
Abstract
Currently there is no consensus regarding the aetiology of the excessive ocular volume that characterizes high myopia. Thus, we aimed to test whether the gene pathways identified by gene set enrichment analysis of RNA-seq transcriptomics refutes the predictions of the Retinal Ion Driven Efflux (RIDE) hypothesis when applied to the induction of form-deprivation myopia (FDM) and subsequent recovery (post-occluder removal). We found that the induction of profound FDM led to significant suppression in the ligand-gated chloride ion channel transport pathway via suppression of glycine, GABAA and GABAC ionotropic receptors. Post-occluder removal for short term recovery from FDM of 6 h and 24 h, induced significant upregulation of the gene families linked to cone receptor phototransduction, mitochondrial energy, and complement pathways. These findings support a model of form deprivation myopia as a Cl− ion driven adaptive fluid response to the modulation of the visual signal cascade by form deprivation that in turn affects the resultant ionic environment of the outer and inner retinal tissues, axial and vitreal elongation as predicted by the RIDE model. Occluder removal and return to normal light conditions led to return to more normal upregulation of phototransduction, slowed growth rate, refractive recovery and apparent return towards physiological homeostasis.
Similar content being viewed by others


Myopia (short-sightedness) is the most common visual disorder worldwide and the greatest risk factor for severe ophthalmic diseases in older individuals especially those with high (-5D) refractive errors1. Myopia is also a public health concern2,3 due to its rapid increase in prevalence (> 80% in young adults in Singapore2, Taiwan4 and China5,6,7). Indeed the global prevalence of myopia has been predicted to rise from 28% (2 billion people) in 2010 to 50% (5 billion people) in 20503. The severity and early onset of myopia7 in many newly urbanized societies implicates both genetics and environment in its induction.
Clinical and experimental myopia share similar morphology and pathophysiology, with the hallmark characteristic being excessively large eyes with abnormal axial elongation especially of the vitreous chamber. As yet, there is no consensus as to why or how this increase in ocular volume is induced though it is well accepted that myopia is a visually driven process and that the retina, and the photoreceptors in particular, are the neural elements primarily sensitive to temporal modulation of luminance8. Many early animal studies exploring the aetiological mechanisms associated with the effects of abnormal light conditions on axial elongation9,10 were concerned with effects on scleral11,12,13,14 and choroidal mechanisms13,14,15. Roles for GABA16, glucagon17,18,19 and dopamine20 in refractive error development have also received extensive investigation, though few studies have explicitly considered how such theories could physiologically explain the large eyes that are the hallmark of clinical myopia or the source of the rapid changes in axial length in humans21,22,23,24,25,26,27 as a result of 30 min of prolonged accommodation or water drinking. In animal models of myopia, rapid axial elongation, refractive change and altered gene expression28 is seen following 6 h of − 10D optical defocus in chicks or within 30mins of removal of form deprivation29; see Fig. 1.

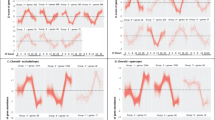
Biometric measurements of normal and form deprived (FD) chicken eyes following induction and recovery from form deprivation myopia. Chickens were visually deprived by occlusion of their right eye then given a variable number of hours of normal visual experience (T = 0 h, 6 h, and 24 h). At each timepoint (n = 8), biometric measures were taken including (a) refraction, (b) axial length, (c) vitreous chamber depth (VCD) and (d) anterior chamber (AC) length. Note: VCD is measured from the posterior surface of the lens to the fibre layer of neural retina. AC length is measured from the anterior surface of the lens to the cornea. For AL measurements of specific chicks utilised in the RNA-seq analysis, refer to Supplementary Table 9.
One such physiological model of myopia development based on very well established retinal/RPE physiology and extensive literature relating to rapid light/dark induced fluid shifts in the retina29,30,31,32,33 has been proposed by Crewther34 as the Retinal Ion Driven Efflux (RIDE) model of myopia. This model postulates that occlusion or acute blur perturbs phototransduction and hence slows the rate of exchange of ions and fluid between photoreceptors, sub-retinal space (SRS) and retinal pigment epithelium (RPE), and consequently attenuates the normal chloride anion driven efflux of fluid from the vitreous across the retina/RPE30,33,35,36,37 to choroid. This model is well supported by ultrastructural evidence38 showing edema in the retina and extensive hyperosmolar stress in the nuclei, mitochondria and basal membranes of the Retinal Pigment Epithelium (RPE)22,29,39. Liang et al.29,39 and Crewther et al.22 have also employed Scanning Electronmicroscopy with elemental microanalysis to demonstrate increased potassium, sodium and chloride ion concentrations across the posterior eye following form deprivation. Thus we aimed to investigate if the biochemical pathways identified by RNAseq transcriptomics during induction of FDM and short term recovery would refute or add any support for the RIDE model theory.
To date, molecular research into gene models of myopia induction have been dominated by human genome wide association studies (GWAS)8,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57 and limited pathway analysis with subsequent transcriptome analyses of single differentially expressed genes (DEGs)58,59,60,61,62,63,64,65,66,67,68,69,70,71 or proteins72,73,74,75,76,77,78,79,80,81,82,83. The lack of consistent results for the above studies could largely arise due to the stability of non-profound refractions of humans in many of the GWASs and the use of differing species, ocular tissues, durations of form deprivation or hours of optical defocus and relative state of refractive compensation. However, more recent use of comparative pathway analysis techniques, and threshold free pathway analysis of multiple gene sets (ie gene set enrichment analysis (GSEA)) in particular have begun to provide more useful insights highlighting dysregulation in several fundamental biological pathways28. In particular, form-deprivation (FD) myopia has now been associated with molecular mechanisms of suppression of neuron structure/growth, signal transduction and inner retinal neurotransmitters associated with Cl− ion transport63,70, while transcriptomic changes associated with early optical induction of myopic and hyperopic refractive errors also highlight altered metabolic pathways in response to lens induced myopia (LIM)69.
The primary aim of this study was to extend our previous transcriptome microarray study of the differentially affected genes and biochemical pathways during prolonged chick FDM and the early hours of recovery, by using next-generation RNA sequencing technology. This technique possesses greater theoretical sensitivity to further elucidate the molecular pathway underpinning the axial length changes in FDM and during recovery than microarray technology. We have aimed to assess expression of a priori identified gene sets and biological pathways associated with myopia using GSEA rather than individual differentially expressed genes (DEGs) (Reviewed in28). The RIDE model and previous molecular work has suggested that gene families associated with cone phototransduction and outer and inner retinal solute transport and energy metabolism, should characterize pathways likely to be associated with increased ocular growth during the induction of form-deprivation myopia (FDMI) and slower growth rates during refractive recovery (FDMR) following reintroduction of normally modulated light conditions.
Material and methods
Animals
Thirty-two male chicks (Leghorn/New Hampshire) were raised with unlimited food and water in a controlled environment on a 12-h light/12-h dark cycle and with the temperature maintained at 30 ± 0.5 °C. Illuminance was maintained at 183 lux during the 12 h day cycle (from 8am to 8 pm) using a 20 W halogen lamp. Twenty-four chicks were monocularly occluded on post-hatch day 5 for 7 days to induce FD myopia. The translucent polystyrene occluders were attached to the periocular feathers of the right eye. On day 12 post-hatch, occluders were removed from FD chicks at 10am and animals were either immediately sacrificed, ie 0 h recovery (n = 8), or sacrificed after 6 h (ie at 4 pm; n = 8) or 24 h (n = 8) of the normal light/dark condition (ie 10 h-light/12 h-dark condition and 2 h light to make up 24 h) following the prolonged form-deprivation. Eight separate chicks were included in the analysis as non-occluded controls at the T = 0 h timepoint.
We chose to use a separate batch of control animals rather than use the within subject contralateral fellow eyes. This decision was based on prior evidence showing binocular changes in choroidal blood flow during monocular occlusion84,85 and similar direction significant changes in refraction and axial length in the fellow eyes14. Binocular interaction effects have been suggested to account for the substantial differences in gene expression as observed by He et al.86 between contralateral eyes and separate controls (see also75,87,88, hence we have chosen to use separate control animals for these reasons. All animal work in this study was approved by the La Trobe University Animal Ethics Committee (Approval number AEC 11/68) and is in accordance with the ARVO Guidelines for Use of Animals in Research, Australian NHMRC Animal Ethics requirements and the ARRIVE guidelines89.
Ocular biometrics analysis
Refractive state (dioptres (D)), vitreous chamber depth (VCD in mm), axial length (AL in mm) and anterior chamber (AC in mm) measures were collected from all animals on day 12 post-hatch and after 0 h recovery (n = 8), 6 h recovery (n = 8) and 24 h recovery (n = 8) while animals were lightly anesthetized with an intramuscular injection of a mixture of ketamine (45 mg/kg) and xylazine (4.5 mg/kg). Refraction in the experimental right eyes were determined by trained ophthalmic practitioners using retinoscopy (Keeler, Vista Diagnostic Instruments) and A-Scan ultrasonography (A-Scan III, TSL; Teknar, Inc. St Louis, USA; 7 MHz probe) was used to measure axial dimensions. Baseline biometric measures were not sought to avoid repeated potential anaesthesia effects on eye growth67, RNA quality and sequencing90,91,92,93. Chicks were only sedated prior to decapitation. Quantitative data were expressed as Means (+ /− Standard Error). Analysis of Variance (ANOVA) measuring group differences was carried out between same age controls, form-deprived and recovery eyes to determine significant changes in biometric measurements followed by post-hoc tests as required.
