
Abstract
In an attempt to improve the oral bioavailability of taxanes, a series of new analogues were synthesized and tested in a panel of human tumor cell lines and cellular permeability assays. Compounds T-13 and T-26 showed potent cytotoxicity and exhibited the highest permeability, so they were selected for pharmacokinetic studies. Here, pharmacokinetics of T-13 and T-26 were studied after intravenous injection (5 mg/kg) and oral administration (60 mg/kg) in male Sprague-Dawley (S.D.) rats, respectively. Plasma concentrations were characterized using liquid chromatography-tandem mass spectrometry (LC-MS/MS). The oral bioavailability of T-13 and T-26 was determined to be 10.71% and 65.79%, respectively. Compounds T-13 and T-26 were both poor substrates of P-glycoprotein (P-gp), and they had a much higher bioavailability than paclitaxel, especially T-26. T-26 with good oral bioavailability represented a potential candidate for potent antitumor activity given oral administration.
Similar content being viewed by others
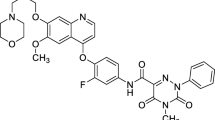


Introduction
Paclitaxel(PTX) has poor aqueous solubility and its intestinal uptake is severely hampered by the ATP-binding cassette (ABC) drug efflux transporter P-glycoprotein (P-gp)1,2,3,4,5,6, which severely limit its oral bioavailability7,8. Therefore, PTX requires to be co-injected with the detergent Cremophor EL, and this detergent frequently causes untoward hypersensitivity reactions, which has created significant problems in developing suitable pharmaceutical formulations suitable for chemotherapy9,10,11.
In an attempt to increase the solubility of taxoids and develop more safe clinical formulations, we previously synthesized a series of novel taxol analogs by modifying the positions of C7, C10, C14 and C3′, which have improved water solubility and oral bioavailability. Among these analogs, T-13, T-15 and T-26 (Fig. 1) exhibited the highest permeability in Caco-2 monolayer cells, with the efflux ratios of less than 2.012. Three taxol analogs demonstrated the potent cytotoxic activities similar to paclitaxel in MCF-7 cancer cell lines. In order to further evaluate the substrate activity of novel taxol analogs with P-gp and whether they are P-gp inhibitors, T-13 and T-26 were selected based on structural characteristics (with or without 1, 14-carbonate moiety) and cytotoxicity in vitro for evaluation of the interaction with P-gp in Madin-Darby canine kidney (MDCK)-multidrug resistance-1 (MDR1) and MDCK-wild type (WT) cells (a more P-gp specific model)13,14 permeability studies, under two sets of conditions: one with and without a P-gp inhibitor (such as verapamil) to look at whether the test compounds were P-gp substrates or not, one with and without a P-gp substrate (such as digoxin) to look at whether the test compounds were P-gp inhibitors or not. The results demonstrated that the interactions of the taxol analogs T-13 and T-26 with P-gp were reduced. Therefore, the permeability of the Caco-2 cells was enhanced in vitro.

Taxanes bearing modifications at the key positions of C-7, C-10, C-14 and C-3′.
The current experiments were performed to study the pharmacokinetic profile and absolute oral bioavailability of T-13, T-26 in S.D. rats after acute intravenous and oral administration at single dose using LC-MS/MS based on highly sensitive and specific analytical methodology. In this paper, the main pharmacokinetic parameters such as elimination half-life (t1/2), total area under the curve (AUC0−∞), and mean residence time (MRT) were estimated. The studies of pharmacodynamic described herein focused on assessing the oral bioavailability of T-13 and T-26.
Methods
All methods were carried out in accordance with the EC Directive 86/609/EEC for animal experiments. The study was approved by Ethics Committee for Animal Experimentation of Mudanjiang Normal University (Mudanjiang, China).
Chemicals
Pure compounds T-13 and T-26 were synthetized and characterized by us as described previously12. T-13 or T-26 solutions for injection were prepared in saline immediately before administration. Paclitaxel was provided as a sample from the national institute for the control of pharmaceutical and biological products (NICPBP). Reagents required for LC-MS/MS assays were purchased from Sigma-Aldrich. Tween 80 and ethyl acetate were purchased from Aladdin reagent, Shanghai.
