
Abstract
The RYR1 gene codes for a ryanodine receptor which is a calcium release channel in the skeletal muscle sarcoplasmic reticulum. It is associated with Malignant Hyperthermia (MH) and congenital myopathies including Central Core Disease (CCD), Multiminicore Disease (MMD) and Congenital Fibre-Type Disproportion (CFTD). There is currently little information on the epidemiology of RYR1 variants in Asians. Our study aims to describe the RYR1 variant landscape in a Singapore cohort unselected for RYR1-associated conditions. Data was retrieved from the SG10K pilot project, where whole genome sequencing was performed on volunteers unselected and undetermined for RYR1-associated conditions. Variants were classified based on pathogenicity using databases ClinVar and InterVar. Allele frequencies of pathogenic variants were compared between Chinese, Indians and Malays. Using databases ExAC, GnomAD and GenomeAsia 100k study, we further compared local allele frequencies to those in Europe, America and Asia. Data was analysed using R Commander. Significant P value was set at p < 0.05. Majority of the RYR1 variants were missense mutations. We identified four pathogenic and four likely pathogenic RYR1 variants. All were related to the aforementioned RYR1-associated conditions. There were 6 carriers of RYR1 pathogenic variants amongst 4810 individuals, corresponding to an allele frequency of 0.06%. The prevalence of pathogenic variants was the highest amongst Indians (4 in 1127 individuals) (p = 0.030). Majority of pathogenic and likely pathogenic mutations were missense and located in mutational hotspots. These variants also occurred at higher frequencies in Asians than globally. This study describes the variant landscape of the RYR1 gene in Singapore. This knowledge will facilitate genetic screening for RYR1-related conditions.
Similar content being viewed by others

Introduction
The RYR1 gene1 codes for a ryanodine receptor found in the skeletal muscle and the receptor serves as a calcium release channel in the sarcoplasmic reticulum. Mutations in RYR1 are strongly associated with Malignant Hyperthermia (MH) and congenital myopathies such as Multiminicore Disease (MMD), Central Core Disease (CCD), and Congenital Fibre-Type Disproportion (CFTD)2 .
MH is a pharmacogenetic disorder of skeletal muscle which may be inherited in an autosomal dominant fashion or arise due to de novo mutations in associated genes, most commonly RYR13,4. The prevalence of MHS is estimated to be 1: 2000–3000 in individuals.
However the incidence of MH during general anaesthesia is estimated to be 1: 5000 to 1: 50,000–100,000 in individuals5. There is little data on the prevalence of MHS and other RYR1 associated myopathies in Singapore6. MH-causative mutations in RYR1 result in increased sensitivity of RYR1 protein to activation7,8,9. In the presence of anesthetic triggering agents, dysregulation of calcium transport results in excessive myoplasmic calcium accumulation. This manifests clinically as a hypermetabolic crisis when an MH-susceptible individual is exposed to a volatile anesthetic agent or depolarizing muscle relaxants. This syndrome is potentially lethal if left untreated. The clinical manifestations of MH include initial hypercarbia, tachypnea, generalised muscle rigidity, masseter muscle rigidity, as well as arrhythmias. Subsequently, hyperthermia and rhabdomyolysis may occur10.
Congenital myopathies are inherited neuromuscular disorders characterised by common histologic features seen on muscle biopsy, such as central cores, nemaline rods, multi-minicores and central nuclei. RYR1 is associated with several such myopathies, such as CCD, certain subtypes of MMD, and CFTD. There is significant genetic, phenotypic and histopathological overlap between these myopathies, as well as considerable phenotypic variability. CCD is inherited in an autosomal dominant fashion and is characterised by hip girdle weakness with orthopaedic complications such as hip dislocation and scoliosis. MMD is inherited in an autosomal recessive fashion and has a variety of clinical phenotypes. The most common subtype leads to spinal rigidity, scoliosis and respiratory dysfunction, and is associated with mutations in the selenoprotein N gene. Other subtypes are associated with RYR1 mutations and can be characterised by hip girdle weakness, external ophthalmoplegia and distal muscle atrophy. Various studies have suggested that these subtypes of MMD may exist on a clinco-pathologic continuum with CCD11,12. CFTD can be inherited in an autosomal dominant or recessive pattern and rarely in an X-linked pattern13. It is characterised by weakness in the proximal muscle groups close to the trunk, such as the shoulder and hips. It can also result in ophthalmoplegia, ptosis (droopy eyelids) and scoliosis. RYR1 mutations account for 10–20% of CFTD14.
