

Abstract
Recent studies have shown that repressive chromatin machinery, including DNA methyltransferases and polycomb repressor complexes, binds to chromosomes throughout mitosis and their depletion results in increased chromosome size. In the present study, we show that enzymes that catalyze H3K9 methylation, such as Suv39h1, Suv39h2, G9a and Glp, are also retained on mitotic chromosomes. Surprisingly, however, mutants lacking histone 3 lysine 9 trimethylation (H3K9me3) have unusually small and compact mitotic chromosomes associated with increased histone H3 phospho Ser10 (H3S10ph) and H3K27me3 levels. Chromosome size and centromere compaction in these mutants were rescued by providing exogenous first protein lysine methyltransferase Suv39h1 or inhibiting Ezh2 activity. Quantitative proteomic comparisons of native mitotic chromosomes isolated from wild-type versus Suv39h1/Suv39h2 double-null mouse embryonic stem cells revealed that H3K9me3 was essential for the efficient retention of bookmarking factors such as Esrrb. These results highlight an unexpected role for repressive heterochromatin domains in preserving transcription factor binding through mitosis and underscore the importance of H3K9me3 for sustaining chromosome architecture and epigenetic memory during cell division.
Similar content being viewed by others


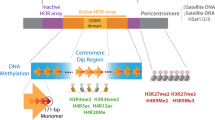
Main
Heterochromatin and euchromatin are defined cytologically as condensed and decondensed regions of the genome, respectively1,2. Constitutive heterochromatin, although gene poor, contains noncoding DNA repeat elements that are often abundantly transcribed3,4. During interphase, heterochromatin-containing domains within different chromosomes cluster together forming structures termed chromocenters5,6,7,8. In mouse cells these dynamic structures are characteristically marked by a high density of histone 3 lysine 9 trimethylation (H3K9me3) and histone histone 4 lysine 20 trimethylation, as well as the H3K9-associated heterochromatin protein 1 (HP1α or Cbx5)1,7. As chromosomes condense and enter mitosis, primary constrictions first become evident within the heterochromatin domains that correspond to centromeric microsatellite arrays, where the mitotic spindles will eventually bind. These centromeric regions are flanked by much larger domains of pericentric noncoding major satellite repeats which, in the mouse, account for approximately 3.6% of the genome9,10,11.
The Suv39h1 and Suv39h2 histone lysine methyltransferases are hallmark enzymes of mammalian heterochromatin that catalyze the trimethylation of histone H3 at lysine 9 (H3K9)12,13,14. They form part of a larger group of enzymes capable of modifying H3K9 that includes G9a (Ehmt2), Glp (Ehmt1)15,16, Setdb1 and Setdb2 (refs. 17,18), which primarily mediate H3K9 mono- and dimethylation, as well as members of the Kdm1, Kdm3 and Kdm4 families of histone demethylases19,20,21,22,23. Although heterochromatin regulation is recognized as being essential in preserving nuclear architecture, genome stability and DNA repair, and for silencing transposon expression in early mouse development24,25,26,27, the underlying repetitiveness of satellite DNA means that factors binding to heterochromatin are often excluded from conventional analyses.
In the present study we set out to examine the impact of repressive H3K9me3 on mitotic chromosome architecture and on the factors that bind chromosomes through mitosis. Several DNA-binding transcription factors (TFs) have been shown to remain bound to chromosomes during mitosis, including Foxa1, Esrrb, Sox2 and Gata1 (refs. 28,29,30,31,32,33), where they occupy a subset of the genomic sites that are present during the interphase. Such demonstrations have raised the interesting possibility that ‘mitotic bookmarking’ by factors such as these could help to convey cellular identity to newly divided daughter cells. Recent studies have also shown that many components of repressive chromatin machinery, including those that characterize constitutive heterochromatin, are also retained at mitotic chromosomes during cell division34,35,36. Although this has prompted speculation of an interplay between mitotic booking factors and repressive chromatin states37, there is currently a paucity of direct evidence to support this. In the present study, we confirm that factors mediating H3K9 trimethylation are indeed retained on native mitotic chromosomes isolated from different cell types. H3K9me3 depletion results in altered chromosome architecture, compensatory changes in the level and distribution of repressive H3K27me3 and discrete and reversible changes in the retention of specific mitotic bookmarking factors.
Results
Suv39h dn mitotic chromosomes are more compact
Previous studies had suggested that Suv39h1, Suv39h2 and HP1 proteins Cbx5 (HP1α), Cbx1 (HP1β) and Cbx3 (HP1γ), as well as other repressive chromatin machinery, remain bound to chromosomes during mitosis34,35,36,38,39,40. Suv39h1, Suv39h2 and HP1 enrichment at mitotic chromosomes was independently confirmed herein using established proteomic approaches36 in mouse embryonic stem cells (ESCs) and mouse embryonic fibroblasts (MEFs) (outlined in Supplementary Fig. 1a,b and depicted in Supplementary Fig. 1c). To investigate the impact of H3K9me3 on mitotic chromosome architecture, native mitotic chromosomes were isolated directly from wild-type (WT) ESCs (Fig. 1a–c) or MEFs (Fig. 1d–f) and compared with mitotic chromosomes isolated from cells lacking both Suv39h1 and Suv39h2 (Suv39h double null or Suv39h dn)13,26. To enable this, dividing cell cultures of ESCs were arrested in metaphase using demecolcine, incubated with polyamine buffer36, and the resulting chromosomes stained with Hoechst 33258 and chromomycin A3. Individual chromosomes were isolated by flow cytometry as described previously36 (Supplementary Fig. 1a provides a schematic of the approach and Supplementary Fig. 1b illustrates the gating strategy). Using purified preparations of chromosomes 19 and X as exemplars, mitotic chromosomes isolated from Suv39h dn ESCs26 were shown to be smaller than their WT counterparts, and a marked increase in compaction at centromeric domains was noted (Fig. 1b,c).

