
Abstract
Glycosylphosphatidylinositol (GPI)-anchored proteins are important for virulence of many pathogenic organisms including the human fungal pathogen, Candida albicans. GPI biosynthesis is initiated by a multi-subunit enzyme, GPI-N-acetylglucosaminyltransferase (GPI-GnT). We showed previously that two GPI-GnT subunits, encoded by CaGPI2 and CaGPI19, are mutually repressive. CaGPI19 also co-regulates CaERG11, the target of azoles while CaGPI2 controls Ras signaling and hyphal morphogenesis. Here, we investigated the role of a third subunit. We show that CaGpi15 is functionally homologous to Saccharomyces cerevisiae Gpi15. CaGPI15 is a master activator of CaGPI2 and CaGPI19. Hence, CaGPI15 mutants are azole-sensitive and hypofilamentous. Altering CaGPI19 or CaGPI2 expression in CaGPI15 mutant can elicit alterations in azole sensitivity via CaERG11 expression or hyphal morphogenesis, respectively. Thus, CaGPI2 and CaGPI19 function downstream of CaGPI15. One mode of regulation is via H3 acetylation of the respective GPI-GnT gene promoters by Rtt109. Azole sensitivity of GPI-GnT mutants is also due to decreased H3 acetylation at the CaERG11 promoter by Rtt109. Using double heterozygous mutants, we also show that CaGPI2 and CaGPI19 can independently activate CaGPI15. CaGPI15 mutant is more susceptible to killing by macrophages and epithelial cells and has reduced ability to damage either of these cell lines relative to the wild type strain, suggesting that it is attenuated in virulence.
Similar content being viewed by others

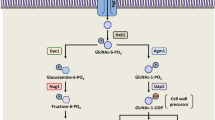
Introduction
GPI anchored proteins play important roles as adhesion molecules, enzymes, activation antigens, differentiation markers, and protozoan coat components. The GPI anchor is synthesized in the endoplasmic reticulum of eukaryotes by the concerted action of multiple enzymes in a pathway that is sequential for the most part1. The pathway is essentially conserved in all eukaryotic organisms and generates a glycolipid anchor of the form NH2CH2CH2PO4H-6Manα1-2Manα1-6Manα1-4GlcNα1-6D-myo-inositol-1-HPO4-lipid, where the lipid can be diacylglycerol, alkylacylglycerol or ceramide. Yet, several species-specific variations in the GPI biosynthetic pathway exist1,2.
The process starts with the transfer of N-acetylglucosamine (GlcNAc) from UDP-GlcNAc to phosphatidylinositol (PI), a reaction that is in all probability catalyzed by Gpi3 (yeast) or Pig-A (mammals). With the exception of Giardia lamblia, in all other eukaryotic systems, Gpi3/Pig-A is assisted by several other proteins that number anywhere between 3 to 6, depending upon the organism, and which together form the GPI-N-acetylglucosaminyl transferase (GPI-GnT) complex in the endoplasmic reticulum3. In humans, the GPI-GnT complex includes, besides Pig-A, five other subunits, Pig-H, Pig-C, Pig-P, Pig-Q and Pig-Y1. Of these, homologs of Pig-C, Pig-P and Pig-Q are present in all eukaryotes. In yeast, they correspond to Gpi2, Gpi19 and Gpi1, respectively. PIG-Y is predicted to be homologous to yeast ERI1, although the latter could not functionally complement Daudi cells defective in PIG-Y4. PIG-H or yeast GPI15 is absent in Caenorhabditis elegans and Entamoeba histolytica3. In yeast, deletion of all other than GPI1 and ERI1, are lethal5,6.
C. albicans is an opportunistic pathogen that causes severe invasive as well systemic infections in immunocompromised individuals often leading to mortality. GPI anchored proteins in this organism are important for yeast-to-hyphae transition as well as for virulence7,8. Disrupting the GPI biosynthetic pathway results in lethality9,10 suggesting that GPI biosynthesis is essential in the organism. In the first set of reports on the GPI-GnT complex of C. albicans, we showed the importance of CaGPI19 in growth, drug response and hyphal morphogenesis of this organism11,12. The CaGPI19 deficient mutant was azole sensitive and hyperfilamentous11. A mutual co-regulation existed between CaGPI19 and CaERG11, an important gene in the ergosterol biosynthetic pathway and the target of azoles12.
We then went on to show that a mutant in a second subunit, CaGPI2, was azole resistant and hypofilamentous. Further, CaGPI2 specifically controlled hyphal morphogenesis via Ras signaling. It was also negatively co-regulated with CaGPI1913.
In the present manuscript, we have explored the role of a third subunit, CaGPI15, in C. albicans. We show that CaGPI15 is important for growth, cell wall integrity and GPI biosynthesis in C. albicans. It also affects response to azole drugs as well as hyphal morphogenesis in C. albicans. It does so because it simultaneously activates both CaGPI2 and CaGPI19 which function downstream of CaGPI15. Thus, here too CaGpi2 controls hyphal morphogenesis via CaRas1 and CaGpi19 controls sensitivity to azoles by regulating CaERG11 levels. The downregulation of CaERG11 in mutants of CaGPI15 as well as CaGPI19 occurs due to decrease in H3 acetylation on the promoter of CaERG11. Both CaGPI2 and CaGPI19 can also independently activate CaGPI15 levels.
Results
Cloning of CaGPI15 gene from C. albicans
The putative CaGPI15 gene was identified using human PIG-H gene as the query sequence for BLAST analysis as well as using the information available at Prof. Eisenhaber’s website as explained in Materials and Methods. The sequence obtained also compared very well with that reported previously14. The putative CaGpi15 protein showed roughly 26.23% and 21.94% identity with Gpi15 sequences from Saccharomyces cerevisiae and Saccharomyces pombe, respectively (Supplementary Fig. 1A). The gene was subsequently cloned from the genomic DNA of C. albicans using gene-specific primers.
CaGPI15 gene complements the S. cerevisiae GPI15 gene
The ScGPI15 gene of YPH500 was placed under the control of the GAL1 promoter. This strain (YPH-pGAL1-ScGPI15) grew well in the presence of galactose but was unable to grow in glucose (Supplementary Fig. 1B). When CaGPI15 was introduced in this strain (YPH-pGAL1-ScGPI15-CaGPI15), the strain was able to grow in glucose (Supplementary Fig. 1B). Growth complementation was accompanied by restoration of GPI-GnT activity (Supplementary Fig. 1C). Thus, despite low sequence conservation, CaGpi15 is the functional homolog of ScGpi15.
Chromosomal disruption of CaGPI15 gene
Heterozygous (CaGPI15Hz) and conditional null (Cagpi15 null) mutants of CaGPI15 were generated in the C. albicans BWP17 strain using a PCR based approach15,16. CaGPI15Hz had one allele of CaGPI15 disrupted with a HIS1 nutritional marker17. Cagpi15 null strain was made in the CaGPI15Hz background with the second CaGPI15 allele placed under the control of the repressible MET3 promoter. Since URA3 is known to alter gene expressions in C. albicans18, one copy of URA3 was inserted at the RPS1 locus in BWP17 (BWP17URA3) as well as in CaGPI15Hz (CaGPI15Hz-URA3) and these were used as controls in studies on all mutants that involved use of URA3 as a selection marker. The downregulation of CaGPI15 expression levels were confirmed by transcript level analysis (Supplementary Fig. 2A).
Depletion of CaGpi15 affects growth of C. albicans
The growth of CaGPI15Hz, on solid and liquid medium was comparable to that of the wild type strain (Fig. 1A(i,ii)). The Cagpi15 null on the other hand, grew slower on solid minimal media containing Met/Cys (Fig. 1A(iii)). Further, in liquid medium, the doubling time for the Cagpi15 null in the presence of 10 mM Met/Cys was found to be higher than in the absence of Met/Cys (Fig. 1A(iv); Supplementary Table 2).

(A) CaGPI15Hz and Cagpi15 conditional null mutant show growth defect. (i) BWP17 and CaGPI15Hz were spotted on YEPD plates. Growth was monitored at 30 °C for 24 h and 72 h. (ii) CaGPI15Hz mutant did not show any growth defect in liquid SD medium. (iii) BWP17URA3 as well as Cagpi15 null were spotted on SD medium plates in the absence or presence of Met/Cys. Growth was monitored at 30 °C for 24 h. (iv) Cagpi15 null mutant shows growth defect in liquid cultures. Cagpi15 null was grown both in absence (p) and presence (r) of 10 mM Met/Cys in liquid medium. For liquid cultures, cell growth for the various strains was monitored by OD600nm at different time points and doubling times are calculated and mentioned in Supplementary Table 2. The experiment was done three times in duplicates; arithmetic mean with standard deviations is shown. For solid media experiments, a 5 µl suspension of cells corresponding to 1 × 107, 2 × 106, 4 × 105, 8 × 104 and 1.6 × 104 numbers were spotted from left to right in each row. The experiments were done thrice using independent cultures. (B) CaGPI15 is required for filamentation. The hyphal growth and quantification of hyphal growth in CaGPI15Hz for up to 120 min in (i,ii) liquid spider media and in (iii,iv) liquid RPMI with 10% serum at 37 °C. A minimum of 100 cells were used for the statistical analysis. The arithmetic mean with standard deviation is plotted. (C) CaGPI15 depletion reduces GPI-GnT activity. GPI-GnT activity was tested in the CaGPI15 mutants as described in Materials and Methods. (D) Hyphal growth in Cagpi15 null and CaGPI15 revertant. The hyphal growth and quantification of hyphal growth in Cagpi15 null and CaGPI15 revertant for up to 90 min in (i,ii) liquid spider media and in (iii,iv) liquid RPMI with 10% serum at 37 °C. A minimum of 100 cells were used for the statistical analysis. The arithmetic mean with standard deviations is plotted. (E) CaRas1 is responsible for the filamentation phenotypes of the CaGPI15 mutants. (i) Cells of the indicated strains were spotted on SD-agar plates and incubated at 30 °C for 48 h after a 10 min heat shock at 48 °C; (ii) and (iii) The hyphal growth and quantification of hyphal growth in various strains for up to 120 minutes in liquid RPMI with 10% serum media at 37 °C. CaRas1 restores filamentation in CaGPI15Hz. These experiments were done twice in duplicates.