Many of the RYR1-associated myopathies are linked to MH-susceptibility, and it is estimated that MH-susceptibility and myopathy co-occur in at least 30% of individuals with RYR1 pathogenic variants. As a result, patients with RYR1-associated myopathies should be considered MH-susceptible unless proven negative by an MH contracture biopsy14.
Genetic testing is a common and non-invasive diagnostic test for many of these conditions. Most of the genetic studies done on RYR1 mutations were performed on patients diagnosed with MH and other myopathies, and this is useful in identifying pathogenic variants. For example, a study done by Wu et al. was conducted on 27 unrelated Japanese patients with CCD, which identified 20 novel and 3 previously reported missense mutations15. Another studied 58 MH-susceptible Japanese patients and found 26 unknown RYR1 pathogenic variants and 7 previously reported variants16. However, to estimate the true population prevalence of these mutations, studies need to be conducted in the general population. Our review of the existing literature revealed one such variant landscape study on the general population, however this was conducted on the American population made up of mainly Caucasians17. There is little information on the epidemiology of RYR1 variants in the Asian population. Our study performed on the multiracial Singapore population will be useful in providing a snapshot of the spectrum of mutations in Asia.
Our study aims to describe the RYR1 variant landscape in a Singaporean cohort unselected for RYR1-associated myopathies and MH. We seek to define the prevalence of pathogenic variants in 3 major ethnic groups in Singapore and compare it to existing databases. This will facilitate genetic screening of these conditions in the Singapore population.
Materials and methods
Data was retrieved from the SG10K pilot project, which is part of a national effort to eventually whole-genome sequence 10,000 Singaporeans. The aim of this study was to provide a snapshot of the genetic diversity in East Asia, Southeast Asia, and South Asia. This is possible given the ethnic diversity in Singapore, which consists mainly of Chinese (74.3%), Malays (13.4%) and Indians (9.1%). The ethnic clusters in Singapore were analysed using principal-components analysis and are further described in the SG10K pilot project. In this study, whole genome sequencing was performed on 4810 samples, including 2780 Chinese, 903 Malays and 1127 Indians. These volunteers include a mixture of healthy and diseased individuals with conditions such as heart and neurodegenerative conditions. Linkage disequilibrium (LD)-based joint calling and quality controls were performed to remove low-quality variants and population-based phasing to obtain haplotypes. After removing relatedness up to the third degree, a subset of 4,441 unrelated individuals were obtained. Further details on the methodology can be obtained from the SG10K paper18.
ClinVar and InterVar were used to classify variants into “benign”, “likely benign”, “uncertain significance”, “likely pathogenic” and “pathogenic”. ClinVar is a database of clinical variations which collects data on genotype–phenotype relationships from various contributors. It aggregates the submissions and reports the clinical significance of variants based on the American College of Medical Genetics (ACMG) and Association for Molecular Pathology (AMP) Guidelines19,20. InterVar is a bioinformatics software which classifies variants via automation of the 2015 ACMG/AMP guidelines21,22. ClinVar serves as our main reference for determining the clinical significance of variants as the classifications are more thoroughly supported with published evidence. The variant positions for RYR1 refer to NM_001042723.
Variants were also classified based on the location and type of mutations. Single Nucleotide Variant (SNV) types include synonymous SNV, missense SNV, stop gain SNV, stop loss SNV and unknown. Indel variant types include frameshift insertion, frameshift deletion, stop gain, stop loss, non-frameshift insertion, non-frameshift deletion and unknown.