a–f, Flow sorting and size measurements of chromosomes (Chr) 19 and X from WT or Suv39h dn mouse ESCs and MEFs. Flow karyotype of mitotic chromosomes is isolated from WT or Suv39h dn ESCs (a) and MEFs (d); gates used to isolate chromosomes 19 and X are indicated. Representative images (right) of mitotic chromosomes 19 (b and e) and X (c and f) from WT or Suv39h dn ESCs (b and c) and MEFs (e and f) are shown, where DAPI stain (gray) and Cenpa label (green) indicate the chromosome body and centromere, respectively. Scale bar, 5 μm. Boxplots (left of images) show area measurements of individual chromosomes and centromeres for WT and Suv39h dn cells. Minimum, lower quartile, median, upper quartile and maximum values are indicated (n = minimum 100 chromosomes analyzed for each cell line over 3 independent experiments). P values of statistically significant changes, measured by unpaired, two-tailed Student’s t-tests, are indicated. g,h, Representative images of WT or Suv39h dn ESC metaphase spreads stained with either chromosome 19 painting probe (green) (g) or chromosome X painting probe (green) (h), in addition to γSat probe (red) and DAPI (blue). Scale bars, 4 μm and 1 μm for the metaphase spread and zoom-in images, respectively. Chromosome and centromere sizes of chromosomes 19 and X were calculated on metaphase spreads of WT and Suv39h dn ESCs. Boxplots show chromosome and centromere area measurements for WT and Suv39h dn spreads. Minimum, lower quartile, median, upper quartile and maximum values are indicated (n = minimum 25 chromosomes analyzed on metaphase spreads for each line over 3 independent experiments). b,c,e–h, P values of statistically significant changes, measured by unpaired, two-tailed Student’s t-tests, are indicated. Source data, including the precise numbers of chromosomes analyzed, are provided.
To determine whether reduced size and enhanced condensation of mitotic chromosomes deficient in H3K9me3 were seen in other cell types, including differentiated cells, mitotic chromosomes from MEFs lacking Suv39h1 and Suv39h2 (ref. 13) were also examined (Fig. 1d). Consistent with results obtained in ESCs, mitotic chromosomes isolated from these fibroblasts were smaller than WT equivalents and showed increased compaction at centromeres (Fig. 1e,f). To exclude these changes being an artifact of the isolation procedure, we also examined conventional metaphase spreads where chromosome-specific probes and DNA–fluorescence in situ hybridization (FISH) was used to label chromosomes 19 and X. These analyses confirmed that mitotic chromosomes from Suv39h dn ESCs were reproducibly smaller than their WT counterparts (Fig. 1g,h). To examine whether deficits in H3K9 mono- and dimethylation affect chromosome compaction, we isolated mitotic chromosomes from a panel of MEFs that lacked Suv39h1 and Suv39h2 (clustered regularly interspaced short palindromic repeats (CRISPR) clone B1) or other H3K9 methyltransferases, specifically deletions of both Setdb1 and Setdb2 (CRISPR clone A4 + 4-hydroxytamoxifen (4-OHT)), or of both G9a and Glp (CRISPR clone H7)41 (Supplementary Fig. 1d). Analysis of isolated native mitotic chromosomes from clones A4 + 4-OHT, H7 and B1 fibroblasts versus WT controls showed that reduced chromosome size was a unique feature of cells lacking H3K9me3 (ref. 41) (Supplementary Fig. 1e).
The compaction of mitotic chromosomes lacking H3K9me3 was unexpected because previous studies had shown that an absence of other repressive chromatin modifiers, such as DNA methylation or polycomb repressive complex 2 (PRC2) activity, produced chromosomes that were larger and more decondensed than equivalent WT mitotic controls36. To investigate the possibility that additional heterochromatin modifications, such as increased H3K27me3, might compensate for deficits in H3K9me3, we examined the distribution of modified histones in Suv39h dn and WT ESCs. As anticipated, H3K9me3 decorated centromeric domains of normal mitotic chromosomes (Fig. 2a and Supplementary Fig. 2a, green, WT top panel), but was absent from equivalent Suv39h dn chromosomes (Fig. 2a and Supplementary Fig. 2a, lower panel, quantified in the graph, left). Instead, we detected increased levels of H3K27me3 across centromeric domains of Suv39h dn chromosomes (Fig. 2b and Supplementary Fig. 2b, pink). Ingress of H3K27me3 at centromeres was consistent with previous studies showing increased H3K27me3 at chromocenters in interphase in the absence of H3K9me3 (refs.42,43,44). We also detected significant increases in histone H3 phospho Ser10 (H3S10ph) on Suv39h dn metaphase chromosomes (Fig. 2c, yellow) compared with WT equivalents. Increased H3K27me3 levels and ingress of this mark at centromeric domains were also seen in mitotic chromosomes isolated from Suv39h dn MEFs (Supplementary Fig. 2c), but not on equivalent chromosomes isolated from either Setdb1/Setdb2 double null or G9a/Glp double null mutant cells (Supplementary Fig. 2c).

