
Abstract
The Entomobryoidea, the largest superfamily of Collembola, encompasses over 2,000 species in the world. However, the lack of high-quality genomes hinders our understanding of the evolution and ecology of this group. This study presents a chromosome-level genome of Entomobrya proxima by combining PacBio long reads, Illumina short reads, and Hi-C data. The genome has a size of 362.37 Mb, with a scaffold N50 size of 57.67 Mb, and 97.12% (351.95 Mb) of the assembly is located on six chromosomes. The BUSCO analysis of our assembly indicates a completeness of 96.1% (n = 1,013), including 946 (93.4%) single-copy BUSCOs and 27 (2.7%) duplicated BUSCOs. We identified that the genome contains 22.16% (80.06 Mb) repeat elements and 20,988 predicted protein-coding genes. Gene family evolution analysis of E. proxima identified 177 gene families that underwent significant expansions, which were primarily associated with detoxification and metabolism. Moreover, our inter-genomic synteny analysis showed strong chromosomal synteny between E. proxima and Sinella curviseta. Our study provides valuable genomic information for comprehending the evolution and ecology of Collembola.
Similar content being viewed by others

Background & Summary
Collembola is one of the most abundant and diverse groups of terrestrial arthropods, containing more than 9,400 known species in the world, with at least 410 million years of evolutionary history1. Their distributions range from the Arctic to Antarctica, and from a 1,760 m deep cave to a 5,000 m high mountain2. Collembola exhibit a wide range of feeding habits, including consuming litter, fungi, pollen, algae, leaves, and roots3,4,5. Furthermore, numerous collembolan species demonstrate sensitivity to environmental factors, making them suitable indicators for assessing soil ecotoxicology and monitoring environmental changes6.
Entomobryoidea is the largest superfamily of Collembola, containing approximately 2,000 species found in various terrestrial ecosystems, and is an essential member of soil communities, mainly occurring in humid forests and grasslands1. This monophyletic group is a key player in collembolan phylogeny and is divided into two families: Orchesellidae and Entomobryidae7, with Entomobrya (Rondani, 1861) being the type genus of Entomobryidae and some species of this group are important pests of mushrooms8,9. To obtain a high-quality genome of Collembola, a combination of second and third-generation sequencing is necessary. However, due to their small body size (<10 mm), a single collembolan individual does not provide sufficient DNA/RNA for library construction, requiring the pooling of multiple individuals for sequencing. Furthermore, several generations of feeding are necessary to reduce heterozygosity, further increasing the challenge of obtaining a high-quality genome. To date, 51 collembolan genomes have been reported (as of July 2023 from NCBI), with only seven species sequenced using third-generation sequencing and four genomes at the chromosome level. However, the majority of these genomes assembled from short reads have poor assembly quality, with scaffold N50 lengths of less than 100 kb.
To enhance our knowledge of the evolution and ecology of Entomobryoidea, we obtained a chromosome-level genome of Entomobrya proxima (Folsom, 1924) through the combination of PacBio long reads, Illumina short reads, and Hi-C data. We annotated repeats, non-coding RNAs (ncRNAs), and protein-coding genes (PCGs), and conducted gene family evolution analysis. Moreover, we explored the interspecific chromosomal variation between the two Entomobryidae species, E. proxima and Sinella curviseta. The high-quality genome of E. proxima is an important milestone in our understanding of Entomobryoidea and will contribute to the study of collembolan evolution and ecology.
Methods
Sample collection and sequencing
The E. proxima samples used in this study were collected in June 2020 from the Purple Mountain in Nanjing, China (32.06 °N, 118.83 °E). Adult individuals were collected by an entomological aspirator, washed in phosphate-buffered saline, and instantly frozen using liquid nitrogen. A total of 100, 30, 50, and 50 individuals were used for PacBio, genome survey, Hi-C, and transcriptome sequencing, respectively. To obtain the requisite amount of DNA/RNA for sequencing, we utilized a pooled extraction approach, in which multiple individuals were combined prior to nucleic acid extraction.
