
Abstract
Malignant hyperthermia (MH) is a pharmacogenetic disorder of skeletal muscle metabolism characterized by generalized muscle rigidity, increased body temperature, rhabdomyolysis, hyperkalemia and severe metabolic acidosis. The underlying mechanism of MH involves excessive Ca2+ release from myotubes via the ryanodine receptor type 1 (RYR1) and the voltage-dependent L-type calcium channel (CACNA1S). As more than 300 variants of unknown significance have been detected to date, we examined whether freely available pathogenicity prediction tools are able to detect relevant MH causing variants. In this diagnostic accuracy study, blood samples from 235 individuals with a history of a clinical malignant hyperthermia or their close relatives were genetically screened for RYR1 variants of all 106 RYR1 exons and additionally for known variants of CACNA1S. In vitro contracture tests were conducted on muscle biopsies obtained from all individuals, independently of whether a pathogenic variant, a variant of unknown significance or no variant was detected. Comparisons were made to three established bioinformatic pathogenicity detection tools to identify the clinical impact of the variants of unknown significance. All detected genetic variants were tested for pathogenicity by three in silico approaches and compared to the in vitro contracture test. Sensitivity and specificity of exon screening of all individuals listed in our MH database was analyzed. Exon screening identified 97 (41%) of the 235 individuals as carriers of pathogenic variants. Variants of unknown significance were detected in 21 individuals. Variants of unknown significance were subdivided into 19 malignant-hyperthermia-susceptible individuals and 2 non-malignant-hyperthermia-susceptible individuals. All pathogenic variants as well as the malignant-hyperthermia-suspectible variants were correctly identified by the bioinformatic prediction tools. Sensitivity of in silico approaches ranged between 0.71 and 0.98 (Polyphen 0.94 [CI 95% 0.75; 0.99]; Sift 0.98 [CI 95% 0.81; 0.99]; MutationTaster 0.92 [CI 95% 0.75; 0.99]). Specificity differed depending on the used tool (Polphen 0.98 [CI 95% 0.32; 0.99]; Sift 0.98 [CI 95% 0.32; 0.99]; MutationTaster 0.00 [CI 95% 0.00; 0.60]). All pathogenic variants and variants of unknown significance were scored as probably damaging in individuals, demonstrating a high sensitivity. Specificity was very low in one of the three tested programs. However, due to potential genotype–phenotype discordance, bioinformatic prediction tools are currently of limited value in diagnosing pathogenicity of MH-susceptible variants.
Similar content being viewed by others

Introduction
Malignant hyperthermia (MH) is a rare pharmacogenetic disorder, resulting in excessive Ca2+ release from the sarcoplasmic reticulum after activation of the abnormal ryanodine receptor (RYR1 encoded by the RYR1 gene). In genetically predisposed individuals, activation of abnormal RYR1 can induce life-threating hypermetabolic events characterized by generalized skeletal muscle rigidity, hyperthermia, cardiac arrhythmia and serve acidosis1,2,3. Volatile anesthetics are potent activators of abnormal RYR11. Moreover, rhabdomyolysis which can be caused even by non-anesthetic drugs has been reported in MH-susceptible individuals with RYR1 variants4. Variants in the CACNA1S gene have also been identified in malignant hyperthermia susceptibility (MHS). Identification of MHS is particularly important given the high mortality rate associated with the syndrome if prompt treatment is not initiated following the onset of clinical signs.
Ca2+ release in muscle is mediated by direct protein–protein interaction between the voltage sensor on the plasmalemma in the region of the t-tubule (Cav1.1 encoded by the CACNA1S gene) and the RYR1 of the sarcoplasmic reticulum5. The RYR1 gene consists of 106 exons with at least 16 non-pathogenic variants of the coding region6,7. Currently more than 400 malignant hyperthermia associated variants of unknown significance in the gene coding for RYR1 have been detected. To date, only about 50 variants have been proven to be pathogenic variants for MH according to the criteria of the European Malignant Hyperthermia Group (www.emhg.org)8. Before being classified as pathogenic, a variant must be both genetically and functionally characterized9. The criteria of the European Malignant Hyperthermia Group recommends that a functional assay with muscle biopsy and in vitro contracture test is required if a variant of unknown significance is detected10,11.