a,b, Representative images (right) of immunofluorescence labeling of histone H3K9me3 (a) (green) or histone H3K27me3 (b) (pink) on chromosome 19 isolated from WT or Suv39h dn ESCs, where DAPI counterstain is shown in light gray. Scale bar, 5 μm. Plots (left of the images) show H3K9me3 (a) or H3K27me3 (b) mean intensities, measured at centromeric regions. c, Representative images of immunofluorescence labeling of histone H3S10ph (yellow) on WT and Suv39h dn metaphase-arrested ESCs, where DAPI counterstain is shown in blue. Scale bar, 4 μm. H3S10ph mean intensities were measured on mitotic chromosomes for each condition (n = minimum 40 chromosomes analyzed for each cell line over 3 independent experiments (a–c)). P values of statistically significant changes, measured by unpaired, two-tailed Student’s t-tests, are indicated. d, Experimental strategy used to measure mitotic chromosome size of Suv39h dn ESCs after treatment with DNA methylation or PRC2 inhibitors (5-Aza or GSK343, respectively). e,f, Representative images of immunofluorescence labeling of histone H3K27me3 (red) on mouse chromosome 19 (e) and chromosome X (f) isolated from Suv39h dn ESCs pretreated with DMSO, 5-Aza or GSK343. DAPI counterstain is shown in light gray. Scale bar, 5 μm. H3K27me3 mean intensities were measured at centromeric regions and whole chromosomes; the mean ± s.d. is shown (n = minimum 50 chromosomes analyzed over 3 independent experiments). P values of statistically significant decreases compared with DMSO treatment, measured by unpaired, two-tailed Student’s t-tests, are indicated. Boxplots show area measurements of individual chromosomes and centromeres for each condition. Minimum, lower quartile, median, upper quartile and maximum values are indicated (n = minimum 100 chromosomes analyzed for each condition over 3 independent experiments). P values of statistically significant increases compared with DMSO treatment, measured by unpaired, two-tailed Student’s t-tests, are indicated. g, Chromatin accessibility profile across chromosome 19 for WT and Suv39h dn asynchronous (Asynch.) and mitotic ESCs and flow-sorted mitotic chromosomes, shown as log2(enrichment of ATAC-seq signal). a–c,e,f, Source data, including the precise numbers of chromosomes analyzed, are provided.
PRC2 inhibition restores Suv39h dn mitotic chromosome size
To investigate whether enhanced compaction of Suv39h dn mitotic chromosomes was the result of compensatory increases in H3K27me3 rather than loss of H3K9me3 as such, Suv39h dn ESCs were treated for 48 hours (h) with drugs that either inhibit PRC2 activity (GSK343, an Ezh2 methyltransferase inhibitor) or block DNA methylation (5-azacytidine (5-Aza)) (Fig. 2d, schematic). Pretreatment with GSK343 reduced global H3K27me3 levels on mitotic chromosomes 19 (Fig. 2e, upper panel) and X (Fig. 2f, upper panel), and effectively restored chromosome size and centromere compaction to that of WT equivalents (Fig. 2e,f, lower panels). Pretreatment of Suv39h dn ESCs with 5-Aza resulted in a smaller increase in mitotic chromosome size and centromere decompaction. In WT ESCs, treatment with either drug resulted in modest increases in mitotic chromosome size (Supplementary Fig. 2d). Collectively these data show that the compact structure of Suv39h dn mitotic chromosomes can be alleviated by inhibiting the activity of PRC2. To exclude chromatin accessibility being grossly altered in Suv39h dn samples, assay for transposase-accessible chromatin using sequencing (ATAC-seq) was performed on WT and Suv39h dn asynchronous ESCs, mitotic-arrested ESCs, as well as sorted ESC mitotic chromosomes. Global ATAC-seq profiles of Suv39h dn and WT chromosomes indicated that they were broadly comparable, as illustrated for chromosome 19 (Fig. 2g) and as shown by differential accessibility analysis (Supplementary Fig. 2e). This suggests that changes in chromosome-scale compaction observed in Suv39h dn ESCs are independent of local changes in chromatin accessibility. We also examined mitotic progression and cell cycle in ESCs lacking Suv39h1/h2 activity using live-cell imaging45 and propidium iodide staining. As shown in Supplementary Fig. 2f,g, dividing cultures of Suv39h dn ESCs produced a similar proportion of cells in G1, S and G2/M phases of the cell cycle because their WT counterparts and mitotic duration were also comparable (Supplementary Fig. 2g, boxplot, right panel). These data exclude any delay in mitotic progression as the cause of increased mitotic chromosome compaction in Suv39h dn cells.
Proteomic analysis of Suv39h dn mitotic chromosomes
To analyze the factors binding to unfixed (native) metaphase chromosomes in Suv39h dn and WT cells (MEFs and ESCs), we performed proteomic analyses using liquid chromatography–tandem mass spectrometry (LC-MS/MS) in which the data were analyzed using the label-free quantification (LFQ) algorithm in the MaxQuant platform, as detailed previously36. For these experiments, an equivalent number of metaphase chromosomes (107) in samples pre- and post-chromosome sorting were compared (schematically shown in Supplementary Fig. 1a), in at least three biological replicates loaded in technical duplicates on the LC-MS/MS (Supplementary Fig. 3a–d, Supplementary Data 1 and 2). After filtering raw MaxQuant data in Perseus as described in Methods, a comparable number of protein hits were identified in WT and Suv39h dn MEF samples (Supplementary Data 1). Among these hits, 4,468 were detected in both mitotic lysate and sorted chromosomes for WT MEFs and 4519 were detected in both mitotic lysate and sorted chromosomes for Suv39h dn MEFs (Supplementary Data 1). To identify proteins that were enriched on mitotic chromosomes (red), depleted (blue) or unaltered in abundance between mitotic lysate pellets and purified chromosomes (gray), data were subjected to multiple Student’s t-tests with permutation-based false discovery rate (FDR) (detailed in Methods) and displayed as volcano plots (Fig. 3a). Both WT and Suv39h dn MEFs showed similar proportions of proteins enriched on sorted mitotic chromosomes (11.9% and 16.3%, respectively). Comparing these enriched hits between WT and Suv39h dn MEFs (based on the corresponding protein IDs) revealed that many of these chromosome-bound proteins were common to both genotypes (464 of 848), although a subset of candidates (287 of 848) was not enriched in Suv39h dn MEF samples compared with WT (Fig. 3b, listed in Supplementary Data 1. These proteins clustered in function, being associated with processes such as transcription, chromosome organization, cell cycle, development and nucleosome organization (Fig. 3c), in addition to H3K9 trimethylation. No obvious functional group was seen among the small group of factors (97 of 848) that were preferentially detected on Suv39h dn MEF mitotic chromosomes, compared with WT.

