
Abstract
Hematologists are frequently involved in the diagnostic pathway of Gaucher disease type 1 (GD1) patients since they present several hematological signs. However, GD1 is mainly underdiagnosed because of a lack of awareness. In this multicenter study, we combine the use of a diagnostic algorithm with a simple test (β-glucosidase activity on Dried Blood Spot) in order to facilitate the diagnosis in a population presenting to the hematologist with splenomegaly and/or thrombocytopenia associated with other hematological signs. In this high-risk population, the prevalence of GD1 is 3.3%. We propose an equation that predicts the probability of having GD1 according to three parameters that are routinely evaluated: platelet count, ferritin, and transferrin saturation.
Similar content being viewed by others


Introduction
Gaucher disease type 1 (GD1) is an autosomal recessive lysosomal storage disorder caused by mutations in the GBA gene resulting in the deficiency of β-glucosidase enzyme. Its prevalence in the non-Ashkenazy Jewish population is estimated at 1:40,000–100,000 subjects, whereas in Ashkenazi Jewish is 1:500–1000.
At diagnosis, patients present with several hematological signs and symptoms, including splenomegaly (86%), anemia (64%), thrombocytopenia (56%), bleeding history, and monoclonal gammopathy of undetermined significance (MGUS), leading them to consult a hematologist on their diagnostic pathway1. However, an international survey showed that only 20% of hematologists include GD1 in the differential diagnosis of a patient with anemia, thrombocytopenia, hepatomegaly, splenomegaly, and bone pain2. As a matter of fact, GD1 is misdiagnosed and underdiagnosed; thus, patients often experience long diagnostic delays, leading to inappropriate procedures, treatments, and complications that often cannot be reversed by the available treatments2.
Moreover, half of the patients are diagnosed through bone marrow biopsy, although the diagnostic gold standard is the activity of β-glucosidase on leucocytes or fibroblasts3. Among the crucial obstacles to diagnosis, physicians mainly identify outsourced testing and, more importantly, the lack of awareness4. Thus, ten years ago, a panel of experts published two diagnostic algorithms, one for the Ashkenazi and one for the non-Ashkenazi Jewish population, to facilitate the diagnosis of GD1 for hematologists5.
The new-born screening has been experimented in some areas, showing an incidence of 1:22,205 in Northern Italy6. However, the large-scale implementation of new-born screening for a disease with high phenotypic heterogeneity, ranging from asymptomatic to severely symptomatic conditions, should be carefully evaluated.
We hypothesized that an approach that combines a diagnostic algorithm and a simple, cheap, and easy-to-do test could facilitate the diagnosis. We designed a multicenter study that aimed at evaluating the prevalence of GD1 in a high-risk population presenting to the hematologist with splenomegaly and/or thrombocytopenia associated with other hematological signs or symptoms suggestive of GD1. Preliminary results of this study on the first 196 patients have been previously published, showing a GD1 prevalence of 3.6% in a high-risk population7.
Materials and methods
Study design
We designed a multicenter study among 35 hematology centers in Italy. According to the feasibility questionnaire, we expected to enroll 500 subjects. The enrolment started in September 2010 and closed in December 2018.
Inclusion and exclusion criteria were based on the published algorithm for the non-Ashkenazi population5:
-
Inclusion criteria: splenomegaly and/or thrombocytopenia and at least one sign or symptom among bone pain history, anemia, MGUS, polyclonal gammopathy in subjects under 30 years of age, splenectomy;
-
Exclusion criteria: onco-hematological diseases, portal hypertension due to liver diseases, hemoglobinopathies, or chronic hemolytic anemias.
Demographic, clinical, and laboratory data were collected at enrolment, gathered in a specific case report form, and collected by the coordinating center at Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy.
Beta-glucosidase activity
The beta-glucosidase activity on Dried Blood Spot (DBS) was centralized at Ospedale Gaslini, Genoa, Italy8,9. Normal values range from 4.4 to 17.7 pmol/punch/h. Subjects showing activity below 4.4 pmol/punch/h were recalled to be assessed with the gold standard assay on nucleated cell homogenates (leucocytes, EBV-lymphoblasts, or fibroblasts). If the enzymatic defect was confirmed, the diagnosis was completed with the molecular GBA analysis.