Depletion of CaGpi15 results in reduced GPI-GnT activity in C. albicans
We previously showed that the GPI-GnT activity was reduced in the CaGPI15Hz strain17. Not only was the GPI-GnT activity significantly lower in the CaGPI15Hz (~50%) with or without the URA3 marker as compared to the wild type controls it was further reduced in the Cagpi15 null strain (28% activity) under repressive conditions of growth (Fig. 1C). Under permissive growth conditions in the Cagpi15 null there is no significant decrease in the GPI-GnT activity (47%) as compared to CaGPI15Hz-URA3 (Fig. 1C). That a drop in GPI-GnT activity of roughly 50% does not seem to cause a corresponding reduction in growth of the strain would suggest that relatively low levels of GPI biosynthetic activity are sufficient for the growth of C. albicans. This has also been reported in other GPI biosynthetic mutants9,11,13,19. However, when GPI biosynthesis drops below a certain threshold, as is seen in the conditional null strain under repressive growth conditions, then it affects the growth of the fungus. GPI-GnT activity was restored in CaGPI15 revertant strain where one allele of CaGPI15 was reintroduced in the CaGPI15Hz using the constitutively active pACT1 promoter (Fig. 1C).
Depletion of CaGpi15 causes cell wall defects
The CaGPI15 knock-down mutants showed several cell wall defects, including increased clumping when grown to near saturation levels and lower chitin and beta glucan levels in the cell wall versus the wild type strain (Supplementary Fig. 2B,C; Supplementary Table 3). The cell wall defects were reversed in the CaGPI15 revertant strain (Supplementary Fig. 2B,C(i–iv)), suggesting that the cell wall defects were specifically due to depletion of CaGpi15.
CaRas1-dependent signaling pathway is altered in the CaGPI15 mutants
As also shown previously17, hyphal growth of CaGPI15Hz was noticeably lesser than that of the wild type BWP17 strain on solid as well as liquid hyphae-inducing media at 37 °C but was restored in the CaGPI15 revertant cells (Fig. 1B(i–iv); Fig. 1D(i–iv); Supplementary Fig. 3A), suggesting that this effect was specific to CaGPI15.
We have previously shown that the Ras/cAMP dependent PKA activity is altered in mutants of the first step of GPI anchor biosynthesis in C albicans12,13,17. Hyperactive Ras mutants are heat shock sensitive13,20 and reduced Ras signaling correlates with heat-shock resistance13. CaGPI15Hz was resistant to heat shock as compared to BWP17 (Fig. 1E(i)), suggesting that Ras-dependent cAMP/PKA signaling was decreased in this mutant. C. albicans has two Ras proteins, CaRas1 and CaRas2 of which CaRas1 is known to be the major determinant of hyphal growth21. Overexpression of CaRAS1 restores filamentation in CaGPI5Hz while overexpression of CaRAS2 does not (Fig. 1E(ii,iii); Supplementary Fig. 3B).
CaGPI15 mutant strains are sensitive to azoles due to compromised ergosterol biosynthesis
CaGPI15Hz as well as Cagpi15 null cells were sensitive to azoles as compared to controls (Fig. 2A(i,ii); Supplementary Fig. 4A(i,ii)). This sensitivity was reversed in the CaGPI15 revertant strain (Fig. 2A(iii); Supplementary Fig. 4A(iii)). Azoles target CaErg11, the lanosterol demethylase in the ergosterol biosynthetic pathway of C. albicans22. Hence, the levels of CaERG11 transcripts in cells of CaGPI15Hz and Cagpi15 null were examined. CaERG11 levels were significantly reduced in both cases (Supplementary Fig. 4B(i)). CaERG11 levels were restored in the CaGPI15 revertant strain (Supplementary Fig. 4B(ii)). The reduction in CaERG11 levels correlated well with the accumulation of lanosterol, the substrate of CaErg11, and a reduction in total ergosterol levels in the CaGPI15Hz and Cagpi15 null strains (Fig. 2B).

Ergosterol biosynthesis pathway is affected in the CaGPI15 mutants. (A) CaGPI15 mutants are sensitive to ketoconazole and the sensitivity is reversed in the CaGPI15 revertant in liquid SD medium. (i) Growth curve analysis of CaGPI15Hz mutant with respect to BWP17 in 0.08 μg/ml ketoconazole. Growth of the strains without ketoconazole are also shown for comparison. (ii) Cagpi15 null mutant was grown both in permissive as well as in repressive liquid medium in the presence of 0.2 μg/ml ketoconazole. (iii) Azole sensitivity is reversed in the CaGPI15 revertant strain in the presence of 0.2 μg/ml ketoconazole. Supplementary Table 2 contains the doubling time for the mutants both in the presence/absence of ketoconazole. The experiments are done twice in duplicates and the arithmetic mean and standard deviations are plotted. (B) CaGPI15 mutants accumulate lanosterol and are deficient in ergosterol. Relative quantification of ergosterol (grey bars) and lanosterol (white bars) was done by GC-MS. (C) Rtt109 regulates CaERG11 transcription in the CaGPI15Hz. (i) Schematic representation of the CaERG11 promoter pairs. Pair 2 shows the binding site for RNA Pol II and Upc2. H3K56Ac sites are also identified on the promoter. (ii) ChIP analysis was done to compare the occupancy of RNA pol II, H3K56Ac and Upc2 at CaERG11 promoter using primer pair 2 in CaGPI15Hz-URA3 relative to BWP17URA3. (D) Levels of CaERG11 are restored in CaGPI15Hz/pACT1-RTT109 strain. (i) Transcript levels of CaERG11 is restored in CaGPI15Hz/pACT1-RTT109 strain. The transcript levels of both RTT109 and CaERG11 were monitored in the mutants relative to BWP17URA3. (ii) The H3K56Ac levels in whole cell lysate were estimated in the mutants relative to BWP17URA3. H3 levels were taken as a loading control. A cropped representative image is shown, and the full-length image is displayed in Supplementary Fig. 5D (iii) The CaERG11 promoter occupancy of RNA pol II, H3K56Ac and H3K9Ac in the mutants relative to BWP17URA3. (E) Azole sensitivity was reversed in CaGPI15Hz/pACT1-RTT109 strain. Growth curve analysis of CaGPI15Hz/pACT1-RTT109 mutant in 0.2 μg/ml ketoconazole. The doubling time of CaGPI15Hz/pACT1-RTT109 in the presence of ketoconazole is almost similar with the wildtype strain grown in the same condition (Supplementary Table 2). (F) CaGPI15Hz/pACT1-RTT109 strain accumulates ergosterol. Relative quantification of ergosterol was done using GC-MS. P-values for CaGPI15Hz were calculated relative to the wild type while for CaGPI15Hz/pACT1-RTT109 it was relative to CaGPI15Hz-URA3.
Upc2 is a transcription factor that controls CaERG11 levels in the cell23. The occupancy of both RNA Pol II (RNAPII) as well as Upc2 were significantly reduced on the promoter of CaERG11 in CaGPI15Hz-URA3 as compared to BWP17URA3 when probed using primer pair 2 (Fig. 2C(i,ii)), suggesting that the promoter had reduced accessibility for transcription. This also correlated well with the fact that acetylation of histone H3, as assessed using the H3K56 antibody, was significantly lower on the promoter of CaERG11 in CaGPI15Hz-URA3 as compared to that in BWP17URA3 (Fig. 2C(ii) & Supplementary Fig. 4D).
The role of histone acetylation in the regulation of CaERG11 expression in C. albicans was further investigated. H3K56 and H3K9 acetylation are mediated by Rtt109, a histone acetyltransferase24,25. As can be seen from (Fig. 2D(i,ii); Supplementary Fig. 5D), the expression of RTT109 at the mRNA as well as protein levels was not significantly altered in the CaGPI15Hz strain. RTT109 was then expressed under the control of pACT1 promoter in the CaGPI15Hz strain and its overexpression was confirmed (Fig. 2D(i)). CaERG11 transcript levels were upregulated in these cells (Fig. 2D(i)). ChIP analysis showed that overexpression of RTT109 in CaGPI15Hz cells increased acetylation of H3, as assessed by anti-H3K56 or anti-H3K9 antibodies, and increased the occupancy of RNAPII on the CaERG11 promoter (Fig. 2D(iii)) Overexpression of RTT109 in CaGPI15Hz cells also reversed the azole sensitivity (Fig. 2E; Supplementary Fig. 4C) and restored ergosterol levels in these cells (Fig. 2F), confirming that acetylation of H3 by Rtt109 regulates the expression of CaERG11 in this strain.