Characterization of T-13 and T-26
The amount of T-13 and T-26 were determined by using an Agilent 1100 series liquid chromatography system equipped with an Agilent G1313A auto-sampler, a binary pump, a reversed-phase C18 Thermo column (150 mm×2.1 mm, 3 μm) with a precolumn (10 mm×2.1 mm, 3 μm) filled with the same material maintained at 30 °C, and a diode-array detector set at 230 nm. The mobile phase was established using a mixture of acetonitrile and water (70:30, v/v) delivered at a flow rate of 0.2 mL/min, and the injection volume was 20 μL. Detection of T-13 and T-26 were performed using a Thermo Finnigan TSQ Quantum triple quadrupole mass spectrometer equipped with an electrospray ionization (ESI) source (San Jose, CA, USA) in positive ion mode. Optimized mass parameters were as follows: ion spray voltage: 4.0 kV, source temperature: 350 °C, sheath gas (nitrogen) 20 psi, auxiliary gas (nitrogen) 5 psi and collision energy: 17 eV for CA, 19 eV for FA and IFA, 15 eV for IS.
Paclitaxel (retention time 3.07 min) used as internal standard, T-13 (retention time 4.2 min) and T-26 (retention time 5.1 min) stock solutions were refrigerated and calibration curves were designed over the range of 5–10,000 ng/mL (γ2 > 0.999). The limit of quantification was calculated to be 5 ng/mL. All data acquired were processed by a computer workstation running Agilent Chemstation Rev.A.09.01 Software. Multiple Reaction Monitoring of T-13 and T-26 and internal standard (PTX) utilized the transitions at m/z 915 → 634, m/z 957 → 901, m/z 876 → 30815, respectively.
Administration of T-13 and T-26 to rats
For the pharmacokinetic study, male S.D. rats (average weight 300 g) were fasted overnight to prevent coprophagia but allowing free access to water. The rats were randomly divided into four groups (6 animals each). The dosages of taxanes used in the pharmacokinetic study were based on the previously published reports15,16,17. For solution, T-13 and T-26 were dissolved in a mixture of Tween 80 and ethanol (50:50, v/v) at 50 mg/mL for intravenous and oral administration, respectively. Group I received the T-13 stock solution diluted with normal saline solution up to 1 mL at a dose of 5 mg/kg for intravenous injection. Group II received T-13 at a dose of 60 mg/kg for oral administration. Group III received the compound T-26 stock solution diluted with normal saline solution up to 1 mL at a dose of 5 mg/kg for intravenous injection. Group IV received T-26 at a dose of 60 mg/kg for oral administration.
Blood samples (~0.20 mL) were collected into heparin coated tubes at 0, 5, 10, 20, 40 min, 1, 2, 4, 6, 8, 12 and 24 h after intravenous injection and 5, 15, 30, 45 min, 1, 2, 4, 6, 8, 12 and 24 h after oral administration. Blood samples were centrifuged at 12,000 rpm for 10 min. The supernatant fractions were transferred into labeled microcentrifuge tubes and stored at −40 °C until LC-MS/MS analysis.
LC-MS/MS quantification of T-13 and T-26 in plasma samples
The concentration of T-13 and T-26 were determined in plasma by LC/MS as described above. Calibration curves were used for the conversion of the T-13/Paclitaxel chromatographic area to the concentration. Calibrator and quality control samples were prepared by adding appropriate volumes of standard T-13 or T-26 acetonitrile solution to drug-free plasma. Calibration curves were designed over the range 5–10,000 ng/mL (γ2 > 0.999). An aliquot (100 μL) of plasma sample was mixed with 100 μL of acetonitrile containing internal standard (PTX, 500 ng/mL), followed by vortex mixing. Then the mixture was added 3 mL of ethyl acetate, followed by vortex gentle agitation for 3 min. The mixture was centrifuged at 4500 rpm for 10 min after liquid-liquid extraction, and then, the organic layer was collected and evaporated until dry. Finally, the residue was reconstituted with 120 μL of a mixture of acetonitrile and water (70:30, v/v) and transferred to a clean vial, followed by centrifugation at 12,000 rpm for 3 min. The supernatant fraction was transferred to auto-sampler vial, and analyzed by HPLC. Under these experimental conditions, the running times of T-13 and T-26 were 6 min and 7 min, respectively.
Pharmacokinetic data analysis
The pharmacokinetic analysis of concentration-time data obtained after the administration of the compounds T-13 and T-26 were analyzed by a noncompartmental model using Xcalibur® (version 1.3) Software (Thermo Finnigan). The following pharmacokinetic parameters were calculated from the plasma data of Group I and Group III using WinNonlin5.2. Software: total area under the plasma concentration versus time curve, determined by extrapolation from 0 to ∞ after intravenous administration (AUCiv), half-life of the terminal phase (t1/2) and the mean residence time (MRT). Other parameters such as maximum plasma concentration (Cmax) and the time to reach Cmax (Tmax) were also analyzed after oral administration in rats.
