
Abstract
Layered Li2RuO3 is an important candidate cathode material in rechargeable lithium ion batteries because of its novel anionic redox process and high reversible capacity. Atomistic scale simulations are used to calculate the intrinsic defect process, favourable dopants and migration energies of lithium ion diffusions together with migration paths in Li2RuO3. The Li Frenkel is calculated to be the most favourable intrinsic defect type. The cation anti-site defect, in which Li and Ru ions exchange their positions is 1.89 eV/defect suggesting that this defect would be observed at high temperatures. Long range vacancy assisted lithium diffusion paths were calculated and it is confirmed that the lowest overall activation energy (0.73 eV) migration path is along the ab plane. Trivalent dopants (Al3+, Co3+, Sc3+, In3+, Y3+, Gd3+ and La3+) were considered to create additional Li in Li2RuO3. Here we show that Al3+ or Co3+ are the ideal dopants and this is in agreement with the experimental studies reported on Co3+ doping in Li2RuO3.
Similar content being viewed by others

Introduction
High energy storage systems needed for the development of electronic vehicles and consumer electronics require high-capacity lithium ion battery cathode materials1,2,3,4,5. The development of such materials has many challenges such as materials being safe, with low cost and high abundance. A variety of new cathode materials6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22 have been studied both experimentally and theoretically though a few of them have been identified as promising. There is a continuous research activity by considering those challenges to find new cathode materials to improve the power density in Li ion batteries.
“Layered” Li2RuO3 has attracted attention because of its novel anionic redox process23. Reversible oxygen redox process is a key feature in Li2RuO3 and enhances the capacity of this material23. Experimental studies24,25,26 demonstrate that extraction of both lithium is possible but one of them can be repeatedly cycleable. Moore et al.24 studied the electrochemical properties of Li2RuO3 and concluded that there are two working plateaus in the first charging process providing a reversible capacity of approximately 270 mAhg−1. A novel hybrid Na+/Li+ battery has been recently made using Li2RuO3 as a cathode material because of its unique structure accommodating both Li+ and Na+ ions27. Li2RuO3 was suggested as an additive to provide high energy lithium-ion capacitors due to its high reversible characteristics for Li+ ion intercalation/de-intercalation and structural stability28. Recently, Arunkumar et al.29 synthesized over-lithiated Li2+xRu1−xCoO3 cathode by aliovalent Co doping on Ru site in Li2RuO3 and concluded that there is an enhancement in the electrochemical lithium reversibility and Li+ extraction compared to those associated in the pristine Li2RuO3.
Electrochemical behaviour of an electrode material by studying its defect properties is important to assess its applicabilty in batteries. Computational modelling can provide useful information of the key issues related to defect processes including cation mixing and doping strategies to increase the Li concentration in this material. In a vast range of oxides including these Li-based systems classical pair potentials do capture the trends and energetics of the defect processes in excellent agreement with experiment30,31,32. For example, the lithium ion migration path calculated in LiFePO4 using classical pair potentials33 was exactly observed later in the neutron diffraction experiment34. Here, we extend our recent simulation studies of the Li5FeO418, Li2CuO222, Li9V3(P2O7)3(PO4)235 and Li2SnO336 electrode materials where we investigated the defects, lithium ion diffusion and dopants. In this study, we have systematically studied the relative energetics of the formation of intrinsic defects, the solution of trivalent dopants (Al3+, Co3+, Sc3+, In3+, Y3+, Gd3+ and La3+), and the possible lithium ion migration pathways in Li2RuO3.
Results and Discussion
Li2RuO3 structure
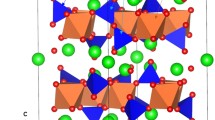
Li2RuO3 is a layered structure and has a monoclinic symmetry with space group C2/c. Its experimental lattice parameters (a = 4.9230 Å, b = 8.7746 Å, c = 9.8776 Å, α = 90°, β = 100.073° and γ = 90°) was reported by Kobayashi et al.26. Figure 1 exhibits this structure, the coordination environments of Ru and Li (both forming octahedrons with six O atoms) and layers in the ab plane with an A-B stacking sequence (P2 type) as classified by Delmas et al.37. First, experimentally observed monoclinic crystal structure was reproduced to assess the quality of the classical pair potentials (potentials parameters are reported in Table S1 in the supplementary information) used in this study. There is a good agreement between the calculated equilibrium lattice constants (tabulated in Table 1) and the experimental values.