a, Volcano plots of proteins significantly enriched (red), depleted (blue) or not significantly changed (gray) on sorted chromosomes relative to mitotic lysate pellets for WT (left) and Suv39h dn (right) MEFs (unpaired, two-tailed Student’s t-test, permutation-based FDR < 0.05, s0 = 0.1 (n = 3 independent experiments each measured in technical duplicate; see Methods for details). Proteins were plotted as log2(fold-change LFQ intensity of sorted chromosome pellets/LFQ intensity of mitotic lysate pellet versus significance) (−log10(P)) using Perseus software). The number of proteins in each category is indicated on the volcano plot. b, Venn diagram showing the overlap of protein IDs enriched on mitotic chromosomes between WT and Suv39h dn MEF samples. c, GO term (biological process) analysis of proteins that lose enrichment on Suv39h dn mitotic MEF chromosomes compared with WT. Analysis was performed at http://geneontology.org using Fisher’s exact test with FDR correction. d, Volcano plots (as in a) of proteins significantly enriched (red), depleted (blue) or not significantly changed (gray) on sorted chromosomes relative to mitotic lysate pellets for WT (left) and Suv39h dn (right) mouse ESCs. e, Venn diagram showing the overlap of protein IDs enriched on mitotic chromosomes between WT and Suv39h dn mouse ESC samples. f, GO term (biological process) analysis (as in c) of proteins that lose enrichment on Suv39h dn mitotic mouse ESC chromosomes compared with WT.
We performed an analogous comparison in ESCs (Fig. 3d–f). After filtering, 4,990 protein hits were detected in both mitotic lysate and sorted chromosome samples for WT ESCs and 4,521 were detected in both mitotic lysate and sorted chromosomes for Suv39h dn ESCs (Supplementary Data 2). Of these, 15.6 and 13.5% were identified as being significantly enriched on mitotic chromosomes for WT and Suv39h dn ESCs, respectively (Fig. 3d). Although a majority (56%) of chromosome-bound candidates were common to both genotypes, a subset (287 of 920) was not detected in the absence of both Suv39h enzymes (Fig. 3e). These candidates showed gene ontology (GO) term assignments that were remarkably similar to those identified previously in MEFs, encompassing chromosome organization, transcription, DNA repair, cellular responses to stress, chromatin silencing and H3K9 trimethylation (compare Fig. 3c,f, Supplementary Data 1 and 2). Taken together, these data highlight a commonality in the functional pathways affected by H3K9me3 removal in very divergent cell types: pluripotent stem cells and differentiated fibroblasts.
Altered retention of TFs on Suv39h dn mitotic chromosomes
To further investigate the factors that require H3K9me3 to remain efficiently bound to mitotic chromosomes in dividing ESCs, we looked in detail at the representation of pluripotency-associated proteins in sorted mitotic chromosomes from WT and Suv39h dn ESC samples. Factors such as Sox2, Utf1, Zfp296, Sall4, Dppa2 and Dppa4, which we previously identified as being bound to unfixed mitotic ESC chromosomes36, showed an equivalent representation in control and mutant samples (Fig. 4a). In the absence of Suv39h enzymes and H3K9me3, however, factors such as Esrrb (estrogen-related receptor-β, a well-characterized mitotic bookmarking factor31,32), Tead4 (TEA domain transcription factor 4) and Tbx3 (T-box transcription factor) were no longer enriched on mitotic chromosomes (Fig. 4a). To exclude such deficits being simply the result of lower levels of expression by mutant cells, we compared our proteomic data with previously published transcriptomic data27. For both genotypes, there was a strong correlation between protein abundance in mitotic lysates and chromosome samples and between protein and transcript levels (Supplementary Fig. 4a). However, differences in the proteome of Suv39h dn mitotic chromosomes, relative to WT, appeared independent of differences in the mitotic lysate or underlying transcriptome (Supplementary Fig. 4b). This suggests that a loss of TF retention in Suv39h dn mitotic chromosomes is unlikely to be explained by changes in their abundance. To explore this in more detail, we examined the retention of a specific candidate, Esrrb. In WT and Suv39h dn ESCs, Esrrb levels were broadly comparable (Supplementary Fig. 4c) and proteomic data confirmed that Esrrb was equivalently represented in WT and Suv39h dn mitotic lysates (Supplementary Fig. 4d). However, sorted mitotic chromosomes from Suv39h dn ESCs showed an underrepresentation of Esrrb (Supplementary Fig. 4e) compared with equivalent WT controls. To validate this result, we isolated native mitotic chromosomes 19 and X from Suv39h dn and WT ESCs and analyzed Esrrb levels by immunofluorescence. As shown in Fig. 4b, Esrrb (red) was significantly reduced in Suv39h dn samples compared with WT ones. This decreased retention of Esrrb was not accompanied by any pronounced loss of chromatin accessibility at Esrrb bookmarking sites32, as judged by ATAC-seq analysis of asynchronous, mitotic and sorted chromosome preparations (Supplementary Fig. 4f). This is consistent with a global similarity in ATAC-seq data derived from WT and Suv39h dn samples (Fig. 2g and Supplementary Fig. 2e). However, direct analyses of Esrrb binding by chromatin immunoprecipitation (ChIP), using a double fixation protocol that preserves DNA–TF interactions in mitotic samples31,32,33, revealed deficits in Esrrb bookmarking in Suv39h dn ESCs (Fig. 4c). We selected candidate sites from previous publications31 as representing sites that were either bookmarked throughout mitosis by Esrrb (Capn2, Esrrb, Jam2-s1, Jam2-s2, Tbx3 and Tet2), bound by Esrrb only in interphase (Mgat3 and Twistnb) or negative control sites that do not bind Esrrb (Esrrb-3′ and Actb)31. A statistically significant decrease in Esrrb binding was observed at five of seven bookmarked sites analyzed in Suv39h dn compared with WT mitotic chromosome samples (upper panel). ChIP analysis of asynchronous samples showed no significant differences in Esrrb binding between these genotypes, suggesting that Esrrb binding loss is selective for (or more evident in) mitosis.