RNA isolation and cDNA library preparation
Tissue samples for RNA isolation and sequencing were collected from right eyes only, from four out of eight chicks per time condition. These four chicks were chosen based on comparable axial length measurements (see Supplementary Table 13). Total RNA was isolated from posterior eye retina/RPE/choroid tissue using the Trizol method and the RNeasy mini kit (Qiagen) with the on-column DNA digest according to manufacturer’s instructions. RNA quality was measured using the Agilent 2100 Bioanalyzer (RNA 6000 Nano Kit; Agilent Technologies, Santa Clara, CA, USA). All samples had a RNA integrity number (RIN) above 8.7. RNA was also quantitated on the Qubit 2.0 Fluorometer (RNA HS kit; Invitrogen, Australia). For library preparation, RNA concentration was calculated using an average measure from the Bioanalyzer and Qubit assays. A total of 2.5 µg of mRNA was purified from total RNA using oligo (dT)-conjugated magnetic beads (Illumina, San Diego, CA, USA). The fragmented mRNA was then subjected to cDNA synthesis using the TruSeq Stranded mRNA kit (Illumina, San Diego, CA, USA) following the manufacturer’s low-sample throughput protocol. All cDNA libraries were assessed on an Agilent 2100 Bioanalyzer (DNA 1000 kit; Agilent Technologies, Santa Clara, CA, USA). Size of the final products were approximately 260 bp. cDNA libraries were quantitated using 3 methods: Bioanalyzer (DNA 1000 kit; Agilent Technologies, Santa Clara, CA, USA), Qubit (dsDNA HS assay; Invitrogen) and qPCR (GeneRead Library Quant Array; Qiagen, Germantown, MD, USA). DNA libraries were normalised to 10 nM with Tris–HCl 10 mM, pH 8.5 with 0.1% Tween 20 and pooled. Libraries were denatured with 0.1 N NaOH. 7 pM of denatured libraries were prepared for cluster generation on the Illumina cBot using the TruSeq SR Cluster Kit V3-cBot (Illumina, San Diego, CA, USA). The sequencing run involved a duel-index, single-end sequencing run of 1 × 100 cycles on an Illumina HiSeq 1500 using the TruSeq SBS Kit V3 (Illumina, San Diego, CA, USA) sequencing reagents. Average mapped counts for each sample were 6,814,421 (Supplementary Table 8). RNA-Seq data for each sample are available at the NCBI Gene Expression Omnibus (accession number GSE80327).
Sequencing data pre-processing
Quality of the sequencing data was assessed with fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequences with Q score < 10 along with adapters were removed using CutAdapt94 and Trimmomatic95. Sequence reads were mapped to the chick genome (GalGal4) using TopHat296,97 and Bowtie298. Htseq-counts were used to count the number of reads that uniquely mapped to a gene (Supplementary Table 10).
Data analysis
Data were analysed to identify differentially expressed genes (DEGs) and enriched pathways in a pairwise manner using a moderated t-test following prolonged occlusion (0 h vs Control) and during the 24 h recovery period (6 h vs 0 h, 24 h vs 6 h, and 24 h vs 0 h). Additionally, DEGs and enriched pathways across the entire recovery period were identified using a moderated F-test and the Gene Set Enrichment Analysis Pearson’s correlation metric, respectively. Specific details for each step in the analysis are outlined below.
Differential gene expression
Data was filtered to only include genes with at least 10 counts (or 1/million) in at least 4 out of 12 samples. DEGs were calculated using EdgeR99 as implemented in Degust (http://degust.erc.monash.edu). We assessed for DEGs after prolonged occlusion (0 h vs Control) and during the 24 h recovery period (6 h vs 0 h, 24 h vs 6 h, and 24 h vs 0 h) using a moderated t-test with a false discovery rate (FDR) of 5%. To determine the number of DEGs over the 24 h recovery period (0 h, 6 h and 24 h), we used a moderated F-test (i.e. ANOVA) with FDR of 5%. Functional classifications of DEGs were characterised using the Protein Analysis Through Evolutionary Relationships (PANTHER) Classification System (v13.1; http://www.pantherdb.org/)100.
Pathway enrichment analysis
Broad Institute’s Gene Set Enrichment Analysis (GSEA) software was used to determine whether an a priori defined set of genes is statistically significant101,102 during the induction and recovery of FD. The ‘CP: Canonical Pathway' gene sets were obtained from the Molecular Signatures Database (MsigDb v6.2; http://software.broadinstitute.org/gsea/msigdb/index.jsp)101,103. This collection of pathways includes curated gene sets from various online pathway databases, the biomedical literature, and contributions from domain experts. The four main databases comprising this collection include KEGG (http://www.genome.jp/kegg/pathway.html), Pathway Interaction Database (http://pid.nci.nih.gov/), Reactome (http://www.reactome.org/) and the Signal Transduction Knowledge Environment (STKE)104. Chick Ensemble Gene IDs were mapped to human gene symbol using BioMart (Ensembl release 89; Supplementary Table 11). Where mapping resulted in no known gene, Chick Ensemble Gene IDs were mapped to human gene orthologues (Homo sapiens GRCh38.p7). The conversion of Chick IDs to human gene symbols was done as the MsigDb and associated genesets that are based on human gene annotations.
The GSEA technique involves ranking all genes within a sample dataset based on their differential expression between two experimental groups using a basic metric e.g. signal-to-noise ratio (S2N), ratio of average expression from two classes (Ratio), T-test statistic (T-test), or the Pearson correlation coefficient for quantitative studies101,102,105. The method then evaluates the general differences in the cumulative distribution in expression of genes in a biological pathway based on a priori knowledge of the gene’s biological function (i.e., gene sets from the Molecular Signatures Database (MSigDb)). An enrichment score (ES) for each gene set is calculated. This ES reflects the degree to which a geneset is overrepresented at the top or bottom of a ranked list of genes. The ES is calculated by walking down the ranked list of genes, increasing a running-sum statistic when a gene is in the gene set and decreasing it when it is not. The ES for each pathway reflects the maximum deviation from zero encountered in walking down the list. A normalised enrichment score (NES) is also calculated by GSEA in which differences in pathway size (i.e. geneset size) are considered, allowing for comparisons between pathways within the analysis101,102. In this study, the default Signal2Noise metric was used to determine significantly enriched pathways following prolonged occlusion (0 h vs Control) and during the 24 h recovery period (6 h vs 0 h, 24 h vs 6 h, and 24 h vs 0 h). This metric uses the difference of means scaled by the standard deviation106. For comparisons purposes, the Pearson’s correlation metric was used as recommended for time-series data106, to assess changes in gene expression associated with the entire recovery period post occluder removal (i.e., 0 h vs 6 h vs 24 h). The analysis involved 1000 gene set permutations with gene sets limited to 15–500. As recommended by The Broad Institute106 for exploratory studies, a FDR of 25% was used for all bioinformatic analyses.
To understand which genes contributed to the gene set’s enrichment signal, we performed leading-edge subset (LES) analysis. This analysis identifies the core genes by creating a ranked list. The genes located at the top of the ranked list are considered to be upregulated and those at the bottom of the list are downregulated101.
Gene validation
As the primary focus of this study was to uncover the molecular pathway underpinnings of FD myopia, validation of the large number (> 300) of core genes identified from this study using qPCR was not feasible to assess. Instead we aimed to validate the large number of core genes using our previously published microarray study of short term refractive recovery in chick eyes following 10 days of form deprivation70. There is also widespread agreement in the literature that highlights good concordance in gene expression between microarray and qPCR methods107,108,109,110,111,112. Briefly, in our previous microarray study70, RNA from retina/RPE/choroid was isolated from the posterior eye cup of form deprived chicks under the same experimental parameters as described above. High quality RNA from each animal (control and experimental) was pooled in equimolar amounts by experimental condition (control (n = 5), 0 h (n = 5), 6 h (n = 5) and 24 h (n = 5) recovery time) and sent to the Australian Genome Research Facility Ltd (Walter and Eliza Hall Institute, Victoria, Australia) for microarray processing (Affymetrix, Inc). Raw data was exported as CEL files containing probe level intensities for preprocessing with Expression Console 1.1 (Affymetrix, Inc). The raw data was summarised and normalised using the Robust Multichip Average (RMA) algorithm to yield log base 2 expression values for each gene transcript. This data can be found on the GEO Database (www.ncbi.nlm.nih.gov/geo/; accession number GSE89325).
Results
Ocular biometrics analysis
Refractive state (dioptres (D)) was assessed using standard retinoscopy while vitreous chamber depth (VCD in mm), axial length (AL in mm) and anterior chamber depth (AC in mm) were assessed using A-scan ultrasonography (Fig. 1). Significant myopia (− 18.6D ± 1.44) was achieved after 7 days of occlusion. Six hours of visual experience induced a significant hypermetropic shift of + 7.3D, reducing the mean level of myopia in FD eyes down to -11.3D ± 0.91. Less refractive compensation was seen after 24 h of visual recovery post-occluder removal (− 16.4 ± 1.18D), presumably due to the circadian impact of the regular night period with this measurement being made 2 h after the first 12 h night/dark period (Fig. 1a).