Statistical analysis
The prevalence of GD1 and its 95% confidence interval (CI) based on the exact method were calculated. Demographic, clinical, and laboratory variables of patients affected by GD1 were compared to those of unaffected patients using Wilcoxon rank-sum (Mann–Whitney U-test) test (for continuous variables) or Fisher’s exact test (for categorical variables). Using univariate and multiple logistic regression models, we analyzed the predictive role of platelets (thousands/mm3), ferritin (μg/L), transferrin saturation (TSAT) (%) in this high-risk population. Furthermore, we considered the three variables jointly, and we calculated the respective areas under the curve (AUC) of the receiver operating characteristic (ROC) curves. Analyses were performed with Stata 16 (StataCorp. 2019. Stata: Release 16. Statistical Software; StataCorp LP, College Station, TX, USA).
Ethical aspects
The study was approved by the ethical review committee of the coordinating center “Comitato Etico Milano Area 2” (Protocol number 714/10) and by all participating Centers, and was carried out in compliance with the principles established in the Helsinki Declaration. Informed consent was obtained from all individual participants included in the study.
Results
The prevalence of GD1 in high risk predominantly Caucasian population is above 3%
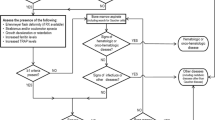
Five hundred subjects have been enrolled in the study. Forty-five have been excluded because they did not fulfill the inclusion and exclusion criteria (Fig. 1). Demographic, clinical, and laboratory characteristics are presented in Table 1. Ninety-one percent (91%) of the subjects were Caucasian. The mean age at enrolment was 46.9 years, and 31.9% (145/455) were females. The majority had splenomegaly (89.7%), and approximately half (47.9%) thrombocytopenia associated with other signs/symptoms. Anemia was the most common adjunctive sign (23.1%) (Table 1).

Results of DBS, β‐glucosidase assay in cell homogenate, and molecular analysis of the enrolled patients. DBS dried blood spot, GD1 Gaucher disease type 1. *They did not fulfill the inclusion and exclusion criteria.
DBS showed normal values in 379 subjects, while 76 (16.7%) had a reduced β-glucosidase activity. These 76 patients and a patient with a family history of GD1 presenting with a β-glucosidase activity slightly above the lower normal range were recalled to test the conventional enzymatic activity. Among the 65 patients tested with β-glucosidase activity on nucleated cell homogenates (12 did not answer), 15 were diagnosed with Gaucher disease type 1, with a prevalence of 3.3% (15/455, 95% CI 1.9–5.4%). In 14, the molecular analysis of the GBA gene identified the mutations. In one patient, no mutations of GBA gene nor PSAP, encoding for saposin C, were identified. Among GD1 patients, 7/15 (46.7%) were female, 14 of Caucasian origin, and the mean age at diagnosis was 43.5 years. They showed a lower platelet count compared to non-GD1 patients (84.000/mm3 vs 131.000/mm3, p = 0.0006), a higher serum ferritin level (551 ng/dL vs 139 ng/dL, p = 0.0002) which was associated to a lower transferrin saturation (20.8% vs 25.7%, p = 0.03).
Platelet count, ferritin, and transferrin saturation predict the probability of having GD1 in this high-risk population
Considering 159 subjects (13 with GD1) with complete data on ferritin, platelets, and TSAT, the best discrimination between GD1 and non-GD1 subjects was provided by platelets (AUC = 0.79), while ferritin and transferrin saturation showed lower AUCs (Fig. 2).

Receiver operating characteristic (ROC) curves of GD1 for platelets (thousands/mm3), ferritin (μg/L), transferrin saturation (%), and for the three variables jointly analyzed in a multiple logistic regression model. AUC area under the curve, CI confidence interval.
The joint analysis of these three variables together in a multiple logistic regression model yielded an AUC of 0.89 (95% CI: 0.82–0.96). The corresponding equation is:
where F = ferritin (μg/L), P = platelets (thousands/mm3), and TS = transferrin saturation (%).
Hence the predicted probability of GD1 in patients presenting with splenomegaly and/or thrombocytopenia plus the abovementioned ancillary signs can be calculated as: 100 × [odds(GD1)/[1 + odds(GD1)].]
When the probability predicted by the equation was < 5%, we observed only one subject with GD1 out of 93 (1.1%). When the predicted probability was 5 to < 10%, the observed frequency was 3.9% (1/26). With predicted probabilities ≥ 10%, the observed GD1 prevalence was substantially higher (11/30 = 27.5%).
The addition of a history of bone pain to this model did not lead to an AUC increase, and thus, this variable was omitted.
