
Abstract
Li2CuO2 is an important candidate material as a cathode in lithium ion batteries. Atomistic simulation methods are used to investigate the defect processes, electronic structure and lithium migration mechanisms in Li2CuO2. Here we show that the lithium energy of migration via the vacancy mechanism is very low, at 0.11 eV. The high lithium Frenkel energy (1.88 eV/defect) prompted the consideration of defect engineering strategies in order to increase the concentration of lithium vacancies that act as vehicles for the vacancy mediated lithium self-diffusion in Li2CuO2. It is shown that aluminium doping will significantly reduce the energy required to form a lithium vacancy from 1.88 eV to 0.97 eV for every aluminium introduced, however, it will also increase the migration energy barrier of lithium in the vicinity of the aluminium dopant to 0.22 eV. Still, the introduction of aluminium is favourable compared to the lithium Frenkel process. Other trivalent dopants considered herein require significantly higher solution energies, whereas their impact on the migration energy barrier was more pronounced. When considering the electronic structure of defective Li2CuO2, the presence of aluminium dopants results in the introduction of electronic states into the energy band gap. Therefore, doping with aluminium is an effective doping strategy to increase the concentration of lithium vacancies, with a minimal impact on the kinetics.
Similar content being viewed by others

Introduction
The requirement for solid-state lithium batteries with better capacity, safety, cycle performance and durability have generated interest to materials with high lithium ion conductivity1,2,3,4,5,6,7,8,9,10,11,12. Oxide materials are actively being considered for energy applications and defect engineering is an effective way to improve their properties and performance13,14.
Li2CuO2 is an attractive material with many interesting characteristics, already considered for CO2 chemisorption15,16. In battery applications, a phase transition during the first circle of charge/discharge is confirmed and attributed to oxygen loss and delithiation. Recently, Ramos-Sanchez et al.17, have reported on this phase transition control and proposed Li2CuO2 for future applications due to its high capacity in pre-charge conditions as well as fast Li+ mobility. The partial pressure chemisorption has been also investigated by Lara-Garcia and Pfeiffer18. In essense, it was found that for low pressures the material’s behavior is not affected while increase of pressure combined with additional oxygen source enhances the CO2 chemisorption18. Furthermore, The Li+ and O2- ion-diffusion can also affect the CO2 chemisorption by means of an extra layer that forms on the surface which limits the process as the CO2 cannot easily reach the bulk material.
In previous studies, the electronic structure of Li2CuO2 has been investigated using the local density approximation (LDA)19, LDA—LCAO20 and the generalized gradient approximation (GGA)21, however, there is lack of clarity in the literature for the exact contributions of each element (and their orbitals) in the valence and conduction band. Ramos-Sanchez et al.17 report on the band gap formation with higher oxygen states close to the Fermi level. To further examine the electronic structure and the defect processes of Li2CuO2, atomistic simulations are required. These can identify the defect engineering processes which are able to lead to the improvement of the material properties and accelerate progress22,23,24,25.
In the present study, we investigate the intrinsic defect processes, electronic structure and lithium vacancy self-diffusion in Li2CuO2 using static atomistic simulations and density functional theory (DFT).
Results and Discussion
Li2CuO2 structure
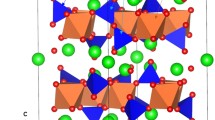
Figure 1(a) presents the crystallographic structure of Li2CuO2. It is characterized by the body centered orthorombic structure of the Immm space group (lattice parameters a = 3.6615 Å, b = 2.7887 Å and c = 9.5734 Å) as reported by Sapiña et al.26 Four O−2 atoms reside around each Cu+2 atom for the bc-plane while the square-shaped CuO2 present a shared edge on the b-axis. The interatomic potentials used in this study accurately reproduce the experimental lattice parameters, angles and bond lengths within an error margin less than 2.6% (refer to Tables S1 and S2 in the Supplementary Information).

(a) Crystal structure of Li2CuO2 (space group Immm) and (b) the possible lithium ion migration paths considered. Li, Cu, and O atoms are shown as green, blue and red spheres respectively.
