
Abstract
Amphibians are particularly sensitive to landscape fragmentation. Potential barriers between breeding sites can negatively influence the dispersal of individuals and increase genetic structure between populations. In this study, we genotyped 10 microsatellites for 334 marbled newts (Triturus marmoratus) at 11 different locations in Western France. Samples were collected in different regions with contrasting agricultural landscapes (low and high proportion of arable land in the north and south, respectively). We found a strong genetic structure between the northern and southern sampling sites. Isolation by distance was recorded after 62 km, but within the northern region, little or no genetic structure was detected over large distances (up to 114 km). Genetic structure at shorter distance (43 km) was found between sites situated in landscapes with larger amounts of arable lands. A significant positive relationship was found between the pairwise genetic distance (Fst) between sites and the amount of arable land together with the distance between sites. Our results suggest that the Loire River might act as a corridor for the marbled newt, while arable land might act as a barrier. Finally, although a large city is located between sampling sites, no effect was detected on population structure.
Similar content being viewed by others


Introduction
Amphibians are one of the most threatened taxa in the world, with 32.5% of the species ranked as vulnerable1. Among the principal causes of decline, habitat loss and fragmentation play an important role2. The latter can be defined as the reduction of overall available habitat, leading to the division of a favourable habitat into smaller patches surrounded by a matrix of non-favourable habitat2,3,4. Habitat fragmentation can be caused by various factors such as urbanisation, transport infrastructures, deforestation and agricultural activities5. It is well recognised that habitat fragmentation negatively affects populations, both at a demographic and a genetic level. For instance, decreasing habitat size has a direct, adverse effect on population size6. Fragmentation also results in lower migration rates between habitat patches7. In an isolated population, the low immigration rate of new individuals from other populations can lead to insufficient gene flow to maintain genetic diversity. This happens when isolation between populations is so strong that inbreeding and genetic drift are no longer counterbalanced by gene flow8. The resulting erosion of genetic diversity will negatively affect fitness and increase extinction risk9. It is therefore important to detect the most isolated populations so as to increase gene flow between populations and to preserve corridors.
Many amphibian species are particularly sensitive to habitat fragmentation because during their life cycle individuals must move between different habitat types. Amphibians can undergo two types of movements: migration and dispersal10. Migrations are the movements within the life cycle of an individual (e.g.: movement from wintering sites to reproduction sites) that happen annually, whereas dispersal involves one-way movements from one habitat patch to another. Because dispersal plays an important part in reducing genetic structure between populations, changes in landscape characteristics that greatly influence the dispersal of individuals can have strong negative impacts on populations2.
In France, the agricultural landscape has changed significantly since the 1950’s. After the Second World War, the growing demand for resources induced a shift from small-scale to large-scale agricultural exploitations that has led to land use modification11. Related agricultural practices such as tillage, drainage of wetland areas, intense grazing and use of large quantities of pesticides have negatively impacted biodiversity by transforming the landscape and making it unsuitable for certain species12. Moreover, the need for high productivity resulted in the transformation of small fields into large open fields at the expense of hedgerows11. Because some animals use hedgerows as habitat and corridors13,14, their destruction induced significant habitat loss and fragmentation for many species13.
The destruction of hedgerows in favour of large fields11 could negatively impact our study species, the marbled newt (Triturus marmoratus), which is mostly found in agricultural landscapes composed of conserved meadows, hedgerows15, and forest areas16,17. Its distribution area includes western France and the northern Iberian peninsula18. Despite being categorised as Least Concern by the IUCN, it is protected by Annex IV of the EU Habitats Directive and Annex III of the Bern Convention. The species is also locally endangered in central France19. The major threats for the marbled newt are degradation of its habitat (due to agricultural intensification, among other things), disease, non-native predators, and illegal capture for the pet trade. Like many amphibian species, marbled newt populations are declining19 and little is known about their population structure. Understanding the influence of landscape on the genetic structure of the marbled newt is important for taking conservation measures to restore gene flow and protect sensitive populations8.
Previous studies on newts reported different results at different scales regarding population structure and dispersal capacities of the marbled newt, or closely related species. Jehle et al.20 found genetic structure for the marbled newt and the crested newt (Triturus cristatus) at a local scale (less than 10 km) whereas Prunier et al.21 found no genetic structure at larger scale (more than 10 km) for another large newt, the alpine newt (Ichthyosaura alpestris). An important dispersal ability was also found for a small-bodied species, Lissotriton helveticus, that could rapidly colonise restored ponds in a forested landscape, resulting in a weak genetic structure22.
Polymorphic genetic markers, such as microsatellites, are particularly useful in observing the influence of landscape characteristics on population structure. They allow us to study the relationships between different sites and estimate the connectivity between populations at different geographical scales2. Depending on the studied species, the time needed to observe a change in population structure between different sites may vary. In our case, the landscape changed 70 years ago. Based on the results of Prunier et al.21 on a closely related species, the alpine newt, we estimate that this is enough time to detect the influence of agricultural landscape on population structure.
In the light of the previously cited results, we defined two main goals for this study: (1) to investigate the population structure of the marbled newt in the west of France at different scales (local, regional and inter-regional), and (2) to test the influence of the agricultural landscape on population structure. Specifically, we expected the spatial genetic structure would be mostly driven by isolation by distance, but also to some extent by landscape structure21,23. We also expected that the agricultural landscape would negatively influence the dispersal of the marbled newt, resulting in a higher population structure.
Results
Genetic diversity
Among the 10 selected loci, no significant linkage disequilibrium was found after Bonferonni correction (see Costanzi et al.24). No potential null alleles were detected, and in total the data set had 4.75% missing data.
In one sampling site (NE1), a significant deviation from the Hardy-Weinberg equilibrium was found after Bonferroni correction for four loci (Tmar 2, Tmar 17, Tmar 20, Tmar 21). No positive correlation was found between Fis and Fst over all loci of sampling site NE1. It seems that the deviation from the Hardy-Weinberg equilibrium is not due to a Whalund effect and therefore sampling site NE1 was kept for the rest of the analysis25.