Genomic DNA and RNA from pooled specimens were extracted using the DNeasy Blood & Tissue Kit and TRIzoTM Reagent, following the manufacturer’s instructions. PCR-free short-read libraries of 150 bp paired-end read with a 350 bp insert size were generated using the Truseq DNA PCR-free Kit. The Hi-C sequencing was carried out by digesting extracted DNA with the Mbol restriction enzyme. We utilized the Illumina NovaSeq 6000 platform to sequence all short-read libraries. A library with a 30 kb-insert size was prepared using the SMRTbell Template Prep Kit 1.0-SPv3 for PacBio sequencing. To generate the library, we used the PacBio Sequel II platform, which employs the PacBio CLR mode. Berry Genomics (Beijing, China) carried out all library construction and sequencing. Finally, we obtained 292.18 Gb of sequencing data, comprising 206.73 Gb (570.50×) of PacBio reads, 45.89 Gb (126.62×) of Illumina reads, 29.83 Gb (82.32×) of Hi-C data, and 9.73 Gb of transcriptome data (Table 1). The raw PacBio long reads had a scaffold N50 and an average length of 29.41 and 24.08 kb, respectively.
Genome assembly
We performed quality control on raw Illumina data using BBTools v38.8210, which included the “clumpify.sh” script to remove duplicate reads. We trimmed low-quality reads using the “bbduk.sh” script, which consisted of base quality trimming of both ends (>Q20), length filtering (>15 bp), polymer A/G/C trimming (>10 bp), and correction of overlapping paired reads.
The primary assembly of PacBio long reads was generated using Flye v2.8.311, with a minimum overlap of 3,000 between reads. We then performed one round of self-polishing on the assembly. NextPolish v1.1.012 was used to polish the primary assembly with Illumina reads in two rounds. Redundant contigs were eliminated using Purge_Dups v1.2.513 with a haploid cutoff set at 50 for identifying contigs as haplotigs (“-s 50”). We used Minimap2 v2.1714 for read mapping during the above “remove haplotigs” and “short-read polishing” steps. We aligned Hi-C reads to the assembly after performing quality control using Juicer v1.6.215. Subsequently, we anchored the contigs onto chromosomes using 3D-DNA v18092216. To ensure accuracy, we manually reviewed and corrected any errors using Juicebox v1.11.0816. We detected possible contaminants using MMseqs. 2 v1117, which performed BLASTN-like searches based on the NCBI nucleotide and UniVec databases with a sequence identity of 0.8 (‘–min-seq-id 0.8’). Additionally, we utilized blastn (BLAST + v2.11.018) against the UniVec database to specifically detect vector contaminants. We considered that sequences with over 90% hits in the aforementioned database likely contained contaminants. Sequences with over 80% hits were checked again via online BLASTN analysis in the NCBI nucleotide database. We then removed potential bacterial and human contamination from the assembled scaffolds. Most of the identified contaminants were bacterial, including Rickettsia and Sphingobacterium. The final chromosome-level genome assembly of E. proxima had a size of 362.37 Mb, comprising 461 scaffolds and 2,007 contigs, with the scaffold and contig N50 sizes of 57.67 Mb and 0.44 Mb, respectively. Among them, 1,542 contigs (97.12%, 351.95 Mb) were anchored into six chromosomes with lengths ranging from 48.13 to 69.80 Mb and the GC content was 35.93% (Table 2; Fig. 1). The genome size and GC content of E. proxima (362.37 Mb and 35.93%) were smaller than that of S. curviseta (363.44 Mb and 37.52%) (Table 3).

Genome-wide chromosomal heatmap of Entomobrya proxima, with each chromosome and contig framed in blue and green, respectively.
Genome annotation
We employed RepeatModeler v2.0.319 and the “-LTRStruct” LTR discovery pipeline to generate a repeat library. We then merged this library with the Repbase-2018102620 and Dfam 3.121 into a custom library. RepeatMasker v4.1.2-p122 was utilized to identify repetitive elements in the E. proxima genome by aligning it against the custom library. RepeatMasker analysis revealed that the E. proxima genome contains approximately 22.16% (80.06 Mb) repetitive elements, comprising unclassified elements (11.88%), LTR elements (5.20%), DNA transposons (1.77%), LINE (1.77%), simple repeats (0.61%), as well as other elements (Table S1). Notably, the percentage of repetitive elements in E. proxima (22.16%) was higher than that in S. curviseta (14.16%) (Table 3).
We used Infernal v1.1.423 and tRNAscan-SE v2.0.924 to identify ncRNAs and tRNAs, respectively. The tRNAscan-SE with the script “EukHighConfidenceFilter” was used to filter low-confidence tRNAs. We identified 391 ncRNAs in the genome of E. proxima, which contained 23 ribosomal RNAs, 23 microRNAs, 42 small nuclear RNAs, 152 transfer RNAs, seven ribozymes, and 144 other ncRNAs (Table S2). The number of ncRNAs in E. proxima (391) was lower than that in S. curviseta (583) (Table 3).