Discussion
This study shows that in a predominantly Caucasian high-risk population presenting to the hematologist with splenomegaly and/or thrombocytopenia associated with other hematological signs, including anemia and MGUS, 3.3% of patients have Gaucher disease. These data confirm our previously published preliminary data7. Similar studies have been reproduced in different regions worldwide, with different prevalence, ranging from no cases detected in a Canadian study on 221 subjects10 to 7.0% in an adult cohort in China11. A similar approach is under evaluation in the Italian pediatric population12, and promising preliminary results have been presented in a Chinese study13. Altogether, these data support the use of the previously published algorithm by Mistry et al.5 associated with a simple first-level diagnostic test to screen high-risk populations. Diagnostic confirmation with β-glucosidase activity on nucleated cell homogenates is necessary to confirm the diagnosis. Of note, given that false negatives in the DBS test may arise due to methodological differences in blood spot drying, transport and storage14,15,16 testing with the gold standard diagnostic exam is warranted when there is any clinical suspicion of Gaucher disease, even in the presence of normal DBS values.
Since lysosomal storage disorders, including GD1, are underdiagnosed, other approaches have been proposed to increase the diagnostic rate. Namely, the new-born screening has been experimented in several regions with different results according to the ethnicity of the tested population6,17,18,19. However, the new-born screening raises unique issues that are primarily related to the inevitable detection of a disease with late-onset phenotypes.
Among the enrolled subjects, the only clearly different parameters between GD1 and non-GD1 patients were platelet count and serum ferritin. Hyperferritinemia with normal transferrin saturation is a common finding in naïve patients with GD120, with prevalence ranging between 63 and 81%21,22. Recently, together with other Italian referral groups for iron disorders, we have proposed a new diagnostic flow-chart23, which enhances hyperferritinemia role when associated with splenomegaly and thrombocytopenia.
Here we propose an equation that predicts the probability of having GD1 according to platelet count and ferritin and TSAT levels and thus may support hematologists when evaluating a subject with splenomegaly and/or thrombocytopenia.
Conclusion
High-risk population testing is effective in identifying Gaucher disease patients who present to the hematologist with splenomegaly and/or thrombocytopenia. The evaluation of the probability of having GD1 according to an equation and the use of DBS as a first-level test are potentially useful tools that can facilitate the diagnostic process.
References
Charrow, J. et al. The Gaucher registry: Demographics and disease characteristics of 1698 patients with Gaucher disease. Arch. Intern. Med. 160, 2835–2843. https://doi.org/10.1001/archinte.160.18.2835 (2000).
Mistry, P. K., Sadan, S., Yang, R., Yee, J. & Yang, M. Consequences of diagnostic delays in type 1 Gaucher disease: The need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am. J. Hematol. 82, 697–701. https://doi.org/10.1002/ajh.20908 (2007).
Thomas, A. S., Mehta, A. B. & Hughes, D. A. Diagnosing Gaucher disease: An on-going need for increased awareness amongst haematologists. Blood Cells Mol. Dis. 50, 212–217. https://doi.org/10.1016/j.bcmd.2012.11.004 (2013).
Mehta, A. et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians. Mol. Genet. Metab. 122, 122–129. https://doi.org/10.1016/j.ymgme.2017.08.002 (2017).
Mistry, P. K. et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am. J. Hematol. 86, 110–115. https://doi.org/10.1002/ajh.21888 (2011).
Burlina, A. B. et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 41, 209–219. https://doi.org/10.1007/s10545-017-0098-3 (2018).
Motta, I. et al. A multicentre observational study for early diagnosis of Gaucher disease in patients with Splenomegaly and/or Thrombocytopenia. Eur. J. Haematol. 96, 352–359. https://doi.org/10.1111/ejh.12596 (2016).
Stroppiano, M. et al. Validity of beta-D-glucosidase activity measured in dried blood samples for detection of potential Gaucher disease patients. Clin. Biochem. 47, 1293–1296. https://doi.org/10.1016/j.clinbiochem.2014.06.005 (2014).
Olivova, P. et al. An improved high-throughput dried blood spot screening method for Gaucher disease. Clin. Chim. Acta 398, 163–164. https://doi.org/10.1016/j.cca.2008.08.024 (2008).
Russell, S. A. et al. Gaucher disease screening at a general adult hematology tertiary care centre: A prospective study. Int. J. Lab. Hematol. 41, e66–e69. https://doi.org/10.1111/ijlh.12960 (2019).
Huang, Y. et al. High risk screening for Gaucher disease in patients with splenomegaly and/or thrombocytopenia in China: 55 cases identified. Clin. Chim. Acta 506, 22–27. https://doi.org/10.1016/j.cca.2020.03.016 (2020).
