
Abstract
Delayed diagnosis and misdiagnosis are frequent in people with amyotrophic lateral sclerosis (ALS), the most common form of motor neuron disease (MND). Neurofilament light chain (NFL) and phosphorylated neurofilament heavy chain (pNFH) are elevated in ALS patients. We retrospectively quantified cerebrospinal fluid (CSF) NFL, CSF pNFH and plasma NFL in stored samples that were collected at the diagnostic work-up of ALS patients (n = 234), ALS mimics (n = 44) and controls (n = 9). We assessed the diagnostic performance, prognostication value and relationship to the site of onset and genotype. CSF NFL, CSF pNFH and plasma NFL levels were significantly increased in ALS patients compared to patients with neuropathies & myelopathies, patients with myopathies and controls. Furthermore, CSF pNFH and plasma NFL levels were significantly higher in ALS patients than in patients with other MNDs. Bulbar onset ALS patients had significantly higher plasma NFL levels than spinal onset ALS patients. ALS patients with C9orf72HRE mutations had significantly higher plasma NFL levels than patients with SOD1 mutations. Survival was negatively correlated with all three biomarkers. Receiver operating characteristics showed the highest area under the curve for CSF pNFH for differentiating ALS from ALS mimics and for plasma NFL for estimating ALS short and long survival. All three biomarkers have diagnostic value in differentiating ALS from clinically relevant ALS mimics. Plasma NFL levels can be used to differentiate between clinical and genetic ALS subgroups.
Similar content being viewed by others


Introduction
Amyotrophic lateral sclerosis (ALS) is an adult onset fatal neurodegenerative syndrome characterized by the insidious onset of progressive motor symptoms and signs secondary to the loss of upper and lower motor neurons and their tracts1. In the majority of patients, symptoms start in a limb, termed spinal onset ALS, or in the head and neck region, named bulbar onset ALS1. The median diagnostic delay is approximately a year2 which is a long time for a condition with a median survival time from onset of paresis of only 30 months3. ALS is heterogeneous, misdiagnoses are frequent, and it is a challenge in clinical practice to determine an ALS diagnosis4. It is essential in the diagnostic process to exclude a number of conditions termed ALS mimics that present with symptoms similar to ALS and may be difficult to differentiate5,6,7. Approximately 10% of ALS patients self-report a family history of ALS (fALS), some also report frontotemporal dementia (FTD), and the other ≈90% are classified as having sporadic ALS (sALS)8. To date, over 40 ALS or ALS-FTD causative genes have been reported, the most common being an intronic hexanucleotide repeat expansion (HRE) in C9orf72 found in 8–12% of ALS patients in Caucasian populations and coding mutations in SOD1 (encoding superoxide dismutase type 1, SOD1) found in 2–5% of ALS patients9. Most of the mutations identified in SOD1 result in an unstable mutant protein with reduced enzymatic activity10. Analysis of SOD1 activity in the blood and SOD1 protein concentration in the cerebrospinal fluid (CSF) of patients were the first biomarkers used for diagnosing ALS and as an endpoint in anti-SOD1 clinical drug trials10,11. The rarity of SOD1 mutations and the finding that seven SOD1 mutants have preserved enzymatic activity limit the clinical use of SOD1 enzymatic analysis, although the finding of reduced SOD1 enzymatic activity may be critical for correctly diagnosing ALS in patients who had been on diagnostic odysseys10,12,13. C9orf72HRE results in the formation of polydipeptides that can be detected in the CSF, but the results have not yet become part of routine clinical use14. Other ALS-causing genes have not resulted in the identification of a specific biomarker for clinical use.
Neurofilaments are neuron-specific cytoskeletal intermediate filament heteropolymers composed of neurofilament light chain (NFL), NF medium chain, and NF heavy chain (NFH) in combination with either α-internexin in the central nervous system or peripherin in the peripheral nervous system15,16. Levels of CSF NFL, CSF phosphorylated NFH (pNFH) and serum NFL levels increase with neuronal injury and axonal damage in several neurological disorders, including ALS17,18,19,20,21. In longitudinal studies of carriers of SOD1 and C9orf72HRE mutations, NFL levels start to increase 1–12 months prior to the onset of paresis (phenoconversion), continues to rise in early symptomatic patients but then remains relatively stable22. NF levels correlate negatively with survival in ALS17,23,24,25. Here, we retrospectively investigated the diagnostic and prognostic value of assaying CSF NFL, CSF pNFH and plasma NFL in samples collected from patients during the diagnostic procedure performed at a specialized university clinic for ALS evaluation and examined whether these biomarkers differed between clinical and genotypic subtypes of ALS.
Materials and methods
Participants
All procedures were performed in accordance with the 1964 Declaration of Helsinki and its later amendments. The study was approved by the Medical Ethical Committee (Forskningsetikkommitten FEK, nr 1994-135 with later amendments in 1998, 2003, 2014, 2017, 2018) and written informed consent was obtained from all patients to obtain and study the samples and publish the results. The participants were referred to the Department of Neurology, University Hospital of Umeå, Sweden, from 1994 to 2016 to be examined for ALS. The patients were evaluated according to the diagnostic guidelines of the European Federation of Neurological Societies for the clinical management of ALS and diagnosed according to the revised El-Escorial criteria26,27. Typically, the diagnostic workup would be performed with the patient in the neurology ward for 3–4 days, and the CSF and plasma samples used in this study were collected at this time. When investigating new patients, since 1994, it has been a custom in our clinic to routinely collect extra blood and CSF samples in case additional material is needed in the diagnostic evaluation after the patient has been discharged from the hospital. These samples are collected and stored with prior written informed consent and may (with approval by the Medical Ethical Committee, FEK) later be used for research purposes should they no longer be needed for further clinical analysis. Patients diagnosed with ALS were followed at regular intervals by a multidisciplinary clinical ALS team. Patients diagnosed with alternative diagnoses were followed by other specialized teams or by local neurologists.
In an initial selection process for inclusion in this study, stored samples from patients referred with a prior medical history of CNS infection, tuberculosis, neuroborreliosis, HIV, syphilis, severe systemic inflammatory disease, severe head trauma or neoplastic conditions or daily medication with anti-inflammatory drugs were excluded. Additionally, patients were excluded if their available CSF and/or plasma samples were insufficient, if they were lost to follow-up (moved abroad or the medical charts were censored for other reasons), participated in a clinical drug trial or their consent forms could not be retrieved.