Separately, in order to ensure that the reduced H3 acetylation was not due to a global reduction in acetylation or due to defects in nucleosome assembly, we checked the H3 acetylation levels at two different intergenic regions (Chromosome 5 and Chromosome R) in BWP17URA3, CaGPI15Hz-URA3 and CaGPI15Hz-pACT1-RTT109 by ChIP (Supplementary Fig. 4E(i,ii). No reduction in H3 acetylation was observed in the two strains at these positions relative to the control, BWP17URA3, suggesting that the ChIP signal observed is not a nucleosome assembly dependent effect and was instead due to a reduction in acetylation levels at the promoter of CaERG11.
The acetylation mediated by Rtt109 is catalyzed with two histone chaperones, Vps75 and Asf126. While the latter has been shown to be important for H3K56Ac alone, the former has been shown to be important for acetylation of H3 at K9 as well K5625. A VPS75Hz mutant should, therefore, mimic a strain defective in acetylation by Rtt109. To confirm that acetylation of H3 at the CaERG11 promoter was important for its regulation in C. albicans, ChIP analysis was done in a VPS75Hz strain generated in the lab. The acetylation of H3 dropped to a significant extent at the promoters of CaERG11, CaGPI2, CaGPI15 and CaGPI19 in this strain when probed using anti-H3K56 antibody (Supplementary Fig. 4F(i–iv)).
CaGPI2 and CaGPI19 levels in the CaGPI15 heterozygous strain can also be restored by RTT109 overexpression
Transcript levels of both CaGPI2 and CaGPI19 were found to be reduced in CaGPI15Hz as compared to BWP17 (Fig. 3A(i)) and were restored in the CaGPI15 revertant strain (Fig. 3A(ii)), suggesting that this effect was specifically linked to CaGPI15 levels. It should be noted that the expression of two housekeeping genes (CaUBC13, CaACT1), two ergosterol biosynthesis genes downstream to CaERG11 (CaERG3, CaERG4) and three other downstream GPI biosynthetic genes (CaGPI12, CaGPI14, CaGPI8) were all found to not be significantly altered in the CaGPI15Hz or the CaGPI15 revertant strains (Supplementary Fig. 5A).

CaGPI2 and CaGPI19 are downstream of CaGPI15. (A) Transcript levels of CaGPI2 and CaGPI19 are controlled by CaGPI15. (i) The transcript levels of both CaGPI2 and CaGPI19 in CaGPI15Hz as compared to BWP17. (ii) The transcript levels of CaGPI15, CaGPI2 and CaGPI19 in the CaGPI15 revertant. P-values were calculated with respect to the wild type controls. (B) H3K56Ac and transcript levels of CaGPI2, CaGPI15 and CaGPI19 are controlled by Rtt109. (i) Schematic representation of the promoters of CaGPI15, CaGPI2 and CaGPI19 along with their H3K56Ac positions. (ii) The relative H3K56Ac levels at the promoters of CaGPI15, CaGPI2 and CaGPI19 in the indicated strains versus wild type. (iii) Transcript levels of CaGPI15, CaGPI19 and CaGPI2 upon RTT109 overexpression in the CaGPI15Hz. (C) Hyphal morphogenesis is restored upon overexpression of RTT109. (i) and (ii) The hyphal growth and quantification of hyphal growth in various strains for up to 60 minutes in liquid spider media at 37 °C. A minimum of 100 cells were used for the statistical analysis. The experiment was repeated twice in duplicates; averages with standard deviations are shown.
Further, the acetylation of histone H3 on the promoter of CaGPI15, CaGPI2 and CaGPI19 in the CaGPI15Hz strain was found to be significantly reduced when probed using anti-H3K56 antibody (Fig. 3B(i,ii)). Overexpression of RTT109 in CaGPI15Hz restored the H3Ac (Fig. 3B(ii)) and the levels of all three genes (Fig. 3B(iii). It also reversed azole sensitivity as described above (Fig. 2E; Supplementary Fig. 4C), and filamentation of the CaGPI15Hz mutant (Fig. 3C(i,ii)). Taken together, these results indicate that CaGPI15 specifically regulates CaGPI19 and CaGPI2 levels.
CaGPI2 and CaGPI19 control the phenotypes of CaGPI15
We recently demonstrated that CaGPI2 functions downstream of CaGPI15 in controlling hyphal morphogenesis17. Filamentation of two strains, CaGPI15Hz/CaGPI2Hz and CaGPI15Hz/pACT1-CaGPI2, confirmed this. In the former, where one allele of CaGPI2 is disrupted in CaGPI15Hz background, filamentation was further reduced (Fig. 4A(i,ii); Supplementary Fig. 3C). In the latter, where CaGPI2 was overexpressed in the CaGPI15Hz background, filamentation was restored (Fig. 4A(i,ii); Supplementary Fig. 3C). Similarly, to confirm that CaGPI19 functions below CaGPI15 in controlling CaERG11 levels and sensitivity to azole drugs, CaGPI15Hz/CaGPI19Hz and CaGPI15Hz/pACT1-CaGPI19 strains were generated. In the former, one allele of CaGPI19 was disrupted and in the latter CaGPI19 was overexpressed in the CaGPI15Hz background. CaERG11 transcripts were reduced in CaGPI15Hz/CaGPI19Hz and upregulated in CaGPI15Hz/pACT1-CaGPI19 as compared to the parent strain (Fig. 4B). The sensitivities of these strains to azoles also correlated with their CaERG11 levels. CaGPI15Hz/CaGPI19Hz was more sensitive to azoles while CaGPI15Hz/pACT1-CaGPI19 was resistant to azoles as compared to CaGPI15Hz (Fig. 4C(i,ii); Supplementary Fig. 5B). Thus, CaGPI19 functions downstream of CaGPI15 in controlling sensitivity to azole drugs.

(A) Hyphal morphogenesis correlates with CaGPI2 expression levels. (i) and (ii) The hyphal growth and quantification of hyphal growth in various strains for up to 120 minutes in liquid RPMI with 10% serum media at 37 °C. A minimum of 100 cells were used for the statistical analysis. The experiment was repeated twice in duplicates; averages with standard deviations are shown. (B) CaERG11 transcript levels correlate with levels of CaGPI19 expression. The transcript levels of CaERG11 were monitored in the mutants strains relative to either BWP17 or BWP17URA3. (C) The azole response of the strains correlates with CaERG11 expression levels. (i) Strains overexpressing CaGPI19 were grown in liquid SD medium containing 0.2 µg/ml ketoconazole. (ii) Depletion of CaGPI19 slows down the doubling time of the CaGPI15Hz/CaGPI19Hz mutant when cultured with 0.08 µg/ml ketoconazole. Supplementary Table 2 contains the doubling time for the mutants both in the presence/absence of ketoconazole. Two independent experiments in duplicates were done for confirmation. (D) Rtt109 regulates CaERG11 transcription and sensitivity to azoles in the CaGPI19 mutant. (i) ChIP analysis was done to compare the H3K56Ac of CaERG11 promoter in the mutant strains relative to BWP17URA3. (ii) Transcript levels of RTT109 and CaERG11 were monitored in the mutant strains relative to BWP17URA3. (iii) Response to azoles was monitored in CaGPI19Hz/pACT1-RTT109 mutant. All strains were grown with or without 0.2 μg/ml ketoconazole and the doubling time was calculated (Supplementary Table 2). The experiments were repeated twice in duplicates. (iv) Relative quantification of ergosterol was done in different mutants of CaGPI19 using GC-MS. P-values for all heterozygous mutants were calculated with respect to the wild type controls while that for overexpression mutants are with respect to CaGPI19Hz-URA3.
ChIP analysis showed that acetylation of H3 was reduced at the promoter of CaERG11 in CaGPI19Hz strain and was restored by overexpression of RTT109 in CaGPI19Hz background (Fig. 4D(i)). Overexpression of RTT109 in CaGPI19Hz background also caused an enhancement in CaERG11 transcripts (Fig. 4D(ii)), reversal of azole sensitivity (Fig. 4D(iii); Supplementary Fig. 5C and restoration in ergosterol levels (Fig. 4D(iv)).
The cross-talk between CaGPI2, CaGPI15 and CaGPI19
The data presented in Fig. 3A, suggested that CaGPI15 is an activator of both CaGPI2 and CaGPI19. To examine the interaction between CaGPI2 and CaGPI19 in the CaGPI15Hz background, we studied two double heterozygous strains, CaGPI15Hz/CaGPI2Hz and CaGPI15Hz/CaGPI19Hz. The levels of CaGPI19 were increased in CaGPI15Hz/CaGPI2Hz while that of CaGPI2 were increased in CaGPI15Hz/CaGPI19Hz. Overexpressing either CaGPI2 or CaGPI19 in CaGPI15Hz resulted in downregulation of the other (Fig. 5A). Thus, the mutually negative regulation between CaGPI2 and CaGPI19 continues to function in the CaGPI15Hz strain.

