
Abstract
Triadin-1, encoded by TRDN, is an important component of the calcium release unit (CRU) in the sarcoplasmic reticulum of cardiac myocytes, interacting both with ryanodine receptors and calsequestrin. This article reviews evidence substantiating a link between TRDN mutations and potentially fatal ventricular arrhythmias.
Evidence from mouse TRDN knockout models indicates structural and functional changes to the cardiac myocyte CRU in the absence of triadin that result in disturbed Ca2+ handling and susceptibility to adrenergic agonist provoked arrhythmias. In particular, cellular electrophysiology experiments have provided evidence of reduced Ca2+ induced inactivation of L-type Ca2+ channels consequent to TRDN knockout. A number of reports since 2012 have identified patients with a CPVT phenotype, some of whom also exhibit prolonged QTc intervals, with TRDN mutations. Symptomatic patients have homozygous or compound heterozygous mutations predicted to result in absent or nonfunctional triadin. Typically they have experienced serious ventricular arrhythmias or cardiac arrest at a very young age against a background of exertion, excitement or stress. Some patients have also shown skeletal muscle weakness. β adrenoceptor blockers, particularly nadolol, have been used successfully in some patients, though others have continued to experience cardiac events. Implantable devices have been critically important in cardioverting episodes of ventricular fibrillation in a number of patients. Flecainide has been successful at reducing the burden on implantable devices in two patients. Potential future additional/alternative pharmacological treatments include L-type Ca2+ channel blockers, the class Ic antiarrhythmic propafenone and the β adrenoceptor blocker carvedilol. The potential value of such strategies should be explored using appropriate in vitro and in vivo models. In conclusion, triadin knockout syndrome is inherited in an autosomal recessive fashion and should be considered in cases of CPVT or Long QT Syndrome in which mutations to ion channels/transporters are not found. It should be particularly, though not exclusively, suspected in cases with concomitant muscle weakness. Further work is required to identify optimal therapeutic strategies.
Similar content being viewed by others

Background
Cardiac excitation-contraction (E-C) coupling is well established to rely on the process of “calcium induced calcium release” (CICR) [1]. CICR is initiated by opening of sarcolemmal L-type calcium channels during the rising phase of the action potential; Ca2+ ions entering via this route trigger the sarcoplasmic reticulum (SR) to release a larger amount of Ca2+ via RyR2 ryanodine receptors [1]. This produces a transient rise in cytosolic [Ca2+] that initiates contraction [1]. RyR2 proteins, together with calsequestrin (Casq2), junctin and triadin form a protein complex in the “junctional” SR (that region of the SR juxtaposed to the t-tubule/plasma membrane containing L-type Ca channels) termed the calcium release complex or calcium release unit (CRC or CRU). Triadin appears to be important to the structural and functional integrity of the complex, influencing intraluminal control of Ca2+ release via RyR2s [2, 3]. Three forms of triadin exist (Trisk 32, Trisk 51, Trisk 95) that are encoded by a single TRDN gene, via alternative splicing [2, 3]. Whilst higher molecular weight isoforms can be detected in the heart, the predominant cardiac isoform is the Trisk 32 (32 kDa) isoform [4]. Accumulating evidence from recent mouse and human studies has implicated loss of functional triadin with heritable ventricular arrhythmia and risk of sudden death [2, 5–8]. Here we summarize briefly the evidence for involvement of TRDN mutations in human ventricular arrhythmia, discuss potential underlying mechanisms of arrhythmogenesis and summarize present and potential future treatment avenues.
The role of triadin-1 in ventricular myocytes
The predominant cardiac triadin (Trisk 32/triadin-1) is comprised of a single transmembrane domain, a cytoplasmic N terminus and luminal C terminus (shown schematically in Fig. 1a). The first 47 amino terminal amino acids of the 286 amino-acid protein are followed by a short transmembrane region (residues 48–68) located in the junctional SR membrane and a long luminal C terminal tail [3, 9]. A conserved region of the luminal domain, containing “KEKE” (lysine-glutamate-lysine-glutamate) motif repeats (which have long been considered to promote interactions between proteins [10]), is involved in binding to Casq2 and RyR2 [3, 9] (Fig. 1a, b). Exons 1–8 of TRDN encode the first 264 residues of both cardiac and skeletal triadin isoforms, raising the possibility that mutations in these exons may affect both cardiac and skeletal muscle function [7].

