99–100% BS, 1 BPP) can be separated. The earliest divergent clade corresponds to the genus Papuahoya (100% BS, 1 BPP), represented by P. urniflora (P.I.Forst.) Rodda & Simonsson and P. neoguineensis Simonsson & Rodda. The following clade includes eight species of Dischidia (100% BS, 1 BPP). Within Dischidia there is one ant-house leaved species, D. milnei, formerly included in the genus Conchophyllum Blume, that froms a clade (99% BS, 1 BPP) with the type of the genus D. nummularia R.Br. and D. albida Griff.
Dischidia is sister to a clade (77% BS, 1 BPP) including all Hoya and Oreosparte taxa. Within Hoya/Oreosparte, the first diverging clade includes four species of Hoya (100% BS, 1 BPP, Clade II) one of which was formerly classified in the genus Clemensiella (C. omlorii, now H. omlorii) and two in Eriostemma, [E. gigas (Schltr.) Kloppenb. & Gilding (now H. gigas Schltr.) and E. coronaria (Blume) Kloppenb. & Gilding (now H. coronaria Blume)]. These correspond to Clade II and Clemensiella in Wanntorp et al.10 and Rodda et al.7, respectively. Together, these three species are sister (100% BS, 1 BPP) to a recently described species of Hoya, H. lithophytica, from NW Thailand. Clade II is sister to a clade (95% BS, 1 BPP) including Oreosparte and the rest of the Hoya species (Hoya s.s.). Oreosparte (100% BS, 1 BPP) includes the type of the genus O. celebica Schltr. and O. parviflora (Ridl.) Rodda & Simonsson. Within Hoya s.s. (99% BS, 1 BPP) there are two larger groups (Group 1 and 2, also recognised in Wanntorp et al.10 and Rodda et al.7) and six well supported clades. Group I (100% BS, 1 BPP) is the earliest divergent group. It includes three clades: Clade IV (100% BS, 1 BPP), with two species from New Guinea; Clade III (100% BS, 1 BPP) includes three Sundaland species, two of which (H. platycaulis Simonsson & Rodda and H. wallichii (Wight) C.M.Burton) are generally non-climbing shrubs; Clade Y (not present in Wanntorp et al.10 and Rodda et al.7) includes H. imperialis and H. obtusifolia, two species from West Malesia characterised by very stout stems and a preference for sunny habitats. Sister to Group 1 is a clade (87% BS, 0.95 BPP) including Group 2 and Clade I.
Group 2 (100% BS, 1 BPP) includes two Clades: Clade V (100% BS, 1 BPP), with four species belonging to the Hoya section Acanthostemma; Clade VI (100% BS, 1 BPP), which includes the type of the genus, H. carnosa as well as some very widespread and variable species such as H. diversifolia and H. verticillata. Clade I (100% BS, 1 BPP), consists of two species from the Pan Himalayan area (H. lanceolata Wall. ex D.Don) and Northern Thailand (H. thailandica Thaithong).
Discussion
With the exception of Wei et al.26, previously published work on plastomes of the Hoya group only resulted in incomplete plastomes16,30 or incorrectly assembled plastomes25. This is not surprising, as assembling plastomes in this group is very challenging: our attempts at automated sequence assembly only resulted in small fragments, which often incorporated mitogenome sequences and required extensive manual corrections. Likely causes of the difficulty in assembling the genomes are the extensive expansion of IRs, and the near-loss of SSC, the large amount of sequences shared by the plastome and the mitogenome and the frequent repetitive elements found in the plastomes.
In our experience, the fastest method of genome assembly is assembly of reads to a reference, followed by manual correction. A highly time-consuming process of manual checking of the entire alignment followed all initial assemblies. As the order of the genes in the plastomes and the placement of IRs was highly conserved in all assembled plastomes, we did not assemble the sequences of the remaining 20 species we sequenced.
The large copy number of plastomes per cell that we observed is not uncommon in Apocynaceae. Similarly high proportions of plastome reads (11.8%) has been reported in Asclepias11. This is much higher than in most angiosperms, where c. 1% of sequencing reads mapping to plastomes is common (pers. obs.). The high copy number helps in part to reduce assembly issues derived from the highly repetitive intergenic parts of the Hoya plastome; however, routine assembly of Hoya plastomes from short sequencing reads is likely to remain challenging. Even with the very high coverage that we attained, some low GC content intergenic regions had very low or even zero sequence coverage, leading to gaps in some of our assemblies. We think this was likely due to heavy degradation of the plastome, which may have occurred as leaves age. Use of younger leaf tissue might help to avoid this issue.
