
Abstract
Long QT Syndrome 3 (LQTS3) arises from gain-of-function Nav1.5 mutations, prolonging action potential repolarisation and electrocardiographic (ECG) QT interval, associated with increased age-dependent risk for major arrhythmic events, and paradoxical responses to β-adrenergic agents. We investigated for independent and interacting effects of age and Scn5a+/ΔKPQ genotype in anaesthetised mice modelling LQTS3 on ECG phenotypes before and following β-agonist challenge, and upon fibrotic change. Prolonged ventricular recovery was independently associated with Scn5a+/ΔKPQ and age. Ventricular activation was prolonged in old Scn5a+/ΔKPQ mice (p = 0.03). We associated Scn5a+/ΔKPQ with increased atrial and ventricular fibrosis (both: p < 0.001). Ventricles also showed increased fibrosis with age (p < 0.001). Age and Scn5a+/ΔKPQ interacted in increasing incidences of repolarisation alternans (p = 0.02). Dobutamine increased ventricular rate (p < 0.001) and reduced both atrioventricular conduction (PR segment-p = 0.02; PR interval-p = 0.02) and incidences of repolarisation alternans (p < 0.001) in all mice. However, in Scn5a+/ΔKPQ mice, dobutamine delayed the changes in ventricular repolarisation following corresponding increases in ventricular rate. The present findings implicate interactions between age and Scn5a+/ΔKPQ in prolonging ventricular activation, correlating them with fibrotic change for the first time, adding activation abnormalities to established recovery abnormalities in LQTS3. These findings, together with dynamic electrophysiological responses to β-adrenergic challenge, have therapeutic implications for ageing LQTS patients.
Similar content being viewed by others


Introduction
Long QT Syndrome (LQTS) is characterised by prolonged electrocardiographic (ECG) QT intervals reflecting increased ventricular action potential durations (APD) and is associated with increased incidences of arrhythmogenesis. The long QT syndrome 3 (LQTS3) variant specifically arises from gain-of-function mutations in the inactivation domain of the SCN5A gene that encodes the cardiac Na+ channel, Nav1.51,2,3. These gain-of-function mutations result in recovery abnormalities attributable to increased late Na+ current (I Na-L) known to enhance both triggers and substrates for arrhythmogenesis. Arrhythmic triggering arises from an occurrence of afterdepolarisation events during or immediately following an AP. Arrhythmic substrate arises from re-entrant processes re-exciting recovered regions resulting from the increased I Na-L 4.
The aim of the present study to investigate the effects of age and adrenergic challenge on the LQTS3 phenotype was prompted by previous clinical and experimental work. Firstly, there is continued discussion concerning effects of effects of ageing in LQTS5 wherein human studies report reduced ages of onset (~50 ± 14 years) for atrial fibrillation (AF)6. More specifically in LQTS3, (a) LQTS3 genotypes were powerful predictors of fatal or near-fatal events after age 40, even compared to LQTS2 and LQTS1 genotypes, with similar risks associated with each LQTS genotype when excluding patients treated with β-blockers from the analysis7. (b) The specific association of LQTS arrhythmias occurring after 40 years of age with LQTS3 phenotypes8. (c) Complementary experimental studies showing increased arrhymogenicity with age in murine models of LQTS39.
Secondly, there are particular clinical paradoxical adrenergic effects in LQTS3. On the one hand, exercise and fright precipitate acute accentuations of QT interval prolongation, T-wave alternans and torsade de pointes accounting for 32% of fatal cardiac events in LQTS310, 11. Adrenergic provocation similarly provokes ECG T wave lability associated with cardiac arrest or syncope in some LQTS3 patients12. β blockade reduces risks of cardiac events specifically in female LQTS3 patients, although the effect in males is yet to be determined due to insufficient data13. On the other hand, in contrast to LQTS1 or LQTS2 patients, 39% of LQTS3 fatalities occur at rest or during sleep, when adrenergic tone is expected to be low and cholinergic activity is expected to be high11,14. A further clinical study associates higher incidences of cardiac events with both the LQTS3 and LQTS2 genotype amongst a cohort of LQTS patients treated with β blockers15. Similarly, contrasts between adrenergic effects have also been shown in experimental studies. Thus, β-adrenergic stimulation was variously pro-arrhythmic16, anti-arrhythmic17, 18 or without effect in LQTS3 models19. Furthermore, β -blockade was also shown to reduce arrhythmogenicity16, 20 and triggering events20. Yet other studies showed β -blockade accentuated reentrant substrate19 and exacerbated transmural gradients of repolarisation with potential pro-arrhythmic effects20.
The present ECG and histological experiments explored the physiological basis for the above effects for the first time, using the Scn5a+/ΔKPQ murine model for LQTS3. This model carries a deletion of three conserved amino acids, Lys 1505, Pro 1506 and Gln 1507, in the Nav1.5 III–IV linker responsible for fast inactivation, known to correspond to a particularly severe clinical LQTS3 phenotype21, 22. Previous studies reported that the Scn5a+/ΔKPQ murine model recapitulated many clinical features of LQTS3, including its ECG abnormalities and arrhythmic tendency18, 19, 23. Previous work had demonstrated the mechanistic basis of possible arrhythmic phenotypes in studies demonstrating Na+ current abnormalities19, triggering events20, 24 and arrhythmic substrate20, 25. The present work uses electrocardiographic and histological studies to examine for independent and interacting effects of ageing and Scn5a+/ΔKPQ genotype on arrhythmic phenotype and electrophysiological properties, before and following β1-adrenergic challenge, and the contribution of fibrotic change to these effects, in intact animals for first time.
Materials and Methods
Animals
A total of 12 wild-type (WT) and 13 Scn5a+/ΔKPQ S129sv mice were studied. They were housed in an animal facility maintained at 21 °C with 12 h light/dark cycles, fed sterile chow (RM3 Maintenance Diet; SDS, Witham, Essex, UK) and had free access to water. All experimental protocols were approved under the UK Home Office regulations (Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012) following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB) and conducted under a designated project license. The experiments also conformed to the Guide for the Care and Use of Laboratory Animals, U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996). The WT and Scn5a+/ΔKPQ mice were each further divided into young (3 ± 1 month) and old (12 ± 1 month) groups.
Electrocardiographic analysis
For the electrocardiographic (ECG) study, the mouse groups consisted of young WT (n = 7), old WT (n = 5), young Scn5a+/ΔKPQ (n = 7) and old Scn5a+/ΔKPQ mice (n = 6). The ECG recordings followed a previously published approach26. To anaesthetise each mouse, avertin (2,2,2 trimethylethanol, Sigma-Aldrich, Poole UK) was administered into the intra-peritoneal space before the ECG recordings were made. Avertin is known to have fewer cardiovascular and electrophysiological conduction effects than other anaesthetic agents, such as isoflurane and xylazine-ketamine27. For example, previous work indicated that ketamine provides reliable relaxation, sedation, and analgesia in mice but exerts bradycardic effects28. All measurements were consistently performed after a 5-min period following establishment of anaesthesia and the recording setup in an attempt to allow autonomic levels to stabilise. The mice were left undisturbed in a dark box until the sedation took effect, weighed, and then placed in a supine position on a heated platform to maintain a 37 °C body temperature. Four 2-mm diameter electrodes (MLA1204; AD Instruments, Colorado Springs, CO, USA) were placed in the limbs and connected to a 4-channel NL844 pre-amplifier. The outputs were then led through 4-channel NL820 isolator and NL135 low-pass filter units (set at a 1.0-kHz cut-off and with a 50-Hz notch) within a NL900D chassis and power supply (Neurolog-Digitimer, Hertfordshire, UK). The signal was sampled at 5 kHz and analogue-to-digital conversion employed a CED 1401c interface (Cambridge Electronic Design, Cambridge, UK). This then conveyed Lead II ECG traces to a computer for display and recording using Spike II software (Cambridge Electronic Design).