Triadin 1 interactions with RyR2 and Casq2 proteins. a Schematic representation of Triadin 1 in relation to sarcoplasmic reticulum (SR) membrane. The cytoplasmic amino terminal is followed by a transmembrane segment, then intracellular segment of the protein. The latter includes the “KEKE” (Lysine-Glutamate-Lysine-Glutamate) motif that interacts with RyR2 and Casq2 proteins. bi Schematic representation of junctional sarcoplasmic reticulum (SR) in relation to surface membrane. The L type Ca channel in the sarcolemmal membrane is juxtaposed to the RyR2. Vertical arrow denotes the direction of Ca2+ flux (influx) through the L type channel. The RyR2 is embedded in the junctional SR membrane. Vertical arrow denotes the direction of Ca2+ flux (efflux) from the SR through the RyR2. Triadin-1 is shown to the right of the RyR2. Transmembrane and KEKE domains are shown in black. In ‘A’ the KEKE domain is shown interacting with the RyR2 channel and with Casq2 proteins (shown as open pentagons). bii Schematic representations of similar structures to those in ‘A’, with one difference. Triadin-1 is now shown as a truncated protein, lacking the KEKE domain (and distal protein). In consequence, triadin-1 can no longer interact with the RyR2 or Casq2 proteins. Diagonal dashed lines indicate that mutations that result in premature stop codons in cytoplasmic, transmembrane or internal segments, upstream of the KEKE motif, can be expected to result in nonfunctional protein
Experiments employing TRDN ‘knockout’ (KO) mice have provided insight into the roles of both skeletal muscle triadin isoforms [5, 11, 12]. Skeletal muscles from two different TRDN KO mice lines were found to show ~30% of triads (structures formed by a t-tubule and adjacent terminal cisterna of the SR, either side of the t-tubule) in a longitudinal or oblique instead of transverse orientation and exhibited reduced Ca2+ release [11, 12]. Some experiments also supported the occurrence of muscle weakness (reduced force) [12]. In cardiac muscle, TRDN deletion has been reported to alter the structure of the CRU [5]. In myocytes lacking triadin, junctional SR proteins (RyR2, Casq2, junctin and junctophilin) were decreased and Casq2 occurred outside of the junctional SR. [5] Additionally, electron micrographs showed reduced frequency of junctional SR cisternae and shorter RyR2 containing contacts, with a ~50% reduction in contacts between the junctional SR and the t tubules [5]. Levels of Cav1.2 and SERCA2a were unaffected, but colocalization between Cav1.2 and RyR2 was reduced. In cellular electrophysiology experiments, the amplitude of L-type Ca2+ current (ICaL) was unaltered in TRDN−/− myocytes, but the timecourse of ICaL inactivation was delayed [5]. This effect was interpreted as resulting from a decreased influence of Ca2+ release through RyRs on the L-type channels, reducing Ca2+-dependent inactivation of ICaL [5]. Further experiments in which SR Ca2+ release was disabled (using the SR inhibitors ryanodine or thapsigargin) abolished differences in ICaL inactivation between wild-type and TRDN−/− myocytes, supporting this explanation [5]. The effects of the β-adrenergic agonist isoprenaline on ICaL amplitude were greater in TRDN−/− than wild-type myocytes, accompanied by slowed ICaL inactivation [5]. Intracellular Ca2+ transients were found to be reduced in amplitude and rise-time in TRDN−/− compared to wild-type myocytes, whilst diastolic [Ca2+] and SR Ca2+ content were increased [5]. The increase in SR Ca2+ content was likely attributable to impaired ICaL inactivation. Fractional SR Ca2+ release was reduced in TRDN−/− compared to wild-type myocytes (presumably reflecting the reduced colocalization between RyRs and L-type channels); this difference was less marked in isoprenaline-stimulated conditions [5]. In electrically paced cells, the number of spontaneous Ca2+ release (SCR) events was low in control conditions for both wild-type and TRDN−/− myocytes; however, in the presence of isoprenaline, TRDN−/− myocytes exhibited a markedly increased frequency of SCRs [5]. Pretreatment with the ICaL inhibitor nifedipine removed the differences in SCR frequency between wild-type and TRDN−/− myocytes. In vivo ECG measurements showed a higher rate of ventricular ectopics and tachycardia in TRDN−/− than wild-type animals, in response to isoprenaline injection. In summary, structural changes to the CRU, together with altered Ca2+ handling, in particular slowed ICaL inactivation, render TRDN−/− mice more susceptible to adrenergic-stress induced ventricular tachycardia [5]. Additionally, a separate recent study has provided evidence that triadin-1 may also be important for recovery of EC coupling following ischaemia: TRDN−/− hearts exhibited impaired contractile recovery following ischaemia/reperfusion through endoplasmic reticulum stress-mediated apoptosis and activation of calpain proteases [13].