The assembly of the mitogenome was even more time consuming, as iterative extension of sequences to bridge gaps was soon interrupted by presence of plastome sequences. Assembling reads from other species to the already assembled mitogenome of H. lithophytica did not help much, suggesting that there is a high level of instability of mitogenomes within Hoya.
The gene order and overall architecture of the ingroup samples is highly similar to that reported by Wei et al.26 in Hoya carnosa, but all our assemblies differed significantly from those reported by Tan et al.25 in H. verticillata and H. diversifolia. We have reported CDSs and/or exons omitted by Wei et al.26, specifically accD, ndhD, ndhH, ycf2 and ycf15, as a corresponding open reading frame was present. Tan et al.25 also omitted the CDSs of accD, ndhH, ycf1, and ycf2.
The dramatic IR–LSC boundary shift reported by Wei et al.26 is shared by the entire Hoya group, including Papuahoya, whose plastome is strongly supported to have diverged before all other ingroup taxa. Since all ingroup taxa included this boundary change which is not seen in any of the outgroups used, it can be considered a synapomorphy for the Hoya group.
The boundary shift was not reported by Tan et al.25, but we believe that this was in error. Our study includes conspecific sequences to those they reported, and in our analyses, they clearly shared the structure with the other Hoya group species. The two species are deeply nested in the phylogeny of the Hoya group.
The genome structure of Hoya has some parallels to other Asclepiads. As reported for Asclepias13 the intergenic regions of Hoya have long repeated regions of very low GC content. These regions make it difficult to map reads of Hoya even to a closely related species, and undoubtedly offer a challenge in use of intergenic reads. However, the boundary-shift observed is unique to the Hoya group.
Outside of Apocynaceae, there are clear parallels between the plastome restructuring in the Hoya group and that of Lamprocapnos spectabilis (L.) Fukuhara (Papaveraceae). Park et al.29 reported the extension of the IR/SSC boundary from ycf1 (outgroup) to ndhF, and Lamprocapnos also has AARs in accD and ycf1 (however, this was not mentioned in ycf2). Unlike in Lamprocapnos, no additional inversions or other changes to gene order were seen in our study in relation to the outgroup used. The SSC observed in Lamprocapnos is the smallest known in any plant, but only slightly smaller than that observed in species in the Hoya group.
The phylogenetic tree obtained is congruent with the most recent phylogeny of the Hoya group7, with the recognition of the monophyletic genera Papuahoya, Oreosparte and Dischidia that are fully supported.
Based on Rodda et al.7 the relationships between Oreosparte, Dischidia and Hoya as well as Group 1 and numerous smaller clades within Hoya are not fully supported (their Fig. 4). Our analysis (Fig. 6) confirms that the earliest divergent genus in the Hoya group is Papuahoya, followed by Dischidia. Dischidia is sister to a clade which is not fully supported (77% BS, 1 BPP) including all species currently attributed to Hoya and Oreosparte. Species of Hoya were segregated in two clades in Rodda et al.7, one (their Clade I) including four species from continental Asia (100% BS) the other containing the rest of the species (80% BS). Basal clades in Group 1 of Hoya were unsupported (53–69% BS, their Fig. 4). In our analysis instead their Clade I is deeply nested in Hoya s.s., while Hoya is still separated into two clades, the first (Clade II) including species formerly included in Eriostemma + Clemensiella (100% BS, 1 BPP) is sister to Oreosparte + Hoya s.s., the second (Hoya s.s., 99% BS, 1 BPP) is sister to Oreosparte. Clade II can therefore be tentatively classified under the already available genus name Clemensiella, here represented by C. omlorii. Hoya coronaria and H. gigas have also been alternatively classified in the genus Eriostemma (type species: Hoya coronaria), which could now be considered a synonym of Clemensiella. This clade also includes H. lithophytica, a rock dwelling species from NW Thailand. The four species in this clade are characterised by terrestrial (or hemi epiphytic) climbing habit and by having pollinia without pellucid margins. These characters are unique to this clade among the species we sampled here, but not unique in the genus as other species can be terrestrial and lack pellucid margins of the pollinium (e.g. Hoya surisana Rodda & S.Rahayu). The second Hoya clade that includes the type of the genus H. carnosa is to be considered as Hoya s.s.
Based on our results either Hoya needs to be separated in two genera, Hoya and Clemensiella, or Hoya needs to be more broadly circumscribed to also include species currently in Oreosparte. In this latter scenario Clemensiella, Oreosparte and Clade I (Group 1 and 2) of Hoya may be allocated to subgeneric rank.