Axial length (AL), vitreous chamber depth (VCD) and anterior chamber (AC) showed similar patterns of biometric growth consistent with previously published diurnal rhythm changes113,114,115. Prolonged occlusion increased axial length from 9.32 mm for controls to 10.30 mm (Fig. 1b), VCD from 5.65 mm to 6.21 mm (Fig. 1c) and AC (Fig. 1d) from 1.53 to 1.83 mm. In recovering eyes, AL decreased by 0.12 mm at both 6 h and 24 h, however these changes were not significantly different to AL at occluder removal (T = 0 h; p > 0.05). The same trend was also seen in VCD and AC at 6 h with VCD reduced by 0.08 mm and AC by 0.04 mm indicating that the excessive ocular growth response to FD is inhibited by occluder removal and that normal light conditions favour re-emmetropisation.
Differentially expressed genes in prolonged FD and FD recovery
To identify the transcriptomic mechanisms involved in the response to, and recovery from FD, single-gene expression changes were analysed. We first assessed DEG after prolonged occlusion (i.e. 7d of FD with 0 h recovery) and during recovery (6 h vs 0 h, and 6 h vs 24 h) using EdgeR with a moderated t-test and a false discovery rate (FDR) of 5%.
Prolonged occlusion, relative to no-lens controls, induced expression changes in 13 genes with an FDR < 5%. BMP2, ALDH1A2, TNC, SHC4, GSN, SIK1 were down-regulated at 7d of FD relative to controls, while LOC417800, HCK, WNT9A, KIAA1199, CLEC3B, SLCO1C1 and RHOB were upregulated in the FD eye compared to controls (Supplementary Table 1). By comparison, recovery from FD identified 439 transcripts at 6 h compared to 0 h, 386 transcripts between 6 and 24 h, and 216 transcripts at 24 h compared to 0 h (Supplementary Tables 2–4). To provide a comparison with past transcriptomic studies on chick FD myopia and early recovery63,70 we analysed all time-points (0 h, 6 h and 24 h) using a moderated F-test which identified a total of 828 transcripts significantly differentially expressed across the 24 h period of normal day/night conditions following 7 days of form deprivation (Supplementary Table 5). The number of overlapping and unique transcripts identified is illustrated in Fig. 2 with the majority of these gene transcripts involved in cellular and metabolic processes (Fig. 3).

Venn diagram of overlapping and unique transcripts for all comparison time points. Significant (p < .05) transcripts were identified using EdgeR. (a) 11 of the 13 differentially expressed transcripts were common in both prolonged occlusion (7d of FD) and during the 24 h recovery period. Only 2 transcripts were uniquely expressed in response to prolonged occlusion and 733 transcripts were uniquely expressed in the 24 h recovery from FD. (b) There were 744 unique transcripts significantly expressed throughout the recovery period with 226 transcripts uniquely expressed after 6 h recovery compared to 0 h, 174 transcripts uniquely expressed after 24 h recovery compared to 6 h, 63 transcripts uniquely expressed after 24 h compared to 0 h and 16 transcripts common across all 3 recovery time-point comparisons.

Gene Ontology (GO) classification of differentially expressed genes (DEG) after prolonged occlusion and during early recovery from form-deprivation myopia. Significant (p < .05) DEGs were identified using EdgeR between (a) No-Lens Control and prolonged form-deprivation myopia (FDM; ie 0 h recovery) (b) 0 h and 6 h recovery, (c) 6 h and 24 h recovery, and (d) overall recovery (0 h, 6 h and 24 h). After 7 days of FD with 0 h recovery, the DEG were mostly involved in cellular processes and response to stimulus. There are few DEGs associated with metabolic and immune system processes after prolonged occlusion compared to controls possibly indicating a new homeostatic state. However, the genes identified during recovery were mostly involved in biological regulation, cellular and metabolic processes along with the reinstatement of immune system processes. For names of individual DEGs see Supplementary Table 8.
Enriched pathways during prolonged FD
Following the identification of the DEGs during induction and recovery from FD, GSEA was employed to identify changes in expression of biological pathways associated with 7 days of FD relative to normal, no lens controls. Prolonged occlusion resulted in suppression of only one pathway, ligand-gated ion channel transport (Table 1; Fig. 4). The leading-edge subset (i.e., core genes) of this pathway implicate suppression of several GABA ionotropic receptors (ie GABAA and GABAA-rho receptors (previously known as the GABAC receptors)) and glycine receptors. Such receptors are associated with Cl− channels116,117 and would be expected to play a role in transretinal fluid movement from vitreous to choroid30,31,34. The core genes identified in the ligand gated pathway show good concordance with gene expression from our previous microarray study using the same experimental paradigm (Supplementary Table 12).

Bubble plots showing all significant pathways from the pairwise GSEA comparisons during the induction of myopia and the first 6 h of recovery from FDM. The significantly enriched pathways are visualised for (a) 0 h versus No-Lens Control, (b) 6 h versus 0 h, and (c) 6 h versus Control. The size of each bubble is proportional to the number of core genes within the pathway. Full details of significant pathways (including core genes driving enrichment) are provided in Supplementary Tables 1–4. NES, Normalised Enrichment Score.
Enriched pathways during recovery from FDM
Pathway changes during recovery period were evaluated using the Signal2Noise metric by comparing gene expression changes between each recovery time-point (6 h vs 0 h, 24 h vs 6 h, and 24 h vs 0 h). The most significantly altered pathways identified in this analysis implicate early cone phototransduction, mitochondrial bioenergetics, retinol metabolism, complement and glucagon in early recovery from FD and in the myopia pathophysiology (Fig. 6). As expected, the data in Fig. 4 suggests that cone receptor phototransduction is still suppressed after only 6 h normal visual experience, i.e. cone phototransduction pathway has not totally recovered its control levels during the first 6 h of visual experience and recovery after occluder removal. An earlier electrophysiological study38 also found reduced cone sensitivity immediately following prolonged FD which would support such gene pathway suppression in early (6 h) and prolonged recovery (24 h). Indeed, our results comparing gene expression changes between each recovery time-point of 6 h and 24 h also indicate that the pathways underlying cone phototransduction were significantly upregulated when evaluated using the Signal2Noise metric as demonstrated in Figs. 4 and 5 and in Table 2. Interestingly, while transcriptomic changes in mitochondrial metabolism are absent during the first 6 h of recovery, increased mitochondrial metabolism is observed over the next 18 h (Fig. 5; Table 2) after a 1 day/night cycle has been completed. These changes occur in parallel to the increase in expression of the high-energy consuming Na+-Ca2+-K+ exchanger (NCKX2, also known as SLC24A2), which is characteristic of the dark-adapted retina118 (Fig. 6).

Bubble plots showing all significant pathways from the pairwise GSEA comparisons following 24 h of recovery from FDM. The significantly enriched pathways are visualised for (a) 24 h versus 0 h, (b) 24 h versus Control and (c) 24 h versus 6 h. The size of each bubble is proportional to the number of core genes within the pathway. Full details of significant pathways (including core genes driving enrichment) are provided in Supplementary Tables 1–4. NES, Normalised Enrichment Score.

Chord diagram showing the core genes of significant pathways in FDMR 0 h versus control, FDMR 6 h versus 0 h and FDMR 24 h versus 6 h as different annuli of genes. Significant pathways are shown on the right, and the fold change of core genes is shown on the left. Left–right connections indicate gene membership in a pathway’s leading-edge subset. PA, phosphatidic acid. Note: Image constructed using GOplot R package157.
To determine if the pathways identified during FD recovery using the Signal2Noise metric produced similar pathway findings to the Pearson’s correlation metric, the data was then analysed (using GSEA) for pathways correlated to recovery time (0 h, 6 h, 24 h). Twenty-nine pathways were found to be significantly altered during the 24 h recovery period with the top 3 pathways involved in the complement cascade, mitochondrial energy metabolism and retinol metabolism (Supplementary Table 7). Other pathways identified by Pearson’s correlation have roles in immunity, lipid metabolism, smooth muscle contraction, apoptosis, protein metabolism, signal transduction, golgi transport, extracellular matrix (ECM), hemostasis and disease. Indeed, the findings from both the Signal2Noise analysis and Pearson’s correlation suggests that these metrics can produce comparable results.
Discussion
This study has utilized pathway enrichment analysis (GSEA) to identify biological pathways shown by RNA seq to be involved in profound myopia induced by 7 days of FD and during the first 24 h of early recovery in a normal modulated light environment. Our findings raise intriguing questions regarding the temporal interaction of transcriptome changes and previously described ionic, physiological and morphological changes associated with induction and recovery from FDM22,29 and theoretical models of myopia.
GABA and glycine mediated chloride ion transport pathways significantly altered in profound FDM
Consistent with the RIDE model, GSEA identified suppression of the GABA- and glycine-mediated chloride ion transport pathway as the only significantly altered biochemical pathway associated with 7 days of excessive ocular growth, thinning of the retina and choroid and altered ion distribution patterns accompanying profound refractive myopia29. The fact that the only pathway showing significant change after 7 days of FDMI was the chloride ion transport pathway is particularly important to understanding development of overly large volume eyes, which is the hallmark sign of myopia. As extensively studied since the early 70s119 and Reviewed in120, chloride channels reside in all plasma membranes and most intracellular organelles and are fundamental to all cell volume regulation, transepithelial transport and regulation of electrical excitability in general. Hence suppression of the Cl- channel pathways across the entire retina/RPE/choroid examined with RNAseq provides an explanation for the ultrastructurally described dehydration seen in FDM tissue in profoundly myopic eyes22,29,39.
