
Abstract
Remora albescens, also known as white suckerfish, recognized for its distinctive suction-cup attachment behavior and medicinal significance. In this study, we produced a high-quality chromosome-level genome assembly of R. albescens through the integration of 23.87 Gb PacBio long reads, 64.54 Gb T7 short reads, and 88.63 Gb Hi-C data. Initially, we constructed a contig-level genome assembly totaling 605.30 Mb with a contig N50 of 23.12 Mb. Subsequently, employing Hi-C technology, approximately 99.68% (603.38 Mb) of the contig-level genome was successfully assigned to 23 pseudo-chromosomes. Through the integration of homologous-based predictions, ab initio predictions, and RNA-sequencing methods, we successfully identified a comprehensive set of 22,445 protein-coding genes. Notably, 96.36% (21,629 genes) of these were effectively annotated with functional information. The genome assembly achieved an estimated completeness of 98.1% according to BUSCO analysis. This work promotes the applicability of the R. albescens genome, laying a solid foundation for future investigations into genomics, biology, and medicinal importance within this species.
Similar content being viewed by others


Background & Summary
Remora albescens, namely white suckerfish or white remora, are in the Echeneidae family, order Carangiformes, and inhabit warm seas (Fig. 1). Similar to other members of the Echeneidae family, white suckerfish have evolved front dorsal fin sucking discs, which extend from the top of the head to the tips of their pectoral fins, consisting of 13-14 plates1. These adaptations enable them to adhere to smooth surfaces through suction, and they spend majority of their lives clinging to a host animal, such as a manta ray or a shark2. They frequently affix themselves to the body, as well as within the gill chamber and the mouth of the host2. The relationship between a white suckerfish and its host is typically considered a form of commensalism, specifically phoresy. Besides their unique biological characteristics, the white suckerfish are used in traditional Chinese medicine for their positive impact on lung and spleen-stomach health3, which grants them considerable medicinal value and commercial benefits.

Morphological characteristics of R. albescens.
High-quality reference genomes are instrumental in facilitating a deep understanding and comprehensive screening of the genetic foundation and variations linked to crucial traits. This knowledge allows us to gain insights into and effectively harness the biological characteristics of the species for various purposes. Currently, the genome of the white suckerfish has not been sequenced, impeding our exploration of genetic basis behind their biological features and behaviours. Overall, a high-quality chromosome-level reference genome will contribute to a profound comprehension of the genetic mechanisms responsible for the medicinal value of R. albescens.
In this study, through the integration of PacBio High fidelity (HiFi) long-reads, T7 paired-end sequencing short-reads and high-throughput chromatin capture (Hi-C) sequencing data (Table 1), we introduce the first chromosomal-level genome assembly of R. albescens. The assembly yielded a genome of 605.30 Mb, composed of 158 contigs, with a contig N50 length of 23.12 Mb. In total, 603.38 Mb, covering 99.68% of the contig-level genome, were accurately mapped onto 23 chromosomes by using Hi-C data. The BUSCO alignment analysis indicated that our ultimate assembly contained 3,571 (98.1%) complete BUSCOs. In conclusion, this high-quality chromosomal-level reference genome establishes a valuable foundation for comprehending the biological characteristics and conducting further research into the medicinal value of the R. albescens.
Methods
Fish sample collection and preparation
A single fish, measuring 18 centimeters in length, was obtained from Northern South China Sea in June 2022 (Fig. 1). The collection of the sampled fish for this study was conducted in accordance with the guidelines and regulations set forth by the Animal Care and Use Committee of Fisheries College of Zhejiang Ocean University, as indicated by Animal Ethics no. 1067. Tissues from the R. albescens were collected and preserved in liquid nitrogen until DNA or RNA extraction. Wherein, muscle and liver tissues were utilized for DNA sequencing to implement the genome assembly. Kidney, spleen, fin, gill and sucker tissues were utilized for RNA sequencing.
WGS BGISEQ library and PacBio library construction, sequencing and contig-level assembly
According to the standard phenol/chloroform extraction instruction, the whole-genome sequencing (WGS) libraries were prepared by extracting genomic DNA from muscle tissues.