Baseline ECGs were recorded for 10- to 15-min, including a 5-min stabilisation period. Dobutamine hydrochloride (0.3 mg/kg: Sigma Aldrich, Poole UK) was then injected intraperitoneally. The ECG recordings continued for at least another 10-min after an observable effect on the ECG trace. Baseline stability in the ECG recordings was optimised by securing the limbs of the mice with adhesive tape to minimize movement artefacts and by performing experiments within a grounded Faraday cage to reduce electrical noise. The ECG record analysis also employed Spike II software and no digital filters were applied during the analysis. The ECG deflections ended with a “D peak” (Fig. 1a), which in some records could be preceded by an additional “C’ peak” (Fig. 1b) before final return of the trace to the isoelectric baseline. All the remaining peaks and troughs were consistent in their occurrence through all the experimental groups that were studied. Thus, there was an internally consistent general ECG waveform, meaning that no traces were excluded. This allowed the application of cursors at the peaks and troughs of the trace as shown in the representative example in Fig. 2a. Only Lead II ECG traces were analysed. Lead I traces were used as references to guide application of cursors. The cursors indicate key reference features in the observed ECGs and were used to subsequently measure interval durations (Fig. 2b).

Representative peaks in the ECG records All peaks shown here, apart from C’peak, marked in red, were consistent across all the ECG records. C’peak was absent in some ECG records (a) but present in others (b). The cursors were then applied accordingly to encompass this difference, by including a QD duration, while allowing the same intervals to be analysed across all records.

Schematic representation of the ECG analysis method (a) This representative lead II ECG trace obtained from an avertin anaesthetized mouse illustrates the cursors (dashed vertical lines) that were applied using Spike II software (Cambridge Electronic Design). Bipolar recordings were obtained with the negative electrode placed on the right forelimb and the positive electrode placed on the left hindlimb. The cursors indicate key reference features in the observed ECGs. Those marked in bold type were the key parameters sought in the automated analysis. Those marked in standard type indicate consistent features in the ECG traces that were used in the signal analysis programme to determine those key parameters. (b) This same example lead II ECG trace now has the intervals sought in the automated analysis programme superimposed. The key below the trace shows the physiological relevance of each of those ECG intervals. This analysis yielded the following ECG intervals which provide indications of different aspects of electrophysiological function: RR interval (the ventricular rate reflecting sino-atrial node pacemaker function); P wave duration (atrial conduction time); PR segment duration (atrioventricular (AV) node conduction time); PR interval (combined atrial and AV node conduction times); QS interval (ventricular activation time); SC and SD intervals (indicators of ventricular recovery time); QC and QD intervals (indicators of the total duration of ventricular excitation).
This analysis yielded the following ECG intervals which provide indications of different aspects of electrophysiological function: RR interval (the ventricular rate reflecting sino-atrial node pacemaker function); P wave duration (atrial conduction time); PR segment duration (atrioventricular (AV) node conduction time); PR interval (summed atrial and AV node conduction times); QS interval (ventricular activation time); SC and SD intervals (indicators of ventricular recovery time); QC and QD intervals (indicators of the total duration of ventricular excitation). Earlier studies comparing recovery intervals at different heart rates used Bazett’s formula29 with an adjustment for mice: (QT/√(RR/100)30. However, the intervals analysed in the present study used uncorrected values. Thus recent reports pointed out that the modified Bazett’s formula can underestimate the QT interval and introduce systematic measurement errors in murine hearts, particularly for recovery diseases such as LQTS31,32,33
The data from the cursor time points was then imported into and analysed with custom-written software in the open-source R programming language (R Core Team, 2015). This was used to calculate the mean and standard errors of the desired ECG interval durations and to quantify the presence of Cpeak alternans. Analysis was performed on 300-sec of ECG data immediately prior to administration of dobutamine and 300-sec post-dobutamine period after having an observable effect. This quantitative analysis was only applied to parts of the trace showing a steady-state ventricular rate.
Histological analysis
The quantification of cardiac fibrosis followed methods used in previous studies in the Scn5a+/− mouse model and therefore permitted the present results to be compared with the previous observations34. The mice were killed by cervical dislocation and the hearts dissected out. Custom made Krebs buffer (containing, in mM, NaCl 119, NaHCO3 25, KCl 4, KH2PO4 1.2, MgCl2 1, CaCl2 1.8, glucose 10 and Na-pyruvate 2, pH 7.4, 95% O2/5% CO2; British Oxygen Company, Manchester, UK) was used to flush the isolated hearts, which were then perfused with 4% buffered formalin for 5-min before being kept in formalin overnight. Following this fixation process, gross transverse sections were taken. This was followed by routine tissue processing and paraffin embedding. The 7 μm thick sections were then stained using a Sirius red protocol, involving a series of immersions: xylene for 2-min; new batch of xylene for 2-min; 95%, 70% and 50% ethanol for 2-min each; Weigerts Haemotoxylin for 8-min; running water for 10-min; picro-sirius red solution for 1 hour; acidified water for 16 dips; 3 changes of 100% ethanol for 1-min each and xylene for 3 dips. The slides were then mounted and subsequently viewed, magnified, and digitally acquired using the Nano Zoomer 2.0 Digital Pathology system (Hamamatsu, Hertfordshire, UK).
Following magnification, a custom-made 17 cm × 30 cm morphometric grid, consisting of square boxes of dimension 1 cm × 1 cm, corresponding to an approximate 0.2 mm × 0.2 mm area of tissue, was then superimposed on each photomicrograph. If a square occupied either completely or partially by cardiac tissue showed the presence of fibrosis, it was counted and then the number of these squares was then expressed as a percentage of total cardiac tissue area for each heart. This analysis was carried out blindly by two investigators independently and then their results were compared for consistency by applying an inter-class correlation coefficient analysis (ICC) to identify if both assessors were in agreement with what counted as fibrotic change. ICC can be interpreted as follows: 0–0.2 indicates poor agreement: 0.3–0.4 indicates fair agreement; 0.5–0.6 indicates moderate agreement; 0.7–0.8 indicates strong agreement; and >0.8 indicates almost perfect agreement.
Statistical analysis
Statistical analyses used Stata SE14 (StataCorp, TX, USA). Each ECG feature or histological parameter was analysed as a separate outcome using regression analysis. We adopted numbers of replicates, n, for each experimental group, that were sufficient for this multivariable regression analysis. Although there are 3 variables (age, genotype and dobutamine challenge), our study involved paired comparisons and was confined to exploring two (and not three) way interactions between variables. We then emphasised only clear and significant associations, reporting conservatively on associations that were narrowly significant, and indicated instances where there was insufficient evidence to support an interaction, rather than inferring its absence. The choice of regression model was determined by the distribution of the parameter under analysis. Thus, the ECG intervals were analysed with multilevel linear regression. Histology results were analysed with a linear regression. The Cpeak alternans parameters included the number of incidences of an alternans episode and the total number of beats involving alternans. Both were analysed with a multilevel negative binomial regression. QC lag and QD lag were dichotomised into an absence or presence of such lag and analysed with a logistic regression.