Di Rocco, M. et al. Early diagnosis of Gaucher disease in pediatric patients: Proposal for a diagnostic algorithm. Pediatr. Blood Cancer 61, 1905–1909. https://doi.org/10.1002/pbc.25165 (2014).
Lei, K. et al. A pilot screening of high-risk Gaucher disease children using dried blood spot methods in Shandong province of China. Orphanet J. Rare Dis. 13, 48. https://doi.org/10.1186/s13023-018-0782-x (2018).
Elbin, C. S. et al. The effect of preparation, storage and shipping of dried blood spots on the activity of five lysosomal enzymes. Clin. Chim. Acta 412, 1207–1212. https://doi.org/10.1016/j.cca.2011.03.012 (2011).
Supriya, M., De, T. & Christopher, R. Effect of temperature on lysosomal enzyme activity during preparation and storage of dried blood spots. J. Clin. Lab. Anal. 32(1), e22220. https://doi.org/10.1002/jcla.22220 (2018).
Mei, J. V., Alexander, J. R., Adam, B. W. & Hannon, W. H. Use of filter paper for the collection and analysis of human whole blood specimens. J. Nutr. 131, 1631S-1636S. https://doi.org/10.1093/jn/131.5.1631S (2001).
Burton, B. K. et al. Newborn screening for lysosomal storage disorders in Illinois: The initial 15-month experience. J. Pediatr. 190, 130–135. https://doi.org/10.1016/j.jpeds.2017.06.048 (2017).
Hopkins, P. V. et al. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 166, 172–177. https://doi.org/10.1016/j.jpeds.2014.09.023 (2015).
Wasserstein, M. P. et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the first 65,000 infants. Genet. Med. 21, 631–640. https://doi.org/10.1038/s41436-018-0129-y (2019).
Stein, P., Yu, H., Jain, D. & Mistry, P. K. Hyperferritinemia and iron overload in type 1 Gaucher disease. Am. J. Hematol. 85, 472–476. https://doi.org/10.1002/ajh.21721 (2010).
Lefebvre, T. et al. Involvement of hepcidin in iron metabolism dysregulation in Gaucher disease. Haematologica 103, 587–596. https://doi.org/10.3324/haematol.2017.177816 (2018).
Lorenz, F. et al. Ferritinemia and serum inflammatory cytokines in Swedish adults with Gaucher disease type 1. Blood Cells Mol. Dis. 68, 35–42. https://doi.org/10.1016/j.bcmd.2016.10.010 (2018).
Marchi, G. et al. Hyperferritinemia and diagnosis of type 1 Gaucher disease. Am. J. Hematol. 95, 570–576. https://doi.org/10.1002/ajh.25752 (2020).
Funding
This work was supported by a grant from Sanofi Genzyme.
Author information
Authors and Affiliations
Consortia
Contributions
M.D.C. designed the research study. I.M., E.C., P.R., L.B., L.F., A.P., W.B. enrolled the patients. M.S., M.F., B.T., F.L. performed the tests. I.M. and C.B. collected the data. D.C., I.M. and C.B. analyzed the data. I.M. and D.C. wrote the paper. All authors contributed to critical revision and final approval of the version to be published.
Corresponding author
Ethics declarations
Competing interests
IM received Lecture Honoraria from Sanofi Genzyme and is a member of Sanofi Genzyme advisory board. WB is a member of Agios, Alexion, Apellis, Biocryst, Bioverativ, Incyte, Momenta, Novartis advisory board, received lecture honoraria from Alexion, Incyte, Novartis, Sanofi and research support from Alexion. MDC is a member of Vifor, Sanofi Genzyme, Celgene, Novartis, and Bluebird advisory board. DC, MS, CB, EC, BT, PR, LB, LF, AP, FL, MF have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Motta, I., Consonni, D., Stroppiano, M. et al. Predicting the probability of Gaucher disease in subjects with splenomegaly and thrombocytopenia. Sci Rep 11, 2594 (2021). https://doi.org/10.1038/s41598-021-82296-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82296-z
- Springer Nature Limited
This article is cited by
-
Blood cytopenias as manifestations of inherited metabolic diseases: a narrative review
Orphanet Journal of Rare Diseases (2024)
-
Development of a rare disease algorithm to identify persons at risk of Gaucher disease using electronic health records in the United States
Orphanet Journal of Rare Diseases (2023)
-
GAU-PED study for early diagnosis of Gaucher disease in children with splenomegaly and cytopenia
Orphanet Journal of Rare Diseases (2023)
-
Confounding factors in the diagnosis and clinical course of rare congenital hemolytic anemias
Orphanet Journal of Rare Diseases (2021)