Intrinsic defect processes
The study of the intrinsic defect processes of energy materials is important in order to understand their electrochemical behavior and their propensity to form intrinsic defects. The intrinsic defect reactions in Kröger-Vink notation are:
The reaction energies for these intrinsic defect processes are reported in Table 1. Typically to most oxides, the formation of all Frenkel and Schottky defects is unfavourable and this suggests that the formation of vacancies and interstitial defects will be hindered at equilibrium conditions. Therefore, these intrinsic defects will be present only at low concentrations in undoped Li2CuO2. The lithium Frenkel is a lower energy process compared to the other Frenkel and Schottky processes considered. The formation enthalpy of Li2O via the Li2O Schottky-like reaction (relation 5) is a processes that requires an energy of 2.57 eV per defect (refer to Table 1). This is a process that can lead to further \(\,{V}_{Li}^{\text{'}}\) and \({V}_{O}^{\bullet \bullet }\) however at elevated temperatures far from room temperature. We also considered the formation of antisite defects (\(L{i}_{Cu}^{\text{'}}\,{\rm{and}}\,C{u}_{Li}^{\bullet }\)) both when the defects are apart (isolated form, relation 6) and when they form a defect pair (cluster, relation 7). For the isolated case, the defect energies of the two antisites were calculated independently in order to calculate the energy of the defect process applying relation 6 (i.e. the effect of defect association is not included), which is 0.88 eV per defect (refer to Table 1). For the antisite pair (relation 7) the energy per defect drops considerably (0.31 eV per defect, refer to Table 1) mainly due to the binding of the oppositively charged defects and relaxation effects.
Lithium self-diffusion
The present static atomistic simulation enabled the examination of possible Li vacancy migration paths and calculated the corresponding activation energies of migration. For the Li vacancy migration, we identified three paths between adjacent Li sites (refer to Fig. 1(b)). The zig-zag diffusion path (path A in Fig. 1(b)) in the ab-plane is the lowest energy process with an activation energy of migration of 0.11 eV (refer to Fig. 2). Here the activation energy of migration is defined as the position of the highest potential energy along the migration path.

Three different energy profiles [as shown in Fig. 2 for Li diffusion paths (A, B and C)] of Li vacancy hopping between two adjacent Li sites in Li2CuO2. Emig corresponds to the activation energy for the Li ion migration.
Trivalent doping
Although there is a very low migration activation energy the diffusion of lithium will be constrained by the high Li Frenkel energies, since there will only be a limited concentration of \(\,{V}_{Li}^{\text{'}}\) that act as vehicles mediating Li self-diffusion via the vacancy mechanism. A defect engineering strategy to introduce a high \(\,{V}_{Li}^{\text{'}}\) is by doping with trivalent ions. This can be described in the Kröger-Vink notation as:
It should be noted that similar defect engineering strategy has been successfully employed in other energy materials such as materials for solid oxide fuel cells27,28. Here we have examined a number of \({R}_{2}{O}_{3}\) oxides, namely those with R = Al, Sc, In, Y, Gd and La, to find the oxide with the minimum solution enthalpy. The results of our calculations are shown in Fig. 3 where it can be observed that the solution energy of Al2O3 is the lowest one (0.97 eV/Al3+). Therefore, doping with Al will result in the formation of \({V}_{Li}^{\text{'}}\,\,\,\)with energies significantly lower than those required by the Li Frenkel reaction. This will lead to a non-equilibrium concentration of \({V}_{Li}^{\text{'}}\). These vacancies will in turn act as vehicles for Li self-diffusion, increasing the Li diffusivity.

Enthalpy of solution of \({{R}}_{2}{{O}}_{3}\) (R = Al, Sc, In, Y, Gd and La) in Li2CuO2.
