
Abstract
Activation of the endothelium by pro-inflammatory stimuli plays a key role in the pathogenesis of a multitude of vascular diseases. Angiogenesis is a crucial component of the vascular response associated with inflammatory signaling. The CD40/CD40 ligand dyad in endothelial cells (EC) has a central role in promoting vascular inflammatory response; however, the molecular mechanism underlying this component of inflammation and angiogenesis is not fully understood. Here we report a novel microRNA mediated suppression of endothelial CD40 expression. We found that CD40 is closely regulated by miR-424 and miR-503, which directly target its 3′ untranslated region. Pro-inflammatory stimuli led to increased endothelial CD40 expression, at least in part due to decreased miR-424 and miR-503 expression. In addition, miR-424 and miR-503 reduced LPS induced EC sprouting, migration and tube formation. Moreover, we found that miR-424 and miR-503 expression is directly regulated by peroxisome proliferator-activated receptor gamma (PPARγ), whose endothelial expression and activity are decreased in response to inflammatory factors. Finally, we demonstrate that mice with endothelial-specific deletion of miR-322 (miR-424 ortholog) and miR-503 have augmented angiogenic response to LPS in a Matrigel plug assay. Overall, these studies identify a PPARγ-dependent miR-424/503-CD40 signaling axis that is critical for regulation of inflammation mediated angiogenesis.
Similar content being viewed by others


Introduction
Endothelial cells (ECs) have unique functions in vascular biology. They can modulate immune response and angiogenesis, and have a key role in the maintenance of vascular homeostasis through protection of the vascular wall from pathological stimuli1, 2. Endothelial dysfunction under conditions of inflammatory stress is known to contribute to the pathogenesis of various diseases, including atherosclerosis, rheumatoid arthritis and tumor growth1, 3,4,5. Pathological angiogenesis is thought to be a key process in the inflammation-induced development of such diseases1, 3,4,5. Thus, it is essential to define the molecular mechanisms underpinning EC activation and consequent angiogenic signaling mediated by inflammatory processes. CD40, a transmembrane receptor of the tumor necrosis factor (TNF) gene superfamily, is largely expressed on antigen presenting cells (APCs), including B cells, macrophages, and monocytes6, 7. While it has been shown to be expressed at low levels in ECs, pro-inflammatory stimuli such as tumor necrosis factor alpha (TNFα) and lipopolysaccharide (LPS) cause rapid induction of CD40 expression in ECs8,9,10. It has already been demonstrated that the CD40/CD40 ligand (CD40L) signaling pathway plays a pivotal role in the immune response in various cells7, 11. Indeed, CD40-CD40L interactions induce various angiogenic factors such as VEGF, suggesting a potential role of CD40 in inflammation-mediated angiogenesis12.
Peroxisome proliferator-activated receptor gamma (PPARγ) receptors, transcription factors belonging to the nuclear hormone receptor superfamily, have been demonstrated to have widespread expression in the vasculature, including in ECs, vascular smooth muscle cells, and monocytes/macrophages13,14,15. It is known that PPARγ has anti-inflammatory properties, making it an important factor in maintaining vascular homeostasis16. As such, pioglitazone, a PPARγ agonist, acts to reduce inflammation in ECs thereby improving endothelial dysfunction14, 17,18,19,20,21,22. In addition, it has been shown that PPARγ agonists inhibit EC tube formation in vitro and VEGF-induced angiogenesis in vivo 23,24,25. Although it was reported that PPARγ agonists can inhibit CD40 expression16, the mechanism of this PPARγ-CD40 signaling axis in ECs remains to be investigated.
In this study, we sought to define the molecular mechanisms that govern inflammation-induced endothelial activation. We demonstrate that endothelial CD40 expression is closely regulated by two microRNAs (miRNAs), miR-424 and miR-503, and this regulatory mechanism becomes disrupted by inflammatory stimuli. Furthermore, we found that PPARγ is a direct transcriptional activator of miR-424 and miR-503, and its activation by pioglitazone leads to suppression of CD40 expression and LPS-induced angiogenesis in a miR-424 and miR-503 dependent manner. Moreover, we demonstrate enhanced angiogenic response to inflammatory stimuli in mice with endothelial specific deletion of miR-322 (miR-424 ortholog) and miR-503. These findings demonstrate a novel mechanism by which PPARγ signaling can augment the expression of homeostatic miRNAs, and likely serves as a key rheostat modulating endothelial inflammatory response.
Results
Pro-inflammatory stimuli induce CD40 and inhibit miR-424 and miR-503 expression in ECs
Previous studies have shown that pro-inflammatory stimuli lead to upregulation of CD40 expression in macrophages26. We assessed whether a similar mechanism exists in ECs. Treatment of human umbilical vein endothelial cells (HUVECs) with LPS (1 μg/mL) or TNFα (10 ng/mL) led to significantly increased CD40 mRNA and protein expression in a time dependent manner (Fig. 1a,b). Given prior studies implicating the involvement of miRNAs in inflammatory processes, as well as studies suggesting that CD40 itself is regulated by miRNAs in other contexts27, we investigated whether inflammation-induced CD40 expression is miRNA dependent. We found that subjecting HUVECs to knockdown of argonaute 2 (AGO2), a key protein of the RNA-induced silencing complex (RISC) complex28,29,30, or DICER, a key protein of miRNA processing, resulted in significant increase in CD40 protein levels (Fig. 1c), suggesting that baseline CD40 expression is miRNA dependent.

Pro-inflammatory stimuli upregulate CD40 and reduce miR-424 and miR-503 expression in HUVECs. (a) CD40 mRNA expression in response to LPS (1 μg/ml) or TNFα (10ng/ml) in a time dependent manner. (b) CD40 protein expression in response to stimulation with either LPS (1 μg/ml for 24 h) or TNFα (10ng/ml for 16 h). (c) CD40 protein expression in HUVECs 72 h after AGO2 or DICER siRNA transfection. (d) Predicted target sequences of CD40 3′ UTRs targeted by miR-424 and miR-503 and mutated sequences (MUT#1, #2) for disrupting miR-424 and miR-503 recognition sequence. (e,f) Quantitative PCR showing expression of the mature and pri forms of miR-424 and miR-503 in response to LPS (1 μg/ml) or TNFα (10ng/ml) in a time dependent manner. (f) *P < 0.05, **P < 0.01, ***P < 0.001 compared to controls. Error bars, s.e.m. Cropped gel images are shown; uncut gels are included in the Supplementary Information.