The increasing availability and decreasing cost of conducting DNA sequencing and next generation sequencing will likely result in the detection of a large number of variants of unknown significance12. To reveal the pathogenic potency of a detected variant of unknown significance, several in silico prediction methods have been developed. Since experimental methods are not a pragmatic approach to evaluate the large quantity of variation data currently generated, the guidelines of the American College of Medical Genetics and Genomics (ACMG) and the European Society of Human Genetics (ESHG) recommend the use of computational predictors as one of several lines of evidence for variant interpretation13,14. The recommendation echoes that of the joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists, in which the use of computational predictors have been advocated as a first line interpretation of genetic variants in cancer15,16. In contrast to cancer or most other genetic associated diseases, MH represents a pharmoco-genetic disorder and therefore needs application of a pharmacological trigger. Therefore such guidelines should either not be used or be used only with extreme caution for the prediction of disease-causing variants responsible for MH. Although prediction tools might offer diagnostic information in disease-causing variants, they currently should not be applied for definitive classification.
To date, several studies have investigated the predictive value of in silico approaches including SIFT, Polyphen or MutationTaster in cancer or neurodegenerative diseases17,18. Whether such in silico approaches might be valuable in diagnosing neuromuscular diseases remains unknown. To evaluate whether these methods may be helpful in identifying MH-susceptible individuals, we compared data from the MH center at the University of Ulm with the prediction results of the three most commonly used and freely available in silico programs: SIFT, Polyphen-2 and MutationTaster. These three in silico methods are based on different algorithm. SIFT predicts whether amino acid substitutions affect protein function, while Polyphen-2 predicts the possible impact of amnio acid substitution on the function and structure of a protein. MutationTaster, in contrast applies a combination of in silico tests. In addition to single amino acid substitution predictions, MutationTaster also determines synonymous and intronic variants, short insertion and deletion variants and even variants spanning the intron–exon borders.
Materials and methods
Individuals
This study is a retrospective analysis of genetic screening and in vitro muscle testing collected from 235 consecutive individuals referred to the malignant hyperthermia center (Ulm, Germany) over a 25-year period (01/1990 to 02/2015). Indications for malignant hyperthermia testing were; (1) an adverse anaesthetic event of the individual or a close relative, (2) a family–history of established MH, or (3) chronic isolated creatine kinase elevation. Patients that had only one diagnostic procedure (in vitro muscle testing or genetic screening) or more than one detected variant were excluded from this study. Written informed consent was obtained from individuals prior to their participation and all procedures were approved by the ethics committee of the University of Ulm, Germany. All methods were carried out in accordance with the guidelines of the University of Ulm Ethic Committee and the guidelines of the European Malignant Hyperthermia Group.
Exon screening
Exon screening was performed on all individuals as described in detail previously19,20. Ethylendiamintetraacetate blood samples of 235 individuals were genetically screened for variants in all 106 exons of the RYR1 gene and for known variants of CACNA1S20. DNA was extracted and amplified by polymerase chain reaction (PCR) for further analysis. PCR samples were mixed with the wild–type amplicons, denatured at 95 °C for 5 min and then cooled at room temperature to allow heteroduplexes to form20. Amplicons with an altered denaturing high-performance liquid chromatography (HPLC) elution profile compared to wild-type amplicons were directly sequenced with dye-terminator chemistry (Applied Biosystems)20.