FDM recovery pathway changes – cone phototransduction first
By comparison with the stable conditions during prolonged occlusion, GSEA of RNAseq data showed rapid upregulation of many cellular processes but particularly upregulation of cone phototransduction and BARD1 expression following occluder removal and reintroduction of only 6 h of normally modulated light conditions. Within 24 h post occlusion, the new molecular state of retina/RPE/choroidal tissue was characterized by further upregulation of cone phototransduction consistent with the predictions of the RIDE model34. Our RNAseq findings also validate previous electrophysiological finding of cone photoreceptor sensitivity38 as well as our previous transcriptome work in chick using FDM70, and optical defocus (+ 10D and -10D) lenses69,83.
Upregulation of BARD1 at 6 h recovery
Upregulation of the BARD1 pathway in the first 6 h of normal light conditions highlights that prolonged occlusion, profound myopia and the morphologically thinned choroid is persistently physiologically stressful to cellular function121,122. Over the last few years the BARD1 pathway has been reported to play a central role in the control of the cell cycle in response to DNA damage in many diseases and its regulation has been shown to mediate the formation of 'Lys-6′-linked polyubiquitin chains and coordinate a diverse range of cellular pathways such as DNA damage repair, histone ubiquitination and transcriptional regulation to maintain genomic stability121,122,123. The presence of BARD1 post profound FDM may be an early indicator of the types of cellular damage observed in human and animal myopia, especially with regard to thinning of the choroid and retina. This may explain why high myopia is a risk factor for secondary ophthalmic diseases later in life.
Implications of suppressed ligand-gated chloride channel pathways for ocular growth
Significant suppression of the ligand gated chloride channel pathway following prolonged FDM is compatible with the notion that increases in axial length and thinning of the retina and choroid in response to FD occur concurrently with changes in the distribution of physiologically important ions including chloride, sodium, potassium and calcium across the posterior eye22. In a review of Ion Channels of the RPE, Wimmers et al.33 similarly to Gallemore et al.30, and Crewther34, have highlighted the role of Cl− and K+ ions as the key drivers of transepithelial water transport and volume regulation in the retina and the interaction of the photoreceptors and the RPE. The established role of GABA and glycine in retinal third order neuronal transmission and in Cl− transport116,117 would also suggest that hydrated Cl− ions could play a particularly important role in axial growth during FD in animals69,70,83 and human myopia8. Indeed suppression of the GABA and glycine ligand-gated chloride ion-transport pathway in response to FD (Table 1) is also consistent with previous reports of significantly reduced retinal concentrations of GABA in chick FDM16,124 and GABA signal following abnormal axial growth in FD guinea pigs71. A number of pharmacological studies have also utilized GABA antagonists (e.g. TPMPA) to inhibit the response to FD by inhibiting axial elongation and vitreous chamber depth16,125,126,127,128. Downregulation of GABA and glycine in retinal tissue has also been seen in normally growing chicks over a 48 h period129, whereas the same study observed an increase in GABA signaling proteins in the same 6–48 h period when chicks wore negative lens wear and during which refractive compensation was achieved129.
Ionotrophic GABA receptor distribution in the posterior eye
Interestingly GABA receptors, that play such an important role in major third order neuronal transmission130, are also found in abundance on RPE cells131,132 suggesting that suppression of such ligand-gated chloride channels in RPE and most retinal cells133 during FD would be expected to reduce the transretinal fluid efflux towards choroid and result in rapid increase of fluid in the vitreous and axial elongation. Furthermore, as the majority of GABA receptors identified in Table 1 are ionotropic, the role of ion homeostasis, particularly K+ and Cl−, in the development of axial elongation and myopia cannot be ignored. This is particularly so given the substantial evidence that fluid flow across the RPE is related to the ionic environment of the retina/RPE/choroid30. Spatial distribution of ions using X-ray microanalysis has revealed significant differences in the distribution of ions of phototransduction (Na, K, Cl, Mg and Ca) as well as other physiologically relevant ions (PO43− and SO42−) known to be important for cellular functioning and structural integrity22. Taken together, these findings offer strong support for the role of light, cone phototransduction, RPE mechanisms, ion regulation and neurotransmission in driving myopia development and in no way refute the predictions of the RIDE model34.
Metabolic recovery following reintroduction of temporally modulated light
Recovery from refractive myopia and reduced axial, vitreal and anterior chamber growth rates seen during the first 24 h following FD removal appear to be associated with both cone-dominated phototransduction and increased mitochondrial metabolism, especially at 6 h and 24 h post FD. These findings are consistent with our earlier FD microarray study70 and RNAseq study of refractively compensated negative lens-induced myopia in chick69,134. Interestingly, mitochondrial respiratory electron transport chain genes have also been identified in a human study of genetic myopia135, highlighting the important contribution of mitochondrial bioenergetics and the phototransduction cascade to ocular function and myopia development. This data is also in line with previous ultrastructural evidence of abnormal photoreceptor elongation, loss of mitochondrial integrity29,39,136, and oxidative stress70 during FD and subsequent reversal of most morphological changes in the 48 h following occluder removal in chick29,137,138. Indeed, our earlier microarray study70 indicated that mitochondrial respiratory complex 1 and 3 genes and mitochondrial reactive oxygen species (mROS)139 are upregulated during FDMI. The release of mROS has previously been suggested to be a response to cellular stress while coincidently acting as a signalling molecule to facilitate cellular adaptation to this stress140. In fact, mROS may upregulate cone photoreceptor pathways and hence neurotransmission in inner retina141, chloride transport and homeostasis142 which are the main processes we have reported previously following termination of occlusion and re-established normal light conditions22,29. Thus, the increase in mitochondrial electron transport chain mRNA seen 24 h after occluder removal (Fig. 5; Table 2) fits with the idea that the dark-adapted (low temporal luminance modulation) retinae require ~ 20% more metabolic activity than the same light-adapted retina143. This is not unexpected given that the FD retina has previously been shown to be in a pseudo dark-adapted state37 and associated with the re-accumulation of K+ in the subretinal space and exclusion of fluid under conditions of low temporal luminance modulation22. Importantly, the ATPase mechanism of the RPE apical surface is electrogenic and modulates the transepithelial potential which is closely related to the control of fluid flow across the RPE and so would be expected to be upregulated during form deprivation, lower in the first 6 h of normal light and then upregulated again during the ensuing night34,144.
Immune pathways in recovery
The presence of immune related pathways in the recovery from the physiologically stressful FD condition and the upregulation of the BARD1 pathway suggest a role for the immune system in refractive compensation. Such an association has recently appeared in regards to the relation of clinical blood counts as measures of immune responses and inflammation and high myopia145,146,147. The identification of the complement pathway during recovery from FD is consistent with our earlier transcriptomic studies in chick70,134 and support previous well-described links between mitochondrial bioenergetics and the complement system in the body and in the eye148,149,150 and in many neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Amyotrophic Lateral Sclerosis (ALS)151.
Complement factors have also been associated with previous reports of human and animal refractive errors70,134,152,153 and should be expected in animal models of FD given the severe choroidal thinning and associated ultrastructural and ionic changes seen in FD eyes post occlusion22,29. These changes have also been demonstrated in choriocapillaris fenestration number, choroidal blood vessel permeability in chicks recovering from FDM154 and seen in oncosis155,156. The previously described increase in extravascular space edema22,29 and lymphatic vessel permeability154 immediately post-occlusion also supports our results here as well as the recent FD transcriptomics70. What was not expected is the involvement of other immune pathways besides complement in FD recovery.
Limitations
Lastly, while our decision to use retina/RPE/choroid may be considered a limitation, we contend that investigation of the combined changes in expression of genes within the entire biological network of the eye is necessary to understand the photoreceptor induced temporal changes in form deprivation myopia seen in animal models. Indeed, when evaluated experimentally in combination with systematic system neuroscience review techniques, the impact of using inner and outer retina, RPE and choroid in large scale genomic and proteomic studies supports the idea that similar biological mechanisms are associated with FD and lens induced defocus, regardless of the varying combinations of tissues used28. Indeed, Fig. 5 illustrates the commonality of the biological processes that underlie the adaptive responses to environmental manipulation of temporal modulation of luminance information across the multiple tissue types of the posterior eye. Thus, we argue that the use of multiple tissues is not an impediment to our GSEA-based interpretation as evidenced by the robustness of our current findings with previous research in human and animals. Furthermore, GSEA has proven a useful tool in identifying significantly enriched biological pathways in large-scale ‘omic’ studies. However we also acknowledge that large-scale discovery-driven studies may produce false positives. Additionally the limited gene sets and the redundancy in the databases currently available in the Molecular Signature database may affect future interpretations of this data. Future studies may benefit from using a wider range of databases as well as re-analysing data periodically to account for updated gene and pathway information. Additionally, we have not considered the role of circadian rhythms of ocular refraction and gene expression, particularly with regard to the differences in refractive normalization seen at 6 h compared to 24 h recovery from FD. We do not have a circadian matched control for our 6 h recovery group and acknowledge that this may be a limitation in our study. Indeed, 2 circadian-related pathways (RORA Activates Circadian Expression and Circadian Repression of Expression by REV-ERBa) were highly expressed at 6 h recovery compared to the no lens control however this result should be taken with the understanding that the no lens control is not a circadian matched control for the 6 h recovery timepoint. As such, we cannot conclude if circadian rhythms have an influence on refractive development and myopia. However, our no lens controls, 7 days induction, and 24 h recovery groups all experienced the same day/night patterns with tissue collected at approximately 10am. No circadian effects were highlighted suggesting that circadian rhythms effects on gene expression will be minimal and as such, may not be a factor in refractive development and myopia. Further studies should incorporate circadian matched controls to minimise circadian rhythm effects on gene expression.