Next, we investigated which specific miRNAs may be involved in regulation of endothelial CD40 expression. Using a combination of target prediction software algorithms (TargetScan, PicTar, and microRNA.org), we found that miR-424 and miR-503, which have highly conserved seed sequences, were predicted to bind to the 3′ untranslated region (UTR) of CD40 (Fig. 1d). These two miRNAs are highly expressed in ECs and are known to have important roles in maintaining vascular homeostasis28,29,30,31,32. We next tested whether pro-inflammatory stimuli can affect endothelial miR-424 and miR-503 expression. We found significant decrease in both the immature pri-forms and the mature forms of miR-424 and miR-503 in response to either LPS or TNFα stimulation, suggesting that these miRs are transcriptionally suppressed in the context of endothelial response to inflammation (Fig. 1e,f).
CD40 is directly targeted by miR-424 and miR-503
We further investigated the relationship between miR-424/503 and CD40. Overexpression of miR-424 and miR-503 led to significant decrease in both the mRNA and protein expression of CD40, whereas inhibition of miR-424 and miR-503 using anti-miRs led to CD40 upregulation (Fig. 2a,b, Sup. Fig. 1a,b). We next determined whether miR-424 and miR-503 regulate CD40 expression via binding directly to the 3′UTR. Co-transfection of the wildtype CD40 3′UTR reporter construct with miR-424 and miR-503 mimics significantly reduced the reporter activity, while reporter activity in cells transfected with mutagenized CD40 3′UTR of two different types (mutant #1 and mutant #2) was unchanged by concurrent transfection with miR-424 and miR-503 mimics (Figs 1d and 2c). Combined, these results show that miR-424 and miR-503 directly regulate CD40 expression.

CD40 is regulated by miR-424 and miR-503 directly. (a) CD40 mRNA expression in response to overexpression of miR-424 or miR-503 mimics, or inhibition of miR-424 or miR-503 with anti-miRs in HUVECs. (b) CD40 protein expression in response to overexpression of miR-424 or miR-503 mimics or inhibition of miR-424 or miR-503 with anti-miRs in HUVECs. (c) Effect of miR-424 or miR-503 overexpression on luciferase reporter containing either the wildtype of mutagenized 3′UTR of human CD40 gene in HeLa cells. Luciferase activity data for constructs with the wild-type (WT) and mutants (#1 and #2) 3′ UTR constructs are shown. *P < 0.05, **P < 0.01, ***P < 0.001 compared to controls. Error bars, s.e.m. Cropped gel images are shown; uncut gels are included in the Supplementary Information.
CD40 inhibition attenuates LPS induced migration and tube formation in ECs
Given the known endothelial angiogenic response when subjected to inflammatory stimuli, we next investigated the function of CD40 signaling in EC tube formation and migration assays. LPS significantly induced EC tube formation, an effect that was abrogated by concurrent CD40 knockdown (Fig. 3a). Furthermore, CD40 knockdown also significantly inhibited LPS-induced HUVEC migration (Fig. 3b). These findings indicate a key role for CD40 signaling in the regulation of angiogenic processes. The finding that miR-424 and miR-503 directly regulate CD40 led us to further investigate the effect of miR-424 and miR-503 overexpression on the regulation of pro-inflammatory stimuli-induced CD40 expression. Transfection of HUVECs with miR-424 and miR-503 mimics prior to treatment with LPS or TNFα led to significantly decreased CD40 protein expression (Fig. 3c). Moreover, overexpression of miR-424 and miR-503 significantly reduced LPS-induced EC migration and tube formation (Fig. 3d,e). In addition, we conducted sprouting bead angiogenesis assays, and found significant inhibition of LPS induced EC sprouting in cells that overexpress miR-424 and miR-503 (Fig. 3f). Lastly, we investigated whether CD40 overexpression could rescue miR-424 and miR-503 effects on EC tube formation and migration. We found abrogation of the effect of miR-424 and miR-503 overexpression by concurrent CD40 expression (Fig. 3g,h). Taken together, these findings support an important role for the miR-424/503-CD40 signaling pathway in pro-inflammatory stimuli-induced angiogenesis.

Inhibition of CD40 abrogates LPS-induced endothelial angiogenic response. (a,b) Tube formation and cell migration assay response to LPS (1 μg/ml) with or without CD40 knockdown. Tubes were assessed at 8 h after cell plating. (c) CD40 protein expression in response to LPS (1 μg/ml), TNFα (10ng/ml) with or without miR-424 and miR-503 overexpression. (d,e) Tube formation and migration assay response to LPS (1 μg/ml) with or without miR-424/503 overexpression. Tubes were assessed at 8 h after cell plating. (f) Sprouting assay using miR-424/503 transfected HUVEC coated beads with or without LPS (1ug/ml) treatment. (g,h) Tube formation and migration assay response to miR-424/503 overexpression with or without CD40 overexpression at 5 h. *P < 0.05, **P < 0.01, ***P < 0.001 compared to controls. # P < 0.05, ## P < 0.01, ### P < 0.001 compared to LPS or miR-424/503. Scale bar, 200 μm. Error bars, s.e.m. Images are representative of three independent experiments. Cropped gel images are shown; uncut gels are included in the Supplementary Information.