Crystal structure of Li2RuO3 (space group C2/c).
Intrinsic defect processes
As defect properties of an electrode material is important to understand its electrochemical behavior, we calculated a series of isolated point defect (vacancy, anti-site and interstitial) energies. Frenkel, Schottky and anti-site defect formation energies were then calculated by combining the isolated point defects. Here we use Kröger-Vink notation38 to represent the reactions involving these defects.
Figure 2 reports the reaction energies for these intrinsic defect processes. The most favorable intrinsic disorder is found to be the Li Frenkel. Formation of other Frenkel and Schottky defects is thermodynamically unfavorable. The second most favorable defect process is the anti-site suggesting that there will be a small percentage of Li on Ru sites \({(\mathrm{Li}}_{{\rm{Ru}}}^{{\prime} {\prime} {\prime} })\) and Ru on Li sites \({(\mathrm{Ru}}_{{\rm{Li}}}^{\bullet \bullet \bullet })\). However, this defect would not be observed at operating temperatures. This defect has been observed experimentally and theoretically in a variety of Li ion battery materials6,9,39,40,41,42. The formation of other Frenkels (Ru and O) and Schottky defects is found to be unfavorable.

Energetics of intrinsic defect process in monoclinic Li2RuO3.
Lithium ion-diffusion
Diffusion of lithium ion diffusion with lower activation energy is a key requirement for a promising cathode materials in lithium ion batteries. Static atomistic simulation allows us to examine various possible Li ion migration paths that are in general diffucly by experimetanl studies alone. For the Li vacancy migration, we have calculated six different local Li hops (refer to Fig. 3). Table 2 reports the migration energies together with the Li-Li separation, whereas energy profile diagrams are shown in Fig. 4. We have constructed long range paths connecting local Li hops with lower overall activation energy. We have identified five long range paths (refer to Fig. 3). The first long range path exhibits a linear path (A → B → A → B) along b axis consisting of a local Li hop with lower activation energy of migration of 0.65 eV (local hop B) but with overall activation energy of 0.76 eV (refer to Table 3). The second path exhibits a zig-zag pattern (C → C → C → C) with an activation energy of 1.09 eV. Both the third and fourth migration paths [D → E → D → E and E → E → E→E] exhibit a linear path along ab plane with the lowest activation energy of 0.73 eV. The fifth migration path (F → F → F → F) is constructed along ac plane and its activation energy is calculated to be 1.13 eV. Here we define the highest potential energy along the migration path as the activation energy.

Possible long range lithium vacancy migration paths considered. Green, grey and red colors correspond to Li, Ru, and O atoms respectively.

Six different energy profiles [as shown in Fig. 3] of Li vacancy hopping between two adjacent Li sites in Li2RuO3.
Trivalent doping
The capacity of a cathode material can be increased by incorporating additional lithium into the as-prepared material. An efficient way to increase the amount of lithium is by doping trivalent cations on Ru site through creating Li interstitials. The experimental study29 on Co3+ doping on Ru site reveals that the resultant Li2.1Ru0.9Co0.1O3 exhibits significant reversible Li+ extraction compared to undoped Li2RuO3. Similar approach has been previously demonstrated computationally in Li2MnSiO4 cathode material12. Here we considered the solution of \({R}_{2}{O}_{3}\) (R = Al, Co, Sc, In, Y, Gd and La) via the following process (in Kröger-Vink notation):
The solution energies of \({R}_{2}{O}_{3}\,\,\)are reported in Fig. 5. The most favorable dopant is found to be Al3+. The solution energy for Co2O3 is higher in energy by only 0.05 eV suggesting that Co3+ is also a candidate dopant to increase the Li concentration in Li2RuO3. Our calculation confirms the experimental investigation29 reported for Co3+ doping and suggests that Al3+ is also a promising dopant for the formation extra lithium into Li2RuO3. The exact composition of the Al incorporated structure should be investigated experimentally. The calculated solution energies are positive for Al2O3 and Co2O3 suggesting that doping can be carried out only at high temperatures. This is further supported by the higher temperature (~1100 °C for 12 hours) used for the synthesis of Co-doped Li2RuO3.