a, Volcano plots as in Fig. 3d, highlighting pluripotency-associated factors that are enriched (red) or not significantly changed (black) on WT (left plot) or Suv39h dn (right plot) ESC mitotic chromosomes. b, Esrrb immunolabeling (red) of WT and Suv39h dn flow-sorted chromosomes 19 (left panel) and X (right panel), where DAPI counterstain is shown in light gray. Scale bar, 5 μm. Esrrb mean intensities were measured across individual chromosomes; the mean ± s.d. is shown (n = minimum 50 chromosomes analyzed over 3 independent experiments). P values of statistically significant changes, measured by unpaired, two-tailed Student’s t-tests, are indicated. c, Esrrb ChIP–qPCR analysis in WT versus Suv39h dn mitotic and asynchronous ESCs. Enrichment (immunoprecipitated as a percentage of input (% IP)) was measured at Esrrb bookmarked sites (Capn2, Esrrb, Jam2-s1, Jam2-s2, Tbx3 and Tet2), Esrrb lost sites (bound only in interphase; Mgat3 and Twistnb) or control sites that do not bind Esrrb (Esrrb-3′ and Actb). The mean + s.d. results are shown. For interphase cells n = 3 biological replicates, for mitotic cells n = 4 biological replicates (except Capn2, Esrrb, Rex1 and Jam2-s1, where n = 5). d, Live-cell imaging of Esrrb–tdTomato mouse ESCs pretreated with DMSO (upper panel) or 100 nM of chaetocin (lower panel) cultured with SiR-DNA (red). Arrows show Esrrb localization to mitotic chromatin. Scale bar, 5 μm. Esrrb–tdTomato mean intensities on mitotic DNA (gated based on SiR-DNA signal) and in interphase nuclei were quantified for each condition; the mean ± s.d. is shown. For mitotic chromosomes: n = 25 (DMSO) or n = 35 (chaetocin) cells analyzed; for interphase nuclei: n = 46 cells analyzed for both DMSO and chaetocin treatments, representing 3 independent experiments. b–d, P values of statistically significant changes, measured by unpaired, two-tailed Student’s t-tests, are indicated. Source data and precise n numbers are provided.
To test the impact of acute H3K9me3 depletion on mitotic retention of Esrrb, we used WT ESCs expressing an endogenous Esrrb–tdTomato fusion protein31. Esrrb–tdTomato ESCs were examined by live-cell imaging with and without addition of chaetocin, a mycotoxin that inhibits Suv39h1 activity46,47 (Supplementary Videos 1 and 2). As anticipated, Esrrb decorated metaphase chromosomes (red) in these ESCs (Fig. 4d, upper panels). However, after chaetocin treatment, Esrrb binding at metaphase chromosomes was substantially lowered (Fig. 4d, lower panel). Live-cell imaging of Esrrb–tdTomato in ESCs pretreated with chaetocin versus dimethylsulfoxide (DMSO) controls confirmed a significant decrease in signal intensity in response to chaetocin, which was evident mitosis, but not interphase (compare histograms, Fig. 4d). In contrast to Esrrb, Sox2 remained unaffected on sorted mitotic chromosomes after chaetocin treatment (Supplementary Fig. 4g,h), indicating that Suv39h1 drug inhibition does not induce a global dissociation of pluripotency factors. Taken together, these data show that both short- and long-term depletion of Suv39h1/Suv39h2 activity in ESCs result in reduced retention of Esrrb on mitotic chromosomes.
Mitotic chromosome size and factor retention depend on Suv39h
Our data raise the intriguing possibility that retention of TFs through mitosis may be sensitive to H3K9me3. As this could reflect a dependency on either H3K9me3 itself or the correct marking and function of constitutive heterochromatin domains, we asked whether other H3K9me3-associated proteins were also enriched or depleted in mitotic samples (Supplementary Fig. 5a). It is interesting that HP1α (or CBx5) binding was retained on Suv39h dn mitotic chromosomes, as were the Swi/Snf chromatin re-modelers Smarcb1 and Atrx, a protein known to bind at heterochromatic repeat elements, including telomeres, ribosomal DNA repeats, endogenous retroviral elements and pericentric domains48,49. In contrast, components of the lysine-specific histone demethylase complex 1A (LSD1 or Kdm1a), which are known to interact with Esrrb in trophoblast stem cells50, were less well retained on H3K9me3-depleted mitotic chromosomes (compare Kdm1a and Rcor1; Supplementary Fig. 5a). Our proteomic comparisons also revealed increases in the representation of certain factors, notably histone H1 variants, at mitotic Suv39h dn chromosomes, relative to WT controls (Supplementary Fig. 5b). This may be relevant because linker histones have been widely implicated in chromatin condensation51,52,53,54,55,56,57,58,59 and an overrepresentation of histone H1 could contribute to the characteristically compact state of Suv39h dn mitotic chromosomes.