ALS patient functional status at sampling was assessed retrospectively using the ALS functional rating scale revised (ALSFRS-R)28 and the disease progression rate (ΔFS) was calculated as described29. Following diagnosis, riluzole was administered to most patients and eventually most patients received a gastrostomy for gastro-enteral feeding and non-invasive ventilation. None of the patients in this study received invasive ventilation through tracheostomy. Survival was defined as the time from onset of paresis to death, usually from respiratory failure due to paresis and/or pneumonia. Disease duration at sampling was defined as the time between onset of paresis and clinical evaluation. Eighteen patients were alive at the time of the study and were excluded from the survival analyses. With separate written informed consent and collection of additional blood for DNA analysis, genetic analyses were performed to evaluate a panel of ALS genes as previously described30,31.
For the data analyses, one patient with C9orf72HRE who was diagnosed with FTD and a patient with C9orf72HRE with FTD and progressive muscular atrophy (PMA) were included in the ALS group. One ALS patient had a SOD1 G93S mutation and a vesicle-associated membrane protein-associated protein B gene (VAPB) mutation and was included in the other SOD1 mutation group. One ALS patient was heterozygous for both C9orf72HRE and SOD1 D90A and was included in the C9orf72HRE mutation group.
Sample collection and measurements
An extra 2–4 mL of CSF was collected by lumbar puncture during the diagnostic workup, aliquoted and immediately stored at −80 °C. The CSF was not centrifuged before storage. Peripheral blood was collected at the same time by standard venipuncture and collected in EDTA-containing tubes, centrifuged at 1500 × g for 15 min, aliquoted and stored at −80 °C. Table 1 summarizes the patient age and disease duration at sampling. The samples included in this study had undergone one freeze–thaw cycle prior to the present analysis. CSF NFL concentrations were analysed with a validated ELISA with intra- and interplate variations of < 8% and < 13%, respectively32. CSF NFL analyses were performed in duplicates. Two control samples were not analysed for CSF NFL due to the limited CSF volume. CSF pNFH concentrations were measured with an in-house-developed ELISA with minor modifications with intra- and interplate variations of < 3.9% and < 9.4%, respectively33. Regarding CSF pNFH analyses, 226 samples were evaluated in singlicates due to the limited CSF volume. Plasma NFL concentrations were measured using a single-molecule array (SIMOA) assay on an HD-1 Analyzer (Quanterix, Billerica, MA, USA) with intra- and interplate variations of < 10% and < 12%, respectively34. Plasma NFL analyses were evaluated in singlicates and in a single batch. Five control samples could not be analysed due to the lack of plasma. CSF NFL, CSF pNFH and plasma NFL analyses were evaluated with the researchers in the laboratory blinded to clinical diagnosis and genotype. The CSF NFL result for one ALS patient was excluded due to a concentration below the calibration curve. For CSF pNFH, biomarker estimates of two ALS patients, one patient from the neuropathies & myelopathies group and one control were excluded because they had concentrations below the calibration curve. For plasma NFL, biomarker estimates of five ALS patients and one patient from the neuropathies & myelopathies group were excluded because they had concentrations below the calibration curve. Patients who had CSF NFL concentrations above the calibration curve (eight ALS patients and one patient in the neuropathies & myelopathies group) were included.
Statistical analysis
Biomarker concentration estimates and patient characteristics were analysed in IBM SPSS Statistics version 26 (International Business Machines Corporation, Armonk, NY, USA). Due to non-normal distributions of estimates, the results are presented as the median and lower and upper quartiles (Q1–Q3) (Table 2). For a prognosis-relevant presentation of biomarkers, ALS patients were also categorized according to the years of survival from symptom onset. The survival times were < 2 years, 2 to < 5 years, 5 to < 10 years and ≥ 10; the biomarker estimates for ALS survival groups are presented as arithmetic mean, standard deviation (SD) and 95% confidence interval (CI) (Table 2). Distributions of biomarker estimates for ALS patients with ≥ 10 years survival are not reported in Table 2 due to the low sample size in this group. Prior to analyses, biomarker estimates were log10-transformed to reduce the influence of inhomogeneous variance, reduce distributional skewness, improve normal approximation and decrease the influence of outliers. To compare different relevant groups, one-way analysis of variance (ANOVA) was performed with planned comparison contrast tests and a 95% CI bias-corrected and accelerated bootstrap initially set at 1000 sample runs. In the statistical comparison of CSF NFL concentrations, sample runs had to be increased to 3000 to achieve convergence (stable p-values above or below < 0.05). For other statistical tests, there was no need to increase the number of samples per run beyond 1000 to achieve convergence. The Welch test was used when homogeneity of variance could not be assumed. Contrast tests were designed to I) test significant differences between ALS patients and all other groups individually; II) test significant differences between patients with other MNDs compared to those with neuropathies & myelopathies, myopathies and controls; III) test significant differences among spinal onset, bulbar onset and truncal onset ALS patients; IV) test significant differences between ALS patients with mutations in SOD1, C9orf72HRE and ALS patients with no known mutation and V) test significant differences in the SOD1 mutation subgroups. The bivariate correlations were investigated using Spearman’s rank-order correlation coefficient (ρ). Kaplan–Meier survival analyses were performed for ALS patients whose total survival from symptom onset to death data were available. The time from self-reported symptom onset to death, rather than the time from sampling or diagnosis to death, was used to generate more accurate survival analyses. Kaplan–Meier survival analyses were performed for I) spinal onset and bulbar onset ALS patients and II) ALS patients with mutations in SOD1, C9orf72HRE and ALS patients with no known mutation. The Mantel–Cox log-rank test was used to test statistical significance for Kaplan–Meier survival analyses. Receiver operating characteristic (ROC) analyses were performed for the original biomarker estimates, and the area under the ROC curve (AUC) was composed for patients with ALS versus ALS mimics for all three biomarkers, where the 95% CI is presented for each AUC. ROC analyses and AUCs for ALS survival are presented for short (< 2 years) and long (≥ 5 years) survival after symptom onset, where patients who were alive during statistical analysis were excluded. AUCs > 0.80 were considered high. Youden’s index highest value (J) was used to determine the optimal cut-off for the biomarker concentration, sensitivity, specificity, positive likelihood ratio (LR +) and negative likelihood ratio (LR-). Biomarker ratios were calculated for CSF NFL and CSF pNFH (NFLCSF/pNFHCSF ratio) and for plasma NFL and CSF NFL (NFLplasma/NFLCSF ratio) using the original biomarker values and are presented as the arithmetic mean ± SD. Statistical differences were considered significant at p < 0.05.