IVCT
In accordance with the recommendations of the European Malignant Hyperthermia Group, MHS was diagnosed using the in vitro contracture test (IVCT) as described in detail in our previous study20,21,22. Muscle samples from all 235 individuals were tested, irrespective of whether a pathogenic, benign, variant of unknown significance or no variant was detected. In brief, the in vitro contracture test determines the contractile sensitivity of surgically excised muscle specimens to halothane and caffeine20. Biopsies were taken from the vastus lateralis muscle under regional anesthesia. Basal tension and the twitch response to supramaximal electrical stimulation (30–70 V, 0.2 Hz, 1 ms) was recorded with a force transducer20. Muscle bundles from malignant hyperthermia susceptible individuals (MHS) have lower contractile thresholds to caffeine and halothane than non- malignant hyperthermia susceptible individuals (MHN)20. A positive result for malignant hyperthermia susceptibility was defined as a contracture force of ≥ 2 mN at a caffeine concentration ≤ 2 mM and a halothane concentration ≤ 2% v/v21. A major limitation by diagnosing malignant hyperthermia using the IVCT is that it results in a categorical classification rather than individual interpretation of continuous contracture data20.
In silico approach
All detected pathogenic variants and variants of unknown significance, including malignant hyperthermia susceptible and non-malignant-hyperthermia-susceptible as determined by the in vitro contracture test, were analyzed by application of three in silico prediction programs. Additionally four previously characterized benign variants (c.5360C < T, synonymous; c.6178G < T, synonymous; c.10747G > C, non-coding; c.11266C > G, synonymous) were tested. In this study Sorting Intolerant from Tolerant (SIFT) (http://sift.jcvi.org), Polymorphism Phenotyping vs. 2 (Polyphen-2) (http://genetics.bwh.harvard.edu/pph2/) [HumDiv dataset] and MutationTaster (http://www.mutationtaster.org/) were applied.
SIFT (sorting intolerant from tolerant) predicts whether an amino acid substitution affects protein function by comparing amino acid alignments from related sequences to calculate a “swift score”. A score between 0.00 and 0.05 is classified as “damaging” while a score between 0.05 and 1.00 is classified as “tolerated”.
Polyphen-2 (Polymorphism Phenotyping vs.-2) predicts the possible impact of amino acid substitution on the structure and function of a human protein. Variants are classified as “benign”, “possibly damaging” or “probably damaging” with scores ranging between 0.0 (benign) and 1.0 (probably damaging).
MutationTaster combines a battery of in silico tests to predict the pathogenicity of variants of unknown significane at the protein and DNA level. Thus, MutationTaster is not limited to substitutions of single amino acids and can determine synonymous and intronic variants, short insertion or deletion variants and variants spanning intron-exons borders.
Statistical analysis
With an estimated prevalence of disease of 80%, sample size calculation (PASS software) revealed that 39 individuals were required to detect a difference of 20% between the in vitro contracture test and the in silico approaches at a significance level of 0.05 and with 80% power. A confusion matrix was used to evaluate the performance of all in silico approaches tested in this study. To this end, true positive was identified when the in vitro contracture test result corresponded with a positive result (pathologic or damaging) of the in silico method. A true negative, in contrast, was identified when the in vitro contracture test result corresponded with a negative result (neutral or benign) of the in silico method. Consequently, we calculated, (1) the sensitivity (proportion of positively identified cases with a true pathogenic variant) and (2) the specificity (proportion of positively identified cases with a true neutral variant) of all three in silico approaches. Positive (3) and negative (4) predictive values were also calculated for each test. Lower (5) and upper (6) 95% confidence interval were also calculated for all tests. Means and standard deviations were calculated for score values (pathogenic, benign and variants of unknown significance) obtained from all three in silico approaches. A alpha level of 0.05 was used for all tests of significance. StatXaxt (Version 5, Cytel Software, Cambridge, MA) and Excel (Version 2010, Microsoft, Redmon, Washington, USA) were used for all statistical procedures.
The following outcome variables were calculated:
-
(1)
Sensitivity: True positive/True positive + false negative
-
(2)
Specificity: True negative/True negative + false positive
-
(3)
Positive predictive value: True positive / True positive + false positive
-
(4)
Negative predictive value: True negative / True negative + false negative
-
(5)
Accuracy: True positives + True negatives / True positives + True negatives + False positives + False negatives
-
(6)
Lower 95% confidence interval: X−1.96 √ [X * (1−X)/n];
-
(7)
Upper 95% conficdence interval: X + 1.96 √ [X * (1−X)/n];
where X represents the sensitivity, specificity, positive predictive value, negative predictive value.