TRDN mutations associated with human ventricular arrhythmia
Catecholaminergic Polymorphic Ventricular Tachycardiac (CPVT) is a polymorphic or bidirectional arrhythmia associated with syncope and risk of ventricular fibrillation and sudden death, brought on by emotional stress or physical exertion [2, 14–16]. Up to 50–65% of CPVT cases are linked to mutations in RYR2 or CASQ2 that lead to leak or spillover of SR Ca2+ during diastole that can result in Ca2+ overload and ventricular arrhythmia under situations of adrenergic stimulation [14, 16]. In 2012, Roux-Buisson et al. performed candidate gene screening on a cohort of 97 CPVT patients negative for mutations in RYR2 or CASQ2, focusing on TRDN and the junctin gene (ASPH) [6]. Two families were identified with TRDN mutations. The proband of the first family was a 2 year old boy who experienced syncope and cardiac arrest whilst playing. ECG analysis post-resuscitation revealed multiple polymorphic or bidirectional extra beats and polymorphic VT [6]. The child died 3 weeks after the initial arrest following severe post-anoxic coma. He was found to be homozygous for a mutation in TRDN exon 2: a deletion of 4 nucleotides (c.del53_56ACAG) led to a frameshift mutation (p.D18Afs*13). This resulted in a premature stop codon at residue 31, before the transmembrane segment of triadin-1 [6]. The parents and sibling of the proband were heterozygous for this mutation and were asymptomatic, consistent with autosomal recessive transmission. In the second family, a 26 year old man who had experienced exercise-associated syncope throughout his life was identified to have compound heterozygous mutations in exon 2 (c.176C > G; p.T59R) and exon 8 (c.613C > T; p.Q205*), the former leading to a missense mutation in the transmembrane segment of triadin and the latter leading to a premature stop codon on the intracellular portion of the protein at a location upstream of the KEKE RyR2/Casq2 interaction site [6]. His brother was similarly affected. Experiments on T59R expressed in a cell line showed that the mutation results in an inability of triadin to exit the endoplasmic reticulum following synthesis (observed using immunofluorescent labelling and confocal microscopy) and that it was subject to degradation by the proteasome pathway [6]. Lack of functional T59R triadin-1 protein was confirmed in cardiac myocytes from TRDN knockout mice transduced to express T59R triadin. The compound mutations in this patient would thus be anticipated to lead to nonfunctional/absent triadin.
A subsequent study in 2015 performed whole exome sequencing on 33 patients with a long QT syndrome phenotype but no identified underlying mutation in normal candidate genes [7]. Five index patients, all of 10 years or younger in age, were identified from different families; they had QTc intervals ranging from 464 to 500 ms and had all experienced exertion/stress induced syncope and cardiac arrest by 3 years of age [7]. All patients exhibited extensive T wave inversion in precordial leads V1 to V4. One patient exhibited the same homozygous p.D18fs*13 (c.del53_56ACAG) mutation reported by Roux-Buisson et al., 2012 [6]. Three patients (from different families) exhibited a homozygous c.del 572_576TAAGA deletion mutation, which resulted in an immediate stop codon (p.K147 fs*0). Parents were asymptomatic and, where DNA was available, were heterozygotic carriers of the K147 fs*0 mutation, consistent with autosomal recessive transmission. For one patient, a cousin had died suddenly at age 6 and DNA from the uncle showed heterozygotic K147 fs*0 (for neither the cousin nor aunt was DNA available, however). For another, an older sister had experienced syncope and cardiac arrest at 2 yrs. of age (though DNA was unavailable) [7]. The fifth case possessed compound heterozygotic mutations: the K147 fs*0 mutation and a c.22 + 29 G > A (p.N9 fs*5) splicing error mutation [7]. All five patients would be anticipated to be essentially triadin-null, leading the authors to classify the condition “triadin knockout syndrome”. No triadin mutations were identified in patients of >10 years of age in the LQT cohort [7].