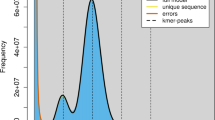
To obtain BGISEQ short reads, the DNA sample underwent evaluation through 1% agarose gel electrophoresis and the Pultton DNA/Protein Analyzer (Plextech). Subsequently, a paired-end library with an insert size of 300 bp to 350 bp was constructed following the BGISEQ standard protocol. Afterward, the DNA sample was purified, quantified, and subjected to sequencing from both ends using the BGISEQ-T7 sequencing platform. BGISEQ sequencing resulted in a total of 66.21 Gb raw reads (Table 1). Following a filtering process utilizing fastp v0.23.24 with default parameters, which aimed to eliminate low-quality, short reads, adapters and redundant sequences, a total of 64.54 Gb clean reads were obtained (Table 1). Then by using GCE v1.0.0 software5, K-mer analysis was performed to estimate the genome size and heterozygosity for R. albescens, which were 563 Mb and 0.63%, respectively (Fig. 2).

K-mer distribution of R. albescens.
To obtain PacBio long reads, the DNA sample was first evaluated using Nanodrop, Qubit and agarose gel electrophoresis. Then, the library with a fragment size of 20 kb was created utilizing the SMRTBell template preparation kit 1.0 following the manufacturer’s instructions. Afterward, the DNA sample was subjected to sequencing using the PacBio Sequel II platform in Circular Consensus Sequence (CCS) mode. After removing low-quality sequences using the CCS v6.0.0 algorithm with default parameters, a sum of 23.87 Gb high-precision reads with an N50 value of 18.88 kb were obtained. With these HiFi reads, the initial contigs were assembled using the Hifiasm v0.16.16 and the purge_haplotigs algorithms7 with the default settings. The assembly yielded a 605.30 Mb genome with a maximum contig size of 51.46 Mb.
Hi-C library preparation, sequencing and chromosomal-level assembly
The contigs obtained in the previous step were anchored onto chromosomes using Hi-C data. In a nutshell, 1 g of liver tissue from R. albescens was treated with 1% formaldehyde for 20 minutes at 20–25 °C temperature to facilitate the coagulation of proteins implicated in chromatin interactions. Next, DNA was digested using MboI and the overhangs of the resulting restriction fragments were labeled with biotinylated nucleotides, after which they were ligated within a confined volume. Following the cross-link reversal, the ligated DNA was purified and fragmented to a size range of 300–500 bp. Following this step, ligation junctions were extracted by streptavidin beads and subjected to sequencing from both ends using the BGISEQ-T7 sequencing platform, producing a total of 88.75 Gb raw data (Table 1). After removing low-quality sequences and adapters, and only retaining paired-end reads, both of which are longer than 50 bp, with fastp v0.23.24 software, a sum of 88.63 Gb clean data were acquired (Table 1). We utilized the HiCUP pipeline8 to obtain credible and nonredundant contigs interaction matrix, and then anchored the contigs onto chromosomes by using 3D-DNA pipeline9. Juicebox Assembly Tools10 was utilized for manual error correction to rectify any occurrences of chromosome inversion and translocation. Finally, 603.38 Mb (~99.63%) of contig-level assembled sequences were positioned onto 23 pseudo-chromosomes (Fig. 3A).

Genome assembly of R. albescens. (A) Hi-C interaction matrix for R. albescens. (B) Circos plot from outer to inner layers depicts the following: (a) GC content; (b) gene density; (c) repeat density; (d) LTR retroelement density; (e) LINE density; and (f) DNA transposons density. a-f were drawn in 500-kb sliding windows.
RNA library construction and sequencing
Total RNA was extracted from the five tissues, including kidney, spleen, fin, gill and sucker, of the R. albescens using TRIzol reagent (Invitrogen). To evaluate RNA quality, we utilized the NanoDrop ND-1000 spectrophotometer (Labtech) and the 2100 Bioanalyzer (Agilent Technologies). The paired-end reads were sequenced using the BGISEQ-T7 Platform. Overall, 6.01 Gb of clean data were obtained following filtering process utilizing fastp v0.23.24 with default settings to eliminate low-quality and short reads, as well as trim adapters and polyG tails (Table 1).