Allele frequencies were compared between the Chinese, Indian and Malay population in Singapore. Allele frequencies in the Singapore population were also compared against European, American and Asian populations using databases ExAC v3 (Exome Aggregation Consortium)23, GnomAD v2.1.1 (Genome Aggregation Database)24, and data from the GenomeAsia 100k study25. The allele frequencies in the ExAC database are based on 60,706 subjects26, that of GnomAD are based on 141,456 subjects27, and that of GnomeAsia 100k are based on 1,739 subjects28 Detailed breakdown of subjects from different ethnicities can be found under the description of Supplementary table S5.
These studies were approved by the Institutional Review Board of the National University of Singapore (Approvals: N-17-030E and H-17-049), SingHealth Centralized Institutional Review Board (Approvals: 2006/612/A, 2014/160/A, 2018/2570, 2018/2717, 2010/196/C, 2015/2194, 2019/2046, 2009/280/D, 2014/692/D, 2002/008/A, 2002/008 g/A, 2013/605/C, 2018/3081 and 2015/2308), National Health Group Domain Specific Review Board (Approvals: 2007/00167, 2016/00269, 2018/00301, TTSH/2014-00040 and 2009/00021). Written informed consent was obtained from all participants of the SG10K pilot project from which data for this study was obtained.
All methods were carried out in accordance with relevant guidelines and regulations.
Statistical analysis
Data was analysed using R statistical software. The statistical difference between the allele frequencies in different populations was calculated based on Fisher’s exact test for count data under the function ‘fisher.test’. The type I error rate to reject the null hypothesis of no association was set at 0.05. One-sided test option was chosen when testing the hypothesis of the odds-ratio being greater or smaller in one population compared to the other.
Ethics approval
These studies were approved by the Institutional Review Board of the National University of Singapore (Approvals: N-17-030E and H-17-049), SingHealth Centralized Institutional Review Board (Approvals: 2006/612/A, 2014/160/A, 2018/2570, 2018/2717, 2010/196/C, 2015/2194, 2019/2046, 2009/280/D, 2014/692/D, 2002/008/A, 2002/008g/A, 2013/605/C, 2018/3081 and 2015/2308), National Health Group Domain Specific Review Board (Approvals: 2007/00167, 2016/00269, 2018/00301, TTSH/2014-00040 and 2009/00021).
Consent to participate
This study was conducted retrospectively using data obtained from the SG10K pilot project. Written informed consent was obtained from all participants of the SG10K pilot project.
Consent for publication
This study was conducted retrospectively using data obtained from the SG10K pilot project. Written informed consent was obtained from all participants of the SG10K pilot project.
Results
Types of mutations found
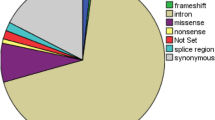
All the RYR1 variants were classified based on mutation type (Fig. 1). All of these mutations occurred in exonic regions. Majority of the mutations were missense, followed by synonymous variants.

Mutation counts and types for RYR1 variants in SG10K pilot data.
Classification of RYR1 variants based on clinical significance
The RYR1 variants found in our study were classified based on their clinical significance given by ClinVar (Supplementary table S1) and InterVar (Supplementary table S2). ClinVar was used as the main reference for defining the clinical significance of the variants. Based on ClinVar, there were 4 pathogenic and 2 likely pathogenic variants for RYR1. Based on InterVar, there were 1 pathogenic and 6 likely pathogenic RYR1 variants. Of these, all overlapped with the pathogenic and likely pathogenic variants found in ClinVar except for 2 likely pathogenic variants. Table 1 summarizes the list of pathogenic and likely pathogenic RYR1 variants found in our study. Table 2 and Supplementary table S3 provide details on the specific diseases associated with each variant. The majority of pathogenic and likely pathogenic RYR1 variants in our study were missense mutations, except for 2 nonsense mutations (Table 1).