A second, independent study in 2015 reported an additional family with compound heterozygotic triadin mutations [17]. One girl had experienced syncope whilst playing at age 5.5; cardiac evaluation found a normal QT interval and a 24 h ECG found only isolated premature ventricular contractions (PVCs) and atrial contractions [17]. An exercise test showed isolated PVCs with intense effort. Subsequently her younger sister (4.5 yrs) underwent sudden death whilst picking fruit under great excitement. She was resuscitated from VF and her resting ECG thereafter showed monomorphic PVCs. An exercise test was unrevealing, but infusion of isoprenaline in both girls showed multiple polymorphic PVCs [17]; neither girl showed signs of muscle weakness. Genetic testing revealed compound heterozygotic TRDN mutations. One was the c.613C > T mutation (originally identified in a separate family by [6]) leading to p.Q205*, with a premature stop codon upstream of the KEKE domain [17]. The second was the same c.22 + 29 G > A splicing mutation as that observed by Altmann and colleagues [7]. These changes were predicted to lead to an absence of functional triadin either through nonsense mediated mRNA decay or production of shortened protein lacking the interaction domain for RyR2/Casq2 [17]. Nadolol treatment was found to be effective in both patients and defibrillators were not implanted on this basis. A younger, asymptomatic sister (3 yrs) was found also to possess both mutations and was given prophylactic β blocker therapy [17].
We recently reported two siblings with compound heterozygotic triadin mutations [8]. A boy presented in 2003 with ventricular fibrillation (VF) at 2 years of age whilst running in a playground. Whilst successfully resuscitated, he experienced significant neurological injury. His QTc interval after resuscitation was 480 ms, but no pre-arrest comparator was available. He was treated with atenolol, but suffered VF again two years later. Screening for known LQT and CPVT mutations was negative [8]. His younger sister had a QTc of 460 ms and received atenolol due to her brother’s history. In 2013 at 2 yrs., she had an out of hospital arrest whilst playing. She received CPR followed by cardioversion from VF, first by paramedics and then in hospital. During resuscitation, biventricular VT was identified [8]. Both siblings were then identified to have the same compound heterozygotic triadin mutations: D18fs*13 and a c502G > T nonsense mutation (p.E168*), which resides in the intracellular portion of the protein upstream of the KEKE RyR2/Casq2 interaction site [8].
Figure 1bi shows schematically the normal interactions between triadin-1, RyR2 and Casq2, whilst Fig. 1bii illustrates that mutations upstream of the KEKE site (assuming they do not prevent protein expression entirely) can be anticipated to lead to non-functional protein, unable to interact with RyR1 and Casq2. Common features across all the above reported cases are autosomal recessive inheritance and manifestation of either exercise or emotional stress/excitement induced arrhythmias at an early age. Some, though not all, cases were associated with persistent or transient QTc prolongation, raising the possibility that triadin knockout syndrome crosses boundaries between CPVT and LQT syndromes [7, 16]. QTc prolongation in this setting presumably reflects delayed ICaL inactivation [5] and so more sustained depolarizing current during the action potential plateau. It is not clear why QTc prolongation has been observed in some but not other patients. Despite the fact that mutations in exons 1–8 would be anticipated to affect both cardiac and skeletal forms of triadin, only some patients presented with clear signs of mild-moderate muscle weakness [6–8, 17]. In some instances, this may have been confounded by neurological damage [8], whilst instances of death at a very young age may also make it difficult to observe [6].