Among all 334 individuals distributed on 11 sites (Fig. 1), the number of alleles per locus ranged from 3 (Tmar 23) to 16 (Tmar 20). Using the rarefaction curves obtained with the R package PopGenkit (PopGenKit v1.0 R package, R Core Team), the average expected number of alleles over all loci per population for 14 individuals varied from 2.59 for NW1 to 4.45 for SE2 (Table 1).

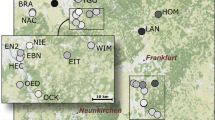
Location of the 11 sampling sites (SE1 to SE5, SW1, SW2, NW1, NW2, NW3 and NE1), the square and dots are the locations of the sites, for southern group and the northern group respectively (groups were assigned with the software STRUCTURE), and each colour represents a different cluster as defined by the software STRUCTURE. The black triangles show the major cities in the area, the black lines represent regions borders and the grey line is the Loire River. The figure was created using ArcMap 10.4 (Esri, Redlands, CA, USA, http://www.esri.com/arcgis/about-arcgis).
Genetic structure
When analysing the whole dataset with STRUCTURE26, the software found that a division into 2 clusters best represented the data (ΔK2 = 131.82). While most of the sampling sites were assigned to a group with an average membership coefficient >0.9, three sampling sites (SW1, SW2 and SE2) presented more admixture than the other sites with an average membership coefficient of 0.80, 0.61 and 0.74 for SW1, SW2 and SE2, respectively. Apart from these three sites a clear separation was revealed by STRUCTURE between the northern sites (NW1, NW2, NW3 and NE1) and most of the southeastern sites (SE1, SE3, SE4 and SE5) (Fig. 2a). Within each cluster, additional levels of substructure were found. For the northern and southwestern sites, three genetic clusters were detected by STRUCTURE (ΔK3 = 58.31). The first cluster grouped the southwestern sites SW1 and SW2, the second gathered the northwestern sites (NW1, NW2, NW3) and the last contained the northeastern site NE1 (Fig. 2b). No substructure was detected within these three genetic clusters when analysed separately. For southeastern sites, three clusters were detected (ΔK3 = 7.72). Sampling sites SE1 and SE2 formed the first sub-cluster, SE3 the second and finally SE4 and SE5 formed the third (Fig. 2c). No clear substructure was detected within these clusters when analysed separately.

Bar plot presenting the results from STRUCTURE for the most likely the number of clusters (K) calculated with the Evanno et al.66 method. (a) Population structure results for all sampling sites, K = 2. (b) Substructure results for the northern and southwestern sampling sites (NW1, NW3, NW2, NE1, SW1, SW2), K = 3. (c) Substructure results for the southeastern sampling sites (SE2, SE1, SE3, SE5, SE4), K = 3.
The results from the DAPC scatter plot presented some similarities with the results from STRUCTURE. There was a clear genetic structure between three clusters. The first cluster gathered four sites (NW1, NW2, NW3 and NE1), the second gathered the sampling sites SW1 and SW2 while the last gathered sites SE1, SE2, SE3, SE4 and SE5 (Fig. 3). A second level of substructure could be observed for the first cluster with NW1 and NW3 being genetically very close, while NE1 and NW2 seem to show some differences (with NE1 being the most different). Regarding the southeast region (SE1, SE2, SE3, SE4 and SE5), the DAPC results mostly show a division in two groups: SE1, SE2 and SE3 for the first group, and SE4 and SE5 for the second group.

Scatterplots of DAPC. Dots are individuals, inertia ellipses and colours represent the different groups.
After Bonferonni correction, Fst analysis showed a significant differentiation between most populations. No significant differentiation was only found in three instances: between sample sites NE1 and NW2 (Fst = 0.03587, p-value > 0.001), SE2 and SE4 (Fst = 0.02695, p-value > 0.001), and SE4 and SE5 (Fst = −0.0016, p-value = 0.55) (Table 2).
Supporting the results of STRUCTURE and DAPC analysis, the pairwise Fst inside the northern group were weak and less than 0.1. The same goes for the pairwise Fst inside the southeastern group, with weak Fst lower than 0.1. The results of STRUCTURE and DAPC show similar patterns as pairwise Fst between the northern group and the southeastern group, with strong Fst always higher than 0.2 (Table 2).
The STRUCTURE analysis revealed that the first cluster associated the northern group more with the southwestern group (SW1 and SW2). However, pairwise Fst results indicate that the southwestern sites present similar genetic distance with the southeastern sites (pairwise Fst for SE2, SE3, SE4, SE5 when compared to SW1, respectively: 0.0519, 0.174, 0.149, 0.131) and with the northern sites (pairwise Fst for NE1, NW2, NW3 when compared to SW1, respectively: 0.142, 0.116 and 0.181) (Table 2). For the DAPC results, the southwestern sites have a genetic identity in-between the northern sites and the southeastern sites. Sampling site NW2 is clearly associated with the other northwestern sites (NW1 and NW3) in the STRUCTURE analysis, while with the pairwise Fst it is genetically closer to the northeastern site (NE1) (Fst = 0.03587, p-value < 0.001). With the DAPC analysis, NW2 has an intermediate genetic identity between the other northwestern sites (NW1 and NW3) and site NE1 (northeastern site).
Regarding the Southern cluster, we detected a population structure at a smaller spatial scale than for the northern cluster. STRUCTURE and DAPC found some genetic structure between SE3 and SE4 + SE5 even though those sites are 43 km apart.
Groups for the analysis of molecular variance (AMOVA) calculation were defined according to the STRUCTURE results by creating six different groups: NW2 and NW3; SW1; NE1; SE2; SE3; SE4 and SE5. The results indicate that 84.01% of the genetic variance can be found within sampling sites, 2.47% among sampling sites but within groups and the remaining 13.52% is attributable to among-groups variability.
The Mantel correlogram showed a significant positive correlation between genetic and geographic distance for the two first classes, 0 to 23 km and 23 to 62 km (p-value: < 0.01), indicating that populations separated by less than 62 km tend to be genetically similar. No significant correlation was detected for class 62 to 139 km. A significant negative correlation was detected for the two last classes, 139 to 177 km and 177 to 216 km (p-value: < 0.05) (Fig. 4).