To ask whether inefficient retention of Esrrb binding during mitosis was reversed by H3K9me3 rescue, we transfected Suv39h dn ESCs with Suv39h1 (ref. 39). The provision of Suv39h1 restored H3K9me3 labeling of centromeric domains, as exemplified for mitotic chromosomes 19 and X (Fig. 5a), and resulted in decreased H3K27me3 across centromeric (DAPI-intense) domains (Fig. 5a). Importantly, Suv39h1 transfection also rescued mitotic chromosome size and centromere compaction (Fig. 5b), and restored efficient Esrrb retention by Suv39h dn mitotic chromosomes, assessed by proteomic profiling (Fig. 5c).

a, Representative images of coimmunolabeling of histone H3K9me3 (blue) and H3K27me3 (red) on chromosomes 19 (upper panel) and X (lower panel) isolated from Suv39h dn ESCs or Suv39h dn ESCs overexpressing Suv39h1-EGFP. The DAPI counterstain is shown in light gray. Scale bar, 5 μm. H3K9me3 and H3K27me3 mean intensities were measured at centromeric regions of chromosomes 19 and X (mean ± s.d. is shown; n = minimum 50 chromosomes analyzed over 3 independent experiments). P values of statistically significant changes, measured by unpaired, two-tailed Student’s t-tests, are indicated. b, Size measurements of chromosomes 19 (upper panel) and X (lower panel) isolated from Suv39h dn ESCs or Suv39h dn ESCs overexpressing Suv39h1-EGFP. Boxplots show area measurements of individual chromosomes and centromeres for each cell line. Minimum, lower quartile, median, upper quartile and maximum values are indicated (n = minimum 100 chromosome measurements for each cell line). P values of statistically significant changes, measured by unpaired, two-tailed Student’s t-tests, are indicated. a,b, Source data and precise n numbers are provided. c,d, Volcano plots highlighting pluripotency-associated factors that are enriched (red) or not significantly changed (black) on mitotic chromosomes versus mitotic lysate pellets for Suv39h dn ESCs and Suv39h dn ESCs expressing Suv39h1-EGFP (c) and Suv39h dn ESCs treated with GSK343 or Suv39h dn ESCs treated with Hesperadin (d). Proteins were plotted as log2(fold-change LFQ intensity of sorted chromosome pellets/LFQ intensity of mitotic lysate pellet and significance) (−log10(P)) using Perseus software (unpaired, two-tailed Student’s t-test, permutation-based FDR < 0.05, s0 = 0.1; n = 3 independent experiments each measured in technical duplicate; see Methods for details).
Impact of H3K9me3 loss on chromatin and TF binding in mitosis
We have shown that H3K9me3 removal results in increased levels of H3K27me3 and H3S10ph on mitotic chromosomes. As these chromatin changes might also contribute to the altered binding of factors such as Tead4 and Esrrb in Suv39h dn cells, we asked whether inhibition of PRC2 activity (GSK343) or aurora kinase b activity (Hesperadin)60 (to reduce H3K27me3 or H3S10ph, respectively) impacts mitotic retention. Proteomic comparisons (Fig. 5d) showed that Esrrb retention by Suv39h dn mitotic chromosomes was increased by inhibiting H3S10ph, whereas Tead4 retention was selectively increased on H3K27me3 inhibition. To understand how Esrrb retention during mitosis might be impacted by the loss of H3K9me3 and altered heterochromatin structure41, we examined the detailed distribution of Esrrb through the cell cycle, relative to heterochromatic and euchromatic chromatin features. Using the tdTomato–Esrrb ESC line31, endogenous Esrrb clearly decorated chromosomes throughout all stages of mitosis (Supplementary Fig. 6a) and also colocalized with DAPI-stained DNA in interphase, as reported previously31. We show in the present study that euchromatic and heterochromatic regions of chromosomes are labeled by Esrrb (Fig. 6a and Supplementary Fig. 6a), with signal also detected at DAPI-intense, pericentromeric domains of isolated mitotic chromosomes (Fig. 6b). Treatment with the Suv39h1 inhibitor chaetocin substantially reduced Esrrb detection throughout mitosis (metaphase and telophase stages) and affected Esrrb distribution at euchromatin and heterochromatin domains (Fig. 6c). As H3K9me3 can regulate the expression of genomic repeat elements27, we examined the expression of euchromatin- and heterochromatin-based repeats in Suv39h dn mitotic ESCs. Cot-1 RNA, which predominately contains LINE-1 (long interspersed nuclear element-1) and SINE (short interspersed nuclear element) repeat elements, was used to probe euchromatin repeat expression61 and gamma-satellite (γSat) RNA to probe heterochromatin-repeat expression62. We observed a marked increase in Cot-1 RNA in mitotic Suv39h dn ESCs compared with WT controls (pink, Fig. 6d), and more specifically LINE-1 expression, as confirmed by quantitative (q)PCR analysis (Supplementary Fig. 6b) and L1 ORF1 protein levels (Supplementary Fig. 6c). In contrast, the expression and distribution of major satellite repeats RNA (yellow, γSat; Fig. 6d) appeared similar in WT and Suv39h dn ESCs through mitosis.

a, Representative images of Esrrb–tdTomato ESC metaphase spreads stained with DAPI (blue). Scale bar, 10 μm. b, Esrrb immunolabeling (red) of flow-sorted chromosomes 19 (top panel) and X (lower panel), where DAPI counterstain is shown in light gray. Scale bar, 5 μm. Linescan analysis (profile plots) is shown of Esrrb (red) and DAPI (gray) intensities across chromosomes 19 and X (right panels). Ab, antibody. c, Representative fluorescence images of Esrrb–tdTomato ESCs at metaphase (left) and telophase (right) stages with and without chaetocin treatment. DAPI stain is in blue. d, Cot-1 (pink) and γSat repeat (yellow) RNA–FISH in WT and Suv39h dn ESCs, with DAPI stain in blue. Scale bar, 5 μm. All the images represent three independent experiments.