Present and potential future treatment options
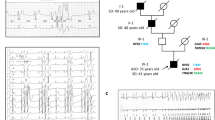
Given the strong association between arrhythmia onset and exercise in patients with triadin-1 mutations, β-blocker therapy would seem to be a reasonable therapeutic option, but has been found to be ineffective at preventing episodes of arrhythmia in a number of cases. On the one hand, the proband and brother with the compound p.T59R and p.Q205* mutations were treated with the nonselective β-blocker nadolol (40 mg/day), though the long term effectiveness of this was not reported [6]. In the subsequent report by the same authors regarding the young girls with the p.Q205* and c.22 + 29 G > A, nadolol (50 mg/m2) was deemed sufficiently effective to obviate the need for an implantable device [17]. On the other hand, patients in the study by Altmann et al. [7] still exhibited cardiac events following β-blocker therapy; some also underwent left cardiac sympathetic denervation (LCSD), but continued to experience arrhythmic episodes [7]. Both patients with the p.D18fs*13 and p.E168* mutations experienced events after receiving atenolol (1–1.5 mg/kg) [8]. These findings indicate mixed effectiveness of β-blockade/LCSD and further work is required to determine the extent to which drug selection/dosing regimen may influence β-blocker effectiveness. All patients in the Altmann et al. [7] and Walsh et al. [8] studies ultimately received implantable cardioverter defibrillators (ICDs). ICD implantation together with β-blockade is likely to have significantly decreased overall risk, especially given the evidence for successful post-implantation shocks by ICDs in these patients [7, 8] (Fig. 2). However, ICD implantation in small children has attendant technical challenges [14]. A question thus arises as to what additional/alternative pharmacological treatments may be beneficial in patients with TRDN mutations? A cardiac sympathectomy is also a potential treatment modality, however considerable expertise is required in performing this procedure.

Arrhythmia during intense emotional state and ICD cardioversion. Ventricular arrhythmia ICD cardioversion in a patient with the compound heterozygotic p.D18fs*13 and p.E168* TRDN mutations. a The left hand box in the upper panel shows the development of increasing ventricular ectopy during a temper tantrum. This deteriorated to a rapid ventricular arrhythmia which degenerated to ventricular fibrillation. The ventricular fibrillation was detected by the implantable defibrillator. b Two shocks were given by the ICD. The first shock (5.1 J) did not terminate the arrhythmia. The second shock (10 J) restored sinus rhythm. Prescribed medications at the time were propranolol (3 × 25 mg/day and flecainide (3 × 25 mg/day)
Multiple subcellular/cellular mechanisms of arrhythmogenesis for CPVT mutations in RyR2 and Casq2 have been proposed (reviewed in [14]), ultimately leading to Ca2+ overload and delayed after-depolarizations (DADs). Although SR Ca2+ leak was not measured directly in experiments on myocytes from TRDN KO mice, key aspects of the cellular phenotype indicate that it was unlikely that SR leak was increased. Both diastolic [Ca2+] and SR Ca2+ content were increased as were SCR events in the presence of isoprenaline compared to control. Thus, the principal mechanism for disrupted Ca2+ handling in TRDN KO mice may be the delayed inactivation of ICaL disrupting Ca2+ homeostasis and indirectly leading to DADs, especially given the beneficial effects in single cell experiments of nifedipine [5]. This may suggest that, in principle at least, clinical administration of L-type Ca2+ channel inhibitors might be beneficial in TRDN mutation patients [5, 7, 16]. To date, no experimental data to test this have been obtained from mice genetically engineered to express clinically observed TRDN mutations. However, as triadin-1 with clinically observed mutations would be expected to be absent or lack RyR/Casq interaction sites, a TRDN deletion model may be a sufficient approximation of the syndrome. One potential limitation, however, arises from the abbreviated nature of the mouse ventricular action potential: SCR in TRDN −/− myocytes were associated with late arrhythmic events consistent with DAD induction. However, the mouse ventricular action potential lacks the high plateau phase present in larger species including humans. Delayed ICaL inactivation in a setting of β-adrenergic drive could potentially both prolong action potential duration and induce early after-depolarizations (EADs). Thus, it is possible (i) that both EADs and DADs could occur in Triadin KO syndrome and (ii) that both proarrhythmic events might be sensitive to ICaL inhibitors such as verapamil.