Pioglitazone ameliorates LPS-induced angiogenesis through inhibition of CD40 expression
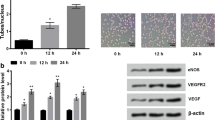
PPARγ has been found to significantly dampen inflammatory responses in ECs, and is a powerful pharmacological target for counteracting inflammatory diseases. In addition, a previous study has suggested indirect regulation of miR-424/503 expression by PPARγ via its induction of the G protein coupled receptor ligand apelin, which positively regulates miR-424 and miR-50333. To determine the molecular mechanism underlying the effect of PPARγ on pro-inflammatory stimuli-induced angiogenesis, we first examined the effect of pioglitazone, a PPARγ agonist, on miR-424 and miR-503 expression. Indeed, we found that treatment of HUVECs with pioglitazone resulted in a significant increase in miR-424 and miR-503 levels (Fig. 4a). In addition, we found that miR-424 and miR-503 levels were restored by pioglitazone when treated in conjunction with LPS (Fig. 4b). We also found that CD40 expression was decreased in response to pioglitazone stimulation (Fig. 4c,d), and that LPS-induced CD40 upregulation was reversed by treatment with pioglitazone (Fig. 4e). We further investigated whether pioglitazone can affect LPS-induced tube formation and migration in HUVECs. Pioglitazone reduced LPS-induced angiogenesis in HUVECs to baseline levels, as assessed by tube formation (Fig. 4f), migration (Fig. 4g), and sprouting angiogenesis assay (Fig. 4h). Taken together, these results suggest that pioglitazone attenuates LPS-induced angiogenesis by engaging the miR-424/503-CD40 signaling axis.

Pioglitazone ameliorates LPS-induced angiogenesis by regulation of CD40 expression. (a) Mature miR-424 and miR-503 expression in response to pioglitazone (10 μM) for 24 h. PIO, Pioglitazone. (b) Mature miR-424 and miR-503 expression in response to LPS (1 μg/ml) with or without concurrent stimulation with pioglitazone (10 μM) for 24 h. (c,d) CD40 mRNA and protein expression in response to LPS (1 μg/ml) with or without pioglitazone (10 μM) for 24 h. (e) CD40 protein expression in response to LPS (1 μg/ml) with or without pioglitazone (10 μM). (f,g) Tube formation and cell migration assay response to LPS (1 μg/ml) with or without concurrent stimulation with pioglitazone (10 μM) at 9 h. (h) Sprouting assay using HUVEC coated beads in response to LPS (1ug/ml) with or without pioglitazone (10 μM). *P < 0.05, **P < 0.01, ***P < 0.001 compared to controls. # P < 0.05, ### P < 0.001 compared to LPS. Error bars, s.e.m. Images are representative of three independent experiments. Cropped gel images are shown; uncut gels are included in the Supplementary Information.
PPARγ regulates miR-424 and miR-503 expression
We further investigated the relationship between PPARγ and miR-424/503. Our in silico analyses of the putative miR-424/503 promoter region (mapper.chip.org) identified two predicted PPARγ binding sites (Fig. 5a). We first examined the effects of PPARγ knockdown on miR-424/503 and CD40 expression. We found both the pri-forms and the mature forms of miR-424 and miR-503 were significantly downregulated by PPARγ knockdown (Fig. 5b). We also found CD40 expression was significantly elevated upon knockdown of PPARγ in HUVECs, further supporting involvement of PPARγ in the regulation of miR-424/503 mediated CD40 expression (Fig. 5c). Chromatin immunoprecipitation (ChIP) assays confirmed that PPARγ binds to the predicted binding sites of the putative miR-424/503 promoter region (Fig. 5d). Next, using a miR-424/503 promoter based luciferase reporter construct, we found significant induction of the luciferase reporter activity by overexpression of PPARγ and retinoid X receptor alpha (RXRα) (which forms a transcriptional complex with PPARγ)34. Moreover, the increased luciferase activity of HUVECs in response to PPARγ and RXRα overexpression was further augmented by concurrent treatment with pioglitazone (Fig. 5e).

PPARγ signaling regulates miR-424/503 expression. (a) Predicted PPARγ binding sites on promoter region of miR-424 and miR-503 and the positions of PCR amplicons used in ChIP assays. (b) Mature and pri forms of miR-424 and miR-503 expression in response to PPARγ knockdown. (c) CD40 protein expression in response to PPARγ knockdown. (d) ChIP assay for investigating the binding sites of PPARγ on miR-424/503 promoter region. The ChIP pull-down PCR products (left) and the inputs (right) are shown. (e) Relative luciferase activity of HUVECs co-transfected with miR-424/503 promoter luciferase construct and PPARγ and RXRα with or without pioglitazone (10 μM) stimulation for 24 h. (f) *P < 0.05, **P < 0.01, ***P < 0.001 compared to controls. ## P < 0.01, ### P < 0.001 compared to PPARγ + RXRα or PIO. n.s.: not significant. Error bars, s.e.m. Cropped gel images are shown; uncut gels are included in the Supplementary Information.
We next investigated whether LPS and TNFα regulate miR-424 and miR-503 expression in a PPARγ dependent manner. Using a luciferase reporter construct driven by the minimal PPAR responsive element (PPRE), we found that LPS stimulation led to a trend towards decreased luciferase reporter activity at baseline, and significant decrease in PPRE reporter activity in response to pioglitazone (Fig. 5f). TNFα stimulation led to significant decrease in the PPRE luciferase reporter activity both at basal state and in conjunction with pioglitazone stimulation (Fig. 5g). Moreover, stimulation of HUVECs with either LPS or TNFα led to significant decrease in both the mRNA and protein levels of PPARγ, suggesting that decreased PPARγ expression in ECs may be a key mechanism by which miR-424 and miR-503 levels are decreased in response to inflammatory stimuli (Fig. 5h–k). Taken together, these results demonstrate that PPARγ is a key factor that controls miR-424 and miR-503 expression in response to endothelial inflammation.