Results
Characterization of individuals
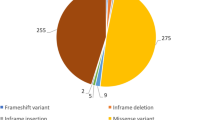
In total, 235 [93 female; 142 male] [median age: 35 (20;51)] individuals underwent in vitro contracture testing during the 25-year study period. According to the in vitro contracture testing protocol of the European Malignant Hyperthermia Group, 163 were classified as malignant hyperthermia susceptible and 72 as non-malignant hyperthermia susceptible. Exon screening identified 122 individuals with a genetic variant. All of them were identified in the RYR1 gene. 99 of these Individuals possessed a known RYR1 pathogenic variant and 23 a variant of unknown significance [Fig. 1]. All variants that were detected are shown in Supplement 1 + 2. Due to missing data from the in vitro contracture test, threshold levels and the results of the exposure to other trigger substance were used for diagnosis in sixteen individuals, in accordance to the European Malignant Hyperthermia Group guidelines.

Results of all tested individuals. A total of 235 individuals with a history of clinical malignant hyperthermia or close relatives were functionally analysed by the in-vitro contracture test (IVCT) and genetically screened. The detected variants were divided into known pathogenic variants and variants of unknown significance. For classification as being pathogenic, each variant has to be fully characterized at the genetic level, including co-segregation of the variant with the disease in the family affected. Moreover, functional assays have to show that a variant in the RyR1 or CACNA1S gene results an effect consistent with the MH phenotype. All other variants are classified as variant of unknown significance.
Comparison of in vitro contracture testing versus exon screening
Of the 122 individuals with an RYR1 variant (99 with diagnostic and 23 with a variant of unknown significance), the in vitro contracture test classified 116 as malignant hyperthermia susceptible, while 6 were classified as non-malignant hyperthermia susceptible. Two of the 6 non-malignant hyperthermia suceptible individuals possessed a pathogenic variant. In 113 individuals no genetic variant was detected. 66 of these individuals were classified as non-malignant hyperthermia susceptible by applying the in vitro contracture test. However, 47 individuals were classified as malignant hyperthermia susceptible (37 were malignant hyperthermia susceptible to both compounds (MHShc; 2 were malignant hyperthermia susceptible to caffeine (MHSc); 8 were malignant hyperthermia susceptible to halothane (MHSh)). Malignant hyperthermia susceptible in vitro contracture test results were significantly different compared to non-malignant hyperthermia susceptible individuals [Supplement data].
In silico analysis of variants on unknown significance
Each of the variants of unknown significance were genetically analyzed and categorized as either a pathogenic variant or a variant of unknown significance. Additionally, muscle specimen was tested using the in vitro contracture test. Some individuals were classified as malignant hyperthermia susceptible due to a positive result in the in-vitro contracture test without the detection of a genetic variant.
All pathogenic and all variants of unknown significance were subsequently tested with SIFT, Polyphen-2 and MutationTaster. All three in silico analysis programs had high sensitivity [Polyphen 0.94 CI 0.75; 0.99; SIFT 0.98 CI 0.81; 0.99; MutationTaster 0.91 CI 0.71; 0.97]. Sift correctly identified all 27 tested variants of unknown significance as “not tolerated”, which means that the tested variant was disease causing. MutationTaster correctly identified 25 of the 27 variants as disease causing. However, two were categorized as a “polymorphism” by MutationTaster, which does not exclude the variant as disease causing. Application of Polyphen2 detected three variants (one pathogenic variant and all malignant hyperthermia susceptible) as possibly rather than probably damaging and one as benign [Tables 1 and 2].
While two of the three in silico approaches correctly detected benign variants of unknown significance, MutationTaster suggested three as disease causing and one as a polymorphism.