Our sampling of the Clemensiella clade is limited and samples of more taxa are needed to verify whether Clemensiella and Eriostemma should be kept separated (either at generic or subgeneric level).
Before making any nomenclatural changes, a more extensive phylogeny should be generated including extensive nuclear data to verify that the topology is congruent and that the observed clades are supported.
Materials and methods
We sequenced 38 species in the Hoya group, and three outgroups (in Marsdenia R.Br. s.l., and Jasminanthes Blume). Outgroups were selected due to their known position as outgroups of Hoya group (Rodda et al.7). The ingroups were selected to represent all the genera of the Hoya group where material is available (Hoya, Dischidia, Oreosparte, Papuahoya). Within Hoya we included at least one sample for each of the main Hoya clades (clades I to VI) of Wanntorp et al.10 and Rodda et al.7. For Dischidia we included eight taxa that represent the morphological variation of the genus, including Dischidia parasita (Blanco) Arshed, Agoo & Rodda, the type of the synonymous genus Dischidiopsis Schltr. Oreosparte and Papuahoya are represented by two species each.
Plant materials and DNA extraction
The leaves were collected from plants cultivated at the Singapore Botanic Gardens or obtained from herbarium specimens. All plant specimens used for this study were collected to the best of our knowledge in compliance with local, institutional, national, or international regulations at the time of collection. All newly prepared voucher specimens were deposited in the Singapore Botanic Gardens Herbarium (SING). Their information is summarised in Table 2. The herbaria acronyms follow Thiers31.
Fresh, silica-dried or herbarium leaf samples were extracted using DNeasy Plant Mini Kit (Qiagen Inc., Valencia, California, U.S.A.). A minimum of 400 ng of total genomic DNA was sent for library preparation and genome skimming sequencing using Illumina HiSeq (AITbiosciences, Singapore). A minimum of 1 Gbp of sequence with a read length of 100 bp were acquired per sample. Sequence quality filtering was done with Geneious 11.1.2 (Biomatters Ltd, New Zealand) trim and filter function, using error probability limit of 0.05, a minimum read length of 70 and removing adapters with a minimum blast alignment score of 16.
Sequencing, assembly and annotation
The plastome of Hoya lithophytica was assembled first, using a combination of GetOrganelle32, and assemblies to several reference genomes in Geneious 11.1.2 and ORG.asm33 without a reference or using a variety of Apocynaceae plastomes as reference. The automated assemblies had a large number of artefacts, mostly due to frequent gene movement between the plastome and the mitogenome. The assemblies were checked by assembling sequencing reads to the initial assembled genomes in Geneious 11.1.2, followed by visual correction of alignment, and extension of gaps using iterative assemblies. The approximate length of the inverted repeat was estimated by observing the part of the genome with high sequencing coverage, and areas of exceptionally low coverage were identified as mitogenome sequences; the position of the inverted repeats was approximately corrected, the mitogenome reads were removed from the assemblies, and further iterative gap filling was carried out, resulting in a full circular plastome.
Twenty one plastomes (19 in the Hoya group and two outgroup taxa) were assembled to Hoya lithophytica in Geneious 11.1.2, with a manual correction of alignment and gaps (with iterative extension of gaps when required), followed by further assemblies to the resulting genome to detect and correct errors. In a few cases, one difficult to sequence region (intergenic region psbA–trnH) was acquired through Sanger Sequencing (AITbiosciences, Singapore). Other gaps were not corrected if present.
For 20 further species (19 in the Hoya group and one outgroup taxon), the assembly of the entire plastome was not attempted, and only exons were assembled by aligning them to the reference.
Final circular plastomes were checked by re-mapping the filtered reads to the plastome using Geneious 11.1.2 (Biomatters Ltd, New Zealand) read mapper using low sensitivity, adjusted to not allowing gaps, and alignments were visually inspected for errors and gaps.
Sequences were annotated by transferring annotation from the published plastome of Hoya liangii (a synonym of Hoya diversifolia) (GenBank accession number NC_04224525), followed by correction of position of CDSs. Sliding window analysis was conducted to generate the nucleotide diversity (Pi) of complete ingroup plastomes. The plastomes were aligned using MAFFT v.7.30934, using scoring matrix 200PAM / k = 2, gap open penalty of 1.53 and offset value of 0.123. The resulting alignment was analysed using DnaSP v. 6.12.0335 to compare levels of nucleotide variation across the plastomes.