Mantel correlogram between pairwise standardised genetic distances (Fst/1 − Fst) and geographic distance (in km). For black squares p < 0.05, and for white squares p > 0.05.
Influence of agricultural landscape on pairwise Fst
Pairwise Fst was best explained by the model including the interaction: “amount of arable land” with “pairwise distance”. The 95% confidence interval did not overlap 0 for the interaction (LCI: 2.12968e-05, UCI: 0.00011), and was therefore considered informative27. The effect of the interaction (Fig. 5) shows that pairwise Fst increased significantly with distance and with the proportion of arable land between sites.

Predicted relationship of pairwise Fst and distance (in km) depending on different proportions of arable land between sites within a 10 km wide corridor (N = 28). The red, green and blue linear regression are for, respectively, 25%, 50% and 75% of arable land.
Discussion
Our study provides insight on the marbled newt population genetic structure at local, inter-regional, and regional scales. Most notably, we found two levels of population structure. Firstly, a strong inter-regional genetic structure between southern and northern sampling sites, and secondly regional genetic structure within those groups. No genetic structure was found at a local scale (<15 km). Moreover, isolation by distance was detected for distance greater than 62 km.
Observed and expected heterozygosity are similar to those found by Jehle et al.23, also in Western France, for the marbled newt. For the expected heterozygosity, Jehle et al.23 found a minimum and maximum of 0.20 and 0.55, respectively, while we found a minimum and maximum of 0.36 and 0.54, respectively. For the observed heterozygosity, the minimum and maximum were respectively 0.15 and 0.47 for Jehle et al.23 and 0.27, 0.53 for our study. These results indicate a similar genetic diversity between the two studies.
At inter-regional scale, over all sampling sites, the STRUCTURE results showed that “two clusters” (ΔK2 = 131.82) was the most likely scenario (Fig. 2a). However, some sampling sites (SW1, SW2 and SE2) presented more admixture than the remaining sites and were assigned to a specific cluster with a membership coefficient <0.9. It is possible that the strong difference between the northern group (NW1, NW2, NW3 and NE1) and most sites of the southeastern group (SE1, SE3, SE4, SE5) might have influenced the STRUCTURE results indicating that two clusters is the most likely scenario, while in fact sites SW1 and SW2 are in a genetically and geographically intermediate position and could therefore represent a third cluster. This is also supported by the DAPC results that displayed three distinct genetic groups (Fig. 3) over all sampling sites: the northern group (NW1, NW2, NW3 and NE1), the southwestern group (SW1 and SW2) and finally the southeastern group (SE1, SE2, SE3, SE4 and SE5). In general, the genetic structure seems to follow the geographic position of the sampling sites. We also notice that site SE2 presents some admixture in the STRUCTURE plot (Fig. 2a), and shares similarities with site SW2 in the DAPC plot (Fig. 3). In light of these results, we speculate that occasional gene flow may have occurred between the three different clusters.
Pairwise Fst between northern and southeastern sampling sites ranged from 0.130 (between NW2 and SE2) to 0.319 (between NW3 and SE4) (Table 2). As these regions are far apart (minimum distance is 79 km between NE1 and SE1, and maximum distance is 236 km between NW1 and SE3), isolation by distance is likely playing a role in the observed structure, as sampling sites separated by more than 62 km tend to be genetically different (Fig. 4). SW1 and SW2 might also be differentiated from the other sampling sites due to isolation by distance, as they are separated by more than 62 km from most of the sites (only NW3 is separated by 53 and 59 km from SW1 and SW2, respectively). This pattern of isolation by distance is relatively high when compared to the results of Prunier et al.21 for the alpine newt. In their study, Prunier et al.21 found evidence of isolation by distance for samples 12 km apart or more. However, our results seem to be closer to those of Emaresi et al.28 who found no isolation by distance pattern over their study area with sampling sites separated by a maximum of 26 km. Our results of isolation by distance suggest some long distance connectivity, which according to Kimura and Weiss29 could indicate that gene flow between sites follows a stepping stone model.
A second set of analyses on each of the previous clusters originally identified by the software STRUCTURE yielded a second level of intra-regional structure, distinguishing 6 different groups (Fig. 2b,c). The AMOVA results were in accordance with the groups defined by STRUCTURE. Indeed, the “among populations within group” variation was very low (2.47%), meaning that individuals from the same group have similar alleles. There was a greater difference between groups (13.52%) corroborating the results found with STRUCTURE and Fst calculation.
The presence of genetic structure between SE3 and the cluster including SE4 + SE5, situated 43 km away from each other, is in contradiction with the Mantel correlogram that showed a significant positive relationship between genetic and geographic distance up to 62 km (Fig. 4). This genetic structure could be explained by the agricultural landscape in the southern part of the southeastern group (SE3, SE4 and SE5) that is mostly composed of arable land used for intensive agriculture (Fig. 6). This type of landscape could have a detrimental influence on newt dispersal (Fig. 5) as cultivated areas and intensive pasture are generally avoided by amphibians30. Boissinot31 found that the presence of cultivated fields around a pond negatively influenced the probability of marbled newt presence in the pond. This is also supported by Trochet et al.32 who found longer travel distances for the marbled newt in forest compared to agricultural land. Our results also suggest that agricultural landscapes seem to increase population structure for the marbled newt. Indeed, we observed that when the proportion of arable land together with the distance between sites were increasing, pairwise Fst was also significantly increasing (Fig. 5). When only pairwise distance was taken into account to explain the variation of Fst in our sampling area, the model did not perform as well as when the interaction, proportion of arable land and distance between sites, was included (ΔAICc = 5.42). This result supports the hypothesis that agricultural landscape could be a barrier to marbled newt dispersal and could increase isolation between sites.

Amount (in percent) of large field crops (cereals, field corn and oil seed) in the total cultivated surface for the sampled area. Darker surfaces represent higer amounts of large field crops, the black dots show the location of the sites, the black triangles show the major citites in the area, the black lines represent region borders and the grey line is the Loire River. Data is from Agreste75. The figure was created using ArcMap 10.4 (Esri, Redlands, CA, USA, http://www.esri.com/arcgis/about-arcgis).