Enthalpy of solution of \({{\rm{R}}}_{2}{{\rm{O}}}_{3}\) (R = Al, Co, Sc, In, Y, Gd and La) with respect to the R3+ ionic radius in Li2RuO3.
A detail information regarding the bond lengths and bond angles of dopant and Ru in the relaxed structure of undoped Li2RuO3 with adjacent oxygens is reported in Fig. 6. The ionic radius of Ru4+ in octahedral environment is 0.62 Å, larger by 0.08 Å than that of Al3+. In the AlO6 unit, all six Al-O bonds are slightly shorter compared to the Ru-O bonds present in the undoped Li2RuO3. This is due to its smaller cation size of Al3+ which strongly polarises the oxygen ions forming strong ionic bonds with O atoms. The ionic radius of Co3+ (0.55 Å) is very close to that of Al3+. This is reflected in the bond lenghts and bond angles. From Sc to La, dopant-oxygen bond distances increase and bond angles decrease gradually indicating the structural distortion and reflecting in the solution enthalpies. The LaO6 unit exhibits approximately the same La-O bond length, but longer by ~0.30 Å than Ru-O bond length present in RuO6. The ionic radius of La3+ is larger by 0.28 Å than that of Ru4+. This reflects in the extremely high solution enthalpy.

Octahedral RuO6 unit in the relaxed structure of undoped Li2RuO3 and the coordination formed by the dopants on the Ru site with neighbor oxygen.
Density of states
Density Functional Theory (DFT) was used to analyze the electronic structure for trivalent doping processes. In particular, the contribution of every element in the Li2RuO3 crystal is visualized through the partial Density of States (PDOS). Figure 7 presents the cases for the (a) Non – defective structure (b) The Li interstitial (c) The Al – doped supercell (d) The Al – doped supercell with one Li interstitial. Overall, the material presents the formation of the valence band governed by the strong O2− p-states at the zero-shifted Fermi level (refer to figures reported in supplementary information). Additionally, the conduction band is characterized by the major contribution of Ru d-states leading to a narrow band gap of approximately 0.2 eV in agreement with other theoretical studies43,44 (refer to Fig. 1 of the SI for the exact contribution of each orbital separately). We point at the presence of in gap states mainly attributed to the Ru4+ d-states and O2− p-states. This should be addressed and experimentally investigated as the properties of electronic conduction have to be controlled for future energy applications. The last in-gap contribution is located at 1.66 from the valence band, however this non-uniformity points to an interesting behavior that originates from electronic configuration parameters and must be considered for the oxidation and reduction reactions. The presence of a Li interstitial does not affect the total DOS in a considerable way. Doping Li2RuO3 with trivalent dopants that substitute the Ru4+ site introduces additional contributions in the electronic structure. Regarding the lowest solution enthalpies, we focus on the Al3+ and Co3+ elements. The dopant is initially introduced as substitutional in a Ru4+ site presenting minor distortions in the crystal even combined with a Li+ ion in an interstitial site. Al3+ doping presents a low contribution at the conduction band with no additional states. However, additional contribution is observed for Co – doping, governed by the Co3+ d – states and O2− p-states whereas a weaker contribution due to the Ru4+ d-states is also observed (Refer to SI for the orbitals profile). Furthermore, doping with elements of bigger radius introduce intense states in the electronic structure except for In3+ (Refer to SI for the extra doping processes considered).

Li2RuO3 PDOS for (a) The non – defective cell (d) The Li interstitial (c) The Al – doped cell (d) The Al – doped cell with one Li interstitial (e) The Co – doped cell (f) The Co – doped cell with one Li interstitial.
Summary
Classical pair potential simulations were employed to provide relevant information about favourable intrinsic disorder, Li diffusion paths together with activation energies and possible dopants that can be substituted on Ru site to introduce additional Li in the layered Li2RuO3. There is a good agreement between the calculated and experimental lattice parameters of Li2RuO3. The Li Frenkel is the lowest energy and thus the dominant defect energy process. Anti-site disorder is calculated to be 1.89 eV/defect suggesting that a small concentration of cation mixing would be observed at high temperatures. The long range Li ion migration path with lowest activation energy (0.73 eV) is found to be along the ab plane. Solution energies of \({R}_{2}{O}_{3}\) (R = Al, Co, Sc, In, Y, Gd and La) were considered to create extra lithium in this material and found that Al2O3 or Co2O3 would be ideal candidates and this is in agreement with the experimental result reported for Co substitution in Li2RuO3. This interesting study stimulates further experimental work on Al doping.