In terms of the location of pathogenic and likely pathogenic RYR1 mutations, all but 2 likely pathogenic variants were found in mutational hotspots. These RYR1 mutational hotspots have been previously identified to include the N-terminal region between codons 34 and 614 (exons 2 to 19), central region between codons 2163 and 2458 (exons 39 to 47), and C-terminal region between codons 4136 and 4973 (exons 85 to 104)29. There was no clear pattern as to the exons in which the pathogenic and likely pathogenic mutations were found. However, we found that most of the pathogenic and likely pathogenic RYR1 variants have mutations occurring in highly conserved regions (Supplementary table S4).
Ethnic distribution of the carriers of the pathogenic RYR1 variants
The ethnic distribution of pathogenic RYR1 variants was analysed. Indians made up the majority (67%) of the individuals who carried pathogenic variants (p = 0.030) (Supplementary figure S1).
On further analysis of the ethnic distribution, each RYR1 variant was found exclusively in a single ethnicity. Of the 4 pathogenic variants identified by ClinVar, 1 was present in Chinese, 2 in Indians, and 1 in a Malay. All variants were present at similar frequencies (only 1 carrier), except c.7268T > A which was present in 3 Indian carriers (Fig. 2).

Ethnic distribution of individual pathogenic RYR1 variants.
The analysis was repeated with both pathogenic and likely pathogenic RYR1 variants. Indians still made up the largest proportion of carriers (50%) (p = 0.009) (Supplementary figure S2). Further analysis similarly revealed that each RYR1 variant was unique to each ethnicity. Of the 8 pathogenic and likely pathogenic variants, 3 were present in the Chinese, 2 were present in the Malays and 3 in the Indians. Three of the variants occurred at higher frequencies—c.7268T > A was found in 3 Indians as previously mentioned, c.1186G > T was found in 3 Malays and c.2654G > A in 3 Indians (Supplementary figure S3).
Allele frequencies across different populations
For the RYR1 gene, allele frequencies of 2 of the pathogenic variants (c.7268T > A and c.14111C > T) found in our study were significantly higher in Singapore than globally (Supplementary tables S5 and S6). c.325C > T occurred at significantly higher frequencies in the Non-Finnish European population based on the ExAC database (p = 0.048). c.6487C > T occurred at significantly higher frequencies in the South Asian population based on the gnomAD database (p = 0.048). Amongst the likely pathogenic variants, 3 variants (c.1186G > T, c.2654G > A and c.8554C > T) occurred at significantly higher allele frequencies in Singapore than globally. Only variant c.742G > T had a significantly higher allele frequency in the South Asian population based on the GenomeAsia 100K study (p = 1.13 × 10–12).
In sum, allele frequencies of the pathogenic and likely pathogenic variants found in our study were generally higher amongst Asians (Singapore population and South Asian population) than globally. Comparing between the Singapore population and South Asian population, different databases have provided different allele frequencies for the South Asian population and therefore we are unable to conclude if these variants occur at higher frequencies in either population. Data from other parts of Asia is also lacking.
Discussion
Due to the low incidence of MH and congenital myopathies, there is no data on the actual prevalence in Asia. There is no readily available genetic testing and gold standard for diagnosis is muscle biopsy which is not available locally. Most of the genetic studies on RYR1 have been conducted on populations selected for RYR1-associated disorders, and there is limited information on the epidemiology of RYR1 variants in the Asian population. This is the first paper that analyses the genetic landscape of RYR1 specifically in a multi-ethnic South-East Asian population. The SG10K study of which our data is retrieved from is a population-based cohort and not a disease-selected cohort, hence reflecting true population prevalence. Whilst some variants corroborate with data from other disease-specific populations, we also identified other variants of which the significance is unknown.
The prevalence of pathogenic RYR1 variants in our study was 6 in 9620, corresponding to an estimate of 1 in 2000 in the Singapore population. Interestingly, we found that the prevalence of pathogenic RYR1 variants in Indians was 4 in 1127, much higher than in Chinese and Malays (1 in 2780 and 1 in 903 respectively). The variant c.7268T > A occurred at the highest frequency out of all the other pathogenic variants—this variant has been previously reported in patients of predominantly European ancestry30,31,32, and interestingly our study shows that this variant was particularly prevalent in those of Indian ethnicity in our population (exclusively found in 3 Indians). The allele frequencies of the pathogenic variants found in our study were generally higher in Asians (Singapore population and South Asian population) than globally. In terms of the location of RYR1 mutations, those found in our study largely occurred within established mutational hotspots.