A further possible treatment avenue is highlighted by the fact that patients with the p.D18fs*13 and p.E168* mutations responded positively to administration of flecainide [8]. Both patients remained arrhythmia-free for ~2 years on flecainide. Although one subsequently experienced appropriate ICD shocks due to episodes of VT/VF, this may have been due to noncompliance with flecainide and occurred when plasma levels were low. Flecainide has previously been found to be effective in patients with RyR2/Casq-linked CPVT [18, 19]. The mechanism by which flecainide exerts this protective effect is a matter of active debate. Some evidence points to a direct effect of the drug on RyR2 channels, either via reducing channel open time or mixed effects on closed/open channel states [20, 21]. However, contradictory evidence suggests that flecainide does not alter luminal to cytosolic Ca2+ flux through RyRs and implicates drug actions on Na channels and cellular Na handling [22–24]. No experimental studies have yet investigated the potential cellular basis for flecainide action in TRDN KO syndrome and such work is required in order better to understand its clinical effects in this setting [8]. If these result, wholly or in part, either from direct effects on RyR2 channels or its Class Ic Na channel actions, then the potential should be explored for another Class Ic drug, propafenone, to be beneficial. In a comparative study of Class I antiarrhythmic drugs (quinidine, procainamide, disopyramide, lignocaine, mexiletine, propafenone, flecainide), only flecainide and propafenone were found to inhibit RyR2 channels in vitro [25]. The RyR2 inhibitory effect of propafenone occurred with both enantiomers of the drug, but R-propafenone exhibited greater potency than did S-propafenone [25]. Propafenone enantiomers significantly reduced arrhythmic Ca2+ waves in single cells from a Casq2 KO mouse model, whilst also preventing exercise-induced CVPT [25]. Moreover, administration of a standard clinical propafenone formulation (phased from 300 mg/day to 900 mg/day) was observed to have a marked effect on arrhythmia incidence in a 22 year old CPVT patient with a RyR2 mutation [25]. An additional feature of propafenone’s pharmacological profile makes it of significant interest in respect of TRDN KO: the drug has been reported to inhibit ICaL at low μM concentrations relevant to the clinical concentration range [26, 27]. Thus, the combination of Class Ic, RyR2 and ICaL effects may make the drug useful in offsetting the consequences of disturbed Ca2+ handling consequent to delayed ICaL inactivation with triadin-1 KO [5]. One potential problem is the current lack of a suitable palatable oral formulation of propafenone, which may make its use in young children more challenging.
The β blocker carvedilol has been shown in vitro to be the only one of fourteen β-blockers tested to inhibit spontaneous Ca2+ waves, an action associated with a direct effect on RyR2 channels by reducing channel open time [28]. The combination of a carvedilol analogue with minimal β-blocking activity with pure β-blockers (metoprolol or bisoprolol) increased the analogue’s efficacy at preventing stress-induced VT in mice with a RyR2 mutation [28]. The potential for carvedilol to protect against TRDN KO associated arrhythmias could be explored in an experimental setting. The benefit of combined α and β-adrenoceptor inhibition in TRDN KO syndrome is not yet established. However, hearts of CASQ2 KO mice with a CPVT phenotype have been shown to express more α1 adrenoceptors than WT controls; moreover, labetolol (which has both α and β-adrenoceptor inhibitory actions) was more effective at suppressing exercise- and adrenaline-induced arrhythmias than were propranolol and metopralol [29]. Similar studies on TRDN −/− mice are warranted to determine whether or not combined α and β-adrenoceptor blockade may be superior to β-blockade alone in this setting.
Conclusions
Accumulating evidence shows TRDN mutations to be responsible for potentially fatal arrhythmias and cardiac arrest, predominantly in very young individuals. The mutations are inherited in an autosomal recessive pattern in either a homozygous or compound heterozygous fashion. Whilst the autosomal recessive transmission in symptomatic patients makes TRDN KO syndrome less likely to occur than conditions with heterozygotic inheritance, the risk of paediatric cardiac arrest at very young ages makes it imperative that cases are identified and managed appropriately. β-blockade has been reported to be successful in some patients, though others have been refractory to this or LCSD. ICD implantation has been essential in preventing sudden death in a number of affected individuals. The data from TRDN KO mice are suggestive that ICaL blockade may be of benefit in patients with TRDN mutations, though this remains to established. Class Ic drugs (flecainide or propafenone) or carvedilol may provide alternative or additional pharmacological approaches. Further work using in vitro and in vivo experimental models should be conducted to interrogate the value and mechanisms of such additional therapeutic strategies.