For 13 of the variants, minor allele frequencies were found using GenomAD. Eight of the variants were pathogenic variants (c.130C > T, p.Arg44Cys; c.742G > A, p.Gly248Arg; c.1841G > T, p.Arg614Leu; c.6617C > T, p.Thr2206Met; c.6614C > G, p.Thr2206Arg; c.7300G > A, p.Gly2434Arg; c.7360C > T, p.Arg2454Cys; c.7361G > A, p.Arg2554His) and four were variants of unknown significance (c.7025A > G, p.Asn2342Ser; c.10616G > A, p.Arg3539His; c.11315G > A, p.Arg3767Pro; c.14928C > G, p.Phe4976Leu) [Table 3].
Discussion
Until now only about 48 variants of the RyR1 have been described as pathogenic for malignant hyperthermia and/or central core disease23www.emhg.org. For classification as being pathogenic each variant has to be fully characterized at the genetic level, including co-segregation of the variant with the disease in the affected family. Moreover, functional assays have to show abnormal calcium release when compared to normal (wild-type) RyR124www.emhg.org. For the other roughly 400 known RYR1 variants, functional data are still necessary for proof of pathogenicity. Several bioinformatic tools have been developed to aid in the prediction of pathogenicity of variants of unknown significance. However, bioinformatic tools are currently not approved for clinical diagnosis of malignant hyperthermia.
99 of the tested individuals in this study possessed a known RyR1 pathogenic variant. In accordance to previously published data, the IVCT identified 97 of these individuals as malignant hyperthermia susceptible. However, two individuals possessed a pathogenic variant but were not identified by the in vitro contracture test. Some variants (e.g. 1021G > A, 1840C > T and 7300G > A) have been reported to show phenotype/genotype discordance which may simply reflect the relative mutation prevalence25. Otherwise, some variants were reported to result in weaker contractures and were therefore expected to show higher discordance rates than those associated with more severe contracture phenotypes25. Thus all three mutations have been shown to be associated with weaker contractions compared to 487C > T, 648C > T, 6488G > A and 7304G > A by analysis of channel mutants in vitro and by comparative analysis of IVCT data in mutation carriers25,26,27,28.
SIFT and Polyphen-2 are the most evaluated methods to predict disease phenotype. Both algorithms are primarily based on the quantification of the constraint on the affected residue from a multiple-sequence alignment29,30,31. However, an amino acid which is not present at the substitution site in the multiple alignments can be replaced by an amino acid with the similar charge and hydrophobicity and be predicted as tolerated29,30,31,32,33. SIFT relies on mere sequence homology and is most accurate if alignments consists on mere orthologous sequences35,36. Polyphen, in contrast, uses UniProt entries to predict whether an amino acid substitution appears within an important structural or functional site of the protein36. Notably, benchmark analysis revealed that specificity and sensitivity vary significantly depending on the algorithm used. In accordance with previous studies involving RYR1 and other genes, SIFT and Polyphen-2 were found to have a high sensitivity for detecting malignant hyperthermia. In accordance with Schiemann et al., we also found the Polyphen-2 approach had a relatively high specifity. Interestingly, both approaches were suggested as being more accurate at predicting loss of function than gain of function mutations36.
Finally, we tested MutationTaster in predicting pathogenicity in RYR1. MutationTaster has been suggested to have greater sensitivity and specificity than earlier in silico approaches32,33,34. Our data tend to confirm these results with sensitivity ranging between 0.92 and 1.0. The heightened sensitivity of MutationTaster may reflect the way which it considers only the loss or decreased strength of splice sites at existing intron–exon borders. MutationTaster automatically detects and categorizes confirmed polymorphisms and known disease variants, which has been suggested to yield a false positive rate of 1% for homozygous alterations. Interestingly, our data tends to contradict this suggestion. None of the four variants previously characterized as benign were identified correctly by MutationTaster. MutationTaster is restricted in characterizing non-exonic alterations and is unable to analyze alterations spanning an intron–exon border which may partly explain the results of this study33.