Repetitive elements annotation
Repeat elements in the R. albescens genome were systematically identified using a dual approach, incorporating both homology-based searches and ab initio predictions. The ab initio prediction of repeat elements was carried out through two tools, namely Tandem Repeat Finder v4.0911 and LTR_FINDER_parallel v1.111 with default parameters. Subsequently, newly discovered repeats were predicted using RepeatMasker v4.0.912, based on the de novo repetitive sequence library that was constructed using LTR_FINDER_parallel and RepeatModeler v2.013, RepeatMasker v4.0.9 and RepeatProteinMask v4.1.0 (http://www.repeatmasker.org) were used to identify known repeat elements with the Repbase v20181026 database14. In total, 18.04% of the R. albescens genome were identified as repetitive sequences (Fig. 3B). Among these repeat elements, DNAs, LTRs, LINEs, and SINEs constituted 6.98%, 2.49%, 5.41%, and 1.69% of the genome, respectively (Table 3).
Gene prediction and annotation
Utilizing the repeat-masked genome as a basis, three strategies, comprising ab initio prediction, homologous prediction and RNA-sequencing method, were employed to predict protein-coding genes within the R. albescens genome. Ab initio prediction was conducted utilizing Augustus v3.3.215 and Genscan16 software. Simultaneously, homologous prediction relied on protein sequences from various annotated species, comprising Seriola lalandi (RefSeq assembly accession: GCF_002814215.2), Seriola dumerili (RefSeq assembly accession: GCF_002260705.1), Echeneis naucrates (RefSeq assembly accession: GCF_900963305.1), Takifugu rubripes (RefSeq assembly accession: GCF_901000725.2), Gasterosteus aculeatus (RefSeq assembly accession: GCF_016920845.1), and Danio rerio (RefSeq assembly accession: GCF_000002035.6). The protein sequences above were retrieved from the NCBI database and then aligned with the R. albescens genome utilizing tblastn tool (e-value ≤ 1e-5). Subsequently, the homologous sequences were aligned with the corresponding proteins with Genewise v2.4.017 to predict detailed gene structures. The RNA-seq dataset were aligned to the assembled genome by using HISAT2 v2.1.018 with default settings, and the predicted transcripts were identified by using StringTie v1.3.519 and TransDecoder v5.1.0 (https://github.com/TransDecoder/TransDecoder) with default settings. Three gene model predictions were merged using MAKER v2.31.1020. Based on that, we further refined the gene set using HiFAP (Wuhan OneMore Tech Co., Ltd., https://www.onemore-tech.com/) with high-quality transcripts and homology annotation results, resulting in a final gene set with a total number of protein-coding genes of 22,445 genes (Fig. 3B and Table 4).
The functional annotation of the predicted protein-coding gene sets was performed using BLASTp (e-value ≤ 1e-5) with the diamond v2.0.8 software21 based on six databases, including Swiss-Prot v2023-03-0122, NCBI nonredundant protein (NR) v2023-04-01, Kyoto Encyclopedia of Genes and Genomes (KEGG) v2023-01-01 (http://www.genome.jp/kegg/), TrEMBL v2023-03-01 (http://www.uniprot.org), eukaryotic orthologous groups of proteins (KOG) v2003-03-0123 and AnimalTFDB v4.0 (http://bioinfo.life.hust.edu.cn/AnimalTFDB4/?#/). Additionally, protein structural domain predictions of gene sets were performed based on InterPro and Pfam databases utilizing InterProScan v5.61-93.024 with parameters “–goterms–pathways -dp”. As a result, 96.36% (21,629 genes) of the total predicted genes were successfully annotated. (Table 5).
Non-coding RNA prediction and annotation
According to the miRBase25 and rfam26 databases, the microRNAs (miRNAs), ribosomal RNAs (rRNAs) and small nuclear RNAs (snRNAs) were annotated utilizing INFERNAL v1.127. The transfer RNAs (tRNAs) were predicted by using tRNAscan-SE v1.3.128. Consequently, 829 miRNAs, 1,832 rRNAs, 820 snRNAs and 7,033 tRNAs were predicted within the R. albescens genome (Table 6).