Methods
Intrinsic defect formation energies and Li migration paths were calculated using GULP code45. This method is based on the classical pair potentials. Ionic crystal lattice is described using Born model and consists of the long-range attractions and short-range repulsive forces in the form of electron-electron repulsion and van der Waals interactions. Buckingham potentials (refer to Table S1) were used to model the short range interactions. Structural optimizations were performed using the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm46. Relaxation around point defects and the diffusing ions were modelled using the Mott-Littleton method47. This method has been well explained in our previous publications18,22. Vacancy assisted Li ion migration was calculated considering seven interstitial Li ions between local Li hops. Activation energy reported in this study is the local maximum energy along the diffusion path. The present calculation is based on the full ionic charge model within the dilute limit. Therefore, the defect energies will be overestimated, however, the relative energies, and the trends will be consistent.
The electronic structure of Li2RuO3 is investigated through the plane wave DFT code CASTEP48,49. We model the perfect and defective supercells with the plane wave basis set with a cut-off energy of 450 eV using a 2 × 2 × 2 Monkhorst-Pack (MP)50 k-point grid within a 96-atomic site supercell. The crystallographic configurations have been initially optimized to the energetically favorable ground state under constant pressure conditions. The exchange and correlation term was modelled using the generalized gradient approximation (GGA) parameterized by Perdew, Burke and Ernzerhof (PBE)51 with the use of ultrasoft pseudopotentials52. The atomic configurations for the doped/undoped and defective supercells were relaxed to the minimum energy for the electronic structure calculations. For the Density of States (DOS) investigation and visualization, we employ the OPTADOS53,54 subcode using a 10 × 10 × 10 k-point grid.
References
Armand, M. & Tarascon, J. M. Building better batteries. Nature 451, 652 (2008).
Winter, M. & Brodd, R. J. What Are Batteries, Fuel Cells, and Supercapacitors? Chem. Rev. 104, 4245–4270 (2004).
Scrosati, B. & Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 195, 2419–2430 (2010).
Simon, P. & Gogotsi, Y. Materials for electrochemical capacitors. Nat. Mater. 7, 845 (2008).
Goodenough, J. B. Rechargeable batteries: challenges old and new. J. Solid State Electrochem 16, 2019–2029 (2012).
Nytén, A., Abouimrane, A., Armand, M., Gustafsson, T. & Thomas, J. O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochem. Commun. 7, 156–160 (2005).
Nishimura, S.-I. et al. Structure of Li2FeSiO4. J. Am. Chem. Soc. 130, 13212–13213 (2008).
Armstrong, A. R., Kuganathan, N., Islam, M. S. & Bruce, P. G. Structure and lithium transport pathways in Li2FeSiO4 Cathodes for Lithium Batteries. J. Am. Chem. Soc. 133, 13031–13035 (2011).
Politaev, V. V., Petrenko, A. A., Nalbandyan, V. B., Medvedev, B. S. & Shvetsova, E. S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 180, 1045–1050 (2007).
Dominko, R. et al. Structure and electrochemical performance of Li2MnSiO4 and Li2FeSiO4 as potential Li-battery cathode materials. Electrochem. Commun. 8, 217–222 (2006).
Masquelier, C. & Croguennec, L. Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chem. Rev 113, 6552–6591 (2013).
Kuganathan, N. & Islam, M. S. Li2MnSiO4 Lithium Battery Material: Atomic-Scale Study of Defects, Lithium Mobility, and Trivalent Dopants. Chem. Mater. 21, 5196–5202 (2009).
Fisher, C. A. J., Kuganathan, N. & Islam, M. S. Defect chemistry and lithium-ion migration in polymorphs of the cathode material Li2MnSiO4. J. Mater. Chem. A 1, 4207–4214 (2013).