To analyse the association between ECG features with dobutamine treatment, age and genotype, regression analyses were conducted in two parts. First a univariable regression analysis was performed to select possible risk factors and confounders. Variables with p < 0.20 were selected for the multivariable analyses. The multivariable regression analysis was performed using backwards elimination. All variables with univariable p < 0.20 were included in the first model and variables with p-value > 0.05 were eliminated from the model. To control for confounding, variables were eliminated one by one from the model. If the coefficients from the remaining variables were changed by >25%, the eliminated variable was then retained in the model as a confounder. Interactions were tested which were thought to be of biological interest. The following interactions were tested: age × genotype, age × treatment and genotype × treatment. Where appropriate, specifically when mice were treated with dobutamine, multilevel analyses were performed to adjust for multiple measurements per mouse.
Positive regression coefficients reflected an increase of the outcome with the named category in comparison with the reference category. Negative coefficients indicated a decrease in the outcome for the named category in comparison to the reference category. The reference categories were pre-dobutamine challenge for treatment, young for age and WT for genotype. A significant interaction means that the risk factor effects are not independent, but are different for various categories. Interpretation of this statistical analysis considered the following limitations. First, a three-way interaction between age, genotype and treatment was not possible due to the available sample size. Secondly, the available sample size and the multiple parameters tested within these needed to consider effects of multiple testing: the larger the numbers of parameters tested, the more likely any given parameter would appear to have an association with at least one category.
Results
Qualitative assessment of ECG records
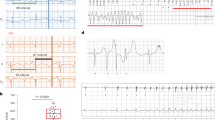
Figure 3 shows examples of different types of ECG lead II waveforms observed in the avertin-anaesthetised mice. The predominant ECG waveform pattern across all groups, whether pre-or post-dobutamine challenge, was one of normal sinus rhythm (Fig. 3a). However, two old Scn5a+/ΔKPQ mice showed arrhythmic episodes post-dobutamine challenge. These patterns were not seen in young Scn5a+/ΔKPQ, consistent with possible influences of age on arrhythmic tendency. Thus, both of these old Scn5a+/ΔKPQ mice showed episodes of isorhythmic AV dissociation (Fig. 3b). During such episodes, the P wave morphology appeared identical to that seen during the previous periods of sinus rhythm and the corresponding mean P–P interval was 178 ms. QRS morphology also appeared unchanged from the earlier periods of the recording and the R–R interval measured 172 ms. However, the PR interval progressively shortened with the P wave eventually merging with the QRS complex, culminating in its disappearance. The arrows in Fig. 3b indicate successive P waves immediately preceding the first five QRS complexes illustrated in the trace, and their disappearance in the next electrocardiographic complex, followed by their return. This return was associated with a prolongation of the R–R interval, with matched P–P and R–R intervals (184 ms) and a fixed PR interval once again (54 ms).

Representative lead II ECG traces following qualitative analysis (a) This trace shows a normal sinus rhythm that was seen throughout the whole recordings, pre- and post-dobutamine challenge, in all young WT (n = 7), all old WT (n = 5) and all young Scn5a+/ΔKPQ (n = 7) but in 4 out of 6 of the old Scn5a+/ΔKPQ. (b) This trace shows isorhythmic AV dissociation that was observed in 2 out of 6 old Scn5a+/ΔKPQ mice post-dobutamine. The arrows are pointing to the P waves, of which there is a noticeable absence in the middle part of the trace.
These observations suggested that in response to dobutamine the P wave rate and QRS rate both increased to similar degrees but were intermittently not identical, giving rise to the time shift of the P wave relative to the QRS complex. During such episodes, the QRS morphology continued to appear narrow and unchanged and identical to that earlier in the recording throughout the episode. This was consistent with a high escape focus, most likely junctional. However, the QRS complexes wherein the P wave is merged or absent could not be driven by sinus node impulse. Furthermore, where the QRS complex occurred in the absence of a sinus node input, there was no R–R delay; this makes it unlikely that this was a sinus pause with an accompanying isolated ventricular escape beat. In contrast, the consistent R - R interval during this period suggested that the ventricles were being driven by an ectopic focus.
The features above are thus consistent with intermittent isorhythmic AV dissociation with an accelerated junctional pacemaker rhythm, such as that reported previously. This could reflect some degree of age-related sinus node dysfunction that may have contributed with a delayed tachycardic response thus being usurped by the junctional focus35, 36.
Analysis of ECG features related to sino-atrial, atrio-ventricular and atrial conduction
Table 1 summarises ECG features representing both activation and recovery events in each cardiac cycle in the atria and ventricles. This mean data was obtained from 5-min intervals both pre- and post-dobutamine challenge. Table 2 shows the results of a multilevel linear regression analysis for independent and interactive associations between these ECG features with dobutamine challenge, increased age, and Scn5a+/ΔKPQ genotype.
Sino-atrial node (SAN) pacemaking increased in frequency following dobutamine challenge in all experimental groups (Table 1). Thus, β-adrenergic stimulation had indistinguishable effects on ventricular rate in Scn5a+/ΔKPQ compared to WT. In both cases, this effect was diminished in old mice, consistent with a reduced chronotropic competence with age. Thus, ventricular rate, assessed from RR intervals and used as a measure for SAN function, increased following dobutamine challenge (p < 0.001). However, there were no independent effects of either age or genotype on ventricular rates whether before or following dobutamine challenge (Table 2). Nevertheless, age acted as a confounding factor (p = 0.08). Furthermore, age and dobutamine challenge together exerted interacting effects, resulting in old mice showing lower ventricular rates post-dobutamine challenge than young mice (p = 0.01). Finally, genotype exerted no interactive effects with either age or dobutamine challenge on ventricular rate
Dobutamine challenge, age and genotype exerted no independent or interactive effects upon atrial activation as reflected in recorded P wave durations. AV node conduction could be assessed from ECG PR segments intervening between the end of the P wave to the start of the QRS complex. AV node conduction was enhanced by dobutamine challenge, but compromised by age to extents indistinguishable between genotypes. Thus, dobutamine challenge independently decreased (p = 0.02), age independently increased (p = 0.001), but genotype exerted no independent effects on PR segments. There were no interactive associations observed for PR segments. Similar inferences emerged from the analysis of PR intervals, which reflect combined times required for atrial activation and AV conduction. Thus, dobutamine challenge independently decreased (p = 0.02), age independently increased (p < 0.001), but genotype exerted no independent effects on PR intervals. There were no interacting effects involving any of the tested variables on such PR intervals.
Analysis of ECG features related to ventricular activation and recovery properties
ECG deflections related to the completion of activation and onset of recovery have previously been correlated with the S trough and the R’ peak. We adopted the QS duration to reflect overall ventricular activation in turn resulting from Na+ channel activation. Previous reports had suggested that including either the entire, or the late component of the R’ wave, partially captures ventricular repolarisation31, 37 overestimating the ventricular activation phase. This finding complements previous reports that had primarily associated LQTS3 only with prolonged repolarisation. Thus, dobutamine challenge, age and genotype exerted no independent effects on QS duration (Table 2). However, age and genotype interacted in increasing QS duration (p = 0.03) with no further interacting effects. Dobutamine challenge did not affect ventricular conduction in contrast to its effects of decreasing PR segment and PR interval, as well decreasing RR interval, in both Scn5a+/ΔKPQ and WT.