The mitogenome of Hoya lithophytica was constructed by filtering sequence reads that completely matched the plastome, and mapping the remaining reads to the mitogenome of Asclepias syriaca (KF541337). While parts of the mitogenome were identical to the plastome, enough reads with a single read error were present to cover all parts of the mitogenome for unambiguous assembly. Only small fragments initially matched the reference genome. The other parts of the mitogenome were assembled by iterative mapping and by identifying the boundaries of mitogenome/plastome overlap by mapping reads to the plastome. Attempts to construct further mitogenomes were abandoned once we identified massive restructuring of the mitogenome even within the ingroup.
Gene movement from plastome to the mitogenome was estimated by cutting the plastome of Hoya lithophytica into 30 bp fragments, and measuring the percentage of resulting fragments that mapped to the mitogenome, using the Geneious mapper in Geneious 11.1.2 (Biomatters Ltd, New Zealand).
Changes in plastomes organisation were compared between major clades in the ingroup and the outgroups as well as published plastomes representing a variety of informal groups of Apocynaceae. The SSC was arranged in the same direction, one of the inverted repeats was removed, and the plastomes were analysed using progessiveMauve in Mauve 2015-02-2536. The following plastomes from GenBank were used in the comparison: Rhazya stricta Decne. (KJ123753), Carissa macrocarpa (Eckl.) A.DC. (NC_033354), Trachelospermum jasminoides (Lindl.) Lem. (MK783315), Cynanchum wilfordii (Maxim.) Hook.f. (KT220733), Asclepias syriaca (NC_022432).
Phylogenetic analysis
For phylogenetic analyses, all exons over 90 bp were extracted from the 41 newly sequenced samples as well as the three Hoya and one Dischidia plastomes available in GenBank16,25,26 and aligned using setting-auto in mafft34, and alignments were checked using setting -automated1 in trimAl37. Shorter exons could not be reliably retrieved for the species for which we did not have a complete plastome. Three protein coding genes (accD, ycf1 and ycf2, all one-exon genes) had long amino acid repeats that could not be aligned unambiguously. These were removed from the phylogenetic analyses. Removing areas with gaps in alignment did not affect the phylogeny or branch support noticeably, and these areas were retained in the final phylogenetic analysis. The exon alignments were partitioned to one exon per partition. A maximum likelihood tree was generated using IQ-TREE 2.0.638, with an independent substitution model test (ModelTest) for each partition. The settings for the maximum likelihood analyses were: -m MFP + MERGE -T 12 with 1000 bootstrap replicates. The models selected were: TVM + F + R3 (atpB, infA, petN, atpE, ndhH, rpoA, psbH, rpl14, rpoB, atpF, ndhJ, rpl16, rpoC1, rps2, trnS-UGA, ndhK, psaI, rpoC1, atpI, petA, psaJ, rpoC2, rps11, rps4, ycf3, psbN, rps14, rps8), TVM + F + R4 (clpP, ndhF, ycf4, clpP), K3Pu + F + I (psbD, ndhI, psbI, ycf3, psbK, ycf3, petB, rpl23, petD, psbB, rpl2, rps12, rps7, petG, psbC, rpl2), K3Pu + F + R2 (psbT, rps15, rps18, rps36, ccsA), K3Pu + F + R2 (rpl32, ndhG, psbE, rpl33, psaA, psbF, rbcL, atpF, ndhA, psaB, rps19, ndhA, psaC, atpH, ndhB, psbJ, ndhB, ndhC, psbA, psbL, ndhD, ndhE), HKY + F + I (rrn16, rrn23, rrn4.5, rrn5) and K3Pu + F + R2 (psbZ, rps16, matK, rpl20, rpl22, rps3, cemA, psbM). For some genes there was more than one exon39. Jasminanthes maingayi (Hook.f.) Rodda and Marsdenia longipedicellata P.I.Forst. were selected as the outgroup, as they were known to be part of a clade that is sister to the other species included.
Baysian support for the nodes was tested with MrBayes 3.2.540. We used 30,000,000 Markov chain Monte Carlo iterations, keeping one tree every 100 generations, with a burn-in of 25% (mcmc ngen = 30,000,000 samplefreq = 100 burnin = 75,000). We used the exon-partitioned sequence alignments generated for the IQ-TREE 2.0.638, and applied the same model, GTR + Gamma (lset nst = 6 rates = gamma) for all partitions, with rates unlinked between datasets (unlink statefreq = (all) revmat = (all) shape = (all) pinvar = (all), prset applyto = (all) ratepr = variable). We accepted the results if the likelihoods had converged and the minimum estimated sample size was over 100 for all parameters by the end of the run. To test the effect of the model used, we also ran the same analysis with all substitution rates set to equal and equal rate variation (lset nst = 1 rates = equal).