Once activated CaGPI19 and CaGPI2 are not sensitive to depletion of CaGPI15. (A) The mutual negative regulation between CaGPI2 and CaGPI19 is maintained in CaGPI15 mutants. Heterozygous deletion of either CaGPI2 or CaGPI19 was made in CaGPI15Hz. Likewise, either CaGPI2 or CaGPI19 was overexpressed in CaGPI15Hz. The transcript levels of CaGPI19 and CaGPI2 were monitored in these mutants. (B) Simultaneous downregulation of CaGPI2 and CaGPI19 downregulates CaGPI15. Transcript levels of CaGPI2, CaGPI19 and CaGPI15 were monitored in double heterozygote strains of CaGPI2 and CaGPI19 along with their respective heterozygous parent strains. (C) Activation of CaGPI15 correlates with activation of either CaGPI2 or CaGPI19. Transcript levels of CaGPI2, CaGPI19 and CaGPI15 were monitored in BWP17 strain overexpressing either CaGPI2, CaGPI19 or CaGPI15 relative to BWP17URA3. (D) Model explaining the regulation of CaERG11 transcription and filamentation by CaGpi15. In C. albicans, CaGPI15 is an activator of CaGPI2 as well as CaGPI19. In turn, both CaGPI2 and CaGPI19 are independently capable of stimulating activation of CaGPI15. CaGPI2 and CaGPI19 are mutually negatively regulated and function downstream of CaGPI15 to control Ras signaling and CaERG11 levels, respectively, in the organism. Azole response via regulation of CaERG11 continues to correlate with CaGPI19 levels while Ras1-dependent hyphal morphogenesis continues to correlate with CaGPI2 levels. (E) CaGPI15 can mediate active GPI-GnT complex formation in CaGPI2Hz and CaGPI19Hz strains. Relative GPI-GnT activity in CaGPI15Hz strain overexpressing either CaGPI2 or CaGPI19 as well as in CaGPI2Hz and CaGPI19Hz strains overexpressing CaGPI15. P-values for heterozygous mutants were calculated with respect to the wild type controls while that for double deletion or overexpression mutants was calculated relative to the heterozygous parent control.
CaGPI15 transcripts are upregulated in CaGPI2Hz13 and CaGPI19Hz (Fig. 5B). We also examined two strains that had simultaneous downregulation of CaGPI2 and CaGPI19. Transcripts of CaGPI15 decreased in both CaGPI19Hz/CaGPI2Hz and CaGPI2Hz/CaGPI19Hz strains (Fig. 5B). Overexpression of CaGPI15 in the wild type strain led to simultaneous upregulation of CaGPI2 and CaGPI19 (Fig. 5C). Similarly, overexpression of either CaGPI2 or CaGPI19 caused overexpression of CaGPI15 while maintaining the negative regulation between CaGPI2 and CaGPI19 (Fig. 5C). Thus, both CaGPI2 and CaGPI19 can independently activate CaGPI15 (Fig. 5D). CaGPI15 acts upstream and serves to activate both CaGPI2 and CaGPI19 (Fig. 5D).
GPI-GnT activity assays also corroborate such a model. Overexpression of CaGPI15 in CaGPI2Hz or CaGPI19Hz pushes up transcript levels of both CaGPI2 and CaGPI19 and causes enhanced GPI-GnT activity in comparison to the parent strains (Fig. 5E). However, due to the negative regulation between CaGPI2 and CaGPI19, overexpressing either CaGPI2 or CaGPI19 in the CaGPI15Hz strain cannot enhance its GPI-GnT activity (Fig. 5E).
CaGPI15 heterozygous strain is more susceptible to killing by MH-S macrophage cells and is less able to kill the macrophage cell line
We tested the effect of CaGPI15 knock-down on C. albicans virulence. For this, a murine alveolar macrophage cell line (MH-S) was co-cultured with BWP17 and CaGPI15Hz strain for 3 h (Fig. 6A,B) and 18 h (Fig. 6C,D). Both strains of Candida formed hyphae when co-cultured with MH-S cells for 18 h. BWP17 co-cultured with MH-S had 55% longer hyphae than CaGPI15Hz co-cultured with MH-S for 18 h (Fig. 6E). The internalization of C. albicans cells by MH-S was roughly similar for the CaGPI15Hz and BWP17 strains (Fig. 6F). This was also the case when phagocytosis was inhibited with the help of cytochalasin D (Cyt D) (Fig. 6F). Thus, mutation of CaGPI15 does not appear to alter the phagocytosis of C. albicans cells by either cytoskeleton-dependent or independent pathways. The colony forming units (CFU) recovered after incubation with the macrophage cells was significantly lower in CaGPI15Hz as compared to BWP17 (Fig. 6G). Hence, more numbers of CaGPI15Hz cells were killed by MH-S in comparison to BWP17. In addition, more live MH-S cells were recovered after co-culture with CaGPI15Hz as compared to BWP17, suggesting that CaGPI15Hz was less virulent (Fig. 6H). There was also no difference in cell death by pyroptosis after co-culturing MH-S with BWP17 and CaGPI15Hz at 1:5 multiplicity of infection (MOI) for 3 h and 18 h (Fig. 6I). But there is a significant difference in pyroptosis between 3 h and 18 h for MH-S co-cultured with either strain. Similarly, there was no difference in cell death by apoptosis (Annexin V staining) when MH-S cells were co-cultured with either with BWP17 or CaGPI15Hz strain at 1:5 MOI for 18 h. Nevertheless, the level of apoptosis was significant in MH-S on infection with either strain of C. albicans for 18 h. So, C. albicans seems to cause MH-S macrophage cell death by apoptosis as well as pyroptosis.

Comparative virulence study of BWP17 and CaGPI15Hz with macrophages. Phagocytosis of yeast (A,B) and Hyphal (C,D) form of BWP17 and CaGPI15Hz, respectively, by MH-S cells. MH-S cells were cultured on coverslips overnight for adherence and then cells were co-cultured with CFSE labelled BWP17 and CaGPI15Hz for 3 h (A,B) & 18 h (C,D) respectively followed by fixation with PFA and were mounted and examined using a confocal laser scanning microscope (Magnification 100X). Scale bar represents 10 µm. (E) Hyphae formation by C. albicans. The hyphae length was quantified using Nikon NIS element software. (F) CFU assay after incubation with MH-S cells. MH-S cells (0.1 million) were seeded in 48 well cell culture plate and kept for adherence followed by addition of the indicated C. albicans strains at MOI 1:5 for 18 h. The MH-S cells were lysed and plated on YEPD plates kept at 30 °C for 24 h. The colony forming units obtained were quantified and plotted. (G) Uptake of BWP17 and CaGPI15Hz by MH-S cells. MH-S cells (0.3 million) were seeded in 24 well cell culture plate and allowed to adhere overnight in CO2 incubator at 37 °C. Cells were pre-treated with 2.5 µg/ml cytochalasin D for 30 min followed by addition of CFSE labelled BWP17 and CaGPI15Hz at MOI 1:5 for 3 h and processed as discussed in Methods. The uptake of the C. albicans strains was calculated and plotted on the basis of percentage of cells taking up yeast form of BWP17 and CaGPI15Hz. (H) Live cell recovery of MH-S. MH-S cells (0.1 million) were seeded in 48 well cell culture plate. After adherence, C. albicans at MOI 1:1 and MOI 1:5 for 18 h were added to these cultures. Cells were trypsinized, washed and live cell recovery was calculated by trypan blue exclusion method. (I) C. albicans induced MH-S macrophage pyroptosis. MH-S cells (50,000) were seeded in 96 well cell culture plate. BWP17 and CaGPI15Hz at MOI 1:5 were added for 3 h and 18 h. 50 µl of supernatant were transferred to another 96 well cell culture plate followed by addition of substrate reagent for 30 min and then the reaction was stopped. (J) C. albicans induced MH-S macrophage apoptosis. MH-S (0.3 million) were seeded in 24 well cell culture plate and allowed to adhere overnight in CO2 incubator at 37 °C. BWP17 and CaGPI15Hz at MOI 1:5 were added for 3 h and 18 h and processed as discussed in Methods. Cells were harvested and 1 µl Annexin V APC was added. Each point represents mean ± SEM of values obtained from three independent assays. *p ≤ 0.05, **p ≤ 0.005 and ***p ≤ 0.0005 represent statistically significant difference between control and treated cells, ns is no statistically significant difference. The significance of any difference was calculated by using one-tailed distribution in a two-sample equal variance student’s t test.
CaGPI15 heterozygous strain is more susceptible to killing by LA-4 epithelial cells and is less able to kill the epithelial cell line
We further investigated the interaction of CaGPI15Hz cells with an epithelial cell line. Murine epithelial cell line LA-4 cells were co-cultured with BWP17 and CaGPI15Hz strain for 3 h (Fig. 7A,B) and 18 h (Fig. 7C,D). Both strains formed hyphae on co-culture with LA-4 cells for 18 h (Fig. 7C,D) but BWP17 exhibited 51% longer hyphae than CaGPI15Hz (Fig. 7E). CaGPI15Hz cells were more susceptible to killing by LA-4 cells as significantly lower CFU were obtained for CaGPI15Hz co-cultured with LA-4 in comparison to that for BWP17 (Fig. 7F). Phagocytosis of BWP17 cells by LA-4 cells was significantly higher than that of CaGPI15Hz cells at MOI 1:5 (Fig. 7G), suggesting a role for fungal GPI anchored proteins in their uptake27. However, the live cell recovery of LA-4 cells was significantly higher for those co-cultured with CaGPI15Hz versus those with BWP17 at MOI 1:5 (Fig. 7H). No significant difference in phagocytosis or live cell recovery was observed in experiments using either of these strains at MOI 1:1. Significantly lower pyroptosis (~30% of wildtype) was seen in LA-4 cells when co-cultured for 18 h with CaGPI15Hz in comparison to those co-cultured with BWP17. No pyroptosis was seen when LA-4 cells were infected with either strain for 3 h. Further, we found a small but significantly higher degree of apoptosis in LA-4 cells infected with BWP17 cells versus CaGPI15Hz after incubation for 18 h at 1:5 MOI (Fig. 7J). Hence, the killing induced by C. albicans infection in epithelial cells also involved apoptosis and pyroptosis. The data from epithelial cell infection studies also supports the notion that the CaGPI15Hz strain is attenuated in virulence.