Abbreviations
- Ca2+ :
-
Calcium
- Casq2:
-
Calsequestrin 2
- CASQ2 :
-
Calsequestrin 2 gene
- CPVT:
-
Catecholaminergic polymorphic ventricular tachycardia
- CRC:
-
Calcium release complex
- CRU:
-
Calcium release unit
- DAD:
-
Delayed after-depolarisation
- EAD:
-
Early after-depolarisation
- E-C:
-
Excitation-contraction
- ECG:
-
Electrocardiogram
- ICaL :
-
L-type calcium channel current
- ICD:
-
Implantable cardioverter defibrillator
- KEKE:
-
Lysine-glutamate-lysine-glutamate
- KO:
-
Knockout
- LQT:
-
Long QT
- PVC:
-
Premature ventricular contractionc
- QTc :
-
Rate corrected QT interval
- RyR2:
-
Ryanodine receptor type 2 Ca2+ release channel
- RYR2 :
-
Ryanodine receptor type 2 Ca2+ release channel gene
- SCR:
-
Spontaneous calcium release
- SR:
-
Sarcoplasmic reticulum
- TRDN −/− :
-
completely lacking (“null” for) the TRDN gene protein product
- TRDN +/− :
-
heterozygotic for the TRDN gene protein product
- TRDN :
-
Triadin gene
- TRDN:
-
Triadin protein
- VF:
-
Ventricular fibrillation
- VT:
-
Ventricular tachycardia
References
Bers DM. Excitation-contraction coupling and cardiac contractile force. 2nd ed. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2001.
Chopra N, Knollmann BC. Triadin regulates cardiac muscle couplon structure and microdomain Ca2+ signalling: a path towards ventricular arrhythmias. Cardiovasc Res. 2013;98(2):187–91.
Marty I. Triadin regulation of the ryanodine receptor complex. J Physiol. 2015;593(15):3261–6.
Marty I, Fauré J, Fourest-Lievin A, Vassilopoulos S, Oddoux S, Brocard J. Triadin: what possible function 20 years later? J Physiol. 2009;587:3117–21.
Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BE, Jones LR, Pessah IN, Allen PD, Franzini-Armstrong C, Knollmann BC. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106(18):7636–41.
Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, Durand P, Guicheney P, Kyndt F, Leenhardt A, Le MH, Lucet V, Mabo P, Probst V, Monnier N, Ray PF, Santoni E, Tremeaux P, Lacampagne A, Faure J, Lunardi J, Marty I. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21(12):2759–67.
Altmann HM, Tester DJ, Will ML, Middha S, Evans JM, Eckloff BW, Ackerman MJ. Homozygous/compound heterozygous Triadin mutations associated with autosomal-recessive long-QT syndrome and pediatric sudden cardiac arrest: elucidation of the Triadin knockout syndrome. Circulation. 2015;131(23):2051–60.
Walsh MA, Stuart AG, Schlecht HB, James AF, Hancox JC, Newbury-Ecob RA. Compound heterozygous Triadin mutation causing cardiac arrest in two siblings. Pacing Clin Electrophysiol. 2016;39(5):497–501.
Guo W, Jorgensen AO, Jones LR, Campbell KP. Biochemical characterization and molecular cloning of cardiac triadin. J Biol Chem. 1996;271(1):458–65.
Realini C, Rogers SW, Rechsteiner M. KEKE Repeats. Proposed roles in protein association and presentation of peptides by MHC class I receptors. FEBS Lett. 1994;348:109–13.
Shen X, Franzini-Armstrong C, Lopez JR, Jones LR, Kobayashi YM, Wang Y, Kerrick WG, Caswell AH, Potter JD, Miller T, Allen PD, Perez CF. Triadins modulate intracellular Ca2+ homeostasis but are not essential for excitation-contraction coupling in skeletal muscle. J Biol Chem. 2007;282(52):37864–74.
Oddoux S, Brocard J, Schweitzer A, Szentesi P, Giannesini B, Brocard J, Faure J, Pernet-Gallay K, Bendahan D, Lunardi J, Csernoch L, Marty I. Triadin deletion induces impaired skeletal muscle function. J Biol Chem. 2009;284(50):34918–29.
Cai WF, Pritchard T, Florea S, Lam CK, Han P, Zhou X, Yuan Q, Lehnart SE, Allen PD, Kranias EG. Ablation of junctin or triadin is associated with increased cardiac injury following ischaemia/reperfusion. Cardiovasc Res. 2012;94(2):333–41.
Sumitomo N. Current topics in catecholaminergic polymorphic ventricular tachycardia. J Arrhythm. 2016;32(5):344–51.
Sumitomo N, Harada K, Nagashima M, Yasuda T, Nakamura Y, Aragaki Y, Saito A, Kurosaki K, Jouo K, Koujiro M, Konishi S, Matsuoka S, Oono T, Hayakawa S, Miura M, Ushinohama H, Shibata T, Niimura I. Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003;89(1):66–70.