We annotated the PCGs using MAKER v3.01.0325 based on three strategies, containing ab initio predictions, transcribed RNA, and homologous proteins. To maximize ab initio predictions, we employed the BRAKER v2.1.626 and GeMoMa v1.7.127 tools, using transcriptome and protein evidence, and combined their results as the ab initio input for MAKER. HISAT2 v2.2.028 was employed for producing transcriptome alignments. BRAKER used Augustus v3.3.429 and GeneMark-ES/ET/EP 4.68_lic30 as predictors and automatically trained them from RNA-seq alignments and reference proteins mined from OrthoDB10 v1 database31. GeMoMa employed protein homology and intron position conservation to predict genes, using the parameters “GeMoMa.c = 0.4 GeMoMa.p = 10” and protein sequences from seven species (Cloeon dipterum (GCA_902829235.1), Daphnia magna (GCF_003990815.1), Drosophila melanogaster (GCF_000001215.4), a sexual strain of Folsomia candida (FCSH, GCA_020923455.1), Rhopalosiphum maidis (GCF_003676215.2), Tribolium castaneum (GCF_000002335.3), and Zootermopsis nevadensis (GCF_000696155.1)) (Table 4). We used RNA-seq alignments produced from HISAT2 to perform genome-guided assembly by StringTie v2.1.632. Additionally, protein sequences from the same seven species employed in GeMoMa were incorporated as evidence of protein homology in MAKER. Finally, we predicted 20,988 PCGs in the E. proxima genome, with an average length of 4,610.4 bp. The average number of exons, introns, and CDS of each gene were 5.9, 4.9, and 5.7, respectively, and their corresponding mean length was 393.1, 481.1, and 281.8 bp, respectively (Table S3). BUSCO completeness of the protein sequences was 96.9% (n = 1,013), including 93.0% (942) single-copy, 3.9% (40) duplicated, 0.7% (7) fragmented, and 2.4% (24) missing BUSCOs, suggesting high-quality predictions.
We conducted the gene functional annotation search against the UniProtKB database using Diamond v2.0.11.14933 in sensitive mode with the parameters “–more-sensitive -e 1e-5”. We further employed eggNOGmapper v2.0.134 and InterProScan 5.53–87.035 to assign Gene Ontology (GO) and (KEGG, Reactome) pathway annotations and to identify protein domains. The InterProScan analyses included five databases: Pfam36, SMART37, Superfamily38, Gene3D39, and CDD40. The results predicted by the above tools were integrated to obtain the final gene function prediction. Genes with 13,129 GO terms, 13,129 KEGG pathways, 14,554 Reactome pathways, 4,393 Enzyme Codes, and 15,211 COG categories were assigned by integrating the InterProScan and eggNOG annotation results (Table S3).
Phylogeny and gene family evolution
We inferred the orthology of PCG sequences across eight arthropod species, comprising one Diplura species (Catajapyx aquilonaris) and seven collembolan species (E. proxima, S. curviseta, Orchesella cincta, a parthenogenetic strain of Folsomia candida (FCDK), FCSH, Tomocerus qinae, and Holacanthella duospinosa) (Table 4). We used OrthoFinder v2.5.241 to infer orthogroups (gene families), after eliminating redundant isoforms. For sequence alignment, we used the Diamond with ultra-sensitive mode (“-S diamond_ultra_sens”). We assigned 143,727 (89.6%) genes to 21,690 orthogroups, of which 3,924 were shared by all eight species and 2,252 were single-copy genes (Table S4). In E. proxima, 19,453 genes (92.7%) were contained in 12,705 gene families, of which 389 families and 1,886 genes were specific to this species.