It is shown here that the Al2O3 solution enthalpy is considerably lower than the enthalpies for Schottky and Frenkel disorder. Therefore, vacancy and interstitial concentrations will be primarily due to the extrinsic impurity compensation processes. Similar processes have been previously considered in ceramic materials for energy applications (solid oxide fuel cells), such as Y2O3 or doped CeO228,29. It should also be noted that the defect enthalpies are expected to be overestimated (although relative energies will be very reliable) as (a) we assume a fully ionic model (refer to Table S1 in the Supplementary Information) and (b) the calculations correspond to the dilute limit.
The introduction of substitutional dopants in the lattice may also impact the migration energies of lithium. Figure 4(a) represents the different energy profiles in the vicinity of an Al dopant atom. It can be deduced that the introduction of dopants in the vicinity of the migrating \({V}_{Li}^{\text{'}}\) will increase the migration energy barriers from 0.22 eV (for the smallest dopant, Al) to 0.88 eV (for the largest dopant, La) (refer to Fig. 4 (b) and Figure S1 in the Supplementary Information). The largest increase of the migration energy barrier is for the largest dopant (i.e. La) and this is consistent with the larger association (i.e. relaxation of the large dopant in the vacant space) of the \(\{L{a}_{Cu}^{\bullet }:{V}_{Li}^{\text{'}}\)}X pairs (−0.65 eV) as compared to the \(\{A{l}_{Cu}^{\bullet }:{V}_{Li}^{\text{'}}\)}X pairs (−0.45 eV). This association in essense introduced an additional energetic barrier that needs to be overcome.

(a) The different energy profiles in the vicinity of an Al atom [effectively path (A) in Fig. 2 but with a nearest neighbour Al substitutional dopant] of Li vacancy hopping between two adjacent Li sites in Li2CuO2. (b) The activation energy of migration, Emig, of Li with respect to the ionic radius of the dopants (R = Al, Sc, In, Y, Gd and La) in the vicinity of the migrating Li ion.
Densities of states
We have performed density of states (DOS) calculations and plotted the individual contribution of each element (partial-DOS) in Fig. 5, for the cases of (a) perfect cell (b) \({{\rm{V}}}_{{\rm{Li}}}^{\text{'}}\) (c) \(A{l}_{Cu}^{\bullet }\,\,\,\)and (d)\(\,\{{{\rm{Al}}}_{{\rm{Cu}}}^{\bullet }:{{\rm{V}}}_{{\rm{Li}}}^{\text{'}}\)}X. The valence band maximum (VBM) is set at zero energy level. For the perfect structure (refer to Fig. 5(a)), the band gap of Li2CuO2 is calculated at 1.05 eV. It is shown that Cu and O mainly contribute to the DOS of the valence band, driven by the Cu-d and O-p orbitals, while Li and Cu are the main contributors to the conduction band, driven by the Li-p and Cu-p orbitals (refer also to Figure S2 in the Supplementary Information). We also observe the high states of Cu and O just below the VBM and the non-uniformity of the band as presented by two separated contributions, in agreement with a previous study17. The effect on the DOS of the introduction of a lithium vacancy in the supercell is minimal (refer to Fig. 5(b)) as no additional states are created into the band gap, wheras only a small increase in the DOS in the conduction band due to Lithium is observed. (for oxygen vacancies refer to supplementary material Figure S3).

The densities of states for Li2CuO2: (a) perfect cell (b) \({{\boldsymbol{V}}}_{{\boldsymbol{Li}}}^{\text{'}}\) (c) \({\boldsymbol{A}}{{\boldsymbol{l}}}_{{\boldsymbol{Cu}}}^{\bullet }\) (d) \({\{{\boldsymbol{A}}{{\boldsymbol{l}}}_{{\boldsymbol{Cu}}}^{\bullet }:{{\boldsymbol{V}}}_{{\boldsymbol{Li}}}^{\text{'}}\}}^{X}\).