Another potentially intriguing result was that STRUCTURE did not detect genetic structure between NW1, NW2 and NW3, even though 91 km separated NW1 from NW2 (Fig. 1). Similarly, the pairwise Fst result between NE1 and NW2, separated by a distance of 114 km, was not significantly different from zero (Table 2) and was in contradiction with the overall trend displayed by the Mantel correlogram (Fig. 4). Fst for NW1 was not computed as too few individuals could be sampled at this locality (n < 20). However, the results of STRUCTURE and the DAPC show that NW1 belongs to the same cluster as NW2 and NW3. Beyond the relatively important distance separating NW1 from the other two sites, the probable connectivity between those localities is interesting due to the presence of potential barriers to dispersal between those sites. A large urbanised area (the city of Nantes) and a large river (the Loire River) (Fig. 1) might both decrease gene flow between the sampling site to the north (NW1) and sampling sites to the south (NW2 and NW3). Indeed, several studies on amphibians have found that urban areas and rivers could act as barriers and thereby increase population structure33,34,35,36. However, in our case, other unsampled areas around the city of Nantes could play the roles of stepping stones and/or reservoirs between the sampling sites. The local agricultural landscape might also play a positive role on the dispersal of individuals, as it features hedgerows, meadows and forest, the habitats favoured by marbled newts for efficient dispersal37, and a low amount of large field crop (Fig. 6). Regarding the Loire River, our results seem to be consistent with the studies of Gascon et al.38, Lougheed et al.39 and Johanet40, which found no significant barrier effect of rivers on gene flow. However, according to our data and the position along the Loire River of our sampling sites, we argue that the river might act as a corridor helping marbled newts dispersal. The Loire River can be relatively narrow during the dry season, with dense vegetation on the water and low stream flow, all of which facilitate the crossing of newts in summer40. Dispersal around the river could also occur during flood periods. In ponds bordering the river, we can also assume that larvae and eggs, which are often attached to vegetation, could be accidentally carried away by strong currents during the flood season40,41,42. Hence, there are good reason to believe the Loire River might not be a barrier to dispersal for the marbled newt and could even positively influence dispersal along its flow.
Overall, the observed connectivity supported by the lack of genetic structure at local scales (<15 km) in this study contrasts with the low migration and dispersal capacity of most amphibians43, and of the marbled newt in particular20,44. For the marbled newt, the maximum observed migration distance was 146 m for Jehle and Arntzen44 and 473 m for Trochet et al.32. This low migration capacity is generally explained by the dependency of amphibians on wetland areas45, as well as their slow terrestrial movements46. For a large newt species like the alpine newt, the maximum recorded dispersal distance was between 200 m47 and 500 m48. Langton et al.49 observed a maximal dispersal distance of approximately 1000 m around the reproduction pond for another large newt species, the crested newt. Regarding genetics, our results are in accordance with Smith and Green50. According to their meta-analysis, in general, anurans and salamanders do not present any structure for distances lower than 10 km. This is also consistent with the results of Isselin-Nondedeu et al.22 who found, based on genetic analysis, that the small palmate newt Lissotriton helveticus was able to rapidly colonise newly restored ponds within a forest.
Our results indicate that the survival and dispersal of marbled newt populations could be largely dependent on landscape type and the associated dispersal corridors available in the surrounding areas. Our study supports the idea that populations far apart might be genetically well connected, provided that good dispersal paths such as rivers51, hedgerows and forests exist. Such connection might facilitate movements and dispersal between reproduction sites, while forests are also used as a shelter against predation and desiccation17. Because of the low dispersal capacity of the marbled newt, a dense pond network could be needed to maintain genetic exchange at distances >10 km in areas with a large amount of arable land. Moreover, large rivers and cities did not seem to represent barriers to dispersal in our study. In fact, the Loire River seems to act as a dispersal corridor for the marbled newt and possibly also for other amphibians. A test to identify the direction of gene flow between our localities could provide an improved understanding of the influence of the river on newt dispersal. On the other hand, genetic structure was found in areas with large amounts of arable land. Therefore, presence of arable land seems to have a greater impact on the marbled newt’s gene flow than do large anthropic constructions, such as cities. In light of our results and of current habitat losses in the west of France, we recommend the following guidelines for the conservation of marbled newt populations: (1) to maintain a dense network of reproduction sites (suitable ponds in proximity of forested areas); (2) to protect hedgerow, meadow and forest landscapes and focus on the restoration of natural areas where intensive agricultural landscapes predominate; (3) finally, it could be beneficial to protect ponds close to rivers in order to promote long distance gene flow.
Material and Methods
Study sites and sampling
Samples were collected on 11 different locations in 34 different ponds distributed at a regional scale in Western France (see Fig. 1). The study areas were selected to provide a diversity of agricultural landscapes (conserved meadows or large field crops) and a variety of situations regarding the presence or absence of nearby forest. Our sampling design provided sites separated by three different levels of geographical distances: inter-region (>100 km), intra-region (between 100 km and 10 km) and local (<10 km). This design allowed us to compare genetic diversity and population structure at different spatial scales. Ponds in an area <4 km2 and unseparated by fragmenting elements were considered as one location in the analysis in order to have a large enough sample size. This threshold was chosen to be more conservative than Smith and Green50, who estimated that population differentiation for amphibians mostly occurs at distances above 10 km.
A total of 334 individuals were sampled for the analysis. Sampling sites were located in different geographical regions. The northern sampling sites (NW1, NW2, NW3 and NE1) were along the Loire Valley. The Loire Valley is dominated by a mix of large and small pastures delimited by hedgerows called “bocage”. This landscape is also characterised by a low proportion of large field crops (arable land): cereals, field corn and oil seed (as defined in Laurent52) (Fig. 6). An exception was site NE1, where the ponds were situated in a forest-dominated landscape. There was a gap of more than 100 km in a straight line without sampling sites between the northwestern sampling sites (NW1, NW2, NW3) and the northeastern sampling site NE1 (Fig. 1). Between the northern and southeastern sampling sites, the landscape was dominated by extensive farming with a weak density of hedgerows and few groves. In the southwest (SW1 and SW2), the density of bocage was high around the sampling sites. Between southwestern (SW1 and SW2) and southeastern (SE1, SE2, SE3, SE4, SE5) sampling groups (Fig. 1), there was extensive farming with large field crops (cereals, corn and oil seed) (Fig. 6) with a low density of hedgerows and few groves. In the southeastern group, SE1 and SE2 were largely dominated by bocage whereas SE3, SE4 and SE5 were surrounded by landscape largely dominated by intensive farming. SE4 and SE5 were in an isolated forest and SE3 was in a small isolated bocage.