Comparative virulence study of BWP17 and CaGPI15Hz with epithelial cells. Phagocytosis of yeast (A,B) and Hyphal (C,D) form of BWP17 and CaGPI15Hz, respectively, by LA-4 cells. LA-4 cells were cultured on coverslips overnight for adherence and then cells were co-cultured with CFSE labelled BWP17 and CaGPI15Hz for 3 h (A,B) & 18 h (C,D) respectively followed by fixation with PFA and were mounted and examined using a confocal laser scanning microscope (Magnification 60X). Scale bar represents 20 µm. (E) Hyphae formation by C. albicans. The hyphae length was quantified using Nikon NIS element software. (F) CFU assay after incubation with LA-4 cells. LA-4 cells (0.1 million) were seeded in 48 well cell culture plate and kept for adherence followed by addition of the indicated C. albicans strains at MOI 1:5 for 18 h. The LA-4 cells were lysed and plated on YEPD plates kept at 30 °C for 24 h. The colony forming units obtained were quantified and plotted. (G) Uptake of BWP17 and CaGPI15Hz by LA-4 cells. LA-4 cells (0.3 million) were seeded in 24 well cell culture plate and allowed to adhere overnight in CO2 incubator at 37 °C. Cells were co-cultured with CFSE labelled BWP17 and CaGPI15Hz at MOI 1:1 (as control) & 1:5 for 3 h & 18 h and processed as discussed in Methods. The uptake of the C. albicans strains was calculated and plotted on the basis of percentage of cells taking up yeast form of BWP17 and CaGPI15Hz. (H) Live cell recovery of LA-4 cells. LA-4 cells (0.3 million) were seeded in 24 well cell culture plate. After adherence, C. albicans at MOI 1:1 & 1:5 for 3 h & 18 h were added to these cultures. Cells were trypsinized, washed and live cell recovery was calculated by trypan blue exclusion method. (I) C. albicans induced LA-4 cells pyroptosis. LA-4 cells (50,000) were seeded in 96 well cell culture plate. BWP17 and CaGPI15Hz at MOI 1:5 were added for 3 h and 18 h. 50 µl of supernatant were transferred to another 96 well cell culture plate followed by addition of substrate reagent for 30 min and then the reaction was stopped. (J) C. albicans induced LA-4 cells apoptosis. LA-4 cells (0.3 million) were seeded in 24 well cell culture plate and allowed to adhere overnight in CO2 incubator at 37 °C. BWP17 and CaGPI15Hz at MOI 1:5 were added for 3 h and 18 h and processed as discussed in Methods. Cells were harvested and 1 µl Annexin V APC was added. Each point represents mean ± SEM of values obtained from three independent assays. *p ≤ 0.05, **p ≤ 0.005 and ***p ≤ 0.0005 represent statistically significant difference between control and treated cells, ns is no statistically significant difference. The significance of any difference was calculated by using one-tailed distribution in a two-sample equal variance student’s t test.
Discussion
The GPI anchor glycolipid is produced in the endoplasmic reticulum in 10–12 sequential biochemical steps. In lower eukaryotes this pathway is essential to the growth and viability of the organism while in higher eukaryotes it is critical only at certain stages of organismal development, such as in embryogenesis. Given their essentiality for eukaryotic pathogens, several steps of the pathway have been the focus of study as probable drug targets28,29. However, isolating and studying the individual enzymes of the GPI biosynthetic pathway is challenging because it involves mostly multi-subunit membrane-bound enzymes of relatively low abundance. No high-resolution X-ray crystallographic data are available for any of the enzymes till date and there are no commercially available substrates for most steps of the pathway. The study of the GPI biosynthetic pathway in C. albicans is made more challenging due to its codon biasness. C. albicans proteins heterologously expressed in another host may at times need to be codon optimised for function. Further, until recently no cell free assay system was available for the GPI biosynthetic pathway in this organism since protocols used to generate microsomes from the closely related yeast, S. cerevisiae, did not yield active microsomes from C. albicans13,19.
Despite the many challenges, studying the GPI anchor biosynthesis of different organisms without depending solely on model organisms can be very rewarding. For example, the first and committed step of the GPI biosynthesis pathway in eukaryotes is an important site for regulation. The presence of poorly conserved accessory subunits to assist the highly conserved catalytic subunit suggests that the regulation of the GPI biosynthetic pathway could be mediated via these proteins. Some evidence in support of such a hypothesis also exists. In S. cerevisiae, Ras2 was shown to inhibit the GPI-GnT complex and vice versa while no such regulation was observed in mammals4,6,30. Similarly, Dpm2 is known to regulate the mammalian GPI-GnT, but an equivalent regulation is not observed in other organisms31. In a series of papers establishing such a link in C. albicans, we showed that the GPI-GnT complex interacts with and regulates two other important pathways, ergosterol biosynthesis and hyphal morphogenesis in C. albicans11,12,13. Specifically, CaGpi19 controls CaERG11 levels and modulates azole drug response while CaGpi2 regulates hyphal morphogenesis by controlling Ras signaling. Moreover, CaRas1, the C. albicans homolog of S. cerevisiae Ras2, activates rather than inhibits the GPI-GnT activity17. In turn, the CaRas1-dependent PKA signaling pathway is activated by CaGpi2, but this is independent of the GPI-GnT activity itself17.
In the present study, we examined a third subunit of the GPI-GnT complex to elucidate its importance for C. albicans. CaGpi15, like CaGpi2 and CaGpi19, is a poorly conserved protein sharing very low sequence homology with its yeast counterpart. Yet, at the functional level it complements a conditionally lethal S. cerevisiae gpi15 mutant. Thus, significant functional similarities exist between the Gpi15 homologs in the two organisms. In C. albicans, as in S. cerevisiae, CaGPI15 appears to be important for cell growth. Gene-dosage effects also operate here since the heterozygous mutants were only marginally affected in doubling times while the conditional null showed significantly longer doubling times as compared to the wild type strains. Cell wall defects and clumping observed in CaGPI15 mutants appear to be a result of GPI anchor deficiency since these have also been observed in other GPI biosynthetic mutants9,11,12,13.
CaGPI15 mutants were ergosterol deficient and sensitive to azoles. Thus, in its response to azoles, it appeared that CaGPI15 mutants mirrored the CaGPI19 mutants. Yet, surprisingly, the hyphal morphogenesis phenotype of CaGPI15 mutant cells was quite unlike the hyperfilamentous phenotype of CaGPI19 mutants11,12. CaGPI15 mutants were hypofilamentous as compared to the wild type strains due to reduced Ras signaling, a feature also observed in CaGPI2 mutants13. Thus, CaGPI15 exhibited phenotypes of a mutant that had both CaGPI2 and CaGPI19 downregulated. A transcript level analysis confirmed this. Significantly reduced histone H3 acetylation was observed on the promoters of CaGPI2, CaGPI15 and CaGPI19 in the CaGPI15 mutant strain. Overexpression of RTT109 could restore H3Ac as well as the transcript levels of the three genes, suggesting that expression levels of these genes are regulated via histone H3 acetylation in the CaGPI15 mutant strain. Overexpressing CaGPI15 could also restore CaGPI2 and CaGPI19 levels. Phenotypic assays as well as GPI-GnT activity assays corroborated these results.
Using double mutants in which one allele of either CaGPI2 or CaGPI19 was disrupted in the heterozygous CaGPI15 background, we discovered that CaGPI2 and CaGPI19 continued to be mutually negatively regulated. Further, the hyphal morphogenesis and azole drug response phenotypes correlated with changes in CaGPI2 and CaGPI19 levels, respectively, in these strains.
Finally, we addressed the question of whether CaGPI2 and CaGPI19 were activators or repressors of CaGPI15. We discovered that simultaneous downregulation of CaGPI2 and CaGPI19 in C. albicans results in downregulation of CaGPI15 and overexpression of either of them activates CaGPI15. In other words, both CaGPI2 and CaGPI19 could independently activate CaGPI15. Thus, we propose a model for how the three subunits of the GPI-GnT complex interact in C. albicans (Fig. 5D): CaGPI15 stimulates activation of CaGPI2 as well as CaGPI19. Downregulating it can simultaneously decrease levels of both CaGPI2 and CaGPI19. In turn, both CaGPI2 and CaGPI19 can independently activate CaGPI15. Disrupting either CaGPI2 or CaGPI19 results in upregulation of the other, which in turn upregulates CaGPI15. Both CaGPI2 and CaGPI19 are mutually negatively regulated and function downstream of CaGPI15 as far as hyphal growth and azole drug response are concerned. Sensitivity to azoles via CaERG11 regulation correlates with CaGPI19 levels while Ras-dependent hyphal morphogenesis correlates with CaGPI2 levels.
Two additional issues needed to be addressed here. The first is the mechanism by which CaERG11 is downregulated in the CaGPI15 mutant. The presence of Upc2, a transcription factor for CaERG11, was found to be reduced on the promoter of the CaERG11 gene in CaGPI15 deficient strain. In exploring the possible reason for this, it was observed that H3Ac on the promoter of CaERG11 is also reduced in the azole sensitive CaGPI15 mutant strain. Since CaGPI19 functions downstream of CaGPI15, this strain was also tested for H3Ac and found to have reduced levels of it on the CaERG11 promoter. This is specifically due to loss of Rtt109 activity in these strains. Overexpressing RTT109 could restore the levels of the GPI-GnT subunits as well as of CaERG11 and reverse the response to azoles for both CaGPI15 and CaGPI19 mutant strains.