Significance of the pathogenic variants found
The pathogenic RYR1 variants found were closely associated with diseases such as MH, MMD, CCD and CFTD.
RYR1 gene encodes the Ca2+-dependent, Ca2+ channel ryanodine receptor 1 (RYR1) found on the sarcoplasmic reticulum of the skeletal muscle33.
MH occurs due to dysregulation of Ca2+ transport caused by abnormal functioning of the skeletal muscle excitation–contraction (EC) coupling complex which comprises of an L-type voltage-gated Ca2+ channel (dihydropyridine receptor, DHPR) and a Ca2+-dependent, Ca2+ channel (RYR1). Excessive Ca2+ release from the sarcoplasmic reticulum causes uncontrolled muscle contraction34.
Mutations in the RYR1 gene are also a common cause of congenital myopathies. Dominant mutations have been associated with CCD, while recessive mutations are associated with MMD and CFTD. Mutations associated with CCD leads to chronic channel dysfunction either through excitation–contraction uncoupling or persistent channel leakiness. However, little is known about recessive mutations and their mechanism of illness35.
Four pathogenic RYR1 variants were found and depicted in Fig. 3. The N-terminal domains (NTD) make key interactions with other domains to stabilise the cytosolic assembly, and these interactions have also been implicated in channel gating and pore opening. c.325C > T is located in NTD-A, and both c.6487C > T and c.7268 T > A are located in the helical domain (B-Sol). Both NTD-A and B-Sol are involved in key interprotomer contacts36. In the context of MH, mutations at these points of contact may weaken the interfaces and facilitate pore opening and spurious Ca2+ leak34. c.6487C > T is a known causative mutation for MH and the amino acid substitution alters the conformational state of RYR1, exposing the receptor’s activating sites to stimulatory agents37. c.325C > T and c.7268T > A occur due to mutations affecting highly conserved amino acid residues and are known to be autosomal recessive in relation to RYR1-related myopathies12,38. However, they have yet to be studied in relation to the MH-susceptibility phenotype.

Schematic diagram of RYR1 protein subunit and pathogenic variants in this study. RYR1 is a homotetramer with each subunit consisting of a large cytoplasmic moiety and a transmembrane domain. This figure depicts one subunit of the RYR1 protein. The cytoplasmic moiety contains (1) the N-terminal domain (NTD) with 3 subdomains (A, B, C), (2) 3 SPRY domains (SPRY 1, 2, 3), of which SPRY 2 and 3 interact with the α1 subunit of DHPR at the II-III loop region, (3) a solenoid region comprising the handle/ junctional solenoid (J-sol) domain, and the helical (HD1, HD2)/ bridging solenoid (B-Sol) domains, (4) 2 tandem repeat domains (R1&2, R3&4), and (5) a Central domain or core solenoid (C-Sol). The transmembrane domain consists of 6 transmembrane helices (S1–6), and a C-terminal domain (CTD). S5 and 6, together with a pore helix in between S5 and S6, make up the pore-forming region (PF)19,25,34. The amino acid residue numbers for the various domains have been labelled, and the pathogenic RYR1 mutations are indicated at their various positions across the protein.
c.14111C > T affects a highly conserved amino acid residue12 located within the transmembrane helices S2-S3. It is hypothesised that this region participates in calcium-sensing and calcium-dependent inactivation of RYR39. This mutation is known to be autosomal recessive in relation to RYR1-related myopathies12. There is conflicting evidence for the clinical significance of this RYR1 variant in relation to MH, with some suggesting that it is likely pathogenic for MH and others suggesting that it is a variant of unknown significance40. Further studies are needed to definitively conclude its relation to MH-susceptibility.