Ethical note
All trapping and handling procedures were in accordance with the relevant guidelines and regulations and were approved by the appropriate authority, the Directions Régionales de l’Environnement, de l′Aménagement et du Logement (DREAL). Only non-invasive methods were used to collect DNA and individuals were released immediately after sampling53. Moreover, chytridiomycosis protocols were followed as advised by the French herpetological society (Société Herpétologique de France). No individuals were injured during capture and handling and all were successfully released after DNA sampling.
DNA sampling and genetic analysis
DNA collection of epithelial cells was realised by buccal swab following Pidancier et al.54. For each pond all captured individuals were sampled at once during the same session in order to avoid replication. Individuals were caught either with landing nets or Ortmann’s funnel traps55.
DNA extractions were performed using the salting out protocol of Sunnucks and Hales56. To compare population structures, 10 microsatellite markers were used in this study. Nine were developed specifically for the marbled newt24 and one (Tcri27) was developed for the crested newt57. PCR and genotyping were performed by the Gentyane INRA platform (Clermont-Ferrand, France). We analysed the genotyped data with Genemapper v4.0 (AppliedBiosystems™).
Data treatment
Deviation from Hardy-Weinberg equilibrium was calculated using GENEPOP v4.258 and significance levels were adjusted with Bonferroni correction. A possible Wahlund effect for populations presenting a deficit in heterozygotes was tested for the correlation between Fis and Fst per locus. Fis and Fst per locus were obtained with the R package hierfstat v0.04-2259 implemented in R v3.3.260, then a linear regression was calculated in order to observe the relationship between both variables25. Expected heterozygosity (Hepx.), observed heterozygosity (Hobs.), and average number of alleles per locus were calculated with the software Genetix v4.05.261. The allelic richness (Ar.) was plotted using allele rarefaction curves, as calculated by the package PopGenKit v1.0 (R package, R Core Team), implemented in R v3.2.262. After visual verification, the minimum sample size was set to 14 in order to have a good estimation of the number of alleles. Pairwise Fst and significance probabilities were obtained with the software Arlequin (Ver 3.5)63. Only sample sites (n = 8) with more than 20 individuals were kept for this analysis. This minimum was set according to Kalinowski64, as it give good results for Fst larger than 0.01. Fst significance was corrected for multiple testing with the Bonferroni method.
Bayesian estimations of population structure were inferred with the software STRUCTURE (2.3.4)26 using a burn-in period of 150,000 followed by 106 Markov chain Monte Carlo (MCMC) iterations. Default settings were used (Admixture model) for all parameters but for the LOCPRIOR option (the sample location was used as a prior), which was enabled as it tends to perform better than models without LOCPRIOR enabled65. For each analysed dataset, 10 replicate runs were performed for each value of the number of clusters (K) from 1 to 10. The most likely number of clusters (K) was determined using the calculation of delta K (ΔK) described in Evanno et al.66, as implemented by the web interface of STRUCTURE HARVESTER67, but also visually directly from STRUCTURE’s bar plots (Fig. 2). Sampling sites were assigned to a specific population according to the highest average membership coefficient. For each cluster detected by STRUCTURE, analysis was run again with the same parameters in order to detect more subtle substructure68 (Fig. 2b,c). When the STRUCTURE bar plot showed that individuals could not be assigned to different clusters and the ΔK was low, the dataset was considered as one cluster.
Discriminant analysis of principal components (DAPC) was run using the dapc function in the R package adegenet69, as a complementary analysis for population structure. This method applies a discriminant analysis on data previously transformed using a principal component analysis (PCA). DAPC optimises the genetic variance between groups while minimising the variance within groups, in order to show better separation of the different groups. For the PCA calculation, after visual interpretation, we decided to retain 35 PCs as it conserved 96% of the variance. Regarding the discriminant analysis, the first two eigenvalues were kept, after visual interpretation, as they were carrying most of the information. The results of the DAPC were gathered in a scatterplot, with individuals represented as dots and the different groups as inertia ellipses.
The smallest clusters detected by STRUCTURE were used as predefined groups for the AMOVA in order to detail our population structure. The AMOVA was calculated with the software Arlequin (Ver 3.5)63.
Isolation by distance among populations was tested with a Mantel correlogram using the matrix of pairwise Euclidian distance (in kilometres) between sample sites and the matrix of pairwise standardised genetic distances between sampling sites (Fst/1 − Fst). The calculation was performed with the R package vegan (vegan v 2.3–3 R package, R Core Team), function mantel.correlog, with 9999 permutations and significance level <0.05 (adjusted for multiple testing with the Holm methods).
Landscape analysis
We tested the influence of the agricultural landscape on genetic distance by analysing habitat types between sampling sites. To do so we created, on Arcgis 10.4 (Esri, Redlands, CA, USA, http://www.esri.com/arcgis/about-arcgis), a 10 km wide corridor between every sampling site. We then used the CORINE (Coordination of Information on the Environment) land cover data from Copernicus land cover monitoring services70 to record the different habitat types within each corridor. The amount (in percent) of different habitat types within each corridor was then calculated with the software FRAGSTATS (v4.2)71. In order to detect the influence of large field crops on genetic distance we used the proportion of arable land (code 21) within each corridor and accounted for the distance between sites. We chose arable land as it is the CORINE land cover habitat that best represents the large field crops and intensive agricultural activities. In our sampling area we only had one type of arable land, non-irrigated arable land (code 211), and so for the rest of the article we will refer to it simply as arable land. We then created a linear model with pairwise Fst as a response variable and proportion (in percent) of arable land, and pairwise distance as explanatory variables. We also included the interaction between the two explanatory variables in order to account for the combined effect of both pairwise distance and amount of arable land. The three candidate models were then compared using Akaike’s Information Criterion corrected for sample size (AICc)72 with the function model.sel from the R package MuMIn73. The model with the lowest AICc score was selected, and if ΔAICc was <4, the most parsimonious model was chosen. Finally, parameters that had zero in their 95% confidence interval were considered non-informative27. A graph presenting the results of the best model was then created using the R package ggplot274 (Fig. 5).