We studied the final electrocardiographic deflections ending each ECG complex as representing the overall duration of electrophysiological activity38. Two intervals following the QS deflection, SC and SD durations, were associated with ventricular recovery and were both analysed. The present findings suggest that SC duration may be an indication of the greater part of ventricular recovery towards the resting potential. However, the absence of changes in SC duration, even when comparing the LQTS3 model with WT, suggests that SC duration may not capture the prolonged QT interval attributable to the increased I Na-L, normally associated with either murine Scn5a+/ΔKPQ hearts or LQTS3 patients. Thus, dobutamine challenge, age and genotype exerted no independent or interactive effects on SC duration. In contrast, Scn5a+/ΔKPQ mice showed longer SD durations than WT mice, under all circumstances. This identifies the SD duration with the LQTS3 phenotype and its associated recovery abnormality. This would in turn suggest that the SD duration is inclusive of I Na-L contributions to the overall AP duration, typical of LQTS3 phenotypes. Thus, the predominant effect was that genotype independently increased SD duration (p < 0.001) but age had no independent effect. The effects of dobutamine challenge were suggestive of an independent borderline effect in decreasing the SD duration (p = 0.04). The latter is consistent with previous reports of the shortening of the QT interval for LQTS3 following β-adrenergic stimulation. This contrasts with the known effects of β-adrenergic stimulation in increasing QT interval in LQTS1 and LQTS239. Furthermore, dobutamine challenge, age, and genotype exerted no interactive effects on the SD duration.
The QC and QD durations provide indications of total ventricular excitation period including both activation and recovery. Both QC and QD durations were analysed due to the uncertainty in the murine model as to which exact ECG interval corresponds accurately to the human QT interval. The statistical analysis of both the QC and QD durations yielded similar associations, but for which there were higher significance values for QD duration. This would be explicable in terms of the QD duration encompassing more of the timecourse of I Na-L than the QC duration. Thus, dobutamine challenge exerted no independent effects on QC and QD duration. However, age independently increased both QC and QD durations (p = 0.03 and p = 0.002 respectively). In addition, genotype independently increased both QC and QD durations (p = 0.001 and p < 0.001 respectively). There were no interactive effects on either QC or QD duration.
Thus, Scn5a+/ΔKPQ mice consistently showed longer QC and QD durations under all circumstances, consistent with the clinical phenotype. However, in the case of QC duration, this difference was not attributable to changes in SC duration because all differences observed concerning SC duration were not significant. Therefore, the differences observed for QC durations were mainly attributable to differences in activation time, as reflected in QS duration, and not differences in recovery time, as reflected in SC duration. Thus, the results bearing on QS duration indicate interacting effects between age and genotype. This reflects the independent non-interacting effects of age and genotype upon the QC duration. Different inferences could be made from measurements of QD duration. Thus, the significant difference in QD duration appears to arise from (a) the significant effect of genotype on SD duration and (b) the interacting effects of age and genotype on QS duration.
Histological analysis of fibrosis
We next sought correlations between the above electrocardiographic changes and the absence (Fig. 4a) or presence (Fig. 4b) of fibrotic change in the atria (i) and ventricles (ii) with similarly stratified groups of picrosirius red stained hearts (Fig. 4). Whereas gain of Na+ channel function would be expected to increase conduction velocity, fibrotic change may compromise conduction velocity40. This may arise through compromised gap junction function41 or fibroblast fusion to cardiac myocytes increasing their membrane capacitance42. However, the resulting effects on conduction velocity need not show simple correlations with these variables.

Representative example of the absence of fibrotic change in young WT (a) and the presence of fibrotic change in an old Scn5a+/∆KPQ mouse (b) showing atrial (i) and ventricular (ii) sections.
Histological assessments of the extent of fibrosis were carried out blindly by two investigators independently. Results were then compared for consistency by applying an intra-class correlation coefficient analysis (ICC). The ICC was 0.979 and 0.989 respectively for the atrial and ventricular fibrosis data, indicating a high level of consistency and permitting mean values for both atria (Fig. 5a) and ventricles (Fig. 5b) to be analysed. It was then possible to assess the extent to which the observed fibrotic change was sufficient to alter electrophysiological parameters.

Mean results for the fibrosis in the atria (a) and ventricles (b) Morphometric squares expressed as a percentage of all squares covering the tissues showing positive evidence of fibrotic changes as detected by picrosirius red staining in the different experimental groups. Numbers of hearts (n) shown below each bar. Linear regression with young and WT as reference categories revealed significant independent effects of (i) age in influencing ventricular (regression coefficient = 16, p < 0.001) but not atrial fibrosis and (ii) Scn5a+/∆KPQ vs WT genotypes on both atrial (regression coefficient = 19, p < 0.001) and ventricular (regression coefficient = 14, p < 0.001) fibrosis. There were no interacting effects arising from age and genotype.
Linear regression was performed to investigate the association between the level of fibrosis with age and genotype in the atria and ventricles. This used young and WT as reference categories. The analysis revealed significant independent effects of (a) age in influencing ventricular (regression coefficient = 16, p < 0.001) but not atrial fibrosis and (b) Scn5a+/∆KPQ vs WT genotypes on both atrial (regression coefficient = 19, p < 0.001) and ventricular (regression coefficient = 14, p < 0.001) fibrosis. There were no interacting effects arising from age and genotype. Thus, in atrial sections, the Scn5a+/ΔKPQ genotype but not age independently increased the level of fibrosis (p < 0.001) and there were no interactive effects between these factors. However, both this level of fibrosis and previous reports of reduced Nav1.5 expression with age in Scn5a+/ΔKPQ, appeared insufficient to modify P wave durations, in agreement with an earlier report9. Thus, it would appear that a lone association of Scn5a+/ΔKPQ genotype with increased fibrosis does not suffice to alter P wave duration. For the ventricles, genotype and age independently increased the level of fibrosis (p < 0.001 for both) and there were no interactive effects. Thus, it could be deduced that the independent effects of Scn5a+/ΔKPQ genotype and old age on increasing the level of fibrotic change match the electrophysiological finding that age and genotype interact to produce reduced conduction in the ventricles, reflected in the prolonged QS duration.
Incidences of repolarisation alternans
Alternans in the ECG is defined as a presence of alternating changes in waveform amplitude43. Alternans reflects electrophysiological instabilities and clinically, repolarisation alternans, specifically T wave alternans, appears to predict clinical arrhythmic events with greater accuracy than other non-invasive markers, such as heart rate variability, signal averaged ECG, or reduced ejection fraction. It has also been implicated in the pathophysiological mechanism of sudden cardiac death (SCD)43. Ventricular repolarisation alternans is known to occur in LQTS44. However, previous reports have found conditions of adrenergic activity exerted paradoxical effects on the incidence of repolarisation alternans in LQTS345,46,47.
The present experiments compared incidences and durations of alternans episodes before and following dobutamine challenge, and any association of the resulting changes with age or genotype. This was accomplished by examining for changes in the amplitude of the largest ECG deflection reflecting ventricular repolarisation, Cpeak over fixed, 5 min, observation periods both before and following dobutamine challenge (Table 3). Thus, the absence (Fig. 6a) or presence (Fig. 6b) of alternans was assessed from the amplitudes of Cpeak wave in each consecutive ECG complex normalised to the corresponding amplitude of the Cpeak in the preceding ECG complex. If the values alternated between being >1 then <1 for a minimum of 12 consecutive beats, then this was counted as alternans. The duration of alternans was assessed from the number of beats over which this occurred. A multilevel negative binomial regression was used to assess association between numbers of incidences and of beats with Cpeak alternans with respect to dobutamine challenge, age and genotype. This used before dobutamine challenge, young and WT as reference categories respectively. The results showed an independent effect of dobutamine challenge decreasing the incidences of and number of beats with alternans (regression coefficients −2.7 and −3.9 giving significances of p < 0.001 and 0.004 respectively), but no independent effects of age or genotype. However, age and Scn5a+/∆KPQ genotype had a significant interactive effect on increasing the incidences of and number of beats with alternans (regression coefficients 3.3 and 6.2 giving p = 0.02 and 0.02 respectively). There were no other significant interactive effects.