The impact of the \(A{l}_{Cu}^{\bullet }\,\,\,\)and \(\{A{l}_{Cu}^{\bullet }:{V}_{Li}^{\text{'}}\)}X pairs on the densities of states on Li2CuO2 was also considered (refer to Fig. 5(c,d)). It is shown that the \({{\rm{Al}}}_{{\rm{Cu}}}^{\bullet }\) introduces a distribution of states in the band gap with a simultaneous minor narrowing of the gap itself (to 0.80 eV). Similar picture is observed in Fig. 5(d) for the \(\{A{l}_{Cu}^{\bullet }:{V}_{Li}^{\text{'}}\)}X case (for \({R}_{Cu}^{\bullet }\) and \(\{{R}_{Cu}^{\bullet }:{V}_{Li}^{\text{'}}\)}X please refer to Figures S4 and S5 in the Supplementary Information). Therefore, the introduction of Al (and other) dopants has an effect on the electronic structure, mainly with the introduction of states in the band gap but also with a minor narrowing of the gap. Although for battery applications, the leading conduction mechanism is the diffusion of ions, the effect of the energy levels in the band gap have to be considered as well.
Summary
In the present study, the intrinsic defect processes and the Li vacancy diffusion in Li2CuO2 were investigated. Lithium vacancy diffusion with a very low migration energy (0.11 eV) along zig-zag diffusion path in the ab-plane is the lowest energy process. The high lithium Frenkel energy in essense implies a low concentration of lithium vacancies. We propose here doping Li2CuO2 with aluminium to introduce a higher concentration of lithium vacancies. The aluminium dopants will impact the activation energy of migration increasing the barrier to 0.22 eV. The combination of low solution enthalpy and low increase in the migration energy of lithium as compared to the intrinsic processes or the other dopants considered here, constitutes doping with aluminium the preferential doping strategy to increase the lithium diffusivity in Li2CuO2. The DOS analysis reveals that the introduction of aluminium will introduce electronic states into the energy band gap. It is anticipated that the present study will motivate further experimental and theoretical work30,31,32 to determine the diffusion properties of Li2CuO2 and its potential application in batteries. Additionally, it will generate further interest in lithium cuprate materials for energy storage applications.
Methods
Atomistic simulations employing interatomic potentials method were used to investigate the relative energetics for the formation of intrinsic defects and the possible pathways for lithium ion migration. In particular Buckingham-type interatomic potentials as implemented in the general utility lattice program (GULP)33 were used, within the Born model of solids. The interactions between ions consist of a long-range Coulombic term and a short-range component, which aims to represent electron-electron repulsion and van der Waals interactions. Short-range interactions were modeled using the Buckingham potentials. The Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm34 was employed to relax the simulation boxes and atomic positions. Lattice relaxation around point defects and the migrating ions were considered using the Mott-Littleton method35. This divides the system into two concentric regions, where the ions within the inner spherical region (>700 ions) surrounding the defect relaxed explicitly. The defect calculations were performed in supercells containing 720 ions.
Li diffusion was investigated by considering two adjacent vacancy sites as initial and final configurations. The Li interstitial ion was placed along the direct pathway between the initial and final vacancy configuration. We have considered seven intermediate (interstitial) positions which were fixed while all other ions were free to relax. The energy difference between the saddle point position and the system in its initial state is effectively the activation energy of migration.
To calculate the DOSs for pure and defective Li2CuO2, density functional theory simulations were employed using the plane wave code CASTEP36,37. The exchange and correlation interactions are modelled by using the corrected density functional of Perdew, Burke and Ernzerhof (PBE)38 in the generalized gradient approximation (GGA), with ultrasoft pseudopotentials39. The kinetic energy cut-off of the plane wave basis is 500 eV, in conjunction with a 3 × 2 × 3 Monkhorst-Pack (MP)40 k-point grid and a 60-atomic site supercell. To consider correlation effects of localized electrons onsite Coulomb repulsions in the range of 4–8 eV is set for the Cu 3d orbitals. We have tested this value to establish that the trends are not affected by the specific choice of U-parameter. The calculations were under constant pressure conditions. The system has been treated as spin polarized. For the PDOS analysis/imaging, the OPTADOS code is employed41,42.
References
Tarascon, J.-M. & Armand, M. Issues and changes facing rechargeable lithium batteries. Nature 414, 359–367 (2001).