Data Availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Change history
19 November 2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Stuart, S. N. Status and Trends of Amphibian Declines and Extinctions Worldwide. Science 306, 1783–1786, https://doi.org/10.1126/science.1103538 (2004).
Cushman, S. A. Effects of habitat loss and fragmentation on amphibians: A review and prospectus. Biol. Conserv. 128, 231–240, https://doi.org/10.1016/j.biocon.2005.09.031 (2006).
Fahrig, L. Effects of habitat fragmentation on biodiversity. Annu. Rev. Ecol. Evol. Syst. 34, 487–515 https://doi.org/10.1146/annurev.ecolsys.34.011802.132419 (2003).
Fischer, J. & Lindenmayer, D. B. Landscape modification and habitat fragmentation: A synthesis. Global Ecol. Biogeogr. 16, 265–280, https://doi.org/10.1111/j.1466-8238.2007.00287.x (2007).
Thompson, J. D. & Ronce, O. Fragmentation des habitats et dynamique de la biodiversité, https://www.sfecologie.org/regard/regards-6-thompson-ronce/ (2010).
Shaffer, M. L. In Viable populations for conservation Vol. 69 (ed. Soulé, M. E.) (Cambridge University Press, 1987).
Brown, J. H. & Kodric-Brown, A. Turnover Rates in Insular Biogeography: Effect of Immigration on Extinction. Ecology 58, 445–449 (1977).
Beebee, T. J. C. Conservation genetics of amphibians. Heredity 95, 423–427, https://doi.org/10.1038/sj.hdy.6800736 (2005).
Allentoft, M. E. & O’Brien, J. Global Amphibian Declines, Loss of Genetic Diversity and Fitness: A Review. Diversity 2, 47–71, https://doi.org/10.3390/d2010047 (2010).
Semlitsch, R. D. Differentiating Migration and Dispersal Processes for Pond-Breeding Amphibians. J. Wildl. Manage. 72, 260–267, https://doi.org/10.2193/2007-082 (2008).
Flatrès, H. & Flatrès, P. Mutations agricoles et transformations des paysages en Europe. Norois 173, 173–193, https://doi.org/10.3406/noroi.1997.6779 (1997).
McLaughlin, A. & Mineau, P. The impact of agricultural practices on biodiversity. Agriculture, Ecosystems & Environment 55, 201–212, https://doi.org/10.1016/0167-8809(95)00609-V (1995).
Forman, R. T. T. & Baudry, J. Hedgerows and hedgerow networks in landscape ecology. Environ. manage. 8, 495–510, https://doi.org/10.1007/BF01871575 (1984).
Hinsley, S. A. & Bellamy, P. E. The influence of hedge structure, management and landscape context on the value of hedgerows to birds: A review. J. Environ. Manage. 60, 33–49, https://doi.org/10.1006/jema.2000.0360 (2000).
Boissinot, A. & Grillet, P. Conservation des bocages pour le patrimoine batrachologique. Le Courrier de la Nature 252, 26–33 (2010).
Schoorl, J. & Zuiderwijk, A. Ecological Isolation in Triturus cristatus and Triturus marmoratus (Amphibia: Salamandridae). Amphibia-Reptilia 1, 235–252, https://doi.org/10.1163/156853881X00357 (1980).
Marty, P., Angélibert, S., Giani, N. & Joly, P. Directionality of pre- and post-breeding migrations of a marbled newt population (Triturus marmoratus): implications for buffer zone management. Aquat. Conserv.: Mar. Freshwat. Ecosyst. 15, 215–225, https://doi.org/10.1002/aqc.672 (2005).
Duguet, R. & Melki, F. Les Amphibiens de France, Belgique et Luxembourg. Parthénope edn, (Biotope, 2003).
Arntzen, J. W. et al. Triturus marmoratus. The IUCN Red List of Threatened Species (2009).
Jehle, R., Burke, T. & Arntzen, J. W. Delineating fine-scale genetic units in amphibians: Probing the primacy of ponds. Conserv. Genet. 6, 227–234, https://doi.org/10.1007/s10592-004-7832-8 (2005).
Prunier, J. G. et al. A 40-year-old divided highway does not prevent gene flow in the alpine newt Ichthyosaura alpestris. Conserv. Genet. 15, 453–468, https://doi.org/10.1007/s10592-013-0553-0 (2014).
Isselin-Nondedeu, F. et al. Spatial genetic structure of Lissotriton helveticus L. following the restoration of a forest ponds network. Conserv. Genet. 18, 853–866, https://doi.org/10.1007/s10592-017-0932-z (2017).
Jehle, R., Wilson, G. A., Arntzen, J. W. & Burke, T. Contemporary gene flow and the spatio-temporal genetic structure of subdivided newt populations (Triturus cristatus, T. marmoratus). J. Evol. Biol. 18, 619–628, https://doi.org/10.1111/j.1420-9101.2004.00864.x (2005).
Costanzi, J.-M. et al. Characterization of nine new microsatellite loci for the marbled newt. Triturus marmoratus. J.Genet. 94, 1–2, https://doi.org/10.1007/s12041-015-0586-x (2015).
Waples, R. S. Testing for Hardy-Weinberg proportions: have we lost the plot? J. Hered. 106, 1–19, https://doi.org/10.1093/jhered/esu062 (2015).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959, https://doi.org/10.1111/j.1471-8286.2007.01758.x (2000).
Arnold, T. W. Uninformative Parameters and Model Selection Using Akaike’s Information Criterion. J. Wildl. Manage. 74, 1175–1178, https://doi.org/10.2193/2009-367 (2010).