Data archiving statement
Raw, demultiplexed sequence reads are available at the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) and can be accessed with the BioProject IDs listed in Table 2. The complete or incomplete plastome sequence data of the 38 species sequenced as well as the complete mitochondrial genome of H. lithophytica obtained for this study have been deposited to the GenBank of NCBI (see Table 2 for accession numbers). The sequence alignment is available in Figshare at https://doi.org/10.6084/m9.figshare.14189021.
References
Rodda, M. Two new species of Hoya R. Br. (Apocynaceae, Asclepiadoideae) from Borneo. PhytoKeys 53, 83–93 (2015).
Bruyns, P. V., Klak, C. & Hanáček, P. A. Phylogenetically-based concept of Ceropegia (Apocynaceae). S. Afr. J. Bot. 112, 399–436 (2017).
Omlor, R. Notes on Marsdenieae (Asclepiadaceae): a new, unusual species of Hoya from Northern Borneo. Novon 6, 288–294 (1996).
Omlor, R. Generische Revision der Marsdenieae (Asclepiadaceae), Aachen: Shaker. (1998).
Wanntorp, L. & Forster, P. I. Phylogenetic relationships between Hoya and the monotypic genera Madangia, Absolmsia, and Micholitzia (Apocynaceae, Marsdenieae): Insights from flower morphology. Ann. Mo. Bot. Gard. 94, 36–55 https://doi.org/10.3417/0026-6493(2007)94[36:PRBHAT]2.0.CO;2 (2007).
Meve, U., Laurente, O., Alejandro, G. J. & Livshultz, T. Systematics of Clemensiella ( Apocynaceae–Asclepiadoideae ). Edinburgh J. Bot. 66, 447–457 (2009).
Rodda, M. et al. Phylogenetic studies in the Hoya group (Apocynaceae, Marsdenieae): the position of Anatropanthus and Oreosparte. Willdenowia 50, 119–138 (2020).
Wanntorp, L., Kocyan, A., van Donkelaar, R. & Renner, S. S. Towards a monophyletic Hoya (Marsdenieae, Apocynaceae): inferences from the chloroplast trnL region and the rbcL-atpB spacer. Syst. Bot. 31, 586–596 (2006).
Wanntorp, L., Gotthardt, K. & Muellner, A. N. Revisiting the wax plants (Hoya, Marsdenieae, Apocynaceae): Phylogenetic tree using the matK gene and psbA-trnH intergenic spacer. Taxon 60, 4–14 (2011).
Wanntorp, L., Grudinski, M., Forster, P. I., Muellner-Riehl, A. N. & Grimm, G. W. Wax plants (Hoya, Apocynaceae) evolution: Epiphytism drives successful radiation. Taxon 63, 89–102 (2014).
Straub, S. C. et al. Building a model: developing genomic resources for common milkweed (Asclepias syriaca) with low coverage genome sequencing. BMC Genom. 12, 1–22 (2011).
Ku, C., Chung, W.-C., Chen, L.-L. & Kuo, C.-H. The complete plastid genome sequence of Madagascar periwinkle Catharanthus roseus (L.) G. Don: Plastid genome evolution, molecular marker identification, and phylogenetic implications in Asterids. PLoS ONE 8, e68518 (2013).
Straub, S. C. K., Cronn, R. C., Edwards, C., Fishbein, M. & Liston, A. Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (Apocynaceae). Genome Biol. Evol. 5, 1872–1885 (2013).
Smith, D. R. Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol. Evol. 3, 743–748 https://doi.org/10.1093/gbe/evr068 (2011).
Straub, S. C. K. et al. Phylogenetic signal detection from an ancient rapid radiation: Effects of noise reduction, long-branch attraction, and model selection in crown clade Apocynaceae. Mol. Phylogen. Evol. 80, 169–185 (2014).
Fishbein, M. et al. Evolution on the backbone: Apocynaceae phylogenomics and new perspectives on growth forms, flowers, and fruits. Am. J. Bot. 105, 495–513 (2018).
Jo, S., Kim, H.-W., Kim, Y.-K., Cheon, S.-H. & Kim, K.-J. The complete plastome sequence of Carissa macrocarpa (Eckl.) A. DC. (Apocynaceae). Mitochondrial DNA Part B 2, 26–28 (2017).