Armand, M. & Tarascon, J.-M. Building better batteries. Nature 451, 652–657 (2001).
Fisher, C. A. J., Hart Prieto, V. M. & Islam, M. S. Lithium battery materials LiMPO4 (M = Mn, Fe, Co, and Ni): Insights into defect association, transport mechanisms, and doping behavior. Chem. Mater. 20, 5907–5915 (2008).
Bruce, P. G., Freunberger, S. A., Hardwick, L. J. & Tarascon, J.-M. Li-O2 and Li-S batteries with high energy storage. Nat. Mater. 11, 19–29 (2012).
Kamaya, N. et al. A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011).
Seino, Y., Ota, T., Takada, K., Hayashi, A. & Tatsumisago, M. A sulphide lithium super ion conductor is superior to liquid ion conductors for use in rechargeable batteries. Energy Environ. Sci. 7, 627–631 (2014).
Shin, D. O. et al. Synergistic multi-doping effects on the Li7La3Zr2O12 solid electrolyte for fast lithium ion conduction. Sci. Rep. 5, 18053 (2015).
Clark, J. M., Nishimura, S.-I., Yamada, A. & Islam, M. S. High-voltage pyrophosphate cathode: Insights into local structure and lithium-diffusion pathways. Angew. Chem. Int. Ed. 51, 13149–13153 (2012).
Fisher, C. A. J., Kuganathan, N. & Islam, M. S. Defect chemistry and lithium-ion migration in polymorphs of the cathode material Li2MnSiO4. J. Mater. Chem. A 1, 4207–4214 (2013).
Jay, E. E., Rushton, M. J. D., Chroneos, A., Grimes, R. W. & Kilner, J. A. Genetics of superionic conductivity in lithium lanthanum titanates. Phys. Chem. Chem. Phys. 17, 178–183 (2015).
Chen, C., Lu, Z. & Ciucci, F. Data mining of molecular dynamics data reveals Li diffusion characteristics in garnet Li7La3Zr2O12. Sci. Rep. 7, 40769 (2017).
He, X., Zhu, Y. & Mo, Y. Origin of fast ion diffusion in super-ionic conductors. Nat. Commun. 8, 15893 (2017).
Steele, B. C. & Heinzel, A. Materials for fuel-cell technologies. Nature 414, 345–352 (2001).
Kang, K., Meng, Y. S., Bréger, J., Grey, C. P. & Ceder, G. Electrodes with high power and high capacity for rechargeable lithium batteries. Science 311, 977–980 (2006).
Palacios-Romero, L. M. & Pfeiffer, H. Lithium cuprate (Li2CuO2): A new possible ceramic material for CO2 chemisorption. Chem. Lett. 37, 862–863 (2008).
Oh-ishi, K., Matsukura, Y., Okumura, T., Matsunaga, Y. & Kobayashi, R. Fundamental research on gas–solid reaction between CO2 and Li2CuO2 linking application for solid CO2 absorbent. J. Solid State Chem. 211, 162–169 (2014).
Ramos-Sanchez, G. et al. Controlling Li2CuO2 single phase transition to preserve cathode capacity and cyclability in Li-ion batteries. Solid State Ionics 303, 89–96 (2017).
Lara-Garcia, H. A. & Pfeiffer, H. High and efficient Li2CuO2-CO2 chemisorption using different partial pressures and enhancement produced by the oxygen addition. Chem. Eng. J. 313, 1288–1294 (2017).
Tanaka, N., Suzuki, M. & Motizuki, K. Electronic band structure of Li2CuO2. J. Magn. Magn. Mater. 196–197, 667–668 (1999).
Xiang, H. J., Lee, C. & Whangbo, M.-H. Absence of a spiral magnetic order in Li2CuO2 containing one-dimensional CuO2 ribbon chains. Phys. Rev. B 76, 220411 (2007).
Rosner, H., Hayn, R. & Drechsler, S. L. The electronic structure of Li2CuO2. Physica B 259–261, 1001–1002 (1999).