Ellis, B. L., Makahnouk, W. R. M., Makimura, Y., Toghill, K. & Nazar, L. F. A multifunctional 3.5 V iron-based phosphate cathode for rechargeable batteries. Nat. Mater. 6, 749 (2007).
Nishimura, S.-I., Nakamura, M., Natsui, R. & Yamada, A. New lithium iron pyrophosphate as 3.5 V class cathode material for lithium ion battery. J. Am. Chem. Soc. 132, 13596–13597 (2010).
Okumura, T., Shikano, M. & Kobayashi, H. Effect of bulk and surface structural changes in Li5FeO4 positive electrodes during first charging on subsequent lithium-ion battery performance. J. Mater. Chem. A 2, 11847–11856 (2014).
Johnson, C. S. et al. Li2O Removal from Li5FeO4: A cathode precursor for lithium-ion batteries. Chem. Mater. 22, 1263–1270 (2010).
Kuganathan, N., Iyngaran, P. & Chroneos, A. Lithium diffusion in Li5FeO4. Sci. Rep. 8, 5832 (2018).
Afyon, S., Wörle, M. & Nesper, R. A. Lithium-rich compound Li7Mn(BO3)3 containing Mn2+ in tetrahedral coordination: A cathode candidate for lithium-ion batteries. Angew. Chemie Inter. Ed. 52, 12541–12544 (2013).
Wang, J. et al. Li3V(MoO4)3 as a novel electrode material with good lithium storage properties and improved initial coulombic efficiency. Nano Energy 44, 272–278 (2018).
Ramos-Sanchez, G. et al. Controlling Li2CuO2 single phase transition to preserve cathode capacity and cycleability in Li-ion batteries. Solid State Ionics 303, 89–96 (2017).
Kordatos, A., Kuganathan, N., Kelaidis, N., Iyngaran, P. & Chroneos, A. Defects and lithium migration in Li2CuO2. Sci. Rep 8, 6754 (2018).
Sathiya, M. et al. Reversible anionic redox chemistry in high-capacity layered-oxide electrodes. Nat. Mater. 12, 827 (2013).
Moore, G. J., Johnson, C. S. & Thackeray, M. M. The electrochemical behavior of xLiNiO2·(1 − x)Li2RuO3 and Li2Ru1−yZryO3 electrodes in lithium cells. J. Power Sources 119–121, 216–220 (2003).
James, A. C. W. P. & Goodenough, J. B. Structure and bonding in lithium ruthenate, Li2RuO3. J. Solid State Chem 74, 287–294 (1988).
Kobayashi, H. et al. Structure and lithium deintercalation of Li2-xRuO3. Solid State Ionics 82, 25–31, (95)00135-S (1995).
Yao, Y. et al. High capacity and rate capability of a layered Li2RuO3 cathode utilized in hybrid Na+/Li+ batteries. J. Mater. Chem. A 3, 18273–18278 (2015).
Park, M.-S. et al. Li2RuO3 as an Additive for High-Energy Lithium-Ion Capacitors. J. Phys. Chem. C 117, 11471–11478 (2013).
Arunkumar, P., Jeong, W. J., Won, S. & Im, W. B. Improved electrochemical reversibility of over-lithiated layered Li2RuO3 cathodes: Understanding aliovalent Co3+ substitution with excess lithium. J. Power Sources 324, 428–438 (2016).
Jay, E. E., Rushton, M. J. D., Chroneos, A., Grimes, R. W. & Kilner, J. A. Genetics of superionic conductivity in lithium lanthanum titanates. Phys,Chem.Chem.Phys 17, 178–183 (2015).
Islam, M. S., Driscoll, D. J., Fisher, C. A. J. & Slater, P. R. Atomic-Scale Investigation of Defects, Dopants, and Lithium Transport in the LiFePO4 Olivine-Type Battery Material. Chem.Mater 17, 5085–5092 (2005).
Parfitt, D., Chroneos, A., Tarancón, A. & Kilner, J. A. Oxygen ion diffusion in cation ordered/disordered GdBaCo2O5+δ. J. Mater. Chem 21, 2183–2186 (2011).