Emaresi, G., Pellet, J., Dubey, S., Hirzel, A. H. & Fumagalli, L. Landscape genetics of the Alpine newt (Mesotriton alpestris) inferred from a strip-based approach. Conserv. Genet. 12, 41–50, https://doi.org/10.1007/s10592-009-9985-y (2011).
Kimura, M. & Weiss, G. H. The Stepping Stone Model of Population Structure and the Decrease of Genetic Correlation with Distance. Genetics 49, 561–576 (1964).
Gray, M. J., Smith, L. M., Miller, D. L. & Bursey, C. R. Influences of agricultural land use on Clinostomum attenuatum metacercariae prevalence in southern Great Plains amphibians, USA. Herpetol. Conserv. Biol. 2, 23–28 (2007).
Boissinot, A. Influence de la structure du biotope de reproduction et de l’agencement du paysage, sur le peuplement d’amphibiens d’une région bocagère de l’ouest de la France. PhD thesis, École Pratique des Hautes Études (2009).
Trochet, A. et al. Postbreeding Movements in Marbled Newts (Caudata, Salamandridae): A Comparative Radiotracking Study in Two Habitat Types. Herpetologica 73, 1–9, https://doi.org/10.1655/Herpetologica-D-15-00072 (2017).
Marsh, D. M. et al. Ecological and genetic evidence that low-order streams inhibit dispersal by red-backed salamanders (Plethodon cinereus). Can. J. Zool. 85, 319–327, https://doi.org/10.1139/Z07-008 (2007).
Hitchings, S. P. & Beebee, T. J. C. Genetic substructuring as a result of barriers to gene flow in urban Rana temporaria (common frog) populations: implications for biodiversity conservation. Heredity 79, 117–127, https://doi.org/10.1038/hdy.1997.134 (1997).
Wagner, R. S., Miller, M. P., Crisafulli, C. & Haig, S. M. Geographic variation, genetic structure and conservation unit designation in the larch mountain salamander (Plethodon larselli). Can. J. Zool. 83, 396–406, https://doi.org/10.1139/z05-033 (2005).
Noël, S., Ouellet, M., Galois, P. & Lapointe, F.-J. Impact of urban fragmentation on the genetic structure of the eastern red-backed salamander. Conserv. Genet. 8, 599–606, https://doi.org/10.1007/s10592-006-9202-1 (2007).
Davies, Z. G. & Pullin, A. S. Are hedgerows effective corridors between fragments of woodland habitat? An evidence-based approach. Landsc. Ecol. 22, 333–351, https://doi.org/10.1007/s10980-006-9064-4 (2007).
Gascon, C., Lougheed, S. C. & Bogart, J. P. Patterns of Genetic Population Differentiation in Four Species of Amazonian Frogs: A Test of the Riverine Barrier Hypothesis. Biotropica 30, 104–119, https://doi.org/10.1111/j.1744-7429.1998.tb00373.x (1998).
Lougheed, S. C., Gascon, C., Jones, D. A., Bogart, J. P. & Boag, P. T. Ridges and rivers: a test of competing hypotheses of Amazonian diversification using a dart-poison frog (Epipedobates femoralis). Proc. R. Soc. Lond. B 266, 1829–1835, https://doi.org/10.1098/rspb.1999.0853 (1999).
Johanet, A. Flux de gènes inter- et intra-spécifiques chez des espèces de vallées alluviales: cas des tritons palmés et ponctués en vallée de la Loire PhD thesis, Université d’Angers (2009).
Hayes, M. P. et al. Dispersion of Coastal Tailed Frog (Ascaphus Truei): An Hypothesis Relating Occurrence of Frogs in Non–fish-bearing Headwater Basins to Their Seasonal Movements. J. Herpetol. 40, 531–543, 10.1670/0022-1511(2006)40[531:DOCTFA]2.0.CO;2 (2006).
Measey, G. J., Galbusera, P., Breyne, P. & Matthysen, E. Gene flow in a direct-developing, leaf litter frog between isolated mountains in the Taita Hills, Kenya. Conserv. Genet. 8, 1177–1188, https://doi.org/10.1007/s10592-006-9272-0 (2007).
Spear, S. F., Peterson, C. R., Matocq, M. D. & Storfer, A. Landscape genetics of the blotched tiger salamander (Ambystoma tigrinum melanostictum). Mol. Ecol. 14, 2553–2564, https://doi.org/10.1111/j.1365-294X.2005.02573.x (2005).
Jehle, R. & Arntzen, J. W. Post-breeding migrations of newts (Triturus cristatus and T. marmoratus) with contrasting ecological requirements. J. Zool. 251, 297–306, https://doi.org/10.1111/j.1469-7998.2000.tb01080.x (2000).
Duellman, W. E. & Trueb, L. Biology of amphibians. (Johns Hopkins University Press, 1994).
Rothermel, B. B. & Semlitsch, R. D. An Experimental Investigation of Landscape Resistance of Forest versus Old-Field Habitats to Emigrating Juvenile Amphibians. Conserv. Biol. 16, 1324–1332, https://doi.org/10.1046/j.1523-1739.2002.01085.x (2002).
Joly, P. & Grolet, O. Colonization dynamics of new ponds, and the age structure of colonizing Alpine newts, Triturus alpestris. Acta Oecol. 17, 599–608 (1996).
Perret, N., Pradel, R., Miaud, C., Grolet, O. & Joly, P. Transience, dispersal and survival rates in newt patchy populations. J. Anim. Ecol. 72, 567–575, https://doi.org/10.1046/j.1365-2656.2003.00726.x (2003).
Langton, T. E. S., Beckett, C. L. & Foster, J. P. Great Crested Newt Conservation Handbook. (2001).
Smith, M. A. & Green, D. M. Dispersal and the metapopulation in amphibian and paradigm ecology are all amphibian conservation: populations metapopulations? Ecography 28, 110–128, https://doi.org/10.1111/j.0906-7590.2005.04042.x (2005).
Brode, J. M. & Bury, B. R. In California Riparian Systems: Ecology, Conservation, and Productive Management. (ed Warner Richard E. & Kathleen M. Hendrix) (Berkeley: University of California Press, 1984).