Conclusions
We conclude that axial myopia is an adaptive response to the environmental perturbation of the visual signal cascade (i.e. Phototransduction). This perturbation in phototransduction consequently affects the ionic environment of the photoreceptors, subretinal space and inner retina as predicted by the RIDE model via GABA and glycine pathway signalling and consequent effects on ligand-gated chloride channels and their role in transretinal fluid flow. Our identification of the different mitochondrial bioenergetic profiles observed between FDMI and FDMR could be considered as a potential molecular hallmark of the myopia condition. Most importantly our findings are also consistent with the predictions of the theoretical implications of the RIDE hypothesis highlighting the importance of light driven osmoadaptive pathways, enhanced mitochondrial bioenergetics and cellular immune responses, during the development of FD myopia and during refractive recovery.
Data availability
The datasets generated during the current study are available in the Gene Expression Omnibus (GEO) repository (https://www.ncbi.nlm.nih.gov/geo/; Accession # GSE80327).
References
Dolgin, E. The myopia boom. Nature 519, 276–278. https://doi.org/10.1038/519276a (2015).
Saw, S. M. et al. Factors related to the progression of myopia in Singaporean children. Optom. Vis. Sci. 77, 549–554 (2000).
Holden, B. A. et al. Global prevalence of myopia and high myopia and temporal trends from 2000 through 2050. Ophthalmology 123, 1036–1042. https://doi.org/10.1016/j.ophtha.2016.01.006 (2016).
Lee, Y. Y., Lo, C. T., Sheu, S. J. & Lin, J. L. What factors are associated with myopia in young adults? A survey study in Taiwan military conscripts. Investig. Ophthalmol. Vis. Sci. https://doi.org/10.1167/iovs.12-10480 (2013).
Sun, J. et al. High prevalence of myopia and high myopia in 5060 Chinese university students in Shanghai. Investig. Ophthalmol. Vis. Sci. 53, 7504–7509. https://doi.org/10.1167/iovs.11-8343 (2012).
Chen, M. et al. The increasing prevalence of myopia and high myopia among high school students in Fenghua city, eastern China: a 15-year population-based survey. BMC Ophthalmol. 18, 159. https://doi.org/10.1186/s12886-018-0829-8 (2018).
Li, Y., Liu, J. & Qi, P. The increasing prevalence of myopia in junior high school students in the Haidian District of Beijing, China: a 10-year population-based survey. BMC Ophthalmol. 17, 88. https://doi.org/10.1186/s12886-017-0483-6 (2017).
Tedja, M. S. et al. Genome-wide association meta-analysis highlights light-induced signaling as a driver for refractive error. Nat. Genet. 50, 834–848. https://doi.org/10.1038/s41588-018-0127-7 (2018).
Wiesel, T. N. & Raviola, E. Myopia and eye enlargement after neonatal lid fusion in monkeys. Nature 266, 66–68 (1977).
Wallman, J., Turkel, J. & Trachtman, J. Extreme myopia produced by modest change in early visual experience. Science 201, 1249–1251 (1978).
McBrien, N. A., Moghaddam, H. O., Reeder, A. P. & Moules, S. Structural and biochemical changes in the sclera of experimentally myopic eyes. Biochem. Soc. Trans. 19, 861–865. https://doi.org/10.1042/bst0190861 (1991).
Wallman, J. Retinal influences on sclera underlie visual deprivation myopia. Ciba Found. Symp. 155, 126–134 (1990) (discussion 135–141).
Wallman, J. et al. Moving the retina: choroidal modulation of refractive state. Vis. Res. 35, 37–50 (1995).
Wildsoet, C. & Wallman, J. Choroidal and scleral mechanisms of compensation for spectacle lenses in chicks. Vis. Res. 35, 1175–1194 (1995).
Nickla, D. L. & Wallman, J. The multifunctional choroid. Prog. Retin. Eye Res. 29, 144–168. https://doi.org/10.1016/j.preteyeres.2009.12.002 (2010).
Stone, R. A. et al. GABA, experimental myopia, and ocular growth in chick. Investig. Ophthalmol. Vis. Sci. 44, 3933–3946 (2003).
Feldkaemper, M. P., Burkhardt, E. & Schaeffel, F. Localization and regulation of glucagon receptors in the chick eye and preproglucagon and glucagon receptor expression in the mouse eye. Exp. Eye Res. 79, 321–329. https://doi.org/10.1016/j.exer.2004.04.009 (2004).
Fischer, A. J., McGuire, J. J., Schaeffel, F. & Stell, W. K. Light- and focus-dependent expression of the transcription factor ZENK in the chick retina. Nat. Neurosci. 2, 706–712. https://doi.org/10.1038/11167 (1999).
Bitzer, M. & Schaeffel, F. Defocus-induced changes in ZENK expression in the chicken retina. Investig. Ophthalmol. Vis. Sci. 43, 246–252 (2002).
Stone, R. A., Lin, T., Laties, A. M. & Iuvone, P. M. Retinal dopamine and form-deprivation myopia. Proc. Natl. Acad. Sci. U. S. A. 86, 704–706 (1989).
Wallman, J. & Winawer, J. Homeostasis of eye growth and the question of myopia. Neuron 43, 447–468 (2004).
Crewther, S. G., Liang, H., Junghans, B. M. & Crewther, D. P. Ionic control of ocular growth and refractive change. Proc. Natl. Acad. Sci. U. S. A. 103, 15663–15668 (2006).
Gupta, P. et al. Peripapillary choroidal thickness in young Asians with high myopia. Investig. Ophthalmol. Vis. Sci. 56, 1475–1481. https://doi.org/10.1167/iovs.14-15742 (2015).
Woodman, E. C., Read, S. A. & Collins, M. J. Axial length and choroidal thickness changes accompanying prolonged accommodation in myopes and emmetropes. Vis. Res. 72, 34–41. https://doi.org/10.1016/j.visres.2012.09.009 (2012).
Mallen, E. A., Kashyap, P. & Hampson, K. M. Transient axial length change during the accommodation response in young adults. Investig. Ophthalmol. Vis. Sci. 47, 1251–1254. https://doi.org/10.1167/iovs.05-1086 (2006).
Drexler, W., Findl, O., Schmetterer, L., Hitzenberger, C. K. & Fercher, A. F. Eye elongation during accommodation in humans: differences between emmetropes and myopes. Investig. Ophthalmol. Vis. Sci. 39, 2140–2147 (1998).
Chakraborty, R., Read, S. A. & Collins, M. J. Monocular myopic defocus and daily changes in axial length and choroidal thickness of human eyes. Exp. Eye Res. 103, 47–54. https://doi.org/10.1016/j.exer.2012.08.002 (2012).
Riddell, N. & Crewther, S. G. Integrated comparison of GWAS, transcriptome, and proteomics studies highlights similarities in the biological basis of animal and human myopia. Investig. Ophthalmol. Vis. Sci. 58, 660–669. https://doi.org/10.1167/iovs.16-20618 (2017).
Liang, H., Crewther, S. G., Crewther, D. P. & Junghans, B. M. Structural and elemental evidence for edema in the retina, retinal pigment epithelium, and choroid during recovery from experimentally induced myopia. Investig. Ophthalmol. Vis. Sci. 45, 2463–2474 (2004).
Gallemore, R. P., Hughes, B. A. & Miller, S. S. Retinal pigment epithelial transport mechanisms and their contributions to the electroretinogram. Prog. Retin. Eye Res. 16, 509–566 (1997).
Marmor, M. & Wolfensberger, T. J. The Retinal Pigment Epithelium: Function and Disease (Oxford University Press, Oxford, 1998).
Li, J. D., Gallemore, R. P., Dmitriev, A. & Steinberg, R. H. Light-dependent hydration of the space surrounding photoreceptors in chick retina. Investig. Ophthalmol. Vis. Sci. 35, 2700–2711 (1994).
Wimmers, S., Karl, M. O. & Strauss, O. Ion channels in the RPE. Prog. Retin. Eye Res. 26, 263–301 (2007).
Crewther, D. P. The role of photoreceptors in the control of refractive state. Prog. Retin. Eye Res. 19, 421–457 (2000).
Fujii, S., Gallemore, R. P., Hughes, B. A. & Steinberg, R. H. Direct evidence for a basolateral membrane Cl− conductance in toad retinal pigment epithelium. Am. J. Physiol. 262, C374-383. https://doi.org/10.1152/ajpcell.1992.262.2.C374 (1992).
Tsuboi, S. Measurement of the volume flow and hydraulic conductivity across the isolated dog retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 28, 1776–1782 (1987).