However, penetrance of malignant hyperthermia is currently under investigation, since there is a clear discordance between genetic prevalence and clinical frequency of malignant hyperthermia. Risk factors for the development of a clinical event include gender (male), age (youth), lack of temperature monitoring, a combination of volatile anesthetics or succinylcholine, hyper-CKemia and most notably, the presence of diagnostic variants or presence of variants of unknown significance.
Finally, malignant hyperthermia susceptibility is unique in that a trigger compound is required and no consistent phenotype can be detected without a trigger compound. Clinical events may be due to incomplete penetrance and variable expressivity and may result in malignant hyperthermia crises even after safe exposure to a trigger anesthetic37.
Although the American and European guidelines currently recommend that the in vitro contracture test should be used to diagnose malignant hyperthermia it is possible that a positive contracture test might be associated with several deficiencies in phenotyping. The IVCT also suffers from poor to unknown inter-laboratory reliability, and an absence of data on the between-day repeatability. Similarly as the number of individuals with a neuromuscular disease that is not associated with malignant hyperthermia but still return a positive in vitro contracture test result is unknown, the true specificity of the test remains undetermined but is potentially knowable. Moreover, the penetrance and expressivity of the in vitro contracture test trait are unknown37. New diagnostic tests are frequently evaluated against “gold standards” which are assumed to classify individuals with unerring accuracy according to presence or absence of disease. Practically, however these “gold standard” tests are rarely perfect predictors of disease and tend to misclassify a small number of individuals38. When an imperfect “gold standard” is used to determine disease status this might subsequently introduce bias into measurements of test performance. For instance, the sensitivity and specificity of a new test will be underestimated, if the new test and imperfect standard test are conditionally independent (i.e. do not err on the same individuals)38.
In the current study, the majority of pathogenic RYR1 variants appeared clustered on the N-terminal amino acid residues, i.e. RYR1 AA 35-614, and the centrally located residues, i.e. AA 2163-2458 [Fig. 2]. All of the RYR1 mutations result in amino acid substitutions in the myoplasmic portion of the protein, with the exception of one mutation in the C-terminus which resides in the transmembrane region. Functional analysis showed inconsistent abnormalities. RYR1 channel kinetics might not simply be changed to be over-reactive to volatile anesthetics, but changes in inactivation kinetics and sensitivity to physiologically channel modifiers such as lipids, ATP and Mg2+ also discussed within the literature might occur. Some variants in the C-terminal region of RyR1 protein are associated with excitation–contraction uncoupling or a partially depleted sacroplasmic reticulum through a constant Ca2+ leak. Long-term myoplasmic Ca2+ overload has been associated with mitochondrial damage resulting in core myopathies. For some RYR1 mutations lower Ca2+ peak levels and lower sensitivity to Ca2+ releasing drugs have been described. Currently, there are insufficient genotype–phenotype associations, to make a definitive statement about clinical risk based on the variant type alone39,40. Therefore evaluation of “functional significance” is not trivial for several RYR1 mutations.

Schematic presentation of the RYR1 gene. Regions with the majority of the pathogenic variants were marked with a red line. Mutations also have been found in the transmembrane domains. In contrast to the mutations in the N-terminal end or in the mid-portion of the receptor, these mutations might only influence the binding of modifying ligands but also directly affect calcium pore flux. Central core disease and multi minicore disease are neuro-muscular disorder which are due to its gene proximity and overlap often associated with malignant hyperthermia.
Conclusion
Sensitivity and specificity of in silico approaches predict potential pathogenicity of variants of unknown significance in malignant hyperthermia is improving. However, based on the findings of the current study, we still recommend following the guidelines of the European Malignant Hyperthermia Group which advises to conduct a further functional test especially if the detected variants is not diagnostic. In vitro contracture testing has been shown to have a high sensitivity (97–99%) and acceptable specificity (approximately 70%) which may be increased to 94% by using two trigger compounds (halothane and caffeine)20. Such values might also be achieved by the application of the combination of bioinformatic based prediction approaches. However, variants of RYR1 or CACNA1S are only detectable in about 50–70% of the malignant hyperthermia families41,42. Five other loci have been identified by linkage analysis41,42. Since the detection of further loci cannot be excluded, functional approaches remain obligatory.