Representative ECG traces to show the absence (a) and presence (b) of Cpeak alternans (a) This typical trace from a young WT animal shows normal sinus rhythm with the absence of Cpeak alternans, with the horizontal dotted line superimposed indicating that the level of the Cpeak remains consistent. (b) This typical trace from an old Scn5a+/∆KPQ animal shows normal sinus rhythm with the presence of Cpeak alternans, with the arrows indicating where Cpeak alternately increases its amplitude higher than the level of the other Cpeak deflections indicated by the horizontal dotted line. The dashed horizontal lines are to show that the QRS complex amplitude is remains consistent through the recording duration.
Thus, the present findings showed that age and genotype had an interactive association with a higher incidence and number of beats with Cpeak alternans indicating the old Scn5a+/ΔKPQ mice show greater electrophysiological instability. In addition, dobutamine challenge reduced the incidence of repolarisation alternans suggestive of anti-arrhythmic effects. This finding parallels clinical observations that LQTS3 does not share the clearcut pro-arrhythmic features following adrenergic stimulation in other LQTS.
Delays between increased ventricular rate and ventricular repolarisation change following dobutamine challenge
Previous reports have implicated cellular abnormalities with LQTS, which delay the QT interval shortening that normally follows an increased heart rate. This would result in an increased transient susceptibility to R-on-T related arrhythmias, where activation events are superimposed on a relatively delayed T wave of the preceding beat and this could degenerate into polymorphic VT48, 49. These delays are also likely to elude straightforward ECG detection. As with LQTS, the focus of ECG interpretation is generally on the prolongation of QT duration rather than the response of this QT duration to rate changes.
The present experiments accordingly explored for an absence (Fig. 7a) or presence (Fig. 7b) of such delayed changes in ventricular repolarisation duration following ventricular rate increases produced by dobutamine challenge (Table 4). The time lags between changes in QC and QD intervals and changes in ventricular rate were measured. A logistic regression model was then applied to the number of mice showing such a QC or a QD interval lag. Statistical testing was not applied to length of QC or QD interval lags due to the large number of mice showing no lag and the consequent large standard errors. The present analysis demonstrated an association between Scn5a+/ΔKPQ and the presence of both a QC and a QD interval lag (regression coefficients = 30 and 36; p = 0.02 and p = 0.004 respectively). Age was a confounding factor only for QC lag (regression coefficient = 8.9; p = 0.08), but this reflected one old WT outlier. Furthermore, as indicated above, it is QD interval that best reflects contributions from I Na-L. These findings would therefore suggest that delays in changes in repolarisation duration in response to rate changes primarily result from effects of I Na-L in turn largely dependent on genotype.

Representative examples showing the absence and presence of the lag in QC duration The dashed lines follow the gradient of the corresponding parts of the traces, with any intersection between two dashed lines indicating a change of gradient and hence an increase or decrease. These changes are marked by the vertical dotted lines. (a) These records represent the absence of a time lag between the ventricular rate increase (top trace) and the QC duration decrease (bottom trace) following dobutamine challenge exemplified in a young WT mouse. Thus, the vertical dotted line marks the same time point on each trace. (b) These records represent the presence of a time lag between the ventricular rate increase (top trace) and the QC duration decrease (bottom trace) following dobutamine challenge exemplified in an old Scn5a +/∆KPQ mouse. Thus, the vertical dotted lines mark different time points on each trace, with the horizontal arrow indicating this difference.
Discussion
The present work extended previous explorations for electrophysiological phenotypes of LQTS3 using electrocardiographic (ECG) methods in intact genetically modified mice carrying the Nav1.5 gain-of-function Scn5a+/ΔKPQ mutation. We extend findings from previous studies through (a) a detailed breakdown of ECG components in terms of their underlying electrophysiological events to demonstrate novel potentially pro-arrhythmic changes in LQTS3. In particular, this revealed age-related changes related to conduction velocity, in addition to the alterations in recovery from excitation previously established in LQTS. (b) The present study demonstrated an underlying age and genotype related fibrotic change for the first time for LQTS3. (c) Furthermore, this study suggested a basis for the paradoxical responses to β-adrenergic stimulation in which we correspondingly demonstrate (d) an increase in the incidence of repolarisation alternans in old Scn5a+/ΔKPQ mice, which reduces with dobutamine challenge. This demonstrates anti-arrhythmic effects of β-adrenergic stimulation. This was accompanied by (e) slowed responses in ventricular repolarisation adaptation in Scn5a+/ΔKPQ mice in response to dobutamine challenge, which could highlight a potentially pro-arrhythmic effect during transient heart rate increases.
Firstly, episodes of ventricular electrophysiological activity in each ECG complex were separated into multiple activation and recovery components for the first time in the murine Scn5a+/ΔKPQ system. This extends previous analysis using only the single indicator of QT interval9, 28. Thus, activation changes were also demonstrated, extending previous results that only examined recovery abnormalities in LQTS models. Two, QC and QD, parameters describing durations of ventricular electrophysiological activity were identified and analysed. Both parameters were prolonged independently both with the presence of Scn5a+/ΔKPQ genotype and age. Prolongations of the QC duration were attributed to increases in the activation, QS, component, in turn arising from interacting effects of age and Scn5a+/ΔKPQ genotype. However, neither age nor Scn5a+/ΔKPQ genotype affected the recovery, SC, component. The SC component is therefore unlikely to include any APD prolongation attributable to the increased I Na-L known to occur in Scn5a+/ΔKPQ 18.
In contrast, prolongation of the QD duration arose from changes in both activation, QS, and recovery, SD, components. Thus, SD was prolonged by the Scn5a+/ΔKPQ genotype but not by age. SD is therefore likely to include APD components specifically related to the increased I Na-L in Scn5a+/ΔKPQ. A fuller breakdown of clinical ECG QT duration into activation and recovery components, than that currently adopted, could therefore reveal that prolonged QT intervals in LQTS reflect not only recovery, but also activation abnormalities.
This combination of both activation and recovery abnormalities observed in the old Scn5a+/ΔKPQ mouse might then account for an appearance of overlap syndromes with age in LQTS3 patients. These syndromes are characterised by a co-existence of more than one of the separate phenotypes expected from individual SCN5A mutations in the same patient50. SCN5A mutations are variously associated with LQTS3, BrS, progressive cardiac conduction defect (PCCD), sick sinus node syndrome (SSS), atrial fibrillation (AF), and even dilated cardiomyopathy (DCM)51. We thus complement previous reports in which an age-dependent penetrance of ECG characteristics was associated with the SCN5A-1795insD mutation. The carriers of this particular mutation showed QT-prolongations and signs of conduction disease both of which were present from birth. They also showed BrS features that developed at a later age52, 53. The presence of overlap syndromes in ageing LQTS3 patients could have potential implications concerning the choice of therapeutic agents.
Secondly, the present experiments went on to explore for possible structural mechanisms for the observed age-dependent phenotypic changes. They complement the previous studies in murine loss-of-function Scn5a+/− hearts that had implicated fibrotic change in the arrhythmogenesis in BrS,40. The present study associated for the first time the gain-of-function Scn5a+/ΔKPQ mutation with increased fibrosis in both atria and ventricles. The observed increases in fibrotic change likely arise from different signalling cascades from those in the loss-of-function BrS murine model, where Na+ channel deficiency manifests as a TGF-β1-mediated fibrosis associated with Scn5a disruption or aging54. For example, the mechanism in the gain-of-function Scn5a+/ΔKPQ could relate to altered Ca2+ homeostasis accompanying the elevated I Na-L 55. The resulting Ca2+ load could then compromise mitochondrial function, increasing a pro-fibrotic ROS production56, 57.