The second is the mechanism by which Ras signaling is altered in the CaGPI15 mutant. Overexpressing CaRAS1, but not CaRAS2, could restore filamentation in CaGPI15 mutant. Further, CaGPI2 functions downstream of CaGPI15 in this. In a recent manuscript, we showed that CaGpi2 physically interacts with CaRas1 in the endoplasmic reticulum and this interaction helps CaRas1 activate GPI-GnT activity17. CaGpi2 also regulates Ras signaling that occurs at the plasma membrane to trigger hyphal morphogenesis, but it does so by its effect on Hsp90 levels in the cell. It is well known that Hsp90 along with its co-chaperone, Sgt1, interacts with Cyr1, the effector of CaRas1, thereby preventing the interaction of GTP-bound CaRas1 with Cyr1 for initiating cAMP-dependent hyphal morphogenesis32. Overexpressing CaGPI2 results in downregulation of Hsp90 and a lifting of the inhibition exerted by Hsp90 on the Ras signaling pathway. Since CaGpi2 seems to control hyphal morphogenesis in the CaGPI15 mutant strain, it is reasonable to expect that this mechanism functions here too.
The CaGPI15 heterozygous mutant was also far more susceptible to killing by macrophages and epithelial cells in our assays. It showed reduced internalization by epithelial cells, and also shorter hyphae length in comparison to BWP17. In addition, its ability to damage macrophages and epithelial cells was significantly lesser than that of the wild type strain. The damage caused to macrophages and epithelial cells by infection with BWP17 and CaGPI15 heterozygous mutant involved apoptosis as well as pyroptosis, but significantly the pyroptosis seen in epithelial cells on infection with CaGPI15 heterozygous mutant was only 30% of that seen on infection with BWP17. This could be an important contributing factor in reduced damage to epithelial cells by this mutant. Hence, we infer that the CaGPI15 mutant strain is attenuated in virulence.
In conclusion, what is evident from our previous and current studies is that alterations in levels of different subunits of the same complex, all of which affect GPI biosynthesis, receive varying responses from ergosterol biosynthesis and hyphal filamentation pathways in C. albicans. Given the importance of GPI biosynthesis for the viability and growth of C. albicans, the multiple modes of interaction and regulation probably allow the GPI biosynthetic pathway to rapidly respond to multiple signals.
The clinical implications of these observations are hard to miss. Hyphal filamentation and invasive growth are important for the establishment of infection by C. albicans and several hyphae-specific factors are known to be GPI anchored7,8. Strains with reduced levels of GPI anchored proteins are known to show attenuated virulence8. Additionally, ergosterol and its biosynthetic pathway continue to be the most important targets of the currently available antifungals. However, one of the major problems in treatment of fungal infections has been the rapid drug resistance that develops upon continued usage of these drugs33. CaGpi15 as an alternative target, a master regulator that simultaneously affects hyphal morphogenesis as well as ergosterol biosynthesis could be an interesting candidate. Indeed, strains in which CaGPI15 levels are reduced are less virulent, as shown here. Additionally, GPI biosynthesis itself is an essential pathway in C. albicans and targeting the first step of GPI biosynthesis via a protein that bears little homology with its mammalian counterpart could be an effective strategy.
Methods
Strains and media
Yeast and C. albicans strains used in this study are described in Table 1. Murine alveolar epithelial type I cell line (LA-4) and Murine alveolar macrophage cell line (MH-S) were procured from the American type tissue culture collection (ATCC), Rockville, MD, USA. All primers used in this study are listed in Supplementary Table 1. Strains were grown in yeast extract-peptone-dextrose (YEPD) media or synthetic dextrose minimal media (SD media). Ura− strains were grown in YEPD or SD media supplemented with 60 µg/ml uridine. Similarly, His− strains or Arg− strains were grown in SD medium supplemented with the appropriate amino acid (85.6 μg/ml His or Arg). Transformations were performed using lithium-acetate method34. RPMI-1640 medium was from Sigma, MO, USA and FBS from Gibco, Life technologies, Grand Island, NY, USA. RNA PolII (sc-21750) from Santa Cruz Biotechnology, Histone H3 (mAb-96C10) from Cell Signaling, H3K56Ac (ab71956) from Abcam and H3K9Ac (mAb-H0913) from Sigma.
Chemicals
Chemicals were purchased from Merck, Qualigens, or Sigma-Aldrich (USA) unless specified otherwise. Enzymes were from Bangalore Genei (India), MBI Fermentas (USA) or New England Biolabs (USA). Gel extraction and PCR purification kits were from Qiagen. Primers were synthesized by GCC Biotech (India).
PCR amplification of CaGPI15 gene
The putative CaGPI15 gene sequence was obtained from Prof. Eisenhaber’s web site (http://mendel.imp.ac.at/SEQUENCES/gpi-biosynthesis/pigs-main.html) and also confirmed by BLASTp analysis using the mammalian PIG-H sequence as query. Forward primer FPCaGPI15-pCaDis and reverse primer RPCaGPI15-pCaDis (Supplementary Table 1) were used to amplify CaGPI15 gene from genomic DNA of C. albicans. The amplified product was visualized on 1% agarose gel.
Chromosomal disruption of CaGPI15 to make heterozygous CaGPI15 mutant
CaGPI15 heterozygote in BWP17 strain of C. albicans (CaGPI15Hz) was made by PCR-mediated disruption using HIS1 as a selection marker15,17.
Construction of regulatable null mutant of CaGPI15 gene
Conditional null mutant of CaGPI15 in BWP17 strain (Cagpi15 null) was made by placing the functional gene in CaGPI15Hz under the MET3 promoter using PCR mediated approach16.
Complementarity between C. albicans and S. cerevisiae genes
ScGPI15 gene in S. cerevisiae YPH500 strain was placed under the glucose responsive GAL1 promoter by a PCR-mediated approach11,35. The transformed colonies were selected on SDUra− plates and confirmed with locus specific PCR amplification. YPH-pGAL1-ScGPI15 was unable grow on glucose media but could easily grow on media containing 4% galactose and 1.5% sucrose. For complementation studies, CaGPI15 gene, placed under the constitutive PMA1 promoter, was cloned between BamHI and MluI site into YEpHIS plasmid and used to transform this strain36.
Preparation of microsomes from yeast and GPI-GnT assay
Primary cultures of yeast strains were grown for 16 h in 4% (w/v) galactose. It was inoculated into 250 ml SD medium containing either 4% galactose or 2% glucose at 30 °C till OD600nm of 2.0 was reached. The cells were harvested and microsomes prepared from it as described previously37.
GPI-GnT activity of the microsomes was assayed as described previously37. The assay produces both [63H]GlcNAc-PI (N-acetyl glucosaminylphosphatidylinositol) and [63H]GlcN-PI (glucosaminylphosphatidylinositol, the product of the next step of the pathway) and were detected using Bioscan AR2000 TLC scanner. The sum of the radioactive counts detected for the two products in the case of the control strain was considered to be 100% and GPI-GnT activity of all strains was calculated relative to it. In all cases averages of data from independent experiments done twice in duplicates along with standard deviations are plotted.
Preparation of microsomes from C. albicans and GPI-GnT assay
Microsomes from C. albicans were prepared using a protocol previously standardized in our lab13 with minor modifications17. GPI-GnT assay was performed using microsomes corresponding to ~1500 μg protein as described previously37. Comparable amount of heat-killed microsomes were used as the negative control for the assay.
Generating the CaGPI15 revertant strain
CaGPI15 revertant strain was previously reported17. The pACT1-CaGPI15 construct was used to transform CaGPI15Hz cells and the positive clones were confirmed by PCR. The control strains, BWP17URA3 and CaGPI15Hz-URA3 were previously reported17.
Construction of other overexpression strains in the heterozygote mutants
CaGPI15Hz/pACT1-CaGPI2 strain was previously reported17. Using the same strategy, plasmid, pACT1-CaGPI19 was used to generate CaGPI15Hz/pACT1-CaGPI19. Plasmid pACT1-CaRAS1 or pACT1-CaRAS2 was used to generate CaGPI15Hz/pACT1-CaRAS1 and CaGPI15Hz/pACT1-CaRAS2, respectively. The plasmid pACT1-RTT109 was used to generate CaGPI15Hz/pACT1-RTT109 and CaGPI19Hz/pACT1-RTT109 strains. BWP17/pACT1-CaGPI2 strain was previously reported17. The overexpression strains BWP17/pACT1-CaGPI19 and BWP17/pACT1-CaGPI15 were similarly made. The control strain CaGPI19Hz-URA3 was previously reported17. Positive colonies were confirmed by PCR using appropriate primers (Supplementary Table 1).
Generation of double heterozygous strains
CaGPI15Hz/CaGPI19Hz and CaGPI15Hz/CaGPI2Hz were generated using the strategy previously described13,17. Confirmation was done by using gene-flanking primers (Table 1).
Generation of VPS75Hz strain
CaVPS75Hz in BWP17URA3 strain of C. albicans (CaVPS75Hz-URA3) was made by PCR-mediated disruption using ARG4 as a selection marker. Confirmation was done by using gene-flanking primers (Table 1).
Monitoring growth rate in liquid cultures
Growth rate of different mutants was monitored by plotting growth curves. Primary cultures were grown overnight in liquid SD medium at 30 °C, 220 rpm. Secondary cultures were set to an initial OD600nm of 0.2 in 60 ml fresh medium and subjected to shaking at 220 rpm, 30 °C. OD600nm was measured at every 2 h interval to assess the growth in each strain till the saturation was reached. Doubling times were calculated by plotting the exponential growth phase of the cells between 4 and 10 h (OD600nm vs time) for each strain. All the experiments were done twice in duplicates.