Results
Study population
The total number of research participants selected for the present study was 287. Participants in the study were categorized as patients with ALS (n = 234) or ALS mimics (n = 44); the latter group consisted of patients with other types of motor neuron diseases (other MNDs) (n = 13), neuropathies & myelopathies (n = 24) or myopathies (n = 7). In addition, controls (n = 9) with no known history of neurological disorders were recruited. The study population is summarized in Fig. 1, and ALS patient characteristics are summarized in Table 3 and Supplementary Table S1. ALS patients were stratified into spinal onset ALS (n = 148), bulbar onset ALS (n = 72), truncal onset ALS (n = 11) and FTD onset ALS (n = 1). ALS patients were also stratified into those with mutations in SOD1 (n = 28), those with C9orf72HRE mutations (n = 28), those with VAPB mutations (n = 3) or other ALS patients without mutations in these genes (n = 175). Patients with SOD1 mutations were further stratified into SOD1 D90A homozygous (n = 14), D90A heterozygous (n = 3), A4V heterozygous (n = 2) and other SOD1 mutation (n = 9) groups.

ALS and ALS mimics patient cohort. The study population consisted of patients who underwent investigation for possible motor neuron disease (MND) according to European Federation of Neurological Societies (EFNS) guidelines. The investigation comprised clinical evaluation, neuroimaging, neurophysiological testing (peripheral nerve conduction studies, central motor nerve conduction studies using transcranial motor evoked potentials analysis, and needle electromyography), cerebrospinal fluid and blood analysis. Patients either received an ALS diagnosis (n = 234) or an ALS mimic diagnosis (n = 44). The ALS mimic cohort consisted of patient with other MNDs (n = 13), neuropathies & myelopathies (n = 24) and myopathies (n = 7). A group of controls with no neurological symptoms was recruited for comparison (n = 9). ALS patients were grouped into spinal (n = 148), bulbar (n = 72), truncal (n = 11) or FTD (n = 1) groups according to the site of first onset. ALS patients were also stratified into carriers of mutations in SOD1 (n = 28), C9orf72HRE (n = 28), VAPB (n = 3) or patients with no mutation in these genes (n = 175).
CSF NFL and CSF pNFH
Concentrations of CSF NFL and CSF pNFH are presented in Figs. 2A,C and 3F and Table 2. ALS patients had significantly higher CSF NFL and CSF pNFH levels than patients with neuropathies & myelopathies (p < 0.01 for both), patients with myopathies (p < 0.01 for both) and controls with no neurological disorder (p < 0.01 for both) (Fig. 2A,C). There was a statistically significant difference when comparing CSF pNFH in ALS patients to that of patients with other MNDs (p < 0.01) (Fig. 2C) but not when comparing CSF NFL (p > 0.05) (Fig. 2A). Patients in the other MNDs group had significantly higher CSF NFL and CSF pNFH levels than patients with neuropathies & myelopathies (p < 0.05 for both), myopathies (p < 0.01 for both) and controls with no neurological disorder (p < 0.01 for both) (Fig. 2A,C). Neither CSF NFL levels nor CSF pNFH levels were significantly different among ALS patients with spinal onset, bulbar onset or truncal onset (p > 0.05 for overall ANOVA).

Neurofilament levels and ROC analyses. (A) Boxplots of CSF NFL concentration estimates of participants in the ALS, ALS mimics and control groups. (B) ROC analysis of participants in the ALS versus ALS mimics groups for CSF NFL. (C) Boxplots of CSF pNFH concentration estimates in participants in the ALS, ALS mimics and control groups. (D) ROC analysis of participants in the ALS versus ALS mimics groups for CSF pNFH. (E) Boxplots of plasma NFL concentration estimates of participants in the ALS, ALS mimics and control groups. (F) ROC analysis of participants in the ALS versus ALS mimics groups for plasma NFL. ALS patients versus participants with other MNDs, participants with neuropathies & myelopathies, participants with myopathies and controls: *p < 0.05, **p < 0.01, ns non-significant. Participants with other MNDs versus participants with neuropathies & myelopathies, participants with myopathies and controls: #p < 0.05, ##p < 0.01.

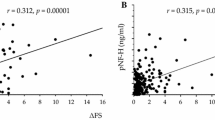
Biomarker correlation to survival, survival analyses in clinical symptom onset and genotype groups, biomarker ratios and ALSFRS-R in ALS patients. (A) Correlation between plasma NFL and survival after symptom onset stratified by spinal and bulbar symptom onset. (B) Kaplan–Meier survival analysis for spinal onset versus bulbar onset ALS patients. (C) Correlation between plasma NFL and survival after symptom onset for participants in the total ALS cohort. (D) Kaplan–Meier survival analysis between patients with no known mutation, SOD1 mutation and C9orf72HRE mutation (p-value presented for overall comparisons). (E) Ratios and correlations between plasma NFL and CSF NFL stratified for spinal onset and bulbar onset ALS patients. (F) Correlation between CSF NFL and CSF pNFH for participants in the total ALS cohort. (G) Correlation between ALSFRS-R and plasma NFL in ALS patients. (H) Correlation between ALSFRS-R and survival after symptom onset in ALS patients.
In patients in the ALS group, the concentrations of CSF NFL and CSF pNFH in patients with no known mutation, SOD1 mutation and C9orf72HRE mutation did not show any statistically significant difference (p > 0.05 for overall ANOVA). When stratifying ALS patients into SOD1 mutation subgroups, ALS patients with SOD1 D90A homozygous or heterozygous mutations combined showed significantly lower levels of CSF pNFH compared to those with SOD1 A4V heterozygous and other ALS SOD1 mutations combined (CSF pNFH; p < 0.01, CSF NFL; p > 0.05). In ALS patients, the concentrations of CSF NFL and CSF pNFH were significantly correlated (ρ = 0.797, p < 0.0001) (Fig. 3F). ALSFRS-R was negatively correlated with CSF NFL levels (ρ = −0.241, p < 0.01) but not with CSF pNFH levels (p > 0.05). Furthermore, ΔFS was correlated with CSF NFL levels (ρ = 0.355, p < 0.0001) and CSF pNFH levels (ρ = 0.346, p < 0.0001). There was no significant correlation between freezer storage time and the concentrations of CSF NFL or between freezer storage time and CSF pNFH in the patients in the ALS or ALS mimics groups (p > 0.05 for both).