We aligned single-copy orthologues using MAFFT v7.45042 with the high-accuracy LINS-I strategy. We then removed alignment gaps using trimAl v1.4.143 with the “automated1” parameter. Finally, we reconstructed the phylogenetic tree on the single-copy orthologs using IQ-TREE v2.0744 with the LG site-homogeneous model. We employed MCMCTree in PAML v4.9j45 to predict species divergence time and nodes were calibrated using two fossils obtained from the PBDB database (https://www.paleobiodb.org/navigator/): the most common ancestor of Diplura and Collembola (407.6–410.8 Ma) and Entomobryomorpha (272.3–279.3 Ma). After filtering out 244 single-copy genes, we performed phylogenetic reconstruction using IQ-TREE based on 2,008 remaining genes. The analysis showed that E. proxima is closely related to S. curviseta, and the divergence time of these species was approximately 47.01‒51.94 Mya (Fig. 2c). Our phylogenetic results were consistent with previous studies, supporting Orchesellidae (O. cincta) as a sister group to Entomobryidae (E. proxima, and S. curviseta)7,46,47 (Fig. 2c).

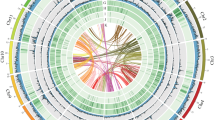
Genome characteristics, phylogeny, and gene family evolution of Entomobrya proxima. (a) From the outer ring to the inner ring are the distributions of chromosome length, GC content, gene density, TEs (DNA, SINE, LINE, and LTR), and simple repeats. (b) The top 20 gene families that significantly expanded in E. proxima. (c) The phylogeny and gene family changes among eight arthropod species, with node values representing the 95% highest probability densities of divergence times (unit, 100 Ma). The values labeled at terminals denote the number of significantly expanded and contracted gene families. “1:1:1” represents universal single-copy genes in all species, “N:N:N” represents multi-copy genes, “others” represents unclassified orthologues, and “unassigned” represents orthologues that cannot be assigned to any orthogroups.
We used CAFÉ v4.2.148 to estimate the gene family evolution (expansion or contraction) based on the generated phylogenetic tree. We identified 1,351 expanded and 1,547 contracted gene families in E. proxima, including 223 gene families that underwent rapid evolution (177 expansions and 46 contractions). The significantly expanded families included the Leucine-rich repeat domain superfamily, zinc finger, Cytochrome P450, and other families that play important roles in the adaptive evolution of E. proxima (Table S5; Fig. 2b). Subsequently, we performed functional enrichment (GO and KEGG) analysis on PCGs from significantly expanded families using ClusterProfiler v3.10.149 with default parameters. The enrichment of GO and KEGG in rapidly expanding families further indicates their function in the regulation of hormone levels, oxidoreductase activity, metabolic, and catabolic processes, among others (Fig. 3a,b). We uploaded the complete enrichment CSV tables to Figshare (https://doi.org/10.6084/m9.figshare.23861901).

Gene enrichment, and chromosomal synteny. (a,b). GO and KEGG function enrichment of significantly expanded gene families. Only the top 30 categories are shown. (c). Chromosomal synteny between Entomobrya proxima (Epro) and Sinella curviseta (Scur).
Chromosome synteny
To investigate interspecific chromosomal evolution between E. proxima and S. curviseta, we used MMseqs. 2 with an e-value of 1e-5. Syntenic blocks were identified using MCScanX50 and the repeat content, gene density, and GC content on individual pseudochromosomes using TBtools51. Comparing the genomes of E. proxima and S. curviseta, we identified 341 syntenic blocks that contained 14,473 collinear genes (45.04%). The average number of genes per block was 42, while a notable 12.02% (41 blocks) contained over 100 collinear genes. We have uploaded the syntenic blocks results containing each block’s details to Figshare (https://doi.org/10.6084/m9.figshare.23861901). Notably, our analysis revealed that the ChrX of S. curviseta shares strong chromosomal syntenic relationships with Chr6 of E. proxima (Fig. 3c). Other chromosomes (1–5) of E. proxima matched with their corresponding chromosomes in S. curviseta, although we did observe some regions lacking homology. These differences may be attributed to the significant divergence time (47.01–51.94 Mya) between the two species.
Data Records
The raw sequencing data and genome assembly of Entomobrya proxima have been deposited at the National Center for Biotechnology Information (NCBI). The PacBio, Illumina, Hi-C, and transcriptome data can be found under identification numbers SRR15910088-SRR1591009252,53,54,55,56. The assembled genome has been deposited in the NCBI assembly with the accession number GCA_029691765.157. Additionally, the results of annotation for repeated sequences, gene structure, and functional prediction have been deposited in the Figshare database58.