Hyphal induction
Hyphal growth was monitored in liquid and solid media using the method described previously12,13. All experiments were done at least thrice in duplicates using independent cultures.
Azole response of mutants
Sensitivity of the strains to azole drugs was studied by spot assays12 as well as by growth in liquid medium in the presence of ketoconazole. Primary and secondary cultures were set as described earlier in ‘Monitoring growth rate in liquid cultures’ section. Final concentration of ketoconazole (0.08 or 0.2 µg/ml) was maintained in 60 ml of 0.2 OD600nm secondary cultures. The way of collecting the samples for growth measurement and calculating the doubling times were same as described earlier in this section. Two independent experiments with duplicates were done for each set of the mutants.
Estimation of sterol levels
The levels of ergosterol and lanosterol were quantified by GC-MS as described previously12. Data in all graphs are averages of two independent experiments done in duplicates along with standard deviations.
Quantification of transcript levels through RT-PCR
RNA extraction, cDNA preparation and quantification of transcript levels was done as described previously using RT primers (Supplementary Table 1)12,13,17,38. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was taken as an internal control. Data in all graphs are averages with standard deviations of two experiments done in duplicates using independent cultures.
Cell Clumping Assay
Cells were grown in minimal media at 30 °C, 220 rpm until saturation. Cells from 500 µl culture were pelleted down at 5000 rpm, 5 min, washed in 1X PBS and resuspended in 50% glycerol. A 5 μl cell suspension was spotted on microscopic slides and observed under Nikon Eclipse Ti Microscope. Cells were quantified for studying cell aggregation.
Calcoflour white (CFW) staining and Congo red (CR) staining in the cell wall
Sample preparation was done as described previously13. Cells were stained with 100 µg/ml CFW or 100 µg/ml CR for 30 min at 30 °C, washed thrice with PBS and observed under a Nikon SMZ TiE fluorescence microscope. Fluorescence intensity was quantified using NIS Elements AR Version 4 software.
Heat sensitivity assays
A 10 min heat shock exposure at 48 °C was given and plate assays done as at 30 °C for 48 h described earlier13.
Chromatin Immunoprecipitation (ChIP) assay
ChIP was carried out as described previously13. Briefly, cells were harvested after cross-linking DNA to protein with 1% (v/v) formaldehyde. Glycine (25 mM) was added and cells lysed by glass beads in lysis buffer (50 mM HEPES, pH 7.4; 140 mM NaCl; 1 mM EDTA; 1% Triton X-100; 1 mM PMSF). After sonication the supernatant was incubated overnight with 1 µg anti-RNA PolII or H3K56Ac antibody. ProteinA-CL agarose beads (20 µl) were added. The beads were spun down after 2 h, washed with lysis buffer, high-salt buffer (lysis buffer with 500 mM NaCl), wash buffer (10 mM Tris-Cl, pH 8.0; 250 mM LiCl; 1 mM EDTA; 0.5% NP-40) and Tris-EDTA buffer. Eluates collected in elution buffer (150 µl Tris-EDTA buffer containing 1.0% SDS) were treated with 20 µg proteinase K. DNA was separated using phenol:chloroform (1:1) and precipitated. The samples were analyzed by PCR using primers for specific regions of promoters.
Cell line and Maintenance of cell line
MH-S and LA-4 cell line were cultured and maintained in RPMI 1640 media, supplement with 25 mM HEPES (Sigma, MO, USA) (N-[2-100 hydroxyethyl]piperazine-N0-[2-ethanesulfonic acid]), 50 µg/ml gentamicin sulfate, 0.05 mM 2-mercaptoethanol, 300 µg/ml L-glutamine and 50 µg/ml and 10% heat inactivate FBS (Gibco, Life technologies, Grand Island, NY, USA) in a humidified atmosphere containing 5% CO2 at 37 °C. Cell lines were maintained as adherent cultures and sub cultured by trypsinization. The cells were then harvested by using 0.25% w/v trypsin from bovine pancreas in 10 mM EDTA disodium salt to detach the monolayer. The harvested cells were collected and pelleted down by centrifugation at 240 g for 5 min at 4 °C. The pelleted cells were suspended in 1 ml media and counted for viability by haemocytometer using trypan blue dye.
Uptake of C. albicans
CFSE labelling of C. albicans
C. albicans cells (100 million) were labelled with 10 μM carboxyfluorescein N-succinimidyl ester (CFSE) as described previously17.
MH-S or LA-4 cells (0.3 million) cultured in 24-well plate were incubated with CFSE labelled BWP17 and CaGPI15Hz at different MOI such as 1:1 and 1:5 for 3 h and 18 h at 37 °C in CO2 incubator. The cells were trypsinized and then harvested with PBS. The harvested cells were fixed in 2% PFA and the uptake of stained BWP17 and CaGPI15Hz by MH-S or LA-4 cells was assessed by using BD FACS Calibur flowcytometer in FL1 channel using Cell Quest software.
Co-culture assay in vitro
Co-culture assay was done as described previously17. MH-S cells or LA-4 cells (0.3 million) were incubated with CFSE labelled fungal cells for 3 h at 1:1 or 1:5 MOI. The phagocytosis-independent uptake of fungal cells by MH-S was monitored using 2.5 µg/ml of Cyt D, an inhibitor of actin polymerization39 as described previously17.
Confocal microscopy
For visualization of uptake of C. albicans by MH-S or LA-4 cells, 0.3 million cells were cultured on glass cover slips overnight. Cells were then co-cultured with CFSE-labelled BWP17 and CaGPI15Hz at MOI (1:5) for 3 h and 18 h at 37 °C. Cells were then washed, fixed with 2% paraformaldehyde (PFA) followed by washing thrice with quencher (ammonium chloride) and examined using a confocal laser scanning microscope (Olympus FluoView FV1000). Five images each were captured having Z sections (depths 0.1 µm)17. The above co-cultured cells of 18 h time point were used to determine the length of hyphae. Nikon NIS element software was used to measure the length of the hyphae.
Macrophage or epithelial cell mediated killing of BWP17 and CaGPI15Hz
The macrophage or epithelial cell mediated killing of C. albicans was studied as described previously17. The number of C. albicans was determined as CFU/ml = number of colonies x dilution factor / volume of culture plate.
Detection of pyroptosis through lactate dehydrogenase (LDH) enzymatic assay
LA-4 and MH-S cells were cultured at a concentration of 50,000 cells/100 µl with complete media in a 96 well cell culture plates. After overnight culture, cells were washed with complete medium to remove debris and dead cells. Cells were co-cultured with BWP17 and CaGPI15Hz for 3 h and 18 h at different MOI (1:5) at 37 °C in CO2 incubator. The co-cultured cells were then subjected for cell cytotoxicity assay. The cell cytotoxicity assay was performed using Cytotoxicity Detection Kit (PierceTM, USA). The standard protocol assay reported here were performed according to the manufacturer’s instructions. The amount of LDH released either in control (Positive control, spontaneous and maximum release) or in the experimental wells was used to calculate the % specific lysis40. The % specific lysis was calculated as: % Specific lysis = (Experimental release − Spontaneous release) × 100/(Maximum release − Spontaneous release).
Assessment of apoptosis
LA-4 and MH-S cells were cultured at a concentration of 0.3 million cells/ml in a 24 well cell culture plates. After overnight culture, cells were washed with complete medium to remove debris and dead cells. Cells were co-cultured with BWP17 and CaGPI15Hz for 18 h at MOI (1:5) at 37 °C in CO2 incubator. The cells were then washed with PBS to remove BWP17 and CaGPI15Hz. The cells were trypsinzed and washed followed by staining with annexin V APC conjugate (Biolegend, San Diego, CA, USA) to assess the apoptotic cells using BD FACS Calibur41 and analyzed through Cell Quest Software.
Statistical significance of data
Unless otherwise stated, statistical significance of the data (p value) was calculated in Sigma plot 8.0 and GraphPad Prism 5.0 using Student’s t-test. The p value > 0.05 is considered not significant and is depicted by n.s., p value ≤ 0.05 is depicted by *p value ≤ 0.005 is depicted by **and p value ≤ 0.0005 is depicted by***.
Data Availability
The datasets and material generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog. Lipid Res. 50, 411–424 (2011).
Komath, S. S., Singh, S. L., Pratyusha, V. A. & Sah, S. K. Generating anchors only to lose them: The unusual story of glycosylphosphatidylinositol anchor biosynthesis and remodeling in yeast and fungi. IUBMB Life 70, 355–383 (2018).
Oswal, N., Sahni, N. S., Bhattacharya, A., Komath, S. S. & Muthuswami, R. Unique motifs identify PIG-A proteins from glycosyltransferases of the GT4 family. BMC Evol. Biol. 8, 168 (2008).
Murakami, Y. The Initial Enzyme for Glycosylphosphatidylinositol Biosynthesis Requires PIG-Y, a Seventh Component. Molecular Biology of the Cell 16, 5236–5246 (2005).
Leidich, S. D. & Orlean, P. Gpi1, a Saccharomyces cerevisiae protein that participates in the first step in glycosylphosphatidylinositol anchor synthesis. J. Biol. Chem. 271, 27829–27837 (1996).