Chroneos, A., Yildiz, B., Tarancón, A., Parfitt, D. & Kilner, J. A. Oxygen diffusion in solid oxide fuel cell cathode and electrolyte materials: mechanistic insights from atomistic simulations. Energy Environ Sci. 4, 2774–2789 (2011).
Lee, J. et al. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries. Science 343, 519–522 (2014).
Zhu, J. J. et al. Intrinsic defects and H doping in WO3. Sci. Rep. 7, 40882 (2017).
Parfitt, D., Kordatos, A., Filippatos, P. P. & Chroneos, A. Diffusion in energy materials: Governing dynamics from atomistic modelling. Appl. Phys. Rev. 4, 031305 (2017).
Sapiña, F. et al. Crystal and magnetic structure of Li2CuO2. Solid State Commun. 74, 779–784 (1990).
Dholabhai, P. P., Adams, J. B., Crozier, P. & Sharma, R. A density functional study of defect migration in gadolinium doped ceria. Phys. Chem. Chem. Phys. 12, 7904–7910 (2010).
Rushton, M. J. D. & Chroneos, A. Impact of uniaxial strain and doping on oxygen diffusion in CeO2. Sci. Rep. 4, 6068 (2014).
Busker, G., Chroneos, A., Grimes, R. W. & Chen, I.-W. Solution mechanisms for dopant oxides in yttria. J. Am. Ceram. Soc. 82, 1553–1559 (1999).
Alexopoulos, K., Lazaridou, M. & Varotsos, P. Activation volumes in lead halides and other solids. Phys. Rev. B 33, 2838–2841 (1986).
Varotsos, P. A. Calculation of point defect parameters in diamond. Phys. Rev. B 75, 172107 (2007).
Saltas, V., Chroneos, A. & Vallianatos, F. Composition and temperature dependence of self-diffusion in Si1-xGex alloys. Sci. Rep. 7, 1374 (2017).
Gale, J. D. & Rohl, A. L. The General Utility Lattice Program (GULP). Molecular Simul. 29, 291–341 (2003).
Gale, J. D. Gulp: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 93, 629–637 (1997).
Mott, N. F. & Littleton, M. J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 34, 485–499 (1938).
Payne, M. C., Teter, M. P., Allan, D. C., Arias, T. A. & Joannopoulos, J. D. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev. Mod. Phys. 64, 1045 (1992).
Segall, M. D. et al. First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 14, 2717 (2002).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892 (1990).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Nicholls, R. J., Morris, A. J., Pickard, C. J. & Yates, J. R. OptaDOS - a new tool for EELS calculations. J. Phys.: Conf. Ser. 371, 012062 (2012).
Morris, A. J., Nicholls, R., Pickard, C. J. & Yates, J. R. OptaDOS: A tool for obtaining density of states, core-level and optical spectra from electronic structure codes. Comp. Phys. Comm. 185, 1477–1485 (2014).
Acknowledgements
Computational facilities and support were provided by High Performance Computing Centre at Imperial College London and Coventry University.
Author information
Authors and Affiliations
Contributions
A.K. and N.K. performed the calculations. All the authors analyzed and discussed the results and contributed to the writing of the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kordatos, A., Kuganathan, N., Kelaidis, N. et al. Defects and lithium migration in Li2CuO2. Sci Rep 8, 6754 (2018). https://doi.org/10.1038/s41598-018-25239-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-25239-5
- Springer Nature Limited
This article is cited by
-
Breaking the energy density limit of LiNiO2: Li2NiO3 or Li2NiO2?
Science China Materials (2022)
-
Defects and dopant properties of Li3V2(PO4)3
Scientific Reports (2019)
-
Defects, dopants and Mg diffusion in MgTiO3
Scientific Reports (2019)
-
Defects, Lithium Mobility and Tetravalent Dopants in the Li3NbO4 Cathode Material
Scientific Reports (2019)
-
Defect Chemistry and Li-ion Diffusion in Li2RuO3
Scientific Reports (2019)