Plasma NFL
Concentrations of plasma NFL are presented in Figs. 2E, and 3A,C,E,G and Table 2. ALS patients had significantly higher plasma NFL concentrations than patients with other MNDs (p < 0.05), patients with neuropathies & myelopathies (p < 0.01), patients with myopathies (p < 0.01) and controls with no neurological disorders (p < 0.01) (Fig. 2E). Patients with other MNDs had significantly higher plasma NFL levels than patients with neuropathies and myelopathies (p < 0.01), patients with myopathies (p < 0.05) and controls with no neurological disorder (p < 0.01) (Fig. 2E).
Patients with bulbar onset ALS had significantly higher plasma NFL concentrations than patients with spinal onset ALS (p < 0.01). There was no statistically significant difference when comparing the levels of plasma NFL of ALS patients with truncal onset to those of ALS patients with spinal onset or bulbar onset (p > 0.05 for both). In ALS patients, the concentrations of plasma NFL were significantly correlated with CSF NFL (ρ = 0.773, p < 0.0001) and CSF pNFH (ρ = 0.696, p < 0.0001). When stratified by the region of symptom onset, patients with spinal onset or bulbar onset ALS both showed a higher correlation between plasma NFL and CSF NFL levels (ρ = 0.790, p < 0.0001 and ρ = 0.762, p < 0.0001, respectively) (Fig. 3E) than all ALS patients as a whole group. The plasma NFL concentration was significantly higher in ALS patients with C9orf72HRE mutations than in patients with SOD1 mutations (p < 0.05). No significant difference in plasma NFL concentration was found by comparing ALS patients with no known mutations to ALS patients with SOD1 or C9orf72HRE mutations (p > 0.05 for both). When stratifying ALS patients into spinal and bulbar onset groups, statistically significant differences in plasma NFL concentrations in spinal onset ALS patients were observed when comparing patients with C9orf72HRE mutations to both patients who were SOD1 mutation carriers (p < 0.01) and ALS patients with no known mutations (p < 0.05); however, there was no statistically significant difference among different genotype groups in bulbar onset ALS patients (p > 0.05 for overall ANOVA). ALS patients homozygous or heterozygous mutation for the D90A SOD1 mutation combined showed significantly lower plasma NFL concentrations compared to patients with SOD1 A4V and other SOD1 mutations combined (p < 0.01). Plasma NFL concentration was negatively correlated with ALSFRS-R (ρ = −0.328, p < 0.0001) (Fig. 3G) and positively correlated with ΔFS (ρ = 0.426, p < 0.0001). There was no statistically significant correlation between plasma NFL concentrations and freezer storage time from samples collected from ALS patients and ALS mimics (p > 0.05 for both).
ROC and survival analysis
ROC analyses showed high AUCs when ALS patients were compared with ALS mimics (Fig. 2B,D,F). CSF pNFH had the highest AUC (AUC: 0.874; 0.803–0.944) (J: 7623 pg/mL, sensitivity: 82.7%, specificity: 83.3%, LR + : 4.96, LR−: 0.21, p < 0.0001) (Fig. 2D), followed by plasma NFL concentration (AUC: 0.833; 0.756–0.910) (J: 95.42 pg/mL, sensitivity: 76.4%, specificity: 83.3%, LR + : 4.59, LR−: 0.28, p < 0.0001) (Fig. 2F) and CSF NFL (AUC: 0.805; 0.719–0.891) (J: 2532.5 pg/mL, sensitivity: 80.3%, specificity: 81.8%, LR + : 4.41, LR−: 0.24, p < 0.0001) (Fig. 2B) when comparing patients with ALS to ALS mimics . Survival after ALS symptom onset was significantly negatively correlated with CSF NFL (ρ = −0.360, p < 0.0001), CSF pNFH (ρ = −0.295, p < 0.0001) and plasma NFL levels (ρ = −0.423, p < 0.0001). Furthermore, disease duration at sampling was significantly positively correlated with total survival in ALS patients (ρ = 0.600, p < 0.0001). There were significant differences in survival between spinal onset ALS and bulbar onset ALS patients (p < 0.0001) (Fig. 3B) and among ALS patient genotype groups (p < 0.0001 for overall comparisons, p < 0.01 for ALS patients with SOD1 mutations versus C9orf72HRE mutations) (Fig. 3D). Due to significant differences in plasma NFL concentrations and survival between spinal onset and bulbar onset ALS patients, a scatterplot revealed a significant negative correlation between plasma NFL levels and survival for patients with spinal onset ALS (ρ = −0.349, p < 0.0001) and bulbar onset ALS (ρ = −0.472, p < 0.0001) (Fig. 3A). ROC analyses of ALS patients with short and long survival times showed the highest AUC for plasma NFL levels (AUC: 0.800; 0.716–0.884) (J: 205.66 pg/mL, sensitivity: 58.9%, specificity: 91.1%, LR + : 6.63, LR−: 0.45) (p < 0.0001), followed by CSF NFL levels (AUC: 0.751; 0.655–0.846) (J: 4265 pg/mL, sensitivity: 70.2%, specificity: 73.9%, LR + : 2.69, LR−: 0.40) (p < 0.0001) and CSF pNFH (AUC: 0.721; 0.618–0.825) (J: 18,537 pg/mL, sensitivity: 56.6%, specificity: 79.1%, LR + : 2.70, LR−: 0.55) (p < 0.0001). ALSFRS-R was significantly positively correlated with survival (ρ = 0.284, p < 0.001) (Fig. 3H) and significantly negatively correlated with disease duration at sampling in ALS patients (ρ = −0.180, p < 0.05). Furthermore, ΔFS was significantly negatively correlated with survival in ALS patients (ρ = −0.659, p < 0.0001).