Mice with endothelial-specific deletion of miR-322 and miR-503 demonstrate increased angiogenesis in response to LPS
To further investigate the relevance of our signaling paradigm in vivo, we evaluated the inflammatory angiogenesis response in mice with conditional, endothelial-specific deletion of miR-322 (mouse ortholog of miR-424) and miR-503 (Mirc24fl/fl;Cdh5-CreERT2, henceforth Mirc24ECKO). Mice with loxP sites surrounding the microRNA cluster 24 (which includes miR-322, miR-503, and miR-351) were crossed to mice with a tamoxifen inducible Cdh5 Cre driver (Cdh5-CreERT2) (Sup. Fig. 2)35, 36. Adult Mirc24ECKO mice injected with tamoxifen displayed no overt phenotype up to 12 weeks after injection. Matrigel plugs containing LPS were implanted subcutaneously, and angiogenesis was assessed 7 days after injection. We found that Mirc24ECKO mice had significantly more neovascularization in the Matrigel plug in response to LPS compared to control mice, as demonstrated by increased blood content on gross assessment of the Matrigel plug (Fig. 6a), greater number of blood cell-filled vessels on H & E staining (Fig. 6b), and significantly increased number of CD31+/SMA+ mature vessels (Fig. 6c,d). This was in the context of increased CD40 expression in the ECs that infiltrated the Matrigel (Fig. 6e). Overall, these findings provide key in vivo genetic evidence supporting the downstream role of miR-424 and miR-503 in determining endothelial response to inflammatory stimuli.

LPS induced angiogenesis is inhibited by Mir24c in vivo. (a) Whole mount images and (b) H&E stained sections of LPS-containing Matrigel implants, showing increased vascularization in mice lacking the Mir 24 cluster that includes miR-322 (mouse miR-424 ortholog), miR-503, and miR-351 (Mirc24ECKO). Scale bar: 100 μm. (c) Immunostaining of LPS-containing Matrigel implant sections for CD31 (green), SMA (red) and DAPI (blue) showing increased vascularization in Mirc24ECKO. Scale bar: 70 μm. (d) Quantification of vascularized/non-vascularized immunostained LPS-containing Matrigel implant sections in control and Mirc24ECKO mice. *P < 0.05. (e) Staining for CD40 (red) in Matrigel implant sections along with CD31 (green) showing increased CD40 expression in infiltrating endothelial cells. Scale bar: 35 μm. (f) Schematic outlining the proposed endothelial mechanism.
Discussion
Studies have reported that dysfunctional ECs are susceptible to pathogenic signals, resulting in various vascular diseases. Angiogenesis is a crucial component of the vascular response associated with activation of inflammatory signaling. The CD40/CD40L pathway is known to be involved in inflammatory response by promoting endothelial dysfunction. Here we identify a miRNA driven mechanism controlling CD40 signaling and show its involvement in the regulation of inflammation-mediated angiogenesis.
Four major conclusions can be drawn from the findings of this study: (i) CD40 and miR-424/503 are conversely regulated by proinflammatory stimuli in HUVECs. (ii) miR-424 and miR-503 directly target CD40 and inhibit inflammation-induced angiogenic phenotypes both in vivo and in vitro. (iii) PPARγ signaling regulates miR-424 and miR-503 expression in HUVECs. (iv) PPARγ agonism by pioglitazone reverses the downregulation of miR-424/503 and upregulation of CD40 caused by proinflammatory stimuli in HUVECs, thus ameliorating inflammation-induced angiogenesis (Fig. 6f).
Earlier studies investigating the regulation of angiogenesis have focused specifically on angiogenic genes and related signaling pathways. In recent years, research efforts have focused on the correlation between miRNA and angiogenesis in ECs. For example, it has been demonstrated that miR-15, 16, 20a, and 20b are anti-angiogenic miRNAs targeting VEGF mRNA37, 38, whereas the miR-17–92 cluster, miR-27b, and let-7f are thought to have pro-angiogenic properties37, 38. The miR-424 and miR-503 transcription unit is highly expressed in ECs and is known to be a key factor in maintaining homeostasis of these cells28,29,30,31,32.
Previous work has demonstrated that upregulation of miR-503 leads to inhibition of tumor angiogenesis through regulation of FGF2 and VEGFA39, while downregulation of miR-503 in ECs has been suggested to lead to improved angiogenesis in diabetes mellitus40. MiR-424 has been shown to target VEGF, VEGFR2 and FGFR1 and inhibit angiogenic activity in HUVECs41. In contrast, another study has shown that miR-424 may promote an angiogenic phenotype in hypoxic ECs42. Taken together, the studies to date suggest a context dependent role for miR-424 and miR-503 in ECs, and further studies are needed to fully elucidate the role of these highly-regulated miRNAs in endothelial function in health and disease.
In our current study, we examined the role of miR-424 and miR-503 in vascular inflammation and found that they control a key molecular mechanism involved in regulation of CD40 expression. Our findings demonstrate that miR-424 and miR-503 can inhibit pro-inflammatory-induced angiogenesis through direct targeting of CD40. Previous studies have implicated activation of CD40-CD40L interaction in atherosclerosis in ECs, smooth muscle cells, and macrophages43. CD40-CD40L signaling induces cell adhesion molecules, chemokines and tissue factor resulting in recruitment of lymphocytes in vascular endothelium44,45,46,47,48. It was also reported that soluble CD40L-induced CD40 activation has proangiogenic effects in vitro and in vivo 12, 49, 50. Our current findings identify a novel mechanism of CD40 signaling pathway regulation, and describe a PPARγ agonist-based pharmacologic strategy that can be used to modulate this signaling pathway.
It has been reported that the PPARγ agonist rosiglitazone suppresses CD40 expression and attenuates inflammation16. In addition, rosiglitazone was shown to attenuate inflammation in LPS-induced peritonitis51. However, the mechanism of PPARγ regulation of CD40 has remained elusive. In this study, we show that CD40 is upregulated in ECs subjected to pro-inflammatory stimuli, and demonstrate a novel PPARγ-miR-424/503-CD40 axis involved in its regulation. Our demonstration of PPARγ binding sequences in the miR-424/503 promoter region, in conjunction with our previous work identifying the transcription factor myocyte enhancer factor 2 (MEF2) as a key cis-acting factor involved in the regulation of miR-424 and miR-50330, point towards the need for future studies to investigate the potential synergy between these and other transcription factors.