Limitations
This study is a retrospective study done using anonymised genetic data from the SG10K study. This study population contains a mixture of healthy and diseased individuals with comorbidities such as eye, neurodegenerative, metabolic, cardiac diseases. We therefore lacked access to the medical histories of the study participants. Hence, we are unable to determine if the carriers of these pathogenic RYR1 variants displayed phenotypes of the associated diseases, and we also cannot determine the penetrance of these mutations.
Application
According to the EMHG guidelines, evaluation of patients for MH-susceptibility involves assessing for risk factors such as family history of MH or unexplained perioperative death, personal adverse reaction to general anaesthesia, postoperative or exertional rhabdomyolysis or exertional heat stroke. For patients with such risk factors, further testing is recommended. The decision between IVCT and genetic testing depends on several factors: the availability of the respective test, the urgency of test, the prior probability of a positive diagnosis and the cost of the tests. Genetic testing offers a less invasive option but is less sensitive than IVCT. Patients with a negative genetic screening result may proceed to do an IVCT to conclusively exclude MH susceptibility. Genetic testing has a major role in family screening and its cost-effectiveness makes it a viable option as the primary investigation of index cases41. The North American MH Genetic Group also adopts a similar approach by incorporating genetic testing into its screening protocol42. Disclosure of incidental findings from MH-susceptibility genetic screening in generally healthy individuals has also been found to reduce cost by USD $900 and increase quality-adjusted life years (QALYs) by 0.0943.
Currently there is no active screening for MH-susceptibility in Singapore due to the fact that MH is a rare condition and that genetic testing is still relatively costly to the individual. Screening for malignant hyperthermia is only done through history-taking. Genetic testing will only be offered for individuals with clinical suspicion of MH-susceptibility. Currently, muscle contracture test which is the gold standard is not available in Singapore. By understanding the variants in our population, it will be valuable in understanding the clinical utility of using these variants in the blood as a screening and diagnostic tool.
Since there is significant overlap between MH susceptibility and congenital myopathies, we recommend genetic screening for malignant hyperthermia in patients with congenital myopathies and vice versa14.
Conclusion
From this study, we conclude that the prevalence of the pathogenic RYR1 variants found in our study is higher amongst Asians than globally. Indians made up the majority of the individuals who carried pathogenic variants, compared to Chinese and Malays. The pathogenic variants were associated with diseases such as MH, MMD, CCD and CFTD. This paper also confirms that the variants found in our population are indeed rare and unique to individuals, suggesting a need to conduct genetic screening using sequencing rather than methods which look at specific mutations known to the database.
Data availability
The accession number for the sequence data reported in this paper is EGA: EGAS00001003875.
Code availability
The accession number for the sequence data reported in this paper is EGA: EGAS00001003875.
Abbreviations
- MH:
-
Malignant hyperthermia
- SNV:
-
Single nucleotide variants
References
NCBI. RYR1 Ryanodine Receptor 1 [Homo sapiens (Human)] (NCBI, 2020).
Todd, J. J., Goldberg, M. F., Dirksen, R. T. & Voermans, N. C. RYR1-Related Diseases. https://rarediseases.org/rare-diseases/ryr-1-related-diseases/ (2021).
Miller, D. M. et al. Genetic epidemiology of malignant hyperthermia in the UK. Br. J. Anaesth. 121, 944–952. https://doi.org/10.1016/j.bja.2018.06.028 (2018).
Rosenberg, H., Pollock, N., Schiemann, A., Bulger, T. & Stowell, K. Malignant hyperthermia: A review. Orphanet J. Rare Dis. 10, 93. https://doi.org/10.1186/s13023-015-0310-1 (2015).
Kim, D.-C. Malignant hyperthermia. Korean J. Anesthesiol. 63, 391–401. https://doi.org/10.4097/kjae.2012.63.5.391 (2012).
Li, D. W. Y., Lai, P. S., Lee, D. W., Yong, R. Y. Y. & Lee, T. L. Malignant hyperthermia and ryanodine receptor type 1 gene (RyR1) mutation in a family in Singapore. Ann. Acad. Med. 46, 60 (2017).