Laurent, F. L’Agriculture de Conservation et sa diffusion en France et dans le monde. Cybergeo: European Journal of Geography [En ligne], Environnement, Nature, Paysage, document 747, https://doi.org/10.4000/cybergeo.27284 (2015).
Prunier, J. et al. Skin swabbing as a new efficient DNA sampling technique in amphibians, and 14 new microsatellite markers in the alpine newt (Ichthyosaura alpestris). Mol. Ecol. Resour 12, 524–531, https://doi.org/10.1111/j.1755-0998.2012.03116.x (2012).
Pidancier, N., Miquel, C. & Miaud, C. Buccal Swabs As a Non-Destructive Tissue Sampling Method for Dna Analysis in Amphibians. Herpetological Journal 13, 175–178 (2003).
Drechsler, A., Bock, D., Ortmann, D. & Steinfartz, S. Ortmann’ s funnel trap – a highly efficient tool for monitoring amphibian species. Herpetology Notes 3, 13–21 (2010).
Sunnucks, P. & Hales, D. F. Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol. Biol. Evol. 13, 510–524, https://doi.org/10.1093/oxfordjournals.molbev.a025612 (1996).
Krupa, A. P. et al. Microsatellite loci in the crested newt (Triturus cristatus) and their utility in other newt taxa. Conserv. Genet. 3, 87–89, https://doi.org/10.1023/A:1014239225553 (2002).
Rousset, F. genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106, https://doi.org/10.1111/j.1471-8286.2007.01931.x (2008).
Goudet, J. HIERFSTAT, a package for R to compute and test hierarchical F -statistics. Mol. Ecol. Notes 5, 184–186, https://doi.org/10.1111/j.1471-8278.2004.00828.x (2005).
R Core Team. R: A language and environment for statistical computing (2016).
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, F. & Bonhomme, N. GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations., (Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II, 2004).
R Core Team. R: A language and environment for statistical computing (2015).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567, https://doi.org/10.1111/j.1755-0998.2010.02847.x (2010).
Kalinowski, S. T. Do polymorphic loci require large sample sizes to estimate genetic distances? Heredity 94, 33–36, https://doi.org/10.1038/sj.hdy.6800548 (2005).
Hubisz, M. J., Falush, D., Stephens, M. & Pritchard, J. K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332, https://doi.org/10.1111/j.1755-0998.2009.02591.x (2009).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620, https://doi.org/10.1111/j.1365-294X.2005.02553.x (2005).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361, https://doi.org/10.1007/s12686-011-9548-7 (2012).
Maletzky, A., Kaiser, R. & Mikulíček, P. Conservation Genetics of Crested Newt Species Triturus cristatus and T. carnifex within a Contact Zone in Central Europe: Impact of Interspecific Introgression and Gene Flow. Diversity 2, 28–46, https://doi.org/10.3390/d2010028 (2010).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405, https://doi.org/10.1093/bioinformatics/btn129 (2008).
Copernicus land monitoring services. Corine Land Cover (CLC), Version 18.5.1, http://land.copernicus.eu/pan-european/corine-land-cover/clc-2012 (2012).
FRAGSTATS v4: Spatial Pattern Analysis Program for Categorical and Continuous Maps. (University of Massachusetts, Amherst, 2012).
Burnham, K. P., Anderson, D. R. & Huyvaert, K. P. AIC model selection and multimodel inference in behavioral ecology: some background, observations, and comparisons. Behav. Ecol. Sociobiol. 65, 23–35, https://doi.org/10.1007/s00265-010-1029-6 (2011).
Barton, K. MuMIn: Multi-Model Inference. R package version 1.15.6. https://CRAN.R-project.org/package = MuMIn (2016).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag, 2016).
Agreste. Recensement agricole, http://agreste.agriculture.gouv.fr/recensement-agricole-2010/ (2010).
Acknowledgements
Financial support for this work came from Région Pays de la Loire, Centre Beautour, Chinon SNB project (stratégie nationale pour la biodiversité 2011, French minister of ecology), Office National de la Chasse et de la Faune Sauvage (Pôle Bocage et Faune Sauvage) and Nouvelle-Aquitaine Region. We thank Alexis Viaud (Bretagne Vivante), Cédric Baudran (réseau herpétofaune de l’Office National des Forêts), Didier Montfort, Mickael Ricordel (réseau herpétofaune de l’Office National des Forêts), Philippe Evrard, Sylvain Courant, Yohann Jaumouillé (Office National des Forêts), Jérémy Souchet, Jérôme Lallemand (Conservatoire des Espaces Naturels Poitou-Charentes), Florian Doré (Deux-Sèvres Nature Environnement) and Fabrice Conort (Office National de la Chasse et de la Faune Sauvage) for their investment in the project and their help in sample collection that made this study possible, also we are grateful to ONF-Centre Val de Loire. We thank all the students for their field contribution to this work, and landowners who let us visit their ponds during our field activities. We also thank Alexander James Briggs, Jasmine Anastasia Hayes, Martin Mayer and Priyank Sharad Nimje for their corrections. And last but not least we thank Théophane You for his support and help at every stage of the project.
Author information
Authors and Affiliations
Contributions
P.D. developed the study design and secured the funding, all authors contributed to the data collection, C.J.M., M.P. and L.P.Q. participated in the DNA extraction, C.J.M. and M.P. performed the statistical analyses, C.J.M. wrote the manuscript. All authors, participated in the draft and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Costanzi, JM., Mège, P., Boissinot, A. et al. Agricultural landscapes and the Loire River influence the genetic structure of the marbled newt in Western France. Sci Rep 8, 14177 (2018). https://doi.org/10.1038/s41598-018-32514-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32514-y
- Springer Nature Limited
Keywords
This article is cited by
-
Fine-scale spatial genetic structure and dispersal among Italian smooth newt populations in a rural landscape
Scientific Reports (2023)
-
Population structure and genetic diversity of the threatened pygmy newt Triturus pygmaeus in a network of natural and artificial ponds
Conservation Genetics (2022)
-
Regional replication of landscape genetics analyses of the Mississippi slimy salamander, Plethodon mississippi
Landscape Ecology (2020)