NFL and pNFH ratios
There was no significant difference in the NFLCSF/pNFHCSF ratio among ALS patients, ALS mimic groups and controls (p > 0.05 for overall ANOVA) or between ALS symptom onset groups (p > 0.05 for overall ANOVA). Bulbar onset ALS patients had a significantly higher NFLplasma/NFLCSF ratio (0.046 ± 0.021) than spinal onset ALS patients (0.034 ± 0.016) (p < 0.01) (Fig. 3E). Although patients with C9orf72HRE mutations had notably higher NFLCSF/pNFHCSF ratios (0.302 ± 0.129) than patients with SOD1 mutations (0.242 ± 0.111) (Fig. 3F), this difference did not reach statistical significance (p > 0.05 for overall ANOVA).
Discussion
Making a correct ALS diagnosis early after symptom onset has become even more important since observations from clinical drug trials show that patients enrolled early after onset of first paresis frequently have better outcomes than patients enrolled later35. The launch of promising bespoken therapy trials targeting SOD1, C9orf72HRE and fused in sarcoma gene (FUS) in symptomatic patients with mutations in these genes further emphasizes the need to be able to diagnose ALS early in the disease course. Additionally, experimental personalized gene therapy in adult asymptomatic carriers of these mutations will begin soon. A major obstacle is determining when to initiate prophylactic personalized medicine; the wide range in age at onset of the first symptom, heterogeneity in the first clinical presentation and the frequent occurrence of reduced disease penetrance in families with mutations pose a challenge10. Reliable biomarkers showing that the neurodegenerative process has begun or not begun are urgently needed. Although there is emerging evidence that in patients with a SOD1 mutation, the mutant SOD1 protein forms cytotoxic prion-like species that propagate through the motor system36, no research group has yet successfully been able to detect such SOD1 prions in in vivo material from ALS patients. Presently, we must use less informative downstream biomarkers for diagnosing early ALS and for differentially diagnosing ALS.
In accordance with reports in smaller cohorts21,37, the present study confirms that ALS patients have significantly higher CSF and plasma levels of NFs than patients with a number of relevant ALS mimics. In this larger study, the overlap between the groups was small, demonstrating the usefulness of assaying NFs in the differential diagnosis of ALS. In particular, the levels of CSF pNFH and plasma NFL were significantly higher in patients in the ALS group than in patients in the other MNDs groups. When discriminating ALS from ALS mimics, CSF pNFH had a higher AUC than CSF NFL and plasma NFL levels, suggesting that CSF pNFH is a better biomarker assay in differentiating ALS from clinically relevant mimics in this study.
It has previously been reported that neurofilament levels in the CSF do not distinguish ALS patients according to the site of ALS symptom onset38. The present study shows similar results in CSF, although CSF NFL and CSF pNFH levels were notably (non-significantly) higher in bulbar onset ALS patients than in spinal onset ALS patients. In a previous study, bulbar onset ALS patients had close to significantly higher plasma pNFH levels than spinal onset ALS patients but without a statistically significant difference in CSF39. In the present larger study, we found that plasma NFL levels were significantly higher in bulbar onset ALS patients than in spinal onset ALS patients. Furthermore, bulbar onset ALS patients showed a significantly higher NFLplasma/NFLCSF ratio than spinal onset ALS patients.
Bulbar onset ALS is associated with a worse prognosis than spinal onset ALS3, and an earlier study suggested that NFL concentrations are related to the volume of damaged neuronal tissue33. The usefulness of NFL and pNFH levels in the prognostication of ALS is controversial24,40,41. Here, we found that ALS patients with lower levels of all three biomarkers survived longer than ALS patients with higher levels. Speculatively, the lower levels of neurofilaments and longer survival in some patients suggest that axonal loss progresses slower in these patients; thus, fewer neurofilaments are displaced into the CFS and plasma. Hence, interventions that markedly lower neurofilament levels in the CSF and plasma slow the progression of neuronal loss. The present finding that the plasma NFL concentration is higher in bulbar onset ALS patients than in spinal onset ALS patients is therefore in accordance with the worse prognosis in bulbar onset ALS patients than in spinal onset ALS patients3.
Hypermetabolism and weight loss correlate with worse prognosis in ALS42,43,44. Weight loss relates to the site of symptom onset and dysphagia in ALS, and survival has been shown to be affected by weight loss in both spinal onset and bulbar onset ALS patients45. However, the effect of weight loss and dysphagia on survival did not differ significantly between spinal or bulbar onset ALS patients45, which might suggest worse survival in bulbar onset patients even if the same relative degree of weight loss was present in both groups. Thus, the significantly higher plasma NFL levels in bulbar onset ALS patients might suggest that the worse prognosis is primarily due to a more aggressive neurodegenerative process. In epidemiological studies, the incidence of bulbar onset ALS is higher in women and in patients as age increases46. This is also the case in our study cohort, making the findings of differences in plasma NFL concentration between spinal and bulbar onset ALS patients clinically applicable.
In this study, a subset of ALS patients were carriers of mutations in SOD1, C9orf72 or VAPB. ALS patients carrying C9orf72HRE mutations have significantly higher CSF pNFH and serum pNFH levels than patients without a mutation in C9orf7247,48. Additionally, SOD1 mutation carriers had significantly lower CSF NFL levels than patients with no SOD1 mutations17. Our new results support these results, showing higher plasma NFL levels and worse survival in ALS patients with C9orf72HRE mutations than in patients with SOD1 mutations. Stratifying ALS patients into spinal and bulbar symptom onset, patients in the spinal onset group with a C9orf72HRE had significantly higher plasma NFL levels than ALS patients with a SOD1 mutation. This finding further supports the importance of evaluating neurodegeneration in ALS patients with regard to both clinical symptoms at onset and genotype.
Since patients with primary lateral sclerosis (PLS) with only upper motor neuron engagement or PMA with only lower motor neuron engagement generally have a better prognosis than ALS patients49,50, it would be unfortunate to enrol patients with PLS or PMA in an ALS intervention trial. It is therefore important to clearly differentiate between early ALS and early PLS or PMA diseases. A study found that ALS patients have significantly higher serum NFL concentrations than PLS patients37. We therefore propose further studies also using upper motor neuron-specific biomarkers (e.g., α-internexin) and lower motor neuron-specific biomarkers (e.g., peripherin).