References
Hopkins, P. M. Malignant hyperthermia: pharmacology of triggering. Br. J. Anaesthes. 107, 48–56 (2011).
Klingler, W., Rueffert, H., Lehmann-Horn, F., Girard, T. & Hopkins, P. M. Core myopathies and risk of malignant hyperthermia. Anesth. Analg. 109, 1167–1173 (2009).
Rosenbaum, H. K. & Miller, J. D. Malignant hyperthermia and myotonic disorders. Anesth. Clin. N. Am. 20, 623–664 (2002).
Brandom, B. W. & Muldoon, S. M. Unexpected MH deaths without exposure to inhalation anesthetics in pediatric patients. Pediatric Anesthesia 23, 851–854 (2013).
Schiemann, A. H. & Stowell, K. M. Comparison of pathogenicity prediction tools on missense variants in RyR1 and CACNA1S associated with malignant hyperthermia. Br. J. Anaesth. 117, 124–128 (2016).
McCarthy, T. V., Quane, K. A. & Lynch, P. J. Ryandoine receptor mutations in malignant hyperthermia and central core disease. Hum. Mut. 15, 410–417 (2000).
Gillard, E. F. et al. Polymorphisms and deduced amino acid substitutions in the coding sequence of the ryanodine receptor (RYR1) gene in individuals with malignant hyperthermia. Genomics 13, 1247–1254 (1992).
Litman, R. S. et al. Malignant hyperthermia susceptible and related disease. Anesthesiology 128, 159–167 (2018).
Stowell, K. M. Malignant hyperthermia: a pharmacogenetic disorder. Pharmocogenetics 9, 1657–1672 (2008).
Hopkins, P. M. et al. European maligant hyperthermia group: the European malignant hyperthermia group guidelines for the investigation of malignant hyperthermia susceptibility. Br. J. Anaesth. 115, 531–539 (2015).
Merritt, A. et al. Assessing the pathogenicity of RyR1 variants in malignant hyperthermia. Br. J. Anaesth. 118, 533–543 (2017).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Richards, S. et al. Standard and guidelines for the interpretation of sequence variants: a joint consesnsus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Matthijs, G. et al. Guidelines for diagnostic next generation sequencing. Eur. J. Hum. Genet. 24, 2–5 (2016).
Li, M. M. et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer. A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 19, 4–23 (2017).
Abhishek, N. & Mauno, V. How good are pathogenicity predictors in detecting benign variants?. PLoS Comput. Biol. 15, 1–17 (2019).
Valdmanis, P. N., Verlaan, D. J. & Rouleau, G. A. The proportion of mutations predicted to have a deleterious effect differs between gain and loss of function genes in neurodegenerative disease. Hum. Mutat. 30, 481–489 (2009).
Doss, C. G. & Sethumadhavan, R. Investigation on the role of nsSNPs in HNPCC genes: a bioinformatics approach. J. Biomed. Sci. 16, 42 (2009).
Zullo, A. et al. Functional characterization of ryanodine receptor (RyR1) sequence variants using a metabolic assay in immortalized B-lymphocytes. Hum. Mutat. 1057, E575–E590 (2009).
Hoppe, K. et al. Hypermetabolism in B-lymphocytes from malignant hyperthermia susceptible individuals. Sci. Rep. 6, 33372 (2016).
Ording, H. In vitro contracture test for the diagnosis of malignant hyperthermia following the protocol of the European MH Group: results of testing patients surviving fulminant MH and unrelated low-risk subjects. The European malignant hyperthermia group. Acta Anaesth. Scand. 41, 955–966 (1997).
Hoppe, K., Lehmann, H., Chaiklieng, S., Jurkat-Rott, K. & Klingler, W. In vitro muscle contracture investigations on malignant hyperthermia like episodes in myotonia congenital. Acta Anesth. Scand. 57, 1017–1023 (2013).