The Mirc24ECKO mice represent a powerful genetic tool that can be used to further investigate the role of this miRNA cluster in endothelial function. The increased angiogenic response to LPS in the Mirc24ECKO mice, in conjunction with increased CD40 expression in ECs, support the hypothesis that unchecked endothelial response to inflammatory stimuli is an important driving force for our findings. Our studies do carry a number of limitations, including the artificial nature of the Matrigel plug assay, as well as potential effect of LPS on endothelial behavior such as maintenance of cell junction and permeability. Future investigation of these mice will provide greater insights into the role of these miRNAs in angiogenesis in both health and disease states.
In conclusion, we demonstrate that downregulation of miR-424 and miR-503 and subsequent upregulation of CD40 is associated with inflammation-induced angiogenesis. Furthermore, the demonstration that PPARγ directly regulates miR-424 and miR-503 expression identifies novel transcriptional targets of this pleiotropic gene that may in part explain its vaso-protective effects.
Methods
Animals
All animal experiments were conducted in compliance with the relevant laws and institutional guidelines and were approved by the Yale University Institutional Animal Care and Use Committee. Mirc24 fl/fl and Cdh5-CreERT2 mice have been previously described35, 36. All animals were maintained on a C57/Bl6 background. Mice were injected intraperitoneously with 1 mg of tamoxifen (Sigma Aldrich, T5648) dissolved in corn oil for five consecutive days. The mice were let to rest for an additional 14 days before experiments were conducted. All mice received tamoxifen.
Cell Culture and Transfection
HUVECs (Lonza and Yale VBT Core) were cultured at 37 °C in a 5% CO2 incubator with EBM-2 basal medium supplemented with EGM-2 (Lonza) with 1% penicillin-streptomycin (Welgene). For experimental treatments, HUVECs (passages 3–7) were grown to 70% to 90% confluence. HeLa cells were cultured in DMEM (Welgene) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin (Welgene). SiRNA (Stealth siRNA, Invitrogen), miRNA mimics and anti-miRs (miRVanaTM, Ambion) were transfected using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Cells were collected 72 h after transfection. HeLa cells were transfected with plasmids and miRNA mimics using Lipofectamine 2000 (Invitrogen). Pioglitazone was used at 10 μM for designated timepoints.
Gene expression analysis
Total RNA was isolated with miRNeasy RNA isolation kit (Qiagen). Purified RNA was reverse transcribed using the TaqMan MiRNA Reverse Transcription Kit (Life Technologies). MiRNA quantitative reverse‐transcriptase PCR (qRT‐PCR) was performed using TaqMan Universal Master Mix II, no UNG (Life Technologies) and MiR-424 and miR-503 were detected with Taqman probes (Life Technologies). Data were normalized to the internal control small RNA (RNU19). For the mRNA, purified RNA was reverse transcribed using qPCRBIO cDNA Synthesis Kit (PCRBIOSYSTEMS). Quantitative RT‐PCR was performed using the qPCRBIO SyGreen Mix Hi-ROX (PCRBIOSYSTEMS) per manufacturer’s instructions. Ribosomal 18S was used as an internal control.
Western Blotting
HUVECs were lysed with RIPA buffer (Gendepot) containing protease and phosphatase Inhibitor cocktail (Roche). Following this, centrifugation was performed at 13000 rpm, 4 °C, for 15 minutes. Protein concentrations were determined by Pierce BCA Protein Assay kit (Thermo Scientific). Equal amounts of total proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and transferred onto polyvinyl difluoride membrane (Millipore). Immunoblotting was performed with the primary antibodies specific to CD40 (1:3000, BD Bioscience), GAPDH (1:5000, Cell signaling), and PPARγ (1:5000, Santa Cruz). Immunodetection was accomplished using HRP-conjugated secondary antibodies (1:3000, Cell Signaling) and developed using an enhanced chemiluminescence detection method (Thermo Scientific).
Luciferase Reporter Assay
For 3′UTR luciferase reporter assay, Human CD40 3′UTR (738p) that included predicted miR-424/503 binding seed sequences was cloned into NotI and XhoI sites of a psiCHECK-2 vector (Promega). The sequence TGCTGCT in the seed sequences of CD40 was mutated to TGTCGCT (Mutant #1), CGCGGCG (Mutant #2) using the QuikChange II Site-Directed Mutagenesis Kit (Agilent). HeLa cells were transfected with the luciferase reporter constructs and either miR-424, miR-503 mimics or negative control miRNAs using lipofectamine 2000 (Invitrogen). At 48hrs after transfection, the cells were lysed, and luciferase activity was measured using the Dual-Luciferase Reporter Assay kit (Promega) according to the manufacturer’s instructions. The human miR-424/503 promoter luciferase construct was previously described31. The PPAR responsive element (PPRE) promoter luciferase construct was a gift from Bruce Spiegelman (Addgene plasmid #1015). HUVECs were transfected with miR-424/503 promoter-luciferase, renilla-luciferase, PPARγ and RXRα constructs (Origene). Luciferase activity was quantified using the Dual-Luciferase Reporter Assay kit (Promega).
EC tube formation assay
HUVECs were seeded on 6-well plates and transfected with siRNA or miRNA mimics using RNAi max (Invitrogen). The next day, 24-well plates were coated with 250ul of Matrigel Matrix (Growth Factor Reduced, phenol-free) (BD Bioscience) and incubated at 37 °C for 30 minutes. Meanwhile, transfected cells were trypsinized and seeded at 3 × 104 cell per well on 24-well plates. The cells were then treated with LPS (Sigma, 1 μg/mL) and Pioglitazone (10 μM). Cells were observed under the microscope 8–9 h after being plated as designated for each experiment. WimTube (ibidi) was used to quantify the EC tube formation length.
Cell migration assay
HUVECs were seeded at 2 × 105 cells per well on 12-well plates and then, were scratched with a P200 pipette tip. HUVECs were incubated with 1% FBS starvation media containing LPS (1 μg/ml) or/and Pioglitazone (10 μM). The cells were allowed to migrate and the gap distance was captured with a camera equipped microscope at the designated timepoints. The width of the gaps was calculated using ImageJ.