Robinson, R., Carpenter, D., Shaw, M. A., Halsall, J. & Hopkins, P. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum. Mutat. 27, 977–989. https://doi.org/10.1002/humu.20356 (2006).
Treves, S., Jungbluth, H., Muntoni, F. & Zorzato, F. Congenital muscle disorders with cores: The ryanodine receptor calcium channel paradigm. Curr. Opin. Pharmacol. 8, 319–326. https://doi.org/10.1016/j.coph.2008.01.005 (2008).
Riazi, S., Kraeva, N. & Hopkins, P. M. Malignant hyperthermia in the post-genomics era: New perspectives on an old concept. Anesthesiology 128, 168–180. https://doi.org/10.1097/ALN.0000000000001878 (2018).
Litman, R. S. in UpToDate (ed Stephanie B Jones) (UpToDate, 2019).
Jungbluth, H. Multi-minicore disease. Orphanet J. Rare Dis. 2, 31–31. https://doi.org/10.1186/1750-1172-2-31 (2007).
Zhou, H. et al. Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain 130, 2024–2036. https://doi.org/10.1093/brain/awm096 (2007).
MedGen. (NCBI).
Litman, R. S., Griggs, S. M., Dowling, J. J. & Riazi, S. Malignant hyperthermia susceptibility and related diseases. Anesthesiology 128, 159–167. https://doi.org/10.1097/ALN.0000000000001877 (2018).
Wu, S. et al. Central core disease is due to RYR1 mutations in more than 90% of patients. Brain 129, 1470–1480 (2006).
Ibarra M, C. A. et al. Malignant hyperthermia in Japan: Mutation screening of the entire ryanodine receptor type 1 gene coding region by direct sequencing. Anesthesiology 104, 1146–1154 (2006).
Gonsalves, S. G. et al. Using exome data to identify malignant hyperthermia susceptibility mutations. Anesthesiology 119, 1043–1053. https://doi.org/10.1097/ALN.0b013e3182a8a8e7 (2013).
Wu, D. et al. Large-scale whole-genome sequencing of three diverse Asian populations in Singapore. Cell 179, 736-749.e715. https://doi.org/10.1016/j.cell.2019.09.019 (2019).
ClinVar and ClinGen. https://www.ncbi.nlm.nih.gov/clinvar/docs/clingen/.
NCBI. ClinVar and ClinGen. https://www.ncbi.nlm.nih.gov/clinvar/docs/clingen/.
Clinical Interpretation of Genetic Variants by ACMG/AMP 2015 Guideline. https://www.ncbi.nlm.nih.gov/clinvar/docs/clingen/.
Li, Q. & Wang, K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 100, 267–280. https://doi.org/10.1016/j.ajhg.2017.01.004 (2015).
ExAC. ExAC Browser. http://exac.broadinstitute.org/.
gnomAD. gnomAD. https://gnomad.broadinstitute.org/.
GenomeAsia. GenomeAsia 100k. https://genomeasia100k.org/.
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. https://doi.org/10.1038/nature19057 (2016).
Laurent Francioli, G. T., Karczewski, K., Solomonson, M. & Watts, N. gnomAD v2.1. https://macarthurlab.org/2018/10/17/gnomad-v2-1/ (2018).
Consortium, G. K. The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature 576, 106–111. https://doi.org/10.1038/s41586-019-1793-z (2019).
Gillies, R. L., Bjorksten, A. R., Davis, M. & Du Sart, D. Identification of genetic mutations in Australian malignant hyperthermia families using sequencing of RYR1 hotspots. Anaesth. Intensive Care 36, 391–403. https://doi.org/10.1177/0310057x0803600311 (2008).
Jungbluth, H. et al. Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene. Neurology 27, 12. https://doi.org/10.1212/01.wnl.0000188870.37076.f2 (2005).
Monnier, N. et al. Null mutations causing depletion of the type 1 ryanodine receptor (RYR1) are commonly associated with recessive structural congenital myopathies with cores. Hum Mutat 29, 670–678. https://doi.org/10.1002/humu.20696 (2008).