Statistical analysis
The data were presented as mean ± standard deviation of at least three experiments. The Mann-Whitney U-test was used to investigate statistical differences at a significance level of P < 0.05. All data processing was performed using the statistical package for social sciences (SPSS) software.
Results
The mean plasma concentration-time profiles and the pharmacokinetic parameters of T-13 and T-26 after intravenous (5 mg/kg) and oral administration (60 mg/kg) in rats were presented in Fig. 2 and Table 1, respectively. As reflected in Table 1, T-13 and T-26 displayed similar values and demonstrated a higher capacity to promote the absorption in vivo. Finally, the oral bioavailability of T-13 and T-26 were calculated to be 10.71% and 65.79%, respectively, which were both higher than 5% for paclitaxel15.

(a) Pharmacokinetics of T-13 after intravenous and oral administration to rats. Animals received an intravenous dose of 5 mg/kg and an oral dose of 60 mg/kg. Data are expressed as mean ± S.D., n = 6 at each time point. (b) Pharmacokinetics of T-26 after intravenous and oral administration to rats. Animals received an intravenous dose of 5 mg/kg and an oral dose of 60 mg/kg. Data are expressed as mean ± S.D., n = 6 at each time point.
Discussions
This study showed that T-13 and T-26 given oral administration in rats have a bioavailability of approximately 11% and 66%, respectively. The compound T-26 bearing 1, 14-carbonate group was remarkably higher than paclitaxel in rats. In the permeability assay of monolayer Caco-2 cells, compounds T-13 and T-26 showed the highest permeability, with efflux ratios better than ortataxel, an oral taxane used in the phase III clinical trial. T-13 and T-26 were both poor substrates of P-gp, possessed inhibiting effects of P-gp mediated efflux, and their bioavailability was much higher than paclitaxel. The experimental results showed that T-13 improved the oral bioavailability to a limited extent, while T-26 was more effective because it overcomes the effect of P-gp mediated efflux on the one hand, and the dihydroxyl groups at C1 and C14 positions greatly improve its water solubility and increase its transport ability through the blood to the site of effect. The finding further supported the view that P-gp was involved in the low bioavailability of paclitaxel. It was thus clear that simultaneous modifications at the C1, C14 positions introducing water-soluble groups and C7, C10 positions overcoming the interaction with P-gp of paclitaxel can significantly improve its oral bioavailability. In view of its good bioavailability after oral administration, the antitumor activity of T-26 after oral administration was under way.
Data availability
All data generated within this study are available from the corresponding author on request.
References
Singh, S. P., Sarwar, B., Farhad, M., Bhupinder, S. & Piyush, T. Novel dietary lipid-based self-nanoemulsifying drug delivery systems of paclitaxel with p-gp inhibitor: implications on cytotoxicity and biopharmaceutical performance. Expert opinion on drug delivery 12(11), 1809–1822, https://doi.org/10.1517/17425247.2015.1060219 (2015).
Tang, B. et al. Lipid-albumin nanoassemblies co-loaded with borneol and paclitaxel for intracellular drug delivery to C6 glioma cells with P-gp inhibition and its tumor targeting. Asian Journal of Pharmaceutical Sciences 10(5), 363–371, https://doi.org/10.1016/j.ajps.2015.04.004 (2015).
Roger, E., Lagarce, F., Garcion, E. & Benoit, J. P. Reciprocal competition between lipid nanocapsules and P-gp for paclitaxel transport across Caco-2 cells. European Journal of Pharmaceutical Sciences 40(5), 422–429, https://doi.org/10.1016/j.ejps.2010.04.015 (2010).
Shi, X. L. et al. Targeting the Bcl-2 family and P-glycoprotein reverses paclitaxel resistance in human esophageal carcinoma cell line. Biomedicine & Pharmacotherapy 90, 897–905, https://doi.org/10.1016/j.biopha.2017.04.043 (2017).
Afrooz, H. et al. Design and characterization of paclitaxel-verapamil co-encapsulated PLGA nanoparticles: Potential system for overcoming P-glycoprotein mediated MDR. Journal of Drug Delivery Science and Technology 41, 174–181, https://doi.org/10.1016/j.jddst.2017.06.020 (2017).