Furthermore, the observed independent actions of both age and genotype upon ventricular fibrotic change correlated with the electrophysiological interacting effects of age and Scn5a+/ΔKPQ genotype in compromising ventricular activation. The mechanistic basis for this could potentially involve gap junction disruption from the fibrotic change, which would then increase resistance to local circuit currents and hinder AP propagation41. Alternatively, fibroblasts or myofibroblasts (Mfbs) could form couplings with cardiomyocytes through Cx43 and Cx45 binding58, 59. The resulting increased cell capacitance would reduce conduction velocity to extents varying with ratio of the numbers of Mfbs to cardiomyocytes42, 60, 61. This would increase arrhythmic tendency through formation of slow-conducting re-entry circuits62, 63. Thus, therapeutic agents for LQTS3 patients, particularly with age, may need to addresses the ventricular activation deficit. One possibility may be through targeting the fibrotic change itself. For example, the use of angiotensin-converting enzyme inhibitors to inhibit RAAS have been shown to reduce myocardial fibrosis in both experimental and clinical studies64, 65.
Thirdly, ECG measurements were compared before and following acute β-adrenergic challenge. Thus dobutamine challenge exerted positive chronotropic and dromotropic effects, reflected in increased ventricular rates and reduced PR segment and PR interval durations, whilst leaving QS intervals unchanged. This could reflect similar and positive effects of β-adrenergic stimulation upon both pacing currents and atrial but not ventricular conduction in both WT and Scn5a+/ΔKPQ.
Fourthly, ventricular repolarisation alternans was compared between experimental groups, and the effect of dobutamine challenge upon this then investigated in the study population. Before pharmacological intervention, old Scn5a+/ΔKPQ mice showed higher incidences and durations of ventricular repolarisation alternans. Clinically, ventricular repolarisation alternans, reflected in T-wave alternans (TWA), is known to occur in LQTS44, with implications for the pathophysiological mechanism of sudden cardiac death (SCD)43. Repolarisation alternans is closely related to the Ca2+ cycling and mechanical alternans43 attributed to variations in intracellular sarcoplasmic reticulum (SR) Ca2+ release66. Thus, both are abolished by reduced [Ca2+]o or ryanodine-mediated RyR2 block44. An increased I Na-L attributed to Scn5a+/ΔKPQ would increase [Ca2+]i through the power function relationship it bears to [Na+]i 67, 68 thereby contributing to development of alternans. The observed interacting effects of Scn5a+/ΔKPQ genotype and age are consistent with increased incidences of alternans in old Scn5a+/ΔKPQ mice producing an underlying Ca2+ cycling alternans given age-related reductions in myocardial SR Ca2+ ATPase (SERCA) density69.
Dobutamine challenge then exerted anti-arrhythmic effects in reducing the incidence and duration of repolarisation alternans. Clinical findings however indicate that adrenergic activity and heart rate exert paradoxical effects on the incidence of TWA in LQTS3. This is reflected in earlier findings of nocturnal episodes of peak TWA in LQTS3 patients that were associated with conditions of rate suppression45, 46. Studies separating the effects of β-adrenergic stimulation and imposed heart rates implicated the increases in heart rate rather than β-adrenergic stimulation in TWA47. The present finding that β-adrenergic stimulation paradoxically reduces repolarisation alternans in LQTS3 might reflect reduction of the normal phospholamban (PLB)-mediated inhibition of SERCA-mediated Ca2+ re-uptake70, 71. Alternatively, β-adrenergic stimulation has been reported to shift the I Na inactivation–voltage relationship to more negative potentials decreasing window current and hence INa-L 72,73,74,75. This would decrease [Na+]i, and in turn [Ca2+]i thereby possibly reducing repolarisation alternans.
Finally, the experiments demonstrated that with the Scn5a+/ΔKPQ genotype, there was a delay in the onset time over which ventricular repolarisation duration changed following corresponding increases in ventricular rate produced by dobutamine challenge. In response to a change in ventricular rate, the QT interval normally shortens immediately and then follows a more gradual mono-exponential reduction to a new steady-state value76. Thus, a discrepancy in the onset time of this shortening could result in a transient increase in ventricular repolarisation time relative to RR interval. This may confer arrhythmic susceptibility during heart rate increases in LQTS3 patients for example48. The present findings thus complement previous reports that had similarly reported paradoxically increased AP duration in mice modelling LQTS3 following sudden rate increases18. The previous studies were performed in isolated ventricular preparations under artificial pacing rather than employing intact animals with normal sympathetic innervations and β-adrenergic stimuli replicating the in vivo situation.
Conclusions
The present study demonstrates for the first time that a gain-of-function Scn5a+/ΔKPQ mutation is associated with an increased level of fibrotic change. This increased fibrosis associated with Scn5a+/ΔKPQ and age slows conduction in the ventricles and could be the cause of the prolonged activation, QS, interval in old Scn5a+/ΔKPQ mice. This has clinical implications in that arrhythmic events in elderly LQTS patients may be due to an activation abnormality as opposed to a sole recovery abnormality. Thus, therapeutic options for LQTS patients may need to be expanded from solely treating the prolonged ventricular repolarisation to treating any activation deficits arising from fibrotic change. Furthermore, therapeutic β-adrenergic agonists or antagonists for LQTS3 patients specifically should consider the paradoxical effects of such administration. The present study demonstrates both pro- and anti-arrhythmic effects of dobutamine challenge, recapitulating the clinical observations of its paradoxical effects. However, the present results suggest that LQTS3 patients are likely to be pro-arrhythmic during the change of heart rate following β-adrenergic stimulation. Moreover, the present results suggest that the anti-arrhythmic effects of β-adrenergic stimulation may be more apparent with age, as there is a reduction in repolarisation alternans and improvement in conduction following acute dobutamine administration. This indicates that age-specific drug choice may be required as the disease phenotype can change with age.
References
Splawski, I. et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102, 1178–1185, doi:10.1161/01.CIR.102.10.1178 (2000).
Wang, Q. et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet 4, 1603–1607 (1995).
Wang, Q. et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80, 805–811 (1995).
Antzelevitch, C. et al. The role of late I Na in development of cardiac arrhythmias. Handb Exp Pharmacol 221, 137–168, doi:10.1007/978-3-642-41588-3_7 (2014).
Jeevaratnam, K. et al. Ion channels, long QT syndrome and arrhythmogenesis in ageing. Clin Exp Pharmacol Physiol, doi:10.1111/1440-1681.12721 (2016).
Johnson, J. N. et al. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm 5, 704–709, doi:10.1016/j.hrthm.2008.02.007 (2008).
Goldenberg, I. et al. Long-QT syndrome after age 40. Circulation 117, 2192–2201, doi:10.1161/CIRCULATIONAHA.107.729368 (2008).
Alders, M. & Christiaans, I. In GeneReviews(R) (eds Pagon R. A. et al.) (1993).
Guzadhur, L. et al. Atrial arrhythmogenicity in aged Scn5a+/DeltaKPQ mice modeling long QT type 3 syndrome and its relationship to Na+ channel expression and cardiac conduction. Pflugers Arch 460, 593–601, doi:10.1007/s00424-010-0851-z (2010).
Jackman, W. M. et al. The long QT syndromes: a critical review, new clinical observations and a unifying hypothesis. Prog Cardiovasc Dis 31, 115–172 (1988).
Schwartz, P. J. et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103, 89–95, doi:10.1161/01.CIR.103.1.89 (2001).
Nemec, J., Hejlik, J. B., Shen, W. K. & Ackerman, M. J. Catecholamine-induced T-wave lability in congenital long QT syndrome: a novel phenomenon associated with syncope and cardiac arrest. Mayo Clin Proc 78, 40–50, doi:10.4065/78.1.40 (2003).