Limitations of the present study include retrospective assessment of ALSFRS-R (the scale did not exist when the first patients were seen in our clinic), some of the CSF pNFH analyses were performed in singlicates and plasma pNFH analyses were not performed. The samples included in the present study were collected over several years and had undergone one freeze–thaw cycle prior to analysis. Arguably, the concentrations of NFL and pNFH could potentially be confounded by these factors. Reportedly, both NFL and NFH can withstand four freeze–thaw cycles without affecting concentrations significantly, and the Arrhenius plot for NFH suggests stable properties for storage at −80 °C for an extended time51. This stability may be due to the phosphorylated carboxy-terminal of pNFH52,53. In our dataset, there was no significant correlation between freezer storage time and concentrations of all three biomarkers, indicating that the samples were not significantly degraded due to storage time and therefore were comparable.
The strengths of the study are the analyses of NFL and pNFH levels in CSF and NFL in plasma samples from a large group of clinically relevant and commonly encountered ALS patients and ALS mimics; all performed in the same laboratory and blinded to clinical data. Moreover, the study extensively characterized patient cohorts with clinical and genotype information from patients from the same site. The long follow-up observation time following the collection of the analysed samples is of importance as it provides enough time for a correct diagnosis of slowly progressing ALS. Finally, 26 of the ALS patients later underwent post-mortem autopsy confirming the diagnosis.
In conclusion, our study results confirm earlier findings on neurofilament and ALS6,18,19,24,25,37,38,41,54,55,56,57,58 but add new knowledge on the comparative performance of plasma and CSF neurofilaments in a clinical context, thus emphasizing the importance of a correct ALS diagnosis early and prognostication. Regarding the diagnostic properties, all three biomarkers are of clinical value in affirming an ALS diagnosis and excluding potential ALS mimics. CSF pNFH showed the highest AUC in terms of differentiating ALS from ALS mimics. Plasma NFL analysis has the advantage that it does not require a lumbar puncture, has only a minimal difference in diagnostic performance compared to CSF NFL levels and shows the highest AUC in terms of prognosticating ALS short and long survival.
Data availability
Data used and analysed in the present study will be available from the corresponding author upon reasonable request from other investigators adhering to the European Union General Data Protection Regulation (EU) 2016/679 (GDPR).
References
Swinnen, B. & Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 10, 661–670. https://doi.org/10.1038/nrneurol.2014.184 (2014).
Paganoni, S. et al. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 453–456. https://doi.org/10.3109/21678421.2014.903974 (2014).
Chio, A. et al. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 10, 310–323. https://doi.org/10.3109/17482960802566824 (2009).
Belsh, J. M. Diagnostic challenges in ALS. Neurology 53, S26-30 (1999) ((discussion S35–26)).
Yedavalli, V. S., Patil, A. & Shah, P. Amyotrophic lateral sclerosis and its mimics/variants: A comprehensive review. J. Clin. Imaging Sci. 8, 53–53. https://doi.org/10.4103/jcis.JCIS_40_18 (2018).
Reijn, T. S., Abdo, W. F., Schelhaas, H. J. & Verbeek, M. M. CSF neurofilament protein analysis in the differential diagnosis of ALS. J. Neurol. 256, 615–619. https://doi.org/10.1007/s00415-009-0131-z (2009).
Traynor, B. J. et al. Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch. Neurol. 57, 109–113 (2000).
Haverkamp, L. J., Appel, V. & Appel, S. H. Natural history of amyotrophic lateral sclerosis in a database population validation of a scoring system and a model for survival prediction. Brain 118, 707–719. https://doi.org/10.1093/brain/118.3.707 (1995).
Zou, Z. Y. et al. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 88, 540–549. https://doi.org/10.1136/jnnp-2016-315018 (2017).
Andersen, P. M. et al. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain 120(Pt 10), 1723–1737. https://doi.org/10.1093/brain/120.10.1723 (1997).
Lange, D. J. et al. Pyrimethamine significantly lowers cerebrospinal fluid Cu/Zn superoxide dismutase in amyotrophic lateral sclerosis patients with SOD1 mutations. Ann. Neurol. 81, 837–848. https://doi.org/10.1002/ana.24950 (2017).
Birve, A. et al. A novel SOD1 splice site mutation associated with familial ALS revealed by SOD activity analysis. Hum. Mol. Genet. 19, 4201–4206. https://doi.org/10.1093/hmg/ddq338 (2010).
Stewart, H. G. et al. Clinicopathological phenotype of ALS with a novel G72C SOD1 gene mutation mimicking a myopathy. Muscle Nerve 33, 701–706. https://doi.org/10.1002/mus.20495 (2006).
Balendra, R., Moens, T. G. & Isaacs, A. M. Specific biomarkers for C9orf72 FTD/ALS could expedite the journey towards effective therapies. EMBO Mol. Med. 9, 853–855. https://doi.org/10.15252/emmm.201707848 (2017).
Fuchs, E. & Cleveland, D. W. A structural scaffolding of intermediate filaments in health and disease. Science 279, 514–519 (1998).
Zhao, J. & Liem, R. K. Alpha-internexin and peripherin: Expression, assembly, functions, and roles in disease. Methods Enzymol. 568, 477–507. https://doi.org/10.1016/bs.mie.2015.09.012 (2016).
Zetterberg, H., Jacobsson, J., Rosengren, L., Blennow, K. & Andersen, P. M. Cerebrospinal fluid neurofilament light levels in amyotrophic lateral sclerosis: Impact of SOD1 genotype. Eur. J. Neurol. 14, 1329–1333. https://doi.org/10.1111/j.1468-1331.2007.01972.x (2007).
Tortelli, R. et al. Elevated cerebrospinal fluid neurofilament light levels in patients with amyotrophic lateral sclerosis: A possible marker of disease severity and progression. Eur. J. Neurol. 19, 1561–1567. https://doi.org/10.1111/j.1468-1331.2012.03777.x (2012).
Gaiottino, J. et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS ONE 8, e75091. https://doi.org/10.1371/journal.pone.0075091 (2013).
Ganesalingam, J. et al. Combination of neurofilament heavy chain and complement C3 as CSF biomarkers for ALS. J. Neurochem. 117, 528–537. https://doi.org/10.1111/j.1471-4159.2011.07224.x (2011).