Technical Validation
Two methods were used to evaluate the quality of the genome assembly. Firstly, we assessed assembly completeness using BUSCO v3.0.259 with the reference arthropod gene set (n = 1,013). The final genome assembly showed a BUSCO completeness of 96.1%, consisting of 946 (93.4%) single-copy BUSCOs, 27 (2.7%) duplicated BUSCOs, 7 (0.7%) fragmented BUSCOs, and 33 (3.2%) missing BUSCOs. Secondly, we calculated the mapping rate as a measure of assembly accuracy. The mapping rates for PacBio, Illumina, and RNA reads were 99.93%, 88.83%, and 86.72%, respectively. These evaluations collectively reflect the high quality of the genome assembly produced in this study.
Code availability
No specific script was used in this work. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.
References
Bellinger, P. F., Christiansen, K. A. & Janssens, F. Checklist of the Collembola of the World. (1996–2023).
Potapov, A. et al. Towards a global synthesis of Collembola knowledge–challenges and potential solutions. Soil Org. 92, 161–188 (2020).
Drift, J. Van Der, J. E. Grazing of springtails on hyphal mats and its influence on fungal growth and respiration. Ecol Bull. 25, 203–209 (1997).
Ponge, J. F. Food resources and diets of soil animals in a small area of Scots pine litter. Geoderma. 49, 33–62 (1991).
Scheu, S. The soil food web: structure and perspectives. Eur. J. Soil Biol. 38, 11–20 (2002).
Hopkin, S. Biology of the springtails (Insecta: Collembola). Oxford University Press. (1997).
Godeiro, N. N. et al. Phylogenomics and systematics of Entomobryoidea (Collembola): marker design, phylogeny and classification. Cladistics. 39, 101–2115 (2023).
Yu, D. et al. Molecular phylogeny and trait evolution in an ancient terrestrial arthropod lineage: systematic revision and implications for ecological divergence (Collembola, Tomocerinae). Mol. Phylogenet. Evol. 154, 106995 (2020).
Sun, X. et al. Collembola associated with edible mushrooms in China. Zool Syst. 46, 1–15 (2021).
Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2022) (2014).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Steinegger, M. & Soding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna. 6, 11 (2015).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–D89 (2016).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2022) (2013–2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Chan, P. P. & Lowe, T. M. TRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962, 1–14 (2019).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinformatics. 12, 491 (2011).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. Nar Genom. Bioinform. 3, lqaa108 (2021).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. Bmc Bioinformatics. 19, 189 (2018).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. Nar Genom. Bioinform. 2, lqaa26 (2020).
Kriventseva, E. V. et al. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Huerta-Cepas, J. et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Finn, R. D. et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37, D380–D386 (2009).
Lewis, T. E. et al. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res. 46, D1282 (2018).
Marchler-Bauer, A. et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 (2017).
Emms, D. M. & Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Katoh, K. & Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25, 1972–1973 (2009).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Luan, Y. X. et al. High-quality genomes reveal significant genetic divergence and cryptic speciation in the model organism Folsomia candida (collembola). Mol. Ecol. Resour. 23, 273–293 (2023).
Yu, D. Y. et al. Phylogenomics of Elongate-Bodied Springtails Reveals Independent Transitions From Aboveground to Belowground Habitats in Deep Time. Syst. Biol. 71, 1023–1031 (2022).
Han, M. V., Thomas, G. W., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Yu, G., Wang, L., Han, Y. & He, Q. Clusterprofiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics. 16, 284–287 (2012).
Wang, Y. et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Chen, C. et al. Tbtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 13, 1194–1202 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR15910088 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR15910089 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR15910090 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR15910091 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR15910092 (2023).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_029691765.1 (2023).
Zhang, F. Genome assembly and annotations of Entomobrya proxima (Collembola: Entomobryidae). figshare https://doi.org/10.6084/m9.figshare.23861901 (2023).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (31970434 and 32270470) and National Science and Technology Fundamental Resources Investigation Program of China (2018FY100300).
Author information
Authors and Affiliations
Contributions
F.Z. and Z.P. contributed to the research design. J.J., Y.Z. and G.Z. collected the samples. J.J., F.Z. and Z.P. analyzed the data. J.J., F.Z. and G.Z. wrote the draft manuscript and revised the manuscript. All co-authors contributed to this manuscript and approved it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jin, J., Zhao, Y., Zhang, G. et al. The first chromosome-level genome assembly of Entomobrya proxima Folsom, 1924 (Collembola: Entomobryidae). Sci Data 10, 541 (2023). https://doi.org/10.1038/s41597-023-02456-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02456-w
- Springer Nature Limited