Wilde, A. A. et al. Clinical Aspects of Type 3 Long-QT Syndrome: An International Multicenter Study. Circulation 134, 872–882, doi:10.1161/CIRCULATIONAHA.116.021823 (2016).
Schwartz, P. J. et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation 92, 3381–3386, doi:10.1161/01.CIR.92.12.3381 (1995).
Priori, S. G. et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA 292, 1341–1344, doi:10.1001/jama.292.11.1341 (2004).
Chinushi, M. et al. Antiarrhythmic vs. pro-arrhythmic effects depending on the intensity of adrenergic stimulation in a canine anthopleurin-A model of type-3 long QT syndrome. Europace 10, 249–255, doi:10.1093/europace/eun002 (2008).
Fabritz, L. et al. Autonomic modulation and antiarrhythmic therapy in a model of long QT syndrome type 3. Cardiovasc Res 87, 60–72, doi:10.1093/cvr/cvq029 (2010).
Nuyens, D. et al. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med 7, 1021–1027, doi:10.1038/nm0901-1021 (2001).
Head, C. E. et al. Paced electrogram fractionation analysis of arrhythmogenic tendency in DeltaKPQ Scn5a mice. J Cardiovasc Electrophysiol 16, 1329–1340, doi:10.1111/j.1540-8167.2005.00200.x (2005).
Thomas, G., Killeen, M. J., Grace, A. A. & Huang, C. L. Pharmacological separation of early afterdepolarizations from arrhythmogenic substrate in DeltaKPQ Scn5a murine hearts modelling human long QT 3 syndrome. Acta Physiol (Oxf) 192, 505–517, doi:10.1111/j.1748-1716.2007.01770.x (2008).
Dumaine, R. et al. Multiple mechanisms of Na+ channel–linked long-QT syndrome. Circ Res 78, 916–924, doi:10.1161/01.RES.78.5.916 (1996).
Clancy, C. E. & Rudy, Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature 400, 566–569, doi:10.1038/23034 (1999).
Stokoe, K. S. et al. Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/Delta murine hearts modelling long QT syndrome 3. J Physiol 578, 69–84, doi:10.1113/jphysiol.2006.117945 (2007).
Thomas, G. et al. Effects of L-type Ca2+ channel antagonism on ventricular arrhythmogenesis in murine hearts containing a modification in the Scn5a gene modelling human long QT syndrome 3. J Physiol 578, 85–97, doi:10.1113/jphysiol.2006.121921 (2007).
Sabir, I. N. et al. Criteria for arrhythmogenicity in genetically-modified Langendorff-perfused murine hearts modelling the congenital long QT syndrome type 3 and the Brugada syndrome. Pflugers Arch 455, 637–651, doi:10.1007/s00424-007-0326-z (2008).
Jeevaratnam, K. et al. Differences in sino-atrial and atrio-ventricular function with age and sex attributable to the Scn5a+/- mutation in a murine cardiac model. Acta Physiol (Oxf) 200, 23–33, doi:10.1111/j.1748-1716.2010.02110.x (2010).
Hart, C. Y., Burnett, J. C. Jr. & Redfield, M. M. Effects of avertin versus xylazine-ketamine anesthesia on cardiac function in normal mice. Am J Physiol Heart Circ Physiol 281, H1938–1945 (2001).
Wu, J. et al. Altered sinoatrial node function and intra-atrial conduction in murine gain-of-function Scn5a+/DeltaKPQ hearts suggest an overlap syndrome. Am J Physiol Heart Circ Physiol 302, H1510–1523, doi:10.1152/ajpheart.00357.2011 (2012).
Bazett, H. C. An Analysis of The Time-Relations Of Electrocardiograms. Annals of Noninvasive Electrocardiology 2, 177–194, doi:10.1111/j.1542-474X.1997.tb00325.x (1997).
Mitchell, G. F., Jeron, A. & Koren, G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol 274, H747–751 (1998).
Boukens, B. J., Rivaud, M. R., Rentschler, S. & Coronel, R. Misinterpretation of the mouse ECG: ‘musing the waves of Mus musculus’. J Physiol 592, 4613–4626, doi:10.1113/jphysiol.2014.279380 (2014).
Salama, G. & London, B. Mouse models of long QT syndrome. J Physiol 578, 43–53, doi:10.1113/jphysiol.2006.118745 (2007).
Speerschneider, T. & Thomsen, M. B. Physiology and analysis of the electrocardiographic T wave in mice. Acta Physiol (Oxf) 209, 262–271, doi:10.1111/apha.12172 (2013).
Jeevaratnam, K. et al. Delayed conduction and its implications in murine Scn5a(+/−) hearts: independent and interacting effects of genotype, age, and sex. Pflugers Arch 461, 29–44, doi:10.1007/s00424-010-0906-1 (2011).
Levy, M. N. & Edflstein, J. The mechanism of synchronization in isorhythmic A-V dissociation. II. Clinical studies. Circulation 42, 689–699 (1970).
Levy, M. N. & Zieske, H. Mechanism of synchronization in isorhythmic dissociation. I. Experiments on dogs. Circ Res 27, 429–443 (1970).
Goldbarg, A. N., Hellerstein, H. K., Bruell, J. H. & Daroczy, A. F. Electrocardiogram of the normal mouse, Mus musculus: general considerations and genetic aspects. Cardiovasc Res 2, 93–99 (1968).
Zhang, Y., Wu, J., King, J. H., Huang, C. L. & Fraser, J. A. Measurement and interpretation of electrocardiographic QT intervals in murine hearts. Am J Physiol Heart Circ Physiol 306, H1553–1557, doi:10.1152/ajpheart.00459.2013 (2014).
Shimizu, W. & Antzelevitch, C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol 35, 778–786, doi:10.1016/S0735-1097(99)00582-3 (2000).
Jeevaratnam, K. et al. Frequency distribution analysis of activation times and regional fibrosis in murine Scn5a+/- hearts: the effects of ageing and sex. Mech Ageing Dev 133, 591–599, doi:10.1016/j.mad.2012.07.006 (2012).
van Veen, T. A. et al. Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation 112, 1927–1935, doi:10.1161/CIRCULATIONAHA.105.539072 (2005).
Davies, L. et al. Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J Am Heart Assoc 3, e000340, doi:10.1161/JAHA.113.000340 (2014).
Walker, M. L. & Rosenbaum, D. S. Repolarization alternans: implications for the mechanism and prevention of sudden cardiac death. Cardiovasc Res 57, 599–614, doi:10.1016/S0008-6363(02)00737-X (2003).
Shimizu, W. & Antzelevitch, C. Cellular and ionic basis for T-wave alternans under long-QT conditions. Circulation 99, 1499–1507, doi:10.1161/01.CIR.99.11.1499 (1999).
Takasugi, N. et al. Prevalence of Microvolt T-Wave Alternans in Patients With Long QT Syndrome and Its Association With Torsade de Pointes. Circ Arrhythm Electrophysiol 9, e003206, doi:10.1161/CIRCEP.115.003206 (2016).
Nishizaki, M., Fujii, H., Sakurada, H., Kimura, A. & Hiraoka, M. Spontaneous T wave alternans in a patient with Brugada syndrome–responses to intravenous administration of class I antiarrhythmic drug, glucose tolerance test, and atrial pacing. J Cardiovasc Electrophysiol 16, 217–220, doi:10.1046/j.1540-8167.2004.40411.x (2005).
Kaufman, E. S., Mackall, J. A., Julka, B., Drabek, C. & Rosenbaum, D. S. Influence of heart rate and sympathetic stimulation on arrhythmogenic T wave alternans. Am J Physiol Heart Circ Physiol 279, H1248–1255 (2000).