Crewther, S. G., Murphy, M. J. & Crewther, D. P. Potassium channel and NKCC cotransporter involvement in ocular refractive control mechanisms. PLoS ONE 3, e2839. https://doi.org/10.1371/journal.pone.0002839 (2008).
Westbrook, A. M., Crewther, D. P. & Crewther, S. G. Cone receptor sensitivity is altered in form deprivation myopia in the chicken. Optom. Vis. Sci. 76, 326–338 (1999).
Liang, H., Crewther, D. P., Crewther, S. G. & Barila, A. M. A role for photoreceptor outer segments in the induction of deprivation myopia. Vis. Res. 35, 1217–1225 (1995).
Hysi, P. G. et al. A genome-wide association study for myopia and refractive error identifies a susceptibility locus at 15q25. Nat. Genet. 42, 902–905. https://doi.org/10.1038/ng.664 (2010).
Li, Y. J. et al. Genome-wide association studies reveal genetic variants in CTNND2 for high myopia in Singapore Chinese. Ophthalmology 118, 368–375. https://doi.org/10.1016/j.ophtha.2010.06.016 (2011).
Li, Z. et al. A genome-wide association study reveals association between common variants in an intergenic region of 4q25 and high-grade myopia in the Chinese Han population. Hum. Mol. Genet. 20, 2861–2868. https://doi.org/10.1093/hmg/ddr169 (2011).
Nakanishi, H. et al. A genome-wide association analysis identified a novel susceptible locus for pathological myopia at 11q24.1. PLoS Genet. 5, e1000660. https://doi.org/10.1371/journal.pgen.1000660 (2009).
Solouki, A. M. et al. A genome-wide association study identifies a susceptibility locus for refractive errors and myopia at 15q14. Nat. Genet. 42, 897–901. https://doi.org/10.1038/ng.663 (2010).
Shi, Y. et al. Genetic variants at 13q12.12 are associated with high myopia in the Han Chinese population. Am. J. Hum. Genet. 88, 805–813. https://doi.org/10.1016/j.ajhg.2011.04.022 (2011).
Fan, Q. et al. Genetic variants on chromosome 1q41 influence ocular axial length and high myopia. PLoS Genet. 8, e1002753. https://doi.org/10.1371/journal.pgen.1002753 (2012).
Meng, W. et al. A genome-wide association study provides evidence for association of chromosome 8p23 (MYP10) and 10q21.1 (MYP15) with high myopia in the French Population. Investig. Ophthalmol. Vis. Sci. 53, 7983–7988. https://doi.org/10.1167/iovs.12-10409 (2012).
Cheng, C. Y. et al. Nine loci for ocular axial length identified through genome-wide association studies, including shared loci with refractive error. Am. J. Hum. Genet. 93, 264–277. https://doi.org/10.1016/j.ajhg.2013.06.016 (2013).
Khor, C. C. et al. Genome-wide association study identifies ZFHX1B as a susceptibility locus for severe myopia. Hum. Mol. Genet. 22, 5288–5294. https://doi.org/10.1093/hmg/ddt385 (2013).
Kiefer, A. K. et al. Genome-wide analysis points to roles for extracellular matrix remodeling, the visual cycle, and neuronal development in myopia. PLoS Genet. 9, e1003299. https://doi.org/10.1371/journal.pgen.1003299 (2013).
Shi, Y. et al. A genome-wide meta-analysis identifies two novel loci associated with high myopia in the Han Chinese population. Hum. Mol. Genet. 22, 2325–2333. https://doi.org/10.1093/hmg/ddt066 (2013).
Stambolian, D. et al. Meta-analysis of genome-wide association studies in five cohorts reveals common variants in RBFOX1, a regulator of tissue-specific splicing, associated with refractive error. Hum. Mol. Genet. 22, 2754–2764. https://doi.org/10.1093/hmg/ddt116 (2013).
Verhoeven, V. J. et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat. Genet. 45, 314–318. https://doi.org/10.1038/ng.2554 (2013).
Simpson, C. L. et al. Genome-wide meta-analysis of myopia and hyperopia provides evidence for replication of 11 loci. PLoS ONE 9, e107110. https://doi.org/10.1371/journal.pone.0107110 (2014).
Miyake, M. et al. Identification of myopia-associated WNT7B polymorphisms provides insights into the mechanism underlying the development of myopia. Nat. Commun. 6, 6689. https://doi.org/10.1038/ncomms7689 (2015).
Fan, Q. et al. Meta-analysis of gene-environment-wide association scans accounting for education level identifies additional loci for refractive error. Nat. Commun. 7, 11008. https://doi.org/10.1038/ncomms11008 (2016).
Hosoda, Y. et al. CCDC102B confers risk of low vision and blindness in high myopia. Nat. Commun. 9, 1782. https://doi.org/10.1038/s41467-018-03649-3 (2018).
Tkatchenko, A. V. et al. Large-scale microRNA expression profiling identifies putative retinal miRNA-mRNA signaling pathways underlying form-deprivation myopia in mice. PLoS ONE 11, e0162541. https://doi.org/10.1371/journal.pone.0162541 (2016).
Tkatchenko, A. V., Walsh, P. A., Tkatchenko, T. V., Gustincich, S. & Raviola, E. Form deprivation modulates retinal neurogenesis in primate experimental myopia. Proc. Natl. Acad. Sci. U. S. A. 103, 4681–4686. https://doi.org/10.1073/pnas.0600589103 (2006).
Tkatchenko, T. V., Shah, R. L., Nagasaki, T. & Tkatchenko, A. V. Analysis of genetic networks regulating refractive eye development in collaborative cross progenitor strain mice reveals new genes and pathways underlying human myopia. bioRxiv https://doi.org/10.1101/530766 (2019).
Tkatchenko, T. V., Troilo, D., Benavente-Perez, A. & Tkatchenko, A. V. Gene expression in response to optical defocus of opposite signs reveals bidirectional mechanism of visually guided eye growth. PLoS Biol. 16, e2006021. https://doi.org/10.1371/journal.pbio.2006021 (2018).
Brand, C., Schaeffel, F. & Feldkaemper, M. P. A microarray analysis of retinal transcripts that are controlled by image contrast in mice. Mol Vis 13, 920–932 (2007).
McGlinn, A. M. et al. Form-deprivation myopia in chick induces limited changes in retinal gene expression. Investig. Ophthalmol. Vis. Sci. 48, 3430–3436. https://doi.org/10.1167/iovs.06-1538 (2007).
Schippert, R., Schaeffel, F. & Feldkaemper, M. P. Microarray analysis of retinal gene expression in chicks during imposed myopic defocus. Mol. Vis. 14, 1589–1599 (2008).
Rada, J. A. & Wiechmann, A. F. Ocular expression of avian thymic hormone: changes during the recovery from induced myopia. Mol. Vis. 15, 778–792 (2009).
Ashby, R. S. & Feldkaemper, M. P. Gene expression within the amacrine cell layer of chicks after myopic and hyperopic defocus. Investig. Ophthalmol. Vis. Sci. 51, 3726–3735. https://doi.org/10.1167/iovs.09-4615 (2009).
Stone, R. A. et al. Image defocus and altered retinal gene expression in chick: clues to the pathogenesis of ametropia. Investig. Ophthalmol. Vis. Sci. 52, 5765–5777. https://doi.org/10.1167/iovs.10-6727 (2011).
Metlapally, R. et al. Genome-wide scleral micro- and messenger-RNA regulation during myopia development in the mouse. Investig. Ophthalmol. Vis. Sci. 57, 6089–6097. https://doi.org/10.1167/iovs.16-19563 (2016).
Riddell, N., Giummarra, L., Hall, N. E. & Crewther, S. G. Bidirectional expression of metabolic, structural, and immune pathways in early myopia and hyperopia. Front. Neurosci. 10, 390. https://doi.org/10.3389/fnins.2016.00390 (2016).
Giummarra, L., Crewther, S. G., Riddell, N., Murphy, M. J. & Crewther, D. P. Pathway analysis identifies altered mitochondrial metabolism, neurotransmission, structural pathways and complement cascade in retina/RPE/ choroid in chick model of form-deprivation myopia. PeerJ 6, e5048. https://doi.org/10.7717/peerj.5048 (2018).
Srinivasalu, N. et al. Gene expression and pathways underlying form deprivation myopia in the guinea pig sclera. Investig. Ophthalmol. Vis. Sci. 59, 1425–1434. https://doi.org/10.1167/iovs.16-21278 (2018).
Barathi, V. A. et al. Involvement of GABA transporters in atropine-treated myopic retina as revealed by iTRAQ quantitative proteomics. J. Proteome Res. 13, 4647–4658. https://doi.org/10.1021/pr500558y (2014).
Bertrand, E. et al. Identification of apolipoprotein A-I as a “STOP” signal for myopia. Mol. Cell. Proteomics 5, 2158–2166. https://doi.org/10.1074/mcp.M600073-MCP200 (2006).
Frost, M. R. & Norton, T. T. Differential protein expression in tree shrew sclera during development of lens-induced myopia and recovery. Mol. Vis. 13, 1580–1588 (2007).
Frost, M. R. & Norton, T. T. Alterations in protein expression in tree shrew sclera during development of lens-induced myopia and recovery. Investig. Ophthalmol. Vis. Sci. 53, 322–336. https://doi.org/10.1167/iovs.11-8354 (2012).