Fisher, C. A. J., Hart Prieto, V. M. & Islam, M. S. Lithium Battery Materials LiMPO4 (M = Mn, Fe, Co, and Ni): Insights into Defect Association, Transport Mechanisms, and Doping Behavior. Chem.Mater 20, 5907–5915 (2008).
Nishimura, S.-i et al. Experimental visualization of lithium diffusion in LixFePO4. Nature Materials 7, 707 (2008).
Kuganathan, N., Ganeshalingam, S. & Chroneos, A. Defects, Dopants and Lithium Mobility in Li9V3(P2O7)3(PO4)2. Sci. Rep. 8, 8140 (2018).
Kuganathan, N., Kordatos, A. & Chroneos, A. Li2SnO3 as a Cathode Material for Lithium-ion Batteries: Defects, Lithium Ion Diffusion and Dopants. Sci. Rep. 8, 12621 (2018).
Delmas, C., Fouassier, C. & Hagenmuller, P. Structural classification and properties of the layered oxides. Physica B + C 99, 81–85 (80)90214-4 (1980).
Kröger, F. A. & Vink, H. J. In Solid State Physics Vol. 3 (eds Frederick Seitz & David Turnbull) 307–435 (Academic Press 1956).
Nyten, A., Kamali, S., Haggstrom, L., Gustafsson, T. & Thomas, J. O. The lithium extraction/insertion mechanism in Li2FeSiO4. J. Mater. Chem. 16, 2266–2272 (2006).
Ensling, D., Stjerndahl, M., Nyten, A., Gustafsson, T. & Thomas, J. O. A comparative XPS surface study of Li2FeSiO4/C cycled with LiTFSI- and LiPF6-based electrolytes. J. Mater. Chem. 19, 82–88 (2009).
Liu, H. et al. Effects of Antisite Defects on Li Diffusion in LiFePO4 Revealed by Li Isotope Exchange. J. Phys. Chem C 121, 12025–12036 (2017).
Kempaiah Devaraju, M., Duc Truong, Q., Hyodo, H., Sasaki, Y. & Honma, I. Synthesis, characterization and observation of antisite defects in LiNiPO4 nanomaterials. Sci. Rep. 5, 11041 (2015).
Johannes, M. D., Stux, A. M. & Swider-Lyons, K. E. Electronic structure and properties of Li-insertion materials: Li2RuO3 and RuO2. Phys. Rev B 77, 075124 (2008).
Foix, D., Sathiya, M., McCalla, E., Tarascon, J.-M. & Gonbeau, D. X-ray Photoemission Spectroscopy Study of Cationic and Anionic Redox Processes in High-Capacity Li-Ion Battery Layered-Oxide Electrodes. J. Phys. Chem. C 120, 862–874 (2016).
Gale, J. D. & Rohl, A. L. The General Utility Lattice Program (GULP). Molec. Simul. 29, 291–341 (2003).
Gale, J. D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 93, 629–637 (1997).
Mott, N. F. & Littleton, M. J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 34, 485–499 (1938).
Payne, M. C., Teter, M. P., Allan, D. C., Arias, T. A. & Joannopoulos, J. D. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev. Mod. Phys. 64, 1045 (1992).
Segall, M. D. et al. First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 14, 2717 (2002).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892 (1990).
Nicholls, R. J., Morris, A. J., Pickard, C. J. & Yates, J. R. OptaDOS - a new tool for EELS calculations. J. Phys.: Conf. Ser. 371, 012062 (2012).
Morris, A. J., Nicholls, R., Pickard, C. J. & Yates, J. R. OptaDOS: A tool for obtaining density of states, core-level and optical spectra from electronic structure codes. Comp. Phys. Comm. 185, 1477–1485 (2014).
Acknowledgements
We acknowledge High Performance Computing Centers at Imperial College London and Coventry University for providing computational facilities and support.
Author information
Authors and Affiliations
Contributions
N.K. and A.K. performed classical and DFT calculations respectively. All authors discussed the results and contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kuganathan, N., Kordatos, A. & Chroneos, A. Defect Chemistry and Li-ion Diffusion in Li2RuO3. Sci Rep 9, 550 (2019). https://doi.org/10.1038/s41598-018-36865-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36865-4
- Springer Nature Limited
This article is cited by
-
Defects, dopants and Mg diffusion in MgTiO3
Scientific Reports (2019)