Sobering, A. K., Romeo, M. J., Vay, H. A. & Levin, D. E. A novel Ras inhibitor, Eri1, engages yeast Ras at the endoplasmic reticulum. Mol. Cell. Biol. 23, 4983–4990 (2003).
Martinez-Lopez, R., Monteoliva, L., Diez-Orejas, R., Nombela, C. & Gil, C. The GPI-anchored protein CaEcm33p is required for cell wall integrity, morphogenesis and virulence in Candida albicans. Microbiology 150, 3341–3354 (2004).
Richard, M. et al. Complete glycosylphosphatidylinositol anchors are required in Candida albicans for full morphogenesis, virulence and resistance to macrophages. Mol. Microbiol. 44, 841–853 (2002).
Grimme, S. J., Colussi, P. A., Taron, C. H. & Orlean, P. Deficiencies in the essential Smp3 mannosyltransferase block glycosylphosphatidylinositol assembly and lead to defects in growth and cell wall biogenesis in Candida albicans. Microbiology 150, 3115–3128 (2004).
Richard, M. et al. GPI7 affects cell-wall protein anchorage in Saccharomyces cerevisiae and Candida albicans. Microbiology 148, 2125–2133 (2002).
Victoria, G. S., Kumar, P. & Komath, S. S. The Candida albicans homologue of PIG-P, CaGpi19p: gene dosage and role in growth and filamentation. Microbiology 156, 3041–3051 (2010).
Victoria, G. S. et al. Mutual co-regulation between GPI-N-acetylglucosaminyltransferase and ergosterol biosynthesis in Candida albicans. Biochem. J. 443, 619–625 (2012).
Yadav, B. et al. First step of glycosylphosphatidylinositol (GPI) biosynthesis cross-talks with ergosterol biosynthesis and Ras signaling in Candida albicans. J. Biol. Chem. 289, 3365–3382 (2014).
Yan, B. C., Westfall, B. A. & Orlean, P. Ynl038wp (Gpi15p) is the Saccharomyces cerevisiae homologue of human Pig-Hp and participates in the first step in glycosylphosphatidylinositol assembly. Yeast 18, 1383–1389 (2001).
Wilson, R. B., Davis, D. & Mitchell, A. P. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J. Bacteriol. 181, 1868–1874 (1999).
Gerami-Nejad, M., Hausauer, D., McClellan, M., Berman, J. & Gale, C. Cassettes for the PCR-mediated construction of regulatable alleles in Candida albicans. Yeast 21, 429–436 (2004).
Jain, P. et al. Ras signaling activates glycosylphosphatidylinositol (GPI) anchor biosynthesis via the GPI-N-acetylglucosaminyltransferase (GPI-GnT) in Candida albicans. J. Biol. Chem. 293, 12222–12238 (2018).
Brand, A., MacCallum, D. M., Brown, A. J. P., Gow, N. A. R. & Odds, F. C. Ectopic expression of URA3 can influence the virulence phenotypes and proteome of Candida albicans but can be overcome by targeted reintegration of URA3 at the RPS10 locus. Eukaryotic Cell 3, 900–909 (2004).
Yadav, U. et al. Characterising N-acetylglucosaminylphosphatidylinositol de-N-acetylase (CaGpi12), the enzyme that catalyses the second step of GPI biosynthesis in Candida albicans. FEMS Yeast Res. 18 (2018).
Feng, Q., Summers, E., Guo, B. & Fink, G. Ras signaling is required for serum-induced hyphal differentiation in Candida albicans. J. Bacteriol. 181, 6339–6346 (1999).
Zhu, Y. et al. Ras1 and Ras2 play antagonistic roles in regulating cellular cAMP level, stationary-phase entry and stress response in Candida albicans. Mol. Microbiol. 74, 862–875 (2009).
Ghannoum, M. A. & Rice, L. B. Antifungal agents: mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin. Microbiol. Rev. 12, 501–517 (1999).
Dunkel, N. et al. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryotic Cell 7, 1180–1190 (2008).
Wurtele, H. et al. Modulation of histone H3 lysine 56 acetylation as an antifungal therapeutic strategy. Nat. Med. 16, 774–780 (2010).
Radovani, E. et al. The carboxyl terminus of Rtt109 functions in chaperone control of histone acetylation. Eukaryotic Cell 12, 654–664 (2013).
Hammond, C. M. et al. The histone chaperone Vps75 forms multiple oligomeric assemblies capable of mediating exchange between histone H3-H4 tetramers and Asf1-H3-H4 complexes. Nucleic Acids Res. 44, 6157–6172 (2016).
Naglik, J. R., König, A., Hube, B. & Gaffen, S. L. Candida albicans-epithelial interactions and induction of mucosal innate immunity. Curr. Opin. Microbiol. 40, 104–112 (2017).
Sütterlin, C. et al. Identification of a species-specific inhibitor of glycosylphosphatidylinositol synthesis. EMBO J. 16, 6374–6383 (1997).
Smith, T. K. et al. Specificity of GlcNAc-PI de-N-acetylase of GPI biosynthesis and synthesis of parasite-specific suicide substrate inhibitors. EMBO J. 20, 3322–3332 (2001).
Sobering, A. K. et al. Yeast Ras regulates the complex that catalyzes the first step in GPI-anchor biosynthesis at the ER. Cell 117, 637–648 (2004).
Watanabe, R. et al. Initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-P and is regulated by DPM2. EMBO J. 19, 4402–4411 (2000).
Shapiro, R. S., Zaas, A. K., Betancourt-Quiroz, M., Perfect, J. R. & Cowen, L. E. The Hsp90 co-chaperone Sgt1 governs Candida albicans morphogenesis and drug resistance. PLoS ONE 7, e44734 (2012).
Pfaller, M. A. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am. J. Med. 125, S3–13 (2012).
Walther, A. & Wendland, J. An improved transformation protocol for the human fungal pathogen Candida albicans. Curr. Genet. 42, 339–343 (2003).
Yadav, A., Singh, S. L., Yadav, B. & Komath, S. S. Saccharomyces cerevisiae Gpi2, an accessory subunit of the enzyme catalyzing the first step of glycosylphosphatidylinositol (GPI) anchor biosynthesis, selectively complements some of the functions of its homolog in Candida albicans. Glycoconj. J. 31, 497–507 (2014).
Figler, R. A., Omote, H., Nakamoto, R. K. & Al-Shawi, M. K. Use of chemical chaperones in the yeast Saccharomyces cerevisiae to enhance heterologous membrane protein expression: high-yield expression and purification of human P-glycoprotein. Arch. Biochem. Biophys. 376, 34–46 (2000).
Ashraf, M. et al. N-acetyl-D-glucosaminylphosphatidylinositol de-N-acetylase from Entamoeba histolytica: metal alters catalytic rates but not substrate affinity. J. Biol. Chem. 286, 2543–2549 (2011).
Wong, M. L. & Medrano, J. F. Real-time PCR for mRNA quantitation. BioTechniques 39, 75–85 (2005).
Hohn, C., Lee, S.-R., Pinchuk, L. M. & Petrie-Hanson, L. Zebrafish Kidney Phagocytes Utilize Macropinocytosis and Ca2+-Dependent Endocytic Mechanisms. PLoS ONE 4, e4314 (2009).
Rayamajhi, M., Zhang, Y. & Miao, E. A. Detection of pyroptosis by measuring released lactate dehydrogenase activity. Methods Mol. Biol. 1040, 85–90 (2013).
Naqvi, N. et al. Role of Mast Cells in clearance of Leishmania through extracellular trap formation. Sci Rep 7, 13240 (2017).
Sikorski, R. S. & Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 (1989).
Acknowledgements
SSK received funding from Department of Biotechnology (DBT) India (BT/PR3749/BRB/10/986/2011), Department of Science and Technology (DST) India (SB/SO/BB-011/2014) as well as from an umbrella grant to the School of Life Sciences from the Department of Science and Technology under Promotion of University Research and Scientific Excellence (DST-PURSE No. SR/PURSE/PHASE 2/11 (C)). PJ, PG, SCS, SLS, BY, PK, UY, NN and R were supported by Senior and Junior Research fellowships from Council of Scientific and Industrial Research or University Grants Commission or Indian Council of Medical Research, India. Two of the strains, CaGPI15Hz and Cagpi15 null were generated by Kalpana Pawar while being employed in a funded research project of SSK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Dr. Aaron P. Mitchell for the BWP17 strain, Dr. Marwan Al-Shawi for the YEpHIS vector, Dr. W.A. Fonzi for pMB7 vector, Prof. P.E. Sudbery for pCaDis vector, Dr. Cheryl Gale for URA3-pMET3-GFP vector and Prof. Alistair JP Brown for pACT1-GFP vector. We thank Dr. Ajai Kumar for the GC-MS runs. The GC-MS data was recorded at the Advanced Instrumentation Research Facility, JNU. Fluorescence microscopy images were recorded at the Central Instrumentation Facility, SLS, JNU.
Author information
Authors and Affiliations
Contributions
P.J., P.G., S.C.S., N.N., B.Y., P.K., S.L.S., U.Y., S.B., R. executed the experiments. S.S.K., R.M., P.J. and P.G. planned the experiments and data analysis. S.S.K., P.J., P.G. wrote the manuscript with inputs from R.M. and N.P. All authors gave final approval for publication.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jain, P., Garai, P., Sethi, S.C. et al. Modulation of azole sensitivity and filamentation by GPI15, encoding a subunit of the first GPI biosynthetic enzyme, in Candida albicans. Sci Rep 9, 8508 (2019). https://doi.org/10.1038/s41598-019-44919-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44919-4
- Springer Nature Limited