Jostrup, R. et al. Identification of myopia-related marker proteins in tilapia retinal, RPE, and choroidal tissue following induced form deprivation. Curr. Eye Res. 34, 966–975. https://doi.org/10.3109/02713680903244138 (2009).
Lam, T. C., Li, K. K., Lo, S. C., Guggenheim, J. A. & To, C. H. Application of fluorescence difference gel electrophoresis technology in searching for protein biomarkers in chick myopia. J. Proteome Res. 6, 4135–4149. https://doi.org/10.1021/pr0701097 (2007).
Li, S. et al. Flicker downregulates the content of crystallin proteins in form-deprived C57BL/6 mouse retina. Exp. Eye Res. 101, 1–8. https://doi.org/10.1016/j.exer.2012.05.003 (2012).
Zhou, X. et al. Changes in protein profiles of guinea pig sclera during development of form deprivation myopia and recovery. Mol. Vis. 16, 2163–2174 (2010).
Wu, Y. et al. Differential retinal protein expressions during form deprivation myopia in albino guinea pigs. Curr. Proteomics 11, 37–47. https://doi.org/10.2174/1570164610666140109002531 (2014).
Yu, F. J. et al. Isotope-coded protein label based quantitative proteomic analysis reveals significant up-regulation of apolipoprotein A1 and ovotransferrin in the myopic chick vitreous. Sci. Rep. 7, 12649. https://doi.org/10.1038/s41598-017-12650-7 (2017).
Zhou, Y. Y. et al. Proteomic analysis of chick retina during early recovery from lensinduced myopia. Mol. Med. Rep. 18, 59–66. https://doi.org/10.3892/mmr.2018.8954 (2018).
Riddell, N. et al. The retina/RPE proteome in chick myopia and hyperopia models: commonalities with inherited and age-related ocular pathologies. Mol. Vis. 23, 872–888 (2017).
Jin, N. & Stjernschantz, J. Regional blood flow in the myopic chick eye during and after form deprivation: a study with radioactively-labelled microspheres. Exp. Eye Res. 71, 233–238. https://doi.org/10.1006/exer.2000.0871 (2000).
Shih, Y. F. et al. Reduction in choroidal blood flow occurs in chicks wearing goggles that induce eye growth toward myopia. Curr. Eye Res. 12, 219–227 (1993).
He, L., Frost, M. R., Siegwart, J. T. Jr. & Norton, T. T. Gene expression signatures in tree shrew choroid during lens-induced myopia and recovery. Exp. Eye Res. 123, 56–71. https://doi.org/10.1016/j.exer.2014.04.005 (2014).
Gao, H., Frost, M. R., Siegwart, J. T. Jr. & Norton, T. T. Patterns of mRNA and protein expression during minus-lens compensation and recovery in tree shrew sclera. Mol. Vis. 17, 903–919 (2011).
Guo, L., Frost, M. R., He, L., Siegwart, J. T. Jr. & Norton, T. T. Gene expression signatures in tree shrew sclera in response to three myopiagenic conditions. Investig. Ophthalmol. Vis. Sci. 54, 6806–6819. https://doi.org/10.1167/iovs.13-12551 (2013).
Percie du Sert, N. et al. Reporting animal research: explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 18, e3000411. https://doi.org/10.1371/journal.pbio.3000411 (2020).
Janssen, B. J. et al. Effects of anesthetics on systemic hemodynamics in mice. Am. J. Physiol. Heart Circ. Physiol. 287, H1618–H1624. https://doi.org/10.1152/ajpheart.01192.2003 (2004).
Langsjo, J. W. et al. Effects of subanesthetic doses of ketamine on regional cerebral blood flow, oxygen consumption, and blood volume in humans. Anesthesiology 99, 614–623. https://doi.org/10.1097/00000542-200309000-00016 (2003).
Luh, C. et al. Influence of a brief episode of anesthesia during the induction of experimental brain trauma on secondary brain damage and inflammation. PLoS ONE 6, e19948. https://doi.org/10.1371/journal.pone.0019948 (2011).
Staib-Lasarzik, I. et al. Anesthesia for euthanasia influences mRNA expression in healthy mice and after traumatic brain injury. J. Neurotrauma 31, 1664–1671. https://doi.org/10.1089/neu.2013.3243 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. https://doi.org/10.14806/ej.17.1.200 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. https://doi.org/10.1038/nprot.2012.016 (2012).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. https://doi.org/10.1186/gb-2013-14-4-r36 (2013).
Langmead, B. Aligning short sequencing reads with Bowtie. Current protocols in bioinformatics Chapter 11, Unit 11 17, Doi: https://doi.org/10.1002/0471250953.bi1107s32 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Mi, H., Muruganujan, A. & Thomas, P. D. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41, D377-386. https://doi.org/10.1093/nar/gks1118 (2013).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Mootha, V. K. et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273. https://doi.org/10.1038/ng1180 (2003).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. https://doi.org/10.1093/bioinformatics/btr260 (2011).
Gough, N. R. Science’s signal transduction knowledge environment: the connections maps database. Ann. N. Y. Acad. Sci. 971, 585–587 (2002).
Garcia-Campos, M. A., Espinal-Enriquez, J. & Hernandez-Lemus, E. Pathway analysis: state of the art. Front. Physiol. 6, 383. https://doi.org/10.3389/fphys.2015.00383 (2015).
The Broad Institute, M. I. o. T. M. Gene Set Enrichment Analysis (GSEA) User Guide, <http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html> (2012).
Etienne, W., Meyer, M. H., Peppers, J. & Meyer, R. A. Jr. Comparison of mRNA gene expression by RT-PCR and DNA microarray. Biotechniques 36, 618–626. https://doi.org/10.2144/04364ST02 (2004).
Morey, J. S., Ryan, J. C. & Van Dolah, F. M. Microarray validation: factors influencing correlation between oligonucleotide microarrays and real-time PCR. Biol. Proced. Online 8, 175–193. https://doi.org/10.1251/bpo126 (2006).
Allanach, K. et al. Comparing microarray versus RT-PCR assessment of renal allograft biopsies: similar performance despite different dynamic ranges. Am. J. Transplant. 8, 1006–1015. https://doi.org/10.1111/j.1600-6143.2008.02199.x (2008).
Talaat, A. M. et al. Genomic DNA standards for gene expression profiling in Mycobacterium tuberculosis. Nucleic Acids Res. 30, e104. https://doi.org/10.1093/nar/gnf103 (2002).
Arikawa, E. et al. Cross-platform comparison of SYBR Green real-time PCR with TaqMan PCR, microarrays and other gene expression measurement technologies evaluated in the MicroArray Quality Control (MAQC) study. BMC Genom. 9, 328. https://doi.org/10.1186/1471-2164-9-328 (2008).
Raymond, F. et al. Comparison of automated microarray detection with real-time PCR assays for detection of respiratory viruses in specimens obtained from children. J. Clin. Microbiol. 47, 743–750. https://doi.org/10.1128/JCM.01297-08 (2009).
Nickla, D. L., Wildsoet, C. & Wallman, J. The circadian rhythm in intraocular pressure and its relation to diurnal ocular growth changes in chicks. Exp. Eye Res. 66, 183–193. https://doi.org/10.1006/exer.1997.0425 (1998).
Nickla, D. L., Wildsoet, C. & Wallman, J. Visual influences on diurnal rhythms in ocular length and choroidal thickness in chick eyes. Exp. Eye Res. 66, 163–181. https://doi.org/10.1006/exer.1997.0420 (1998).
Chakraborty, R. et al. Circadian rhythms, refractive development, and myopia. Ophthalmic Physiol. Opt. 38, 217–245. https://doi.org/10.1111/opo.12453 (2018).
Takeuchi, H., Watanabe, K. & Tamura, H. Penetrable and impenetrable anions into the GABA-activated chloride channel on the postsynaptic neuromembrane of an identifiable giant neurone of an African giant snail (Achatina fulica Ferussac). Comp. Biochem. Physiol. C 61 C, 309–315 (1978).
Lynch, J. W. Molecular structure and function of the glycine receptor chloride channel. Physiol. Rev. 84, 1051–1095. https://doi.org/10.1152/physrev.00042.2003 (2004).
Schnetkamp, P. P. The SLC24 Na+/Ca2+–K+ exchanger family: vision and beyond. Pflugers Arch. 447, 683–688. https://doi.org/10.1007/s00424-003-1069-0 (2004).
Armstrong, C. M. & Bezanilla, F. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70, 567–590. https://doi.org/10.1085/jgp.70.5.567 (1977).
Jentsch, T. J., Stein, V., Weinreich, F. & Zdebik, A. A. Molecular structure and physiological function of chloride channels. Physiol. Rev. 82, 503–568. https://doi.org/10.1152/physrev.00029.2001 (2002).
Uckelmann, M. & Sixma, T. K. Histone ubiquitination in the DNA damage response. DNA Repair (Amst) 56, 92–101. https://doi.org/10.1016/j.dnarep.2017.06.011 (2017).
Wu-Baer, F., Lagrazon, K., Yuan, W. & Baer, R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 278, 34743–34746. https://doi.org/10.1074/jbc.C300249200 (2003).
Wu-Baer, F., Ludwig, T. & Baer, R. The UBXN1 protein associates with autoubiquitinated forms of the BRCA1 tumor suppressor and inhibits its enzymatic function. Mol. Cell. Biol. 30, 2787–2798. https://doi.org/10.1128/MCB.01056-09 (2010).