Klein, A. et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum Mutat 33, 981–988. https://doi.org/10.1002/humu.22056 (2012).
Beam, T. A., Loudermilk, E. F. & Kisor, D. F. Pharmacogenetics and pathophysiology of CACNA1S mutations in malignant hyperthermia. Physiol. Genom. 49, 81–87. https://doi.org/10.1152/physiolgenomics.00126.2016 (2016).
Santulli, G., Lewis, D., van Georges, A., Marks, A. R. & Frank, J. Ryanodine receptor structure and function in health and disease. Subcell Biochem. 87, 329–352. https://doi.org/10.1007/978-981-10-7757-9_11 (2018).
Amburgey, K. et al. Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J. Rare Dis. 8, 117. https://doi.org/10.1186/1750-1172-8-117 (2013).
Wang, Q. et al. Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a Chinese family. J. Mol. Med. (Berl) 83, 203–208. https://doi.org/10.1007/s00109-005-0638-4 (2005).
Treves, S. et al. Ryanodine receptor 1 mutations, dysregulation of calcium homeostasis and neuromuscular disorders. Neuromuscul. Disord. 15, 577–587. https://doi.org/10.1016/j.nmd.2005.06.008 (2005).
Yuchi, Z., Kimlicka, L. & Van, F. Structural Insights into Disease Mutations of the Ryanodine Receptor (2013).
Zalk, R. et al. Structure of a mammalian ryanodine receptor. Nature 517, 44–49. https://doi.org/10.1038/nature13950 (2015).
NCBI. (NCBI, 2019).
Girard, T. Testing for MH Susceptibility. https://www.emhg.org/testing-for-mh (2015).
Nelson, T. E., Rosenberg, H. & Muldoon, S. M. Genetic testing for malignant hyperthermia in North America. Anesthesiol. J. Am. Soc. Anesthesiol. 100, 212–214 (2004).
Bennette, C. S., Gallego, C. J., Burke, W., Jarvik, G. P. & Veenstra, D. L. The cost-effectiveness of returning incidental findings from next-generation genomic sequencing. Genet. Med. 17, 587–595. https://doi.org/10.1038/gim.2014.156 (2015).
ClinGen. Variant: NM_001042723.2(RYR1):c.2654G>A (p.Arg885His). https://erepo.clinicalgenome.org/evrepo/ui/interpretation/75f6a216-0fff-441e-a01f-0c9a73ffa399.
Acknowledgements
We thank all the individuals who have contributed to this study.
Funding
Support was provided by the Department of Anaesthesia, National University Health System Singapore.
Author information
Authors and Affiliations
Contributions
C.T.Y.F.: first author, prepared the manuscript, figures and tables. Y.H.T.: first author, prepared the manuscript, figures and tables. A.I.: experienced in genetic research, provided guidance and supervision to first authors in their research and data analysis. A.Y.-J.N.: provided technical support in interpreting genetic data retrospectively obtained from the SG10K project. B.Y.: provided technical support in interpreting genetic data retrospectively obtained from the SG10K project. S.T.H.C.: utilised her clinical expertise to provide guidance and supervision to first authors in their research and data analysis. J.L.: provided guidance and supervision to first authors. L.K.T.: conceptualised this study, provided guidance and supervision to first authors.
Corresponding author
Ethics declarations
Competing interests
A.I. is an employee of Nalagenetics Pte Ltd. A.I. and J.L. has financial holdings in Nalagenetics Pte Ltd. The rest of the authors do not have any conflict of interest to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Foo, C.T.Y., To, Y.H., Irwanto, A. et al. Variant landscape of the RYR1 gene based on whole genome sequencing of the Singaporean population. Sci Rep 12, 5429 (2022). https://doi.org/10.1038/s41598-022-09310-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-09310-w
- Springer Nature Limited
This article is cited by
-
Function of a mutant ryanodine receptor (T4709M) linked to congenital myopathy
Scientific Reports (2023)