Kim, J. H., Jarvik, G. P. & Browning, B. L. Exome sequencing reveals novel rare variants in the ryanodine receptor and calcium channel genes in malignant hyperthermia families. Anesthesiology 119, 1054–1065 (2013).
Tammaro, A. et al. Scanning for mutations of the ryandine receptor (RYR1) gene by denaturing HPLC: detection of three novel malignant hyperthermia alleles. Clin. Chem. 49, 761–768 (2003).
Robinson, R. L. et al. Recent advances in the diagnosis of malignant hyperthermia susceptibility: how confident can we be of genetic testing?. Eur. J. Hum. Gen. 11, 342–348 (2003).
Tong, J. et al. Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J. Biol. Chem. 272, 26332–26339 (1997).
Robinson, R., Brooks, C. & Brown, S. L. RYR1 mutations causing central core disease are associated with more severe malignant hyperthermia in vitro contracture test phenotypes. Hum. Mutat. 20, 88–97 (2002).
Girad, T. et al. Genotype–phenotype comparison of the Swiss malignant hyperthermia population. Hum. Mutat. 18, 357–364 (2001).
Ng, P. C. & Henikoff, S. SIFT: predicting amnio acid changes that affect protein function. Nucleic Acids Res. 31, 13 (2003).
Kumar, P., Henikoff, S. & Ng, P. C. Prediciting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–361 (2014).
Masica, D. L. & Karchin, R. Towards Increasing the clinical relevance of in silico methods to predict pathogenic missense variants. PLoS Comp. Biol. 12, e1004725 (2016).
Ritchie, G. R., Dunham, I., Zeggini, E. & Flicek, P. Functional annotation of noncoding sequence variants. Nat. Methods 11, 294–296 (2014).
Flanagan, S. E., Patch, A. M. & Ellard, S. Using SIFT and polyphen to predict loss-of-function and gain-of-function mutations. Genet. Test. Mol. Biomark. 14, 533–537 (2010).
Riazi, S., Kraeva, N. & Hopkins, P. M. Malignant Hyperthermia in the post-genomics Era: new perspectives on an old concept. Anesthesiology 128, 168–180 (2018).
Valenstein, P. N. Evalating diagnostic tests with imperfect standards. Am. J. Clin. Pathol. 93, 252–258 (1990).
Klingler, W., Rueffert, H. & Lehmann-Horn, F. Core myopathies and risk of malignant hyperthermia. Anesth. Anlag. 109, 1167–1173 (2009).
McCarthy, T. V., Quane, K. A. & Lynch, P. J. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum. Mutat. 15, 410–417 (2000).
Robinson, R., Carpenter, D., Shaw, M. A., Halsall, J. & Hopkins, P. Mutation in RyR1 in malignant hyperthermia and central core disease. Hum. Mutat. 27, 977–989 (2009).
Jurkat-Rott, K., McCarthy, T. & Lehmann-Horn, F. Genetics and pathogenesis of malignant hyperthermia. Muscle Nerve 23, 4–17 (2000).
Acknowledgements
The Authors thank Jens Dreyhaupt, Institut for Biostatistics, University of Ulm for skilful assistance.
Funding
Open Access funding enabled and organized by Projekt DEAL. Funding was provided solely from institutional and or departmental sources.
Author information
Authors and Affiliations
Contributions
W.K., K.J.R., S.W. and K.H. were responsible for intellectual Content and the study design. S.M., K.J.R., W.K., S.H., S.K. and K.H. were responsible of Collection, Analysis and Interpretation of the data. K.H., W.K., S.M., S.K. and S.H. were responsible for drafting the manuscript and graphical representation of the data. W.K., K.J.R., S.K. and S.W. were responsible for critical Evaluation of the manuscript. All authors approved the final version of the manuscript submitted for publication. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hoppe, K., Jurkat-Rott, K., Kranepuhl, S. et al. Relevance of pathogenicity prediction tools in human RYR1 variants of unknown significance. Sci Rep 11, 3445 (2021). https://doi.org/10.1038/s41598-021-82024-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82024-7
- Springer Nature Limited