Matsuzawa, T. & Obara, Y. Amino acid synthesis from ornithine: enzymes and quantitative comparison in brain slices and detached retinas from rats and chicks. Brain Res. 413, 314–319 (1987).
Schmid, K. L., Strasberg, G., Rayner, C. L. & Hartfield, P. J. The effects and interactions of GABAergic and dopaminergic agents in the prevention of form deprivation myopia by brief periods of normal vision. Exp. Eye Res. 110, 88–95. https://doi.org/10.1016/j.exer.2013.02.017 (2013).
Chebib, M. et al. Novel, potent, and selective GABAC antagonists inhibit myopia development and facilitate learning and memory. J. Pharmacol. Exp. Ther. 328, 448–457. https://doi.org/10.1124/jpet.108.146464 (2009).
Cheng, Z. Y., Wang, X. P., Schmid, K. L. & Han, X. G. Inhibition of form-deprivation myopia by a GABAAOr receptor antagonist, (1,2,5,6-tetrahydropyridin-4-yl) methylphosphinic acid (TPMPA), in guinea pigs. Graefes Arch. Clin. Exp. Ophthalmol. 252, 1939–1946. https://doi.org/10.1007/s00417-014-2765-5 (2014).
Cheng, Z. Y. et al. GABAB receptor antagonist CGP46381 inhibits form-deprivation myopia development in guinea pigs. Biomed. Res. Int. 2015, 207312. https://doi.org/10.1155/2015/207312 (2015).
Riddell, N., Faou, P. & Crewther, S. G. Short term optical defocus perturbs normal developmental shifts in retina/RPE protein abundance. BMC Dev. Biol. 18, 18. https://doi.org/10.1186/s12861-018-0177-1 (2018).
Lukasiewicz, P. D., Eggers, E. D., Sagdullaev, B. T. & McCall, M. A. GABAC receptor-mediated inhibition in the retina. Vis. Res. 44, 3289–3296. https://doi.org/10.1016/j.visres.2004.07.023 (2004).
Cheng, Z. Y. et al. GABAAalpha1 and GABAArho1 subunits are expressed in cultured human RPE cells and GABAA receptor agents modify the intracellular calcium concentration. Mol. Vis. 21, 939–947 (2015).
Cheng, Z. Y., Wang, X. P., Schmid, K. L. & Liu, L. Identification of GABA receptors in chick retinal pigment epithelium. Neurosci. Lett. 539, 43–47. https://doi.org/10.1016/j.neulet.2013.01.038 (2013).
Yang, X. L. Characterization of receptors for glutamate and GABA in retinal neurons. Prog. Neurobiol. 73, 127–150. https://doi.org/10.1016/j.pneurobio.2004.04.002 (2004).
Riddell, N. & Crewther, S. G. Novel evidence for complement system activation in chick myopia and hyperopia models: a meta-analysis of transcriptome datasets. Sci. Rep. 7, 9719. https://doi.org/10.1038/s41598-017-10277-2 (2017).
Wang, B. et al. A novel potentially causative variant of NDUFAF7 revealed by mutation screening in a Chinese family with pathologic myopia. Investig. Ophthalmol. Vis. Sci. 58, 4182–4192. https://doi.org/10.1167/iovs.16-20941 (2017).
Beresford, J. A., Crewther, S. G. & Crewther, D. P. Anatomical correlates of experimentally induced myopia. Aust. N. Z. J. Ophthalmol. 26(Suppl 1), S84-87 (1998).
Liang, H., Crewther, S. G., Crewther, D. P. & Pirie, B. Morphology of the recovery from form deprivation myopia in the chick. Aust. N. Z. J. Ophthalmol. 24, 41–44 (1996).
Junghans, B. M. et al. Lymphatics in the chick choroid?. Aust. N. Z. J. Ophthalmol. 24, 47–49 (1996).
Murphy, M. P. How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. https://doi.org/10.1042/BJ20081386 (2009).
Sena, L. A. & Chandel, N. S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167. https://doi.org/10.1016/j.molcel.2012.09.025 (2012).
Wilson, C. & Gonzalez-Billault, C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: implications for neuronal development and trafficking. Front. Cell. Neurosci. 9, 381. https://doi.org/10.3389/fncel.2015.00381 (2015).
Liu, H. T., Akita, T., Shimizu, T., Sabirov, R. Z. & Okada, Y. Bradykinin-induced astrocyte-neuron signalling: glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J. Physiol. 587, 2197–2209. https://doi.org/10.1113/jphysiol.2008.165084 (2009).
Ames, A. 3rd., Li, Y. Y., Heher, E. C. & Kimble, C. R. Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. J. Neurosci. 12, 840–853 (1992).
Murphy, M. J. & Crewther, S. G. Ouabain inhibition of Na/K-ATPase across the retina prevents signed refractive compensation to lens-induced defocus, but not default ocular growth in young chicks. F1000Res 2, 97. https://doi.org/10.12688/f1000research.2-97.v1 (2013).
Icel, E. et al. The relation of neutrophil to lymphocyte ratio and platelet to lymphocyte ratio with high axial myopia. Ocul. Immunol. Inflamm. 28, 396–401. https://doi.org/10.1080/09273948.2019.1588334 (2020).
Feske, S., Wulff, H. & Skolnik, E. Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 33, 291–353. https://doi.org/10.1146/annurev-immunol-032414-112212 (2015).
Freeman, S. A. et al. Lipid-gated monovalent ion fluxes regulate endocytic traffic and support immune surveillance. Science 367, 301–305. https://doi.org/10.1126/science.aaw9544 (2020).
Black, S. M. et al. IL-4 induces protection of vascular endothelial cells against killing by complement and melittin through lipid biosynthesis. Eur. J. Immunol. 40, 803–812. https://doi.org/10.1002/eji.200939488 (2010).
Lu, K. L. et al. Association of mitochondrial dysfunction with oxidative stress and immune suppression in Blunt Snout Bream Megalobrama amblycephala fed a high-fat diet. J. Aquat. Anim. Health 26, 100–112. https://doi.org/10.1080/08997659.2014.893460 (2014).
Nashine, S. et al. Differential expression of complement markers in normal and AMD transmitochondrial cybrids. PLoS ONE 11, e0159828. https://doi.org/10.1371/journal.pone.0159828 (2016).
Wilkins, H. M., Weidling, I. W., Ji, Y. & Swerdlow, R. H. Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front. Immunol. 8, 508. https://doi.org/10.3389/fimmu.2017.00508 (2017).
Long, Q., Ye, J., Li, Y., Wang, S. & Jiang, Y. C-reactive protein and complement components in patients with pathological myopia. Optom. Vis. Sci. 90, 501–506. https://doi.org/10.1097/OPX.0b013e31828daa6e (2013).
Gao, T. T., Long, Q. & Yang, X. Complement factors C1q, C3 and C5b–9 in the posterior sclera of guinea pigs with negative lens-defocused myopia. Int. J. Ophthalmol. 8, 675–680. https://doi.org/10.3980/j.issn.2222-3959.2015.04.06 (2015).
Junghans, B., Crewther, S. & Crewther, D. Fine structure of lymphatics in the avian choroid. Investig. Ophthalmol. Vis. Sci. 39, 854–855 (1998).
Loh, K. Y., Wang, Z. & Liao, P. Oncotic cell death in stroke. Rev. Physiol. Biochem. Pharmacol. https://doi.org/10.1007/112_2018_13 (2018).
Buja, L. M. Myocardial ischemia and reperfusion injury. Cardiovasc. Pathol. 14, 170–175. https://doi.org/10.1016/j.carpath.2005.03.006 (2005).
Walter, W., Sanchez-Cabo, F. & Ricote, M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914. https://doi.org/10.1093/bioinformatics/btv300 (2015).
Funding
2007 NHMRC Development Grant and ARC Discovery Project (DP110103784).
Author information
Authors and Affiliations
Contributions
L.G.V. as a Ph.D. student was involved in conception and design of the experiments, contributed to molecular aspects of data collection, analyzed the data, prepared figures and/or tables, authored and reviewed the paper, approved the final draft. S.C. as Senior Author, conceived and designed the experiments, provided funding. contributed to data collection and analyses, guided theoretical interpretation, co-authored and reviewed drafts of the paper, approved the final draft. N.R. contributed to data collection, data analysis and preparation of figures and/or tables, reviewed drafts of the paper, approved the final draft. N.E.H. did first analysis of the RNAseq data and approved the final draft. M.M. contributed to biometric data collection and reviewed drafts of the paper, approved the final draft. D.C. as co-Senior Author conceived and funded the project, contributed to theoretical interpretation, reviewed drafts of the paper, approved the final draft.
The views expressed in this manuscript are our own and not an official position of the institution or funder.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vocale, L.G., Crewther, S., Riddell, N. et al. RNA-seq and GSEA identifies suppression of ligand-gated chloride efflux channels as the major gene pathway contributing to form deprivation myopia. Sci Rep 11, 5280 (2021). https://doi.org/10.1038/s41598-021-84338-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-84338-y
- Springer Nature Limited
This article is cited by
-
Diurnal retinal and choroidal gene expression patterns support a role for circadian biology in myopia pathogenesis
Scientific Reports (2024)
-
The Role of Retinal Dysfunction in Myopia Development
Cellular and Molecular Neurobiology (2023)