Oeckl, P. P. et al. Multicenter validation of CSF neurofilaments as diagnostic biomarkers for ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. 17, 404–413. https://doi.org/10.3109/21678421.2016.1167913 (2016).
Benatar, M., Wuu, J., Andersen, P. M., Lombardi, V. & Malaspina, A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann. Neurol. 84, 130–139. https://doi.org/10.1002/ana.25276 (2018).
Brettschneider, J., Petzold, A., Sussmuth, S. D., Ludolph, A. C. & Tumani, H. Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology 66, 852–856. https://doi.org/10.1212/01.wnl.0000203120.85850.54 (2006).
Lu, C. H. et al. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology 84, 2247–2257. https://doi.org/10.1212/wnl.0000000000001642 (2015).
McCombe, P. A. et al. Serial measurements of phosphorylated neurofilament-heavy in the serum of subjects with amyotrophic lateral sclerosis. J. Neurol. Sci. 353, 122–129. https://doi.org/10.1016/j.jns.2015.04.032 (2015).
Andersen, P. M. et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)-revised report of an EFNS task force. Eur. J. Neurol. 19, 360–375. https://doi.org/10.1111/j.1468-1331.2011.03501.x (2012).
Andersen, P. M. et al. EFNS task force on management of amyotrophic lateral sclerosis: Guidelines for diagnosing and clinical care of patients and relatives. Eur. J. Neurol. 12, 921–938. https://doi.org/10.1111/j.1468-1331.2005.01351.x (2005).
Cedarbaum, J. M. et al. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. J. Neurol. Sci. 169, 13–21. https://doi.org/10.1016/S0022-510X(99)00210-5 (1999).
Kimura, F. et al. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 66, 265–267. https://doi.org/10.1212/01.wnl.0000194316.91908.8a (2006).
Synofzik, M. et al. Mutant superoxide dismutase-1 indistinguishable from wild-type causes ALS. Hum. Mol. Genet. 21, 3568–3574. https://doi.org/10.1093/hmg/dds188 (2012).
Akimoto, C. et al. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J. Med. Genet. 51, 419–424. https://doi.org/10.1136/jmedgenet-2014-102360 (2014).
Gaetani, L. et al. A new enzyme-linked immunosorbent assay for neurofilament light in cerebrospinal fluid: Analytical validation and clinical evaluation. Alzheimers Res. Ther. 10, 8. https://doi.org/10.1186/s13195-018-0339-1 (2018).
Pujol-Calderón, F. et al. Neurofilament changes in serum and cerebrospinal fluid after acute ischemic stroke. Neurosci. Lett. 698, 58–63. https://doi.org/10.1016/j.neulet.2018.12.042 (2019).
Kuhle, J. et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin. Chem. Lab. Med. 54, 1655–1661. https://doi.org/10.1515/cclm-2015-1195 (2016).
Mora, J. S. et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Frontotemporal Degener. 21, 5–14. https://doi.org/10.1080/21678421.2019.1632346 (2020).
Ekhtiari Bidhendi, E. et al. Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol. 136, 939–953. https://doi.org/10.1007/s00401-018-1915-y (2018).
Gille, B. et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 45, 291–304. https://doi.org/10.1111/nan.12511 (2019).
Rossi, D. et al. CSF neurofilament proteins as diagnostic and prognostic biomarkers for amyotrophic lateral sclerosis. J. Neurol. 265, 510–521. https://doi.org/10.1007/s00415-017-8730-6 (2018).
Boylan, K. B. et al. Phosphorylated neurofilament heavy subunit (pNF-H) in peripheral blood and CSF as a potential prognostic biomarker in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 84, 467–472. https://doi.org/10.1136/jnnp-2012-303768 (2013).
Gong, Z. Y. et al. Neurofilament subunit L levels in the cerebrospinal fluid and serum of patients with amyotrophic lateral sclerosis. Neurodegener. Dis. 18, 165–172. https://doi.org/10.1159/000488681 (2018).
Steinacker, P. et al. Neurofilaments in the diagnosis of motoneuron diseases: A prospective study on 455 patients. J. Neurol. Neurosurg. Psychiatry 87, 12. https://doi.org/10.1136/jnnp-2015-311387 (2016).
Steyn, F. J. et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 89, 1016–1023. https://doi.org/10.1136/jnnp-2017-317887 (2018).
Dupuis, L., Oudart, H., Rene, F., Gonzalez de Aguilar, J. L. & Loeffler, J. P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. U S A 101, 11159–11164. https://doi.org/10.1073/pnas.0402026101 (2004).
Moglia, C. et al. Early weight loss in amyotrophic lateral sclerosis: Outcome relevance and clinical correlates in a population-based cohort. J. Neurol. Neurosurg. Psychiatry 90, 666–673. https://doi.org/10.1136/jnnp-2018-319611 (2019).
Janse van Mantgem, M. R. et al. Prognostic value of weight loss in patients with amyotrophic lateral sclerosis: A population-based study. J. Neurol. Neurosurg. Psychiatry 91, 867–875. https://doi.org/10.1136/jnnp-2020-322909 (2020).
Wijesekera, L. C. & Leigh, P. N. Amyotrophic lateral sclerosis. Orphanet J. Rare. Dis. 4, 3. https://doi.org/10.1186/1750-1172-4-3 (2009).
Gendron, T. F. et al. Phosphorylated neurofilament heavy chain: A biomarker of survival for C9ORF72-associated amyotrophic lateral sclerosis. Ann. Neurol. 82, 139–146. https://doi.org/10.1002/ana.24980 (2017).
Falzone, Y. M. et al. Serum phosphorylated neurofilament heavy-chain levels reflect phenotypic heterogeneity and are an independent predictor of survival in motor neuron disease. J. Neurol. 267, 2272–2280. https://doi.org/10.1007/s00415-020-09838-9 (2020).
Tartaglia, M. C. et al. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis: Examination of symptoms and signs at disease onset and during follow-up. Arch. Neurol. 64, 232–236. https://doi.org/10.1001/archneur.64.2.232 (2007).
Kim, W. K. et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology 73, 1686–1692. https://doi.org/10.1212/WNL.0b013e3181c1dea3 (2009).