Fibrin gel bead assay
Cytodex 3 microcarriers (Amersham Pharmacia Biotech) were swollen in PBS for 3 h and sterilized by autoclaving. HUVECs were trypsinized and mixed with Cytodex 3 microcarriers at a concentration of 400 HUVECs per bead in 1 ml of EGM-2. Beads and HUVECs were incubated for 4 h and shaken every 20 min at 37 °C and 5% CO2. After incubation, beads with HUVECs were supplemented with 5 ml of EGM-2 and incubated overnight at 37 °C and 5% CO2. The following day, beads were washed twice with EGM-2 and resuspended in a solution of 2 mg/ml fibrinogen (Sigma-Aldrich) containing 0.15units/ml aprotinin (Sigma-Aldrich) at a concentration of 150 beads/ml. 500ul of fibrinogen/bead solution was added in a well containing 0.625units thrombin (Sigma-Aldrich). The fibrinogen/bead solution was allowed to clot for 5 min at RT and then at 37 °C and 5% CO2 for 15 min. During this time, fibroblasts were trypsinized and resuspended in EGM-2 and then plated on the top of the clot at a concentration of 20,000 cells per well. The medium was changed every other day. Average sprout length was analyzed using ImageJ.
Chromatin Immunoprecipitation
HUVEC were plated on 100mm culture dishes and grown for 24 h before native protein-DNA complexes were crosslinked by treatment with 1% formaldehyde for 15 minutes. Simple ChIP Plus Enzymatic Chromatin IP kit (Cell Signaling) was used per the manufacturer’s protocol. Briefly, equal aliquots of isolated chromatin were subjected to immunoprecipitation with anti-PPARγ antibody or rabbit IgG control. PCR reactions of immunoprecipitated DNA were performed to validate PPARγ binding on the miR-424/503 promoter. PCR primers used: #1: FWD: GAGGTGGCTTTTTAGGGGT and REV: CGAGCCATCATGTCAGAAGT; #2: FWD: AGAGGCGTATTCTTTGGCTC and REV: TCCCTTCCAGATTGCTCTTG; #3: FWD: TCCAAGAGCAATCTGGAAGG and REV: TCTCCCAACATTTTGTTCCA. PCR products were separated by gel electrophoresis and visualized by SYBRsafe (Invitrogen).
Lentivirus production
For CD40 overexpression in HUVECs, a lentivirus bearing CD40 was obtained from Origene. The Lenti-X HTX Packaging System (Clontech) with Lenti-X Concentrator was used to generate the lentivirus particles for in vitro cellular transduction.
Matrigel plug angiogenesis assay
Liquid Matrigel (8–10 mg of protein/ml, Corning, 356231) was mixed at 4 °C with LPS (Sigma Aldrich, L2630) at a final concentration of 10 μg/ml and Heparin (Sigma Aldrich, H3393) at a final concentration of 20U/ml and injected subcutaneously (0.5 ml/injection) into the left and right groins of 5–6 month old Mirc24 fl/fl or Mirc24 fl/fl mir24fl/fl;Cdh5-CreERT2 female mice. The mice were sacrificed 7 d after injection and the plugs were fixed in 4% PFA for 2 h at RT, incubated in 30% sucrose and finally embedded in Tissue Tec OCT (Sakura). The Matrigel plugs were sectioned using a Leica CM1950 cryotome at 10 μm, and processed for immunohistochemistry or immunofluorescence.
Immunohistochemistry and immunofluorescence
Matrigel sections were washed in 1x PBS, blocked for 1 h at RT in blocking buffer (1% BSA, 10% goat serum, 1xPBS) and incubated with anti-CD31 (BD Pharmigen, 553370), anti-smooth muscle actin conjugated to Cy3 (C6198, Sigma), and anti-CD40 antibody (R&D Systems, AF440) overnight at 4 °C. The next day the sections were incubated with Alexa Fluor secondary goat antibodies for 1 h at RT and counterstained with DAPI (Thermo Scientific) to visualize the nuclei. Imaging was performed using a Zeiss confocal microscope with a 10x objective. Vascularization was evaluated in 5 random fields per plug, 2 plugs per mouse, using ImageJ. H&E staining was performed using standard methods and visualized with a light microscope (Nikon Eclipse 80i).
Statistical Analysis
All experiments were performed at least three times and analyses were performed with GraphPad Prism 5.0 software. When only two groups were compared, statistical differences were assessed with unpaired two-tailed Student’s t-test. Otherwise, statistical significance was determined using one-way analysis of variance followed by Bonferroni’s multiple comparison test. Relationships between variables were determined by the Pearson correlation coefficient. For the Matrigel plug analysis, two-tailed Fisher’s exact test was used to compare vascularization frequency in the mice of different genotypes. P < 0.05 was considered statistically significant.
References
Deanfield, J. E., Halcox, J. P. & Rabelink, T. J. Endothelial function and dysfunction - Testing and clinical relevance. Circulation 115, 1285–1295, doi:10.1161/Circulationaha.106.652859 (2007).
Simons, M. et al. State-of-the-Art Methods for Evaluation of Angiogenesis and Tissue Vascularization: A Scientific Statement From the American Heart Association. Circ Res 116, e99–132, doi:10.1161/RES.0000000000000054 (2015).
Folkman, J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med 333, 1757–1763, doi:10.1056/NEJM199512283332608 (1995).
Carmeliet, P. & Jain, R. K. Angiogenesis in cancer and other diseases. Nature 407, 249–257, doi:10.1038/35025220 (2000).
Chiodoni, C. et al. Triggering CD40 on endothelial cells contributes to tumor growth. J Exp Med 203, 2441–2450, doi:10.1084/jem.20060844 (2006).
Aloui, C. et al. The Signaling Role of CD40 Ligand in Platelet Biology and in Platelet Component Transfusion. Int J Mol Sci 15, 22342–22364, doi:10.3390/ijms151222342 (2014).
Chatzigeorgiou, A., Lyberi, M., Chatzilymperis, G., Nezos, A. & Kamper, E. CD40/CD40L signaling and its implication in health and disease. Biofactors 35, 474–483, doi:10.1002/biof.62 (2009).