Gray B, Behr ER. New insights into the genetic basis of inherited arrhythmia syndromes. Circ Cardiovasc Genet. 2016;9(6):569–77.
Rooryck C, Kyndt F, Bozon D, Roux-Buisson N, Sacher F, Probst V, Thambo JB. New family with Catecholaminergic polymorphic ventricular tachycardia linked to the Triadin gene. J Cardiovasc Electrophysiol. 2015;26(10):1146–50.
Khoury A, Marai I, Suleiman M, Blich M, Lorber A, Gepstein L, Boulos M. Flecainide therapy suppresses exercise-induced ventricular arrhythmias in patients with CASQ2-associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10(11):1671–5.
Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15(4):380–3.
Hilliard FA, Steele DS, Laver D, Yang Z, Le Marchand SJ, Chopra N, Piston DW, Huke S, Knollmann BC. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol. 2010;48(2):293–301.
Mehra D, Imtiaz MS, van Helden DF, Knollmann BC, Laver DR. Multiple modes of ryanodine receptor 2 inhibition by flecainide. Mol Pharmacol. 2014;86(6):696–706.
Sikkel MB, Collins TP, Rowlands C, Shah M, O'Gara P, Williams AJ, Harding SE, Lyon AR, MacLeod KT. Flecainide reduces Ca2+ spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovasc Res. 2013;98(2):286–96.
Bannister ML, Varez-Laviada a, Thomas NL, Mason SA, Coleman S, du Plessis CL, Moran AT, Neill-hall D, Osman H, Bagley MC, KT ML, George CH, Williams AJ. Effect of flecainide derivatives on sarcoplasmic reticulum calcium release suggests a lack of direct action on the cardiac ryanodine receptor. Br J Pharmacol. 2016;173(15):2446–59.
Bannister ML, Thomas NL, Sikkel MB, Mukherjee S, Maxwell C, MacLeod KT, George CH, Williams AJ. The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ Res. 2015;116(8):1324–35.
Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, Yin H, Knollmann BC. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2011;4(2):128–35.
Fei L, Gill JS, McKenna WJ, Camm AJ. Effects of propafenone on calcium currents in single ventricular myocytes of guinea-pig. Br J Pharmac. 1993;109:178–82.
Hancox JC, Mitcheson JS. Inhibition of L-type calcium current by propafenone in single myocytes isolated from the rabbit atrioventricular node. Br J Pharmacol. 1997;121:7–14.
Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, Smith CD, Xie C, Chen W, Zhang J, Tian X, Jones PP, Zhong X, Guo A, Chen H, Zhang L, Zhu W, Yang D, Li X, Chen J, Gillis AM, Duff HJ, Cheng H, Feldman AM, Song LS, Fill M. Back TG, Chen SR. carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med. 2011;17(8):1003–9.
Kurtzwald-Josefson E, Hochhauser E, Bogachenko K, Harun-Khun S, Katz G, Aravot D, Seidman JG, Seidman CE, Eldar M, Shainberg A, Arad M. alpha Blockade potentiates CPVT therapy in calsequestrin-mutant mice. Heart Rhythm. 2014;11(8):1471–9.
Acknowledgements
None.
Funding
No specific funding was involved in the preparation of this review. JCH and AFJ receive research funding from the British Heart Foundation and JCH acknowledges the award of a University of Bristol Research Fellowship.
Availability of data and materials
Data sharing not applicable to this review article as no datasets were generated or analysed.
Author information
Authors and Affiliations
Contributions
JCH conceived and drafted the article and provided Fig. 1. AFJ helped draft and revise the article critically for important intellectual content and provided Fig. 1. AGS and MAW helped draft and revise the article critically for important intellectual content and provided Fig. 2. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Review article; not applicable.
Consent for publication
Parental consent was obtained to use the recording from the patient from whom the ICD recording in Fig. 2 was obtained.
Competing interests
AGS is Editor-in-Chief of the Journal of Congenital Cardiology.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hancox, J.C., James, A.F., Walsh, M.A. et al. Triadin mutations - a cause of ventricular arrhythmias in children and young adults. J Congenit Heart Dis 1, 9 (2017). https://doi.org/10.1186/s40949-017-0011-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40949-017-0011-9