Zhang, F. F. et al. Microvesicles mediate transfer of P-glycoprotein to paclitaxel-sensitive A2780 human ovarian cancer cells, conferring paclitaxel-resistance. European Journal of Pharmacology 738, 83–90, https://doi.org/10.1016/j.ejphar.2014.05.026 (2014).
He, L., Zhong, G. H. & Zhao, X. M. Rational design of hybrid nanomicelles integrating mucosal penetration and P-glycoprotein inhibition for efficient oral delivery of paclitaxel. Colloids and Surfaces B: Biointerfaces 155, 429–439, https://doi.org/10.1016/j.colsurfb.2017.04.045 (2017).
Wang, X. Y. et al. Amphiphilic carboxymethyl chitosan-quercetin conjugate with P-gp inhibitory properties for oral delivery of paclitaxel. Biomaterials 35(26), 7654–7665, https://doi.org/10.1016/j.biomaterials.2014.05.053 (2014).
Campos, F. C. et al. Systemic toxicity induced by paclitaxel in vivo is associated with the solvent cremophor EL through oxidative stress-driven mechanisms. Food and Chemical Toxicology 68, 78–86, https://doi.org/10.1016/j.fct.2014.03.013 (2014).
Kim, H. S. et al. A Prospective Phase II Study of Cisplatin and Cremophor EL-Free Paclitaxel (Genexol-PM) in Patients with Unresectable Thymic Epithelial Tumors. Journal of Thoracic Oncology 10(12), 1800–1806, https://doi.org/10.1097/JTO.0000000000000692 (2015).
Ilinskaya, A. N., Clogston, J. D., McNeil, S. E. & Dobrovolskaia, M. A. Induction of oxidative stress by Taxol® vehicle Cremophor-EL triggers production of interleukin-8 by peripheral blood mononuclear cells through the mechanism not requiring de novo synthesis of mRNA. Nanomedicine: Nanotechnology, Biology and Medicine 11(8), 1925–1938, https://doi.org/10.1016/j.nano.2015.07.012 (2015).
Jing, Y. R., Zhou, W., Li, W. L., Zhao, L. X. & Wang, Y. F. The Synthesis of Novel Taxoids for Oral Administration. Bioorganic & Medicinal Chemistry 22(1), 194–203, https://doi.org/10.1016/j.bmc.2013.11.037 (2014).
Mease, K., Sane, R., Podila, L. & Taub, M. E. Differential Selectivity of Efflux Transporter Inhibitors in Caco-2 and MDCK–MDR1 Monolayers: A Strategy to Assess the Interaction of a New Chemical Entity with P-gp, BCRP, and MRP2. Journal of Pharmaceutical Sciences 101(5), 1888–1897, https://doi.org/10.1002/jps.23069 (2012).
Sun, Y. et al. Establishment and characterization of an MDCK cell line stably-transfected with chicken Abcb1 encoding P-glycoprotein. Research in Veterinary Science 106, 37–44, https://doi.org/10.1016/j.rvsc.2016.03.004 (2016).
Jin, J. et al. Comparative pharmacokinetics of paclitaxel after oral administration of Taxus yunnanensis extract and pure paclitaxel to rats. Fitoterapia 90, 1–9, https://doi.org/10.1016/j.fitote.2013.06.013 (2013).
Jang, Y. et al. Effect of paclitaxel content in the DHP107 oral formulation on oral bioavailability and antitumor activity. Journal of Drug Delivery Science and Technology 48, 183–192, https://doi.org/10.1016/j.jddst.2018.09.014 (2018).
Hou, J. et al. Improved oral bioavailability and anticancer efficacy on breast cancer of paclitaxel via Novel Soluplus®—Solutol® HS15 binary mixed micelles system. International Journal of Pharmaceutics 512(1), 186–193, https://doi.org/10.1016/j.ijpharm.2016.08.045 (2016).
Acknowledgements
The authors acknowledge the Grant support from Heilongjiang Human Resources and Social Security Bureau.
Author information
Authors and Affiliations
Contributions
Yunrong Jing and Wei Zhou designed experiments; Yunrong Jing and Xiangyang Wang carried out experiments; Wei Zhou analyzed experimental results; Yunrong Jing wrote the main manuscript text and prepared Figs. 1 and 2.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jing, YR., Zhou, W. & Wang, XY. T-13 and T-26, the novel taxanes with improved oral bioavailability in rats. Sci Rep 10, 3211 (2020). https://doi.org/10.1038/s41598-020-60184-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60184-2
- Springer Nature Limited