Attwell, D. & Lee, J. A. A cellular basis for the primary long Q-T syndromes. Lancet 1, 1136–1139 (1988).
Smirk, F. H. R waves interrupting T waves. Br Heart J 11, 23–36 (1949).
Napolitano, C., Rivolta, I. & Priori, S. G. Cardiac sodium channel diseases. Clin Chem Lab Med 41, 439–444, doi:10.1515/CCLM.2003.066 (2003).
Ruan, Y., Liu, N. & Priori, S. G. Sodium channel mutations and arrhythmias. Nat Rev Cardiol 6, 337–348, doi:10.1038/nrcardio.2009.44 (2009).
Remme, C. A., Wilde, A. A. & Bezzina, C. R. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med 18, 78–87, doi:10.1016/j.tcm.2008.01.002 (2008).
Beaufort-Krol, G. C. et al. Developmental aspects of long QT syndrome type 3 and Brugada syndrome on the basis of a single SCN5A mutation in childhood. J Am Coll Cardiol 46, 331–337, doi:10.1016/j.jacc.2005.03.066 (2005).
Hao, X. et al. TGF-beta1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ Arrhythm Electrophysiol 4, 397–406, doi:10.1161/CIRCEP.110.960807 (2011).
Belardinelli, L., Giles, W. R., Rajamani, S., Karagueuzian, H. S. & Shryock, J. C. Cardiac late Na(+) current: proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm 12, 440–448, doi:10.1016/j.hrthm.2014.11.009 (2015).
Judge, S., Jang, Y. M., Smith, A., Hagen, T. & Leeuwenburgh, C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J 19, 419–421, doi:10.1096/fj.04-2622fje (2005).
Wu, J., Xia, S., Kalionis, B., Wan, W. & Sun, T. The role of oxidative stress and inflammation in cardiovascular aging. Biomed Res Int 2014, 615312, doi:10.1155/2014/615312 (2014).
Camelliti, P., Green, C. R., LeGrice, I. & Kohl, P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res 94, 828–835, doi:10.1161/01.RES.0000122382.19400.14 (2004).
Chilton, L., Giles, W. R. & Smith, G. L. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol 583, 225–236, doi:10.1113/jphysiol.2007.135038 (2007).
Xie, Y. et al. Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: A computational study. Heart Rhythm 6, 1641–1649, doi:10.1016/j.hrthm.2009.08.003 (2009).
Francis Stuart, S. D., De Jesus, N. M., Lindsey, M. L. & Ripplinger, C. M. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol 91, 114–122, doi:10.1016/j.yjmcc.2015.12.024 (2016).
King, J. H., Huang, C. L. & Fraser, J. A. Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front Physiol 4, 154, doi:10.3389/fphys.2013.00154 (2013).
Mandapati, R., Skanes, A., Chen, J., Berenfeld, O. & Jalife, J. Stable microreentrant sources as a mechanism of atrial fibrillation in the isolated sheep heart. Circulation 101, 194–199, doi:10.1161/01.CIR.101.2.194 (2000).
Stein, M. et al. Reduction of fibrosis-related arrhythmias by chronic renin-angiotensin-aldosterone system inhibitors in an aged mouse model. Am J Physiol Heart Circ Physiol 299, H310–321, doi:10.1152/ajpheart.01137.2009 (2010).
Brilla, C. G., Funck, R. C. & Rupp, H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 102, 1388–1393, doi:10.1161/01.CIR.102.12.1388 (2000).
Lab, M. J. & Lee, J. A. Changes in intracellular calcium during mechanical alternans in isolated ferret ventricular muscle. Circ Res 66, 585–595, doi:10.1161/01.RES.66.3.585 (1990).
Eisner, D. A., Lederer, W. J. & Sheu, S. S. The role of intracellular sodium activity in the anti-arrhythmic action of local anaesthetics in sheep Purkinje fibres. J Physiol 340, 239–257 (1983).
Eisner, D. A., Lederer, W. J. & Vaughan-Jones, R. D. The quantitative relationship between twitch tension and intracellular sodium activity in sheep cardiac Purkinje fibres. J Physiol 355, 251–266 (1984).
Cain, B. S. et al. Human SERCA2a levels correlate inversely with age in senescent human myocardium. J Am Coll Cardiol 32, 458–467 (1998).
Sampson, K. J. & Kass, R. S. Molecular mechanisms of adrenergic stimulation in the heart. Heart Rhythm 7, 1151–1153, doi:10.1016/j.hrthm.2010.02.013 (2010).
Campbell, A. S., Johnstone, S. R., Baillie, G. S. & Smith, G. beta-Adrenergic modulation of myocardial conduction velocity: Connexins vs. sodium current. J Mol Cell Cardiol 77, 147–154, doi:10.1016/j.yjmcc.2014.09.030 (2014).
Windisch, H. & Tritthart, H. A. Isoproterenol, norepinephrine and phosphodiesterase inhibitors are blockers of the depressed fast Na + -system in ventricular muscle fibers. J Mol Cell Cardiol 14, 431–434 (1982).
Hisatome, I., Kiyosue, T., Imanishi, S. & Arita, M. Isoproterenol inhibits residual fast channel via stimulation of beta-adrenoceptors in guinea-pig ventricular muscle. J Mol Cell Cardiol 17, 657–665 (1985).
Schubert, B., Vandongen, A. M., Kirsch, G. E. & Brown, A. M. Inhibition of cardiac Na+ currents by isoproterenol. Am J Physiol 258, H977–982 (1990).
Ono, K., Kiyosue, T. & Arita, M. Isoproterenol, DBcAMP, and forskolin inhibit cardiac sodium current. Am J Physiol 256, C1131–1137 (1989).
Seethala, S., Shusterman, V., Saba, S., Mularski, S. & Nemec, J. Effect of beta-adrenergic stimulation on QT interval accommodation. Heart Rhythm 8, 263–270, doi:10.1016/j.hrthm.2010.10.012 (2011).
Acknowledgements
KC was funded by the Physiological Society, United Kingdom at the University of Surrey. SA is funded by a Medical Research Council Research Fellowship (MR/M001288/1). HV is funded by the Wellcome Trust Research Training Fellowship (105727/Z/14/Z) and Sudden Arrhythmic Death Syndrome (SADS), UK. AG is funded by the McVeigh Benefaction and Sudden Arrhythmic Death Syndrome (SADS), UK. CLHH is funded by the Medical Research Council (MR/M001288/1), Wellcome Trust (105727/Z/14/Z), British Heart Foundation (PG/14/79/31102), the McVeigh Benefaction and SADS UK. KJ is funded by the Fundamental Research Grant Scheme (FRGS/2/2014/SKK01/PERDANA/02/1), Ministry of Education, Malaysia and the Research Support Fund, Faculty of Health and Medical Science, University of Surrey.
Author information
Authors and Affiliations
Contributions
K.C. undertook the experiments, analysed all data, drafted the initial manuscript and edited subsequent drafts. S.A., H.V., I.D.U. and A.B.A. analysed data and edited subsequent drafts. I.D.U. provided additional statistical support. S.S. undertook experiments and edited subsequent drafts of the manuscript. A.A.G. provided the murine models and conceptualised the study. C.L.H. conceptualized the study, drafted the initial manuscript and edited subsequent drafts. K.J. conceptualized the study, undertook experiments, analysed the data, help draft the initial manuscript and edited the subsequent drafts;
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chadda, K.R., Ahmad, S., Valli, H. et al. The effects of ageing and adrenergic challenge on electrocardiographic phenotypes in a murine model of long QT syndrome type 3. Sci Rep 7, 11070 (2017). https://doi.org/10.1038/s41598-017-11210-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11210-3
- Springer Nature Limited