Karmann, K., Hughes, C. C., Schechner, J., Fanslow, W. C. & Pober, J. S. CD40 on human endothelial cells: inducibility by cytokines and functional regulation of adhesion molecule expression. Proc Natl Acad Sci USA 92, 4342–4346, doi:10.1073/pnas.92.10.4342 (1995).
Omari, K. M. & Dorovini-Zis, K. CD40 expressed by human brain endothelial cells regulates CD4+ T cell adhesion to endothelium. J Neuroimmunol 134, 166–178, doi:10.1016/S0165-5728(02)00423-X (2003).
Yang, L. et al. SIRT1 regulates CD40 expression induced by TNF-alpha via NF-kB pathway in endothelial cells. Cell Physiol Biochem 30, 1287–1298, doi:10.1159/000343318 (2012).
Elgueta, R. et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 229, 152–172, doi:10.1111/j.1600-065X.2009.00782.x (2009).
Reinders, M. E. J., Sho, M., Robertson, S. W., Geehan, C. S. & Briscoe, D. M. Proangiogenic function of CD40 ligand-CD40 interactions. J Immunol 171, 1534–1541, doi:10.4049/jimmunol.171.3.1534 (2003).
Hsueh, W. A., Jackson, S. & Law, R. E. Control of vascular cell proliferation and migration by PPAR-gamma: a new approach to the macrovascular complications of diabetes. Diabetes Care 24, 392–397, doi:10.2337/diacare.24.2.392 (2001).
Sasaki, M. et al. Troglitazone, a PPAR-gamma activator prevents endothelial cell adhesion molecule expression and lymphocyte adhesion mediated by TNF-alpha. BMC Physiol 5, 3, doi:10.1186/1472-6793-5-3 (2005).
Reddy, R. C. et al. Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-{gamma}. Blood 112, 4250–4258, doi:10.1182/blood-2007-12-128967 (2008).
Sun, H. et al. Peroxisome proliferator-activated receptor gamma agonist, rosiglitazone, suppresses CD40 expression and attenuates inflammatory responses after lithium pilocarpine-induced status epilepticus in rats. Int J Dev Neurosci 26, 505–515, doi:10.1016/j.ijdevneu.2008.01.009 (2008).
Diep, Q. N. et al. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats - Role of peroxisome proliferator-activated receptor-gamma. Circulation 105, 2296–2302, doi:10.1161/01.CIR.0000016049.86468.23 (2002).
Marx, N. et al. Peroxisome proliferator-activated receptor-gamma activators inhibit IFN-gamma-induced expression of the T cell-active CXC chemokines IP-10, Mig, and I-TAC in human endothelial cells. J Immunol 164, 6503–6508, doi:10.4049/jimmunol.164.12.6503 (2000).
Pasceri, V., Wu, H. D., Willerson, J. T. & Yeh, E. T. Modulation of vascular inflammation in vitro and in vivo by peroxisome proliferator-activated receptor-gamma activators. Circulation 101, 235–238, doi:10.1161/01.CIR.101.3.235 (2000).
Jackson, S. M. et al. Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arterioscler Thromb Vasc Biol 19, 2094–2104, doi:10.1161/01.ATV.19.9.2094 (1999).
Chen, N. G. et al. PPARgamma agonists enhance human vascular endothelial adhesiveness by increasing ICAM-1 expression. Biochem Biophys Res Commun 263, 718–722, doi:10.1006/bbrc.1999.1437 (1999).
Verrier, E. et al. PPARgamma agonists ameliorate endothelial cell activation via inhibition of diacylglycerol-protein kinase C signaling pathway: role of diacylglycerol kinase. Circ Res 94, 1515–1522, doi:10.1161/01.RES.0000130527.92537.06 (2004).
Kim, K. Y. & Cheon, H. G. Antiangiogenic effect of rosiglitazone is mediated via peroxisome proliferator-activated receptor gamma-activated maxi-K channel opening in human umbilical vein endothelial cells. J Biol Chem 281, 13503–13512, doi:10.1074/jbc.M510357200 (2006).
Murata, T. et al. Peroxisome proliferator-activated receptor-gamma ligands inhibit choroidal neovascularization. Invest Ophthalmol Vis Sci 41, 2309–2317 (2000).
Xin, X., Yang, S., Kowalski, J. & Gerritsen, M. E. Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. J Biol Chem 274, 9116–9121, doi:10.1074/jbc.274.13.9116 (1999).
Suttles, J. & Stout, R. D. Macrophage CD40 signaling: a pivotal regulator of disease protection and pathogenesis. Semin Immunol 21, 257–264, doi:10.1016/j.smim.2009.05.011 (2009).
Cheng, G. H., Sun, S. L., Wang, Z. F. & Jin, S. Z. Investigation of the interaction between the MIR-503 and CD40 genes in irradiated U937 cells. Radiat Oncol 7 (2012).
Kim, J. Apelin-APJ signaling: a potential therapeutic target for pulmonary arterial hypertension. Mol Cells 37, 196–201, doi:10.14348/molcells.2014.2308 (2014).
Kim, J. D. et al. Epigenetic modulation as a therapeutic approach for pulmonary arterial hypertension. Experimental & molecular medicine 47, e175, doi:10.1038/emm.2015.45 (2015).
Kim, J. et al. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 131, 190–199, doi:10.1161/circulationaha.114.013339 (2015).
Kim, J. et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med 19, 74–82, doi:10.1038/nm.3040 (2013).
Lee, A. et al. Therapeutic implications of microRNAs in pulmonary arterial hypertension. BMB reports 47, 311–317, doi:10.5483/BMBRep.2014.47.6.085 (2014).
Bertero, T. et al. The MicroRNA-130/301 Family Controls Vasoconstriction in Pulmonary Hypertension. J Biol Chem 290, 2069–2085, doi:10.1074/jbc.M114.617845 (2015).
Plutzky, J. The PPAR-RXR transcriptional complex in the vasculature: energy in the balance. Circ Res 108, 1002–1016, doi:10.1161/CIRCRESAHA.110.226860 (2011).