Discussion
To gain a broad understanding of the impact that H3K9 methylation loss has during mitosis, we examined the size and epigenetic features of chromosomes isolated from ESCs and fibroblasts that lacked Suv39h1/Suv39h2, Setdb1/Setdb2 or G9a/Glp. Mitotic chromosomes from cells devoid of H3K9me3 (Suv39h1/Suv39h2 double null) were distinct in being significantly more compact than WT equivalents, with highly condensed centromeres decorated by H3K27me3 (rather than H3K9me3). These mitotic chromosomes also showed a twofold enrichment in H3S10ph and significantly increased representation of several histone H1 variants, as determined by proteomic analysis (histones H1.1, H1.2, H1.3, H1.4 and H1.5; Supplementary Fig. 5b). Increased levels of histone H1 variants, H3S10ph and H3K27me3, have been independently shown to result in enhanced chromosome compaction in other settings36,51,52,53,63,64,65 and we show in the present study that inhibition of H3K27me3 activity can reverse the compaction of Suv39h dn mitotic chromosomes. These results contribute to an extensive catalog of structural and organizational defects that have previously been described in interphase mammalian cells on H3K9me3 withdrawal66,67. This includes altered heterochromatin organization, extended telomere length, DNA-repair pathway activation, repeat element re-expression and genomic instability, which manifests as H3K9me3-deficient cells transit mitotic or meiotic division13,41. Our data, which are focused on mitotic events, reveal that H3K9me3 is also required to sustain binding of a rich cadre of proteins to mitotic chromosomes. These proteins collectively influence DNA-templated transcription, chromatin silencing, DNA repair, cellular stress and chromosome organization, processes that are likely to be particularly important during and immediately after cell division. Although we did not observe mitotic defects in our cultured ESC lines, it is worth noting that Suv39h1/Suv39h2-deficient mice show severely impaired viability and aneuploidy with increased tumor incidence, and also fail to generate mature functional sperm13.
In the present study, we showed that H3K9me3 is required for the efficient retention of some TFs such as Esrrb on mitotic chromosomes during ESC division. Genetic ablation of Suv39h1 and Suv39h2 resulted in a reduced representation of Esrrb on native sorted mitotic chromosomes, as revealed by quantitative proteomics, as well as reduced Esrrb protein detected by cellular fluorescence-based labeling. This dependency of Esrrb was supported independently by studies of acute H3K9me3 depletion targeted by pharmacological exposure to chaetocin, and demonstrations that Esrrb representation on Suv39h dn mitotic chromosomes was restored by transfection of Suv39h1 and H3K9me3 rescue. These results highlight a previously unrecognized role for H3K9me3 in retaining TFs, including Esrrb, Tead4, Tbx3 and other proteins (involved in transcription, DNA repair, chromatin silencing and chromosome organization) on condensed mitotic chromosomes. The binding of Tead4, a factor involved in Hippo signaling68, was diminished in Suv39h dn mitotic ESC chromosomes, as was Tbx3, a factor expressed early in the developing intracellular matrix and implicated in regulating extraembryonic endoderm69 and the mitotic bookmarking factor Esrrb31,32. In contrast, the pluripotency factor Sox2, which is implicated in regulating target genes in association with either Oct4 or Esrrb70,71,72, was equivalently represented on mitotic chromosomes with or without H3K9me3. Previous studies have shown that Sox2 and Esrrb normally remain chromosome bound in mitosis30,31 and these results reveal an intriguing selective sensitivity of Esrrb to H3K9me3 depletion.
ATAC-seq comparisons of WT and Suv39h dn asynchronous ESCs, mitotic cells and isolated native mitotic chromosomes showed that the profiles of mutant and control samples were surprisingly similar, indicating that canonical binding sites for this protein remain largely accessible. However, ChIP analysis at selected target genes clearly showed that Esrrb binding was diminished in Suv39h dn mitotic chromosome samples at several ‘bookmarked’ target sites31. It is possible that changes in Esrrb binding could reflect chromatin events that are downstream of H3K9me3 loss, such as increased phosphorylation of H3S10. In this regard, we showed that increased H3S10ph correlated with reduced representation of Esrrb and the Esrrb-associated factors Rfeb1, Tfeb50 and Tcf7l1. Furthermore, inhibition of aurora kinase B activity in Suv39h dn ESCs fully restored the representation of Esrrb on mitotic chromosomes, highlighting a functional overlap between H3K9me3 loss and H310Sph gain. At least two other mechanisms should also be considered. The first acknowledges that, in mitosis, extreme chromosome condensation could elicit liquid–liquid phase separation and the formation of local condensates of DNA-binding factors that may coat chromosomes. This may be impacted by Suv39h loss and the resulting changes of heterochromatin. Second, it remains formally possible that Suv39h1 and Suv39h2 have nonhistone substrates that could impact Esrrb and chromosome function73. For example, in B cells, Suv39h1 has been shown to methylate RAG-2 (recombination activating gene 2 protein) and is implicated in class switch recombination74, and also methylates Dot1L and the mycobacterial protein HupB75. Likewise, Suv39h2 can trimethylate the histone H3K4 demethylating enzyme lysine-specific demethylase 1 (LSD1) and methylates H2AX during damage repair76,77.