Wang, H.-X., Cheng, X.-L., Chen, W.-S., Li, L.-M. & Chen, L. Complete plastome sequence of Trachelospermum Jasminoides (Lindley) Lemaire (Apocynaceae). Mitochondrial DNA Part B 4, 2086–2087 (2019).
da Silva, O. E. J., Marques, A. & Almeida, C. The chloroplast genome of Hancornia speciosa Gomes: structural organization and phylogenomic studies in Rauvolfioideae (Apocynaceae). Braz. J. Bot. 42, 449–455 (2019).
Jin, L., Yang, J., Liu, C. & He, M. Complete plastome of the medicinally important plant, Alstonia scholaris (Apocynaceae). Mitochondrial DNA Part B 4, 2896–2897 (2019).
Abba, A., Alzahrani, D., Yaradua, S. & Albokhari, E. B. Complete plastome genome of Pergularia tomentosa L. (Asclepiadoideae, Apocynaceae). Mitochondrial DNA Part B 5, 566–567 (2020).
Ali, M. A. Complete chloroplast genome of medicinally important poisonous shrub Adenium obesum (Forssk) Roem & Schult. (Apocynaceae). Mitochondrial DNA Part B 5, 568–569 (2020).
Wang, D.-L., Liu, Y.-Y., Tian, D., Yu, L.-Y. & Gui, L.-J. Characterization of the complete chloroplast genome of Plumeria rubra cv. Acutifolia (Apocynaceae). Mitochondrial DNA Part B 5, 927–928 (2020).
Chen, W., Liang, W., Li, A. & Ma, J. Characterization of the complete plastid genome of Rauvolfia verticillata (Apocynaceae), with its phylogenetic analysis. Mitochondrial DNA Part B 4, 4190–4191 (2019).
Tan, X.-H., Wang, J.-H., Zhao, K.-K., Zhu, Z.-X. & Wang, H.-F. Complete plastome sequence of Hoya pottsii Traill and Hoya liangii Tsiang (Apocynaceae). Mitochondrial DNA Part B 3, 1176–1177 (2018).
Wei, X.-F., Zeng, S.-J., Zhang, G.-Q., Tang, G.-D. & Huang, J.-X. Complete plastome sequence of Hoya carnosa (L. f.) R. Br. (Apocynaceae). Mitochondrial DNA Part B 5, 522–523 (2020).
Park, S. et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics 15, 1–18 (2014).
Kim, C.-K. & Kim, Y.-K. The multipartite mitochondrial genome of Cynanchum auriculatum (Gentianales: Apocynaceae). Mitochondrial DNA Part B 4, 3360–3361 (2019).
Park, S., An, B. & Park, S. Reconfiguration of the plastid genome in Lamprocapnos spectabilis: IR boundary shifting, inversion, and intraspecific variation. Sci. Rep. 8, 1–14 (2018).
Crook, T. Plastome Assembly in the wax plants (Hoya) and near relatives (Marsdenieae, Apocynaceae). Published online at https://repository.upenn.edu/morrisarboretum_internreports/16 (2017).
Thiers, B. M. Index Herbariorum: A global directory of public herbaria and associated staff. New York Botanical Garden’s Virtual Herbarium. http://sweetgum.nybg.org/ih/ (continously updated).
Jin, J.-J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31 (2020).
ORG.asm. https://git.metabarcoding.org/org-asm/org-asm/wikis/home.
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 https://doi.org/10.1093/molbev/msx248 (2017).
Darling, A. E., Mau, B. & Perna, N. T. Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5, e11147 https://doi.org/10.1371/journal.pone.0011147 (2010).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic Era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 https://doi.org/10.1093/bioinformatics/btl446 (2006).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Acknowledgements
We thank the National Parks Board Singapore for supporting our research. We thank Dr Gillian Khew for the use of the Molecular Laboratory at the Singapore Botanic Gardens.
Author information
Authors and Affiliations
Contributions
M.R. conceived the experiment, M.R. obtained the data, M.N. analysed the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rodda, M., Niissalo, M.A. Plastome evolution and organisation in the Hoya group (Apocynaceae). Sci Rep 11, 14520 (2021). https://doi.org/10.1038/s41598-021-93890-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93890-6
- Springer Nature Limited
This article is cited by
-
Mitochondrial genome of Hancornia speciosa gomes: intergenic regions containing retrotransposons and predicted genes
Molecular Biology Reports (2024)