Data Records
The raw sequencing data for R. albescens in this study is available from the Sequence Read Archive (SRA) under Bioproject number PRJNA1036795, which includes WGS T7 sequencing data (SRR2683110029), Pacbio HiFi sequencing data (SRR2683109930), Hi-C sequencing data (SRR2683109831), and RNA sequencing data (SRR2853758732). The assembled genome of R. albescens has been deposited in GenBank under accession JAXCVL00000000033. Additionally, files contained the assembled genome, protein-coding gene annotation, non-coding RNA prediction, and repeat annotation of R. albescens have been made available in the Figshare database34.
Technical Validation
Our initial assessment of the continuity of the R. albescens genome assembly was conducted using QUAST v5.2.035. The contig N50 reaches 23.12 Mb and the genome displays a minimal number of gaps (1.75 per 100 kbp), which exhibits better assembly performance than closely related species (Echeneis naucrates: GCA_900963305.1) (Table 2). Next, we remapped T7 clean short reads and PacBio clean long reads to the R. albescens genome using BWA36 and Minimap237, yielding mapping rates of 99.83%, 99.96% and coverage rates (at least 4X) of 99.61%, 99.76%, respectively (Table 7). Furthermore, the completeness of the R. albescens genome was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO, v5.1.0)38 with the actinopterygii_odb10 database. The analysis revealed that the genome assembly contained 3,571 (98.1%) complete BUSCO genes, comprising 3,551 (97.55%) single-copy BUSCO genes, 20 (0.55%) duplicated BUSCO genes, and 11 (0.3%) fragmented BUSCO genes (Table 8). Collectively, the comprehensive assessment indicates that the R. albescens genome serves as a high-quality reference genome.
Code availability
No specific custom codes were developed in this study. Data analyses were conducted following the guidelines and protocols provided by the developers of the respective bioinformatics tools, as detailed in the methods section.
References
Schwartz, F. J. Five species of sharksuckers (family Echeneidae) in North Carolina. JNCAS 120, 44–49 (2004).
O’Toole, B. Phylogeny of the species of the superfamily Echeneoidea (Perciformes: Carangoidei: Echeneidae, Rachycentridae, and Coryphaenidae), with an interpretation of echeneid hitchhiking behaviour. Can J Zool 80, 596–623 (2002).
Tang, W. C. Chinese medicinal materials from the sea. (1987).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant Biol 35, 62–67 (2013).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460 (2018).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res 4, 1310 (2015).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3, 95–98 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, Unit 4 10 (2004).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110, 462–467 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, 435–439 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 268, 78–94 (1997).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res 14, 988–995 (2004).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11, 1650–1667 (2016).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18, 188–196 (2008).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31, 365–370 (2003).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41 (2003).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. miRBase: from microRNA sequences to function. Nucleic Acids Res 47, 155–162 (2019).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, 121–124 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26831100 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26831099 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26831098 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28537587 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc:JAXCVL000000000 (2023).
Wang, D. Y. et al. Chromosome-level genome assembly and annotation of Remora albescens. figshare https://doi.org/10.6084/m9.figshare.24624144.v1 (2024).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Waterhouse, R. M. et al. BUSCO Applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35, 543–548 (2018).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (41976083).
Author information
Authors and Affiliations
Contributions
D.W. and T.G. conceived and designed the study. C.Z. and T.G. conducted animal work and prepared biological samples. C.Z., Q.L. and Y.Q. performed the genome assembly and analysis. D.W., T.G. and C.Z. wrote the paper. D.W., T.G., C.Z. Q.L. and Y.Q. revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, C., Liu, Q., Qu, Y. et al. The first chromosomal-level genome assembly and annotation of white suckerfish Remora albescens. Sci Data 11, 523 (2024). https://doi.org/10.1038/s41597-024-03363-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03363-4
- Springer Nature Limited