Koel-Simmelink, M. J. A. et al. The impact of pre-analytical variables on the stability of neurofilament proteins in CSF, determined by a novel validated SinglePlex Luminex assay and ELISA. J. Immunol. Methods 402, 43–49. https://doi.org/10.1016/j.jim.2013.11.008 (2014).
Pant, H. C. Dephosphorylation of neurofilament proteins enhances their susceptibility to degradation by calpain. Biochem. J. 256, 665–668. https://doi.org/10.1042/bj2560665 (1988).
Rao, M. V., Yuan, A., Campbell, J., Kumar, A. & Nixon, R. A. The C-terminal domains of NF-H and NF-M subunits maintain axonal neurofilament content by blocking turnover of the stationary neurofilament network. PLoS ONE 7, e44320–e44320. https://doi.org/10.1371/journal.pone.0044320 (2012).
Rosengren, L. E., Karlsson, J. E., Karlsson, J. O., Persson, L. I. & Wikkelso, C. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J. Neurochem. 67, 2013–2018 (1996).
Menke, R. A. et al. CSF neurofilament light chain reflects corticospinal tract degeneration in ALS. Ann. Clin. Transl. Neurol. 2, 748–755. https://doi.org/10.1002/acn3.212 (2015).
Weydt, P. et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann. Neurol. 79, 152–158. https://doi.org/10.1002/ana.24552 (2016).
Gaiani, A. et al. Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis: Neurofilament light chain levels in definite subtypes of disease. JAMA Neurol. 74, 525–532. https://doi.org/10.1001/jamaneurol.2016.5398 (2017).
Poesen, K. et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 88, 2302–2309. https://doi.org/10.1212/wnl.0000000000004029 (2017).
Acknowledgements
We are indebted to all the patients who made this study possible. The authors thank the laboratory technicians in the Mölndal laboratory, as well as Ann-Charloth Nilsson, Helena Alstermark and Eva Jonsson for excellent technical assistance.
Funding
Open access funding provided by Umea University. The study was sponsored by the Swedish Brain Foundation (grants nr. 2012-0262, 2012-0305, 2013-0279, 2016-0303, and 2020-0353), the Swedish Research Council (grants nr. 2012-3167 and 2017-03100), the Knut and Alice Wallenberg Foundation (grants nr. 2012.0091, 2014.0305, and 2020.0232), the Bertil Hållsten Foundation, the Ulla-Carin Lindquist Foundation, the Neuroförbundet Association, the Torsten and Ragnar Söderberg Foundation, Umeå University Insamlingsstiftelsen (223-2808-12, 223-1881-13, and 2.1.12-1605-14), the Västerbotten County Council (grant nr 56103–7002829), Swedish Brain Power, King Gustaf V:s and Queen Victoria’s Freemason’s Foundation. K.B. was supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF agreement (#ALFGBG-715986), and the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236). H.Z. is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018–02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF-21-831376-C, #ADSF-21-831381-C and #ADSF-21-831377-C), the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019-0228), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. A.B. was supported by a research grant from Västernorrland County Council (LVNFOU939000). A.E.T. was supported by Västerbotten County Council. F.P.C. was funded by the MIROCALS project from the European Union’s Horizon 2020 research and innovation program under grant agreement No. 633413.
Author information
Authors and Affiliations
Contributions
A.B.: major role in data acquisition; statistical analysis of the data; interpretation of the data; drafted and revised the manuscript for intellectual content. F.P.C.: major role in data acquisition; interpretation of the data; drafted and revised the manuscript for intellectual content. A.E.T.: statistical analysis of the data; interpretation of the data; drafted and revised the manuscript for intellectual content. A.W.: statistical analysis of the data; interpretation of the data; revised the manuscript for intellectual content. K.H.: major role in data acquisition; interpretation of the data; revised the manuscript for intellectual content. K.F.: conceptualization and design of the study; major role in data acquisition; interpretation of the data; revised the manuscript for intellectual content. E.P.: major role in data acquisition; interpretation of the data; revised the manuscript for intellectual content. K.B.: major role in data acquisition; interpretation of the data; revised the manuscript for intellectual content. H.Z.: conceptualization and design of the study; major role in data acquisition; interpretation of the data; revised the manuscript for intellectual content. P.M.A.: principal investigator; conceptualization and design of the study; funding acquisition; patient enrolment and genotyping; major role in data acquisition; interpretation of the data; drafted and revised the manuscript for intellectual content.
Corresponding author
Ethics declarations
Competing interests
A.B. declares no disclosures relevant to the manuscript. F.P.C. declares no disclosures relevant to the manuscript. A.E.T. declares no disclosures relevant to the manuscript. A.W. declares no disclosures relevant to the manuscript. K.H. declares no disclosures relevant to the manuscript. K.F. declares no disclosures relevant to the manuscript. E.P. declares no disclosures relevant to the manuscript. K.B. served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. H.Z. reports that he has served at scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics and CogRx, has given lectures in symposia sponsored by Fujirebio, Alzecure and Biogen, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. P.M.A. reports grants from the Swedish Research Council, the Swedish Brain Foundation, the Knut and Alice Wallenberg Foundation, the Ulla-Carin Lindquist Foundation; has been/is a consultant for Biogen, Orphazyme A/S, Hoffman la Roche/Genentech and Regeneron on matters regarding ALS genetics and clinical trials.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Behzadi, A., Pujol-Calderón, F., Tjust, A.E. et al. Neurofilaments can differentiate ALS subgroups and ALS from common diagnostic mimics. Sci Rep 11, 22128 (2021). https://doi.org/10.1038/s41598-021-01499-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-01499-6
- Springer Nature Limited
This article is cited by
-
Fluid biomarkers for amyotrophic lateral sclerosis: a review
Molecular Neurodegeneration (2024)
-
Diagnostic value of neurofilaments in differentiating motor neuron disease from multifocal motor neuropathy
Journal of Neurology (2024)
-
Comparison of CSF and plasma NfL and pNfH for Alzheimer’s disease diagnosis: a memory clinic study
Journal of Neurology (2023)
-
Biomarkers: Role and Scope in Neurological Disorders
Neurochemical Research (2023)
-
Roadmap for C9ORF72 in Frontotemporal Dementia and Amyotrophic Lateral Sclerosis: Report on the C9ORF72 FTD/ALS Summit
Neurology and Therapy (2023)