Park, C. Y. et al. A resource for the conditional ablation of microRNAs in the mouse. Cell Rep 1, 385–391, doi:10.1016/j.celrep.2012.02.008 (2012).
Wang, Y. et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486, doi:10.1038/nature09002 (2010).
Kuehbacher, A., Urbich, C. & Dimmeler, S. Targeting microRNA expression to regulate angiogenesis. Trends Pharmacol Sci 29, 12–15, doi:10.1016/j.tips.2007.10.014 (2008).
Wang, S. & Olson, E. N. AngiomiRs–key regulators of angiogenesis. Curr Opin Genet Dev 19, 205–211, doi:10.1016/j.gde.2009.04.002 (2009).
Zhou, B. et al. MicroRNA-503 targets FGF2 and VEGFA and inhibits tumor angiogenesis and growth. Cancer Lett 333, 159–169, doi:10.1016/j.canlet.2013.01.028 (2013).
Caporali, A. et al. Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation 123, 282–291, doi:10.1161/CIRCULATIONAHA.110.952325 (2011).
Chamorro-Jorganes, A. et al. MicroRNA-16 and microRNA-424 regulate cell-autonomous angiogenic functions in endothelial cells via targeting vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1. Arterioscler Thromb Vasc Biol 31, 2595–2606, doi:10.1161/ATVBAHA.111.236521 (2011).
Ghosh, G. et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-alpha isoforms and promotes angiogenesis. J Clin Invest 120, 4141–4154, doi:10.1172/JCI42980 (2010).
Mach, F. et al. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci USA 94, 1931–1936, doi:10.1073/pnas.94.5.1931 (1997).
Lievens, D., Eijgelaar, W. J., Biessen, E. A., Daemen, M. J. & Lutgens, E. The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb Haemost 102, 206–214, doi:10.1160/TH09-01-0029 (2009).
Bavendiek, U. et al. Induction of tissue factor expression in human endothelial cells by CD40 ligand is mediated via activator protein 1, nuclear factor kappa B, and Egr-1. J Biol Chem 277, 25032–25039, doi:10.1074/jbc.M204003200 (2002).
Wagner, A. H., Guldenzoph, B., Lienenluke, B. & Hecker, M. CD154/CD40-mediated expression of CD154 in endothelial cells: consequences for endothelial cell-monocyte interaction. Arterioscler Thromb Vasc Biol 24, 715–720, doi:10.1161/01.ATV.0000122853.99978.b1 (2004).
Stout, R. D. & Suttles, J. The many roles of CD40 in cell-mediated inflammatory responses. Immunol Today 17, 487–492, doi:10.1016/0167-5699(96)10060-I (1996).
Kotowicz, K., Dixon, G. L., Klein, N. J., Peters, M. J. & Callard, R. E. Biological function of CD40 on human endothelial cells: costimulation with CD40 ligand and interleukin-4 selectively induces expression of vascular cell adhesion molecule-1 and P-selectin resulting in preferential adhesion of lymphocytes. Immunology 100, 441–448, doi:10.1046/j.1365-2567.2000.00061.x (2000).
Deregibus, M. C., Buttiglieri, S., Russo, S., Bussolati, B. & Camussi, G. CD40-dependent activation of phosphatidylinositol 3-kinase/Akt pathway mediates endothelial cell survival and in vitro angiogenesis. J Biol Chem 278, 18008–18014, doi:10.1074/jbc.M300711200 (2003).
Hristov, M., Gumbel, D., Lutgens, E., Zernecke, A. & Weber, C. Soluble CD40 ligand impairs the function of peripheral blood angiogenic outgrowth cells and increases neointimal formation after arterial injury. Circulation 121, 315–324, doi:10.1161/CIRCULATIONAHA.109.862771 (2010).
Zhang, Y. F., Zou, X. L., Wu, J., Yu, X. Q. & Yang, X. Rosiglitazone, a Peroxisome Proliferator-Activated Receptor (PPAR)-gamma Agonist, Attenuates Inflammation Via NF-kappa B Inhibition in Lipopolysaccharide-Induced Peritonitis. Inflammation 38, 2105–2115, doi:10.1007/s10753-015-0193-2 (2015).
Acknowledgements
We thank C. Hwangbo, R. Webber, and N. Copeland for assistance with mouse breeding and colony management, and D. Defilippo for administrative assistance. We thank R. Adams for the Cdh5-CreERT2 mice. This research was supported by NIH/NHLBI (HL113005 to H.J.C.), American Heart Association (Established Investigator Award to H.J.C.), American Diabetes Association (1-14-BS-035 to H.J.C.), the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the minister of Education, Science and Technology (NRF-2016R1A5A1011974 and NRF-2016R1C1B2006591 to J.K), and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI13C1372 to J.K.).
Author information
Authors and Affiliations
Contributions
A.L., J.K. and H.J.C. designed the research. A.L., I.P., J.C., Y. Park., H.N.J., H.K., H.N.J., J.K. and H.J.C. performed the experiments. A.L., I.P., J.K. and H.J.C. prepared the figures. A.L., I.P., J.K. and H.J.C. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, A., Papangeli, I., Park, Y. et al. A PPARγ-dependent miR-424/503-CD40 axis regulates inflammation mediated angiogenesis. Sci Rep 7, 2528 (2017). https://doi.org/10.1038/s41598-017-02852-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02852-4
- Springer Nature Limited
This article is cited by
-
Peroxisome proliferator-activator receptor γ and psoriasis, molecular and cellular biochemistry
Molecular and Cellular Biochemistry (2022)
-
Urinary miRNA profiles discriminate between obstruction-induced bladder dysfunction and healthy controls
Scientific Reports (2021)
-
Long non-coding RNA NEAT1 promotes lipopolysaccharide-induced acute lung injury by regulating miR-424-5p/MAPK14 axis
Genes & Genomics (2021)
-
miR-322/-503 rescues myoblast defects in myotonic dystrophy type 1 cell model by targeting CUG repeats
Cell Death & Disease (2020)