Cells lacking Suv39h1 and Suv39h2 also display changes in the repertoire of factors bound to mitotic chromosomes, providing important primary evidence that chromatin is critical for TF retention through mitosis. Suv39h dn fibroblasts and ESCs showed reduced representation of several TFs on H3K9me3-depleted mitotic chromosomes, in addition to proteins involved in DNA repair, nucleosome organization, chromatin silencing and chromosome organization. It is interesting to speculate that these groups of proteins might be relevant for protecting the genetic and epigenetic state of daughter cells that inherit a cargo of chromosome-associated proteins after cytokinesis, ahead of de novo gene expression beginning in G1. In this setting, both WT and Suv39h dn mitotic chromosomes showed an enrichment in core PRC1, PRC2 components and DNA-methylation machinery, relative to mitotic lysates. In addition, although retention of certain TFs was impaired after H3K9me3 loss, many pluripotency-associated factors, such as Dppa2, Dppa4, Mpp8 and Sox2, remained chromosome associated in Suv39h1/Suv39h2 mutants through mitosis. Dppa2 and Dppa4 are small heterodimerizing nuclear proteins that are known to regulate zygotic genome activation. In ESCs, these proteins have also been shown to be critical for maintaining the functionally primed state of a subset of bivalent genes in ESCs, protecting them from de novo DNA methylation and irreversible silencing78,79. M-phase phosphoprotein 8 (Mpp8 or Mphosph8) is an essential player in safeguarding ground-state pluripotency and stem-cell renewal, through interactions with a plethora of epigenetic silencing proteins, including Dnmt3a, Setdb1 and Sirt1. It is of interest that Mpp8 has also been implicated in repressing LINE-1 and L1-ORF2 expression, together with the human silencing hub (HUSH) complex80. Although we do not yet know the basis of mitotic factor sensitivity or resistance to H3K9me3 loss, it is possible that chromosome-bound Dppa2, Dppa4 and Mpp8 could act as safeguards, protecting the epigenetic fidelity and pluripotent state of newly established daughter cells, particularly in the context of de novo DNA methylation conveyed by chromosome-bound Dnmt3a. Our analyses also showed that ESCs that lack Suv39h1/Suv39h2 overexpress LINE-1 and L1-ORF2, consistent with previous studies27, and we detected an increased representation of L1-OF2 protein on H3K9me3-depleted mitotic chromosomes (Supplementary Fig. 6b,c). As H3K9me3 is not strictly required for Mpp8-mediated repression of LINE-1 elements80, it is likely that the overexpression of LINE-1 elements in mutant Suv39h1h2-null cells may stem from failures to recruit and maintain Setdb1 and the HUSH complex at appropriate targets.
Our study asked whether repressive chromatin has a role in retaining chromosome-associated factors through mitosis. This important question was addressed using an approach that enables unfixed ‘native’ mitotic chromosomes to be directly isolated from dividing cells36. By examining purified mitotic chromosomes either from cells at different stages of development or from cells that lack specific chromatin features, it was possible to compile a comprehensive and quantitative catalog of proteins retained by mitotic chromosomes in the absence of Suv39h1/Suv39h2-mediated H3K9me3. We showed that repressive chromatin is critical for maintaining the efficient binding of certain proteins to mitotic chromosomes. Therefore, in addition to the important role of H3K9me3 in regulating gene and chromosome function during interphase, H3K9me3 is important for mitotic chromosome structure and the efficient retention of a cohort of TFs during mitosis.
Methods
Cells
Mouse ESCs used were WT (WT26), Suv39h dn (DN57)26, Suv39h1-EGFP (Suv39h dn ESCs overexpressing full-length Suv39h1)39 and Esrrb–tdTomato31 (gift from N. Festuccia). ESC lines were grown on 0.1% gelatin (Sigma-Aldrich)-coated dishes in KnockOut Dulbecco’s modified Eagle’s medium (KO-DMEM) supplemented with 15% fetal bovine serum (FBS), l-glutamine, nonessential amino acids, 2-mercaptoethanol, antibiotics and leukemia inhibitory factor (made in-house). MEFs used in the present study were WT (W8), Suv39h dn (D5)13, WT (Eset25 control), Suv39h1/2−/− (B1), Setdb1/2−/− (A4 + 4-OHT) and G9a/Glp−/− (H7)41. MEFs were maintained in DMEM supplemented with 10% FBS, l-glutamine, antibiotics, 2-mercaptoethanol, nonessential amino acids and sodium pyruvate. For A4 cells, Setdb1 deletion was induced by plating cells in 2 µM 4-OHT for 2 d, followed by 2 days of recovery in complete medium without tamoxifen. All cell lines used in the present study are male and equivalent passage between WT and mutants.
Drug treatments
After passaging, cells were treated for 24 h with 1 µM GSK343 (Ezh2 inhibitor, Sigma-Aldrich), 100 nM 5-Aza (DNA methylation inhibitor, Sigma-Aldrich), 100 nM chaetocin (Suv39h1 inhibitor, Enzo-LifeScience) or 100 nM Hesperadin (Aurkb inhibitor, Sigma-Aldrich), from stock solutions prepared in DMSO.
Mitotic chromosome preparation and flow sorting
Mitotic chromosomes were prepared using a polyamine-based method36,81. Mitotic arrest, chromosome preparation and flow sorting were performed as previously described36. Cells were arrested using 0.1 μg ml−1 of demecolcine (Sigma-Aldrich) for 6 h at 37 °C. Mitotic cells were collected by mitotic shake off, pelleted at 289g for 5 minutes (min) and resuspended in 10 ml of hypotonic solution (75 mM KCl, 10 mM MgSO4, 0.5 mM spermidine trihydrochloride and 0.2 mM spermine tetrahydrochloride, adjusted to pH 8) for 20 min at room temperature (RT). Swollen cells were centrifuged at 300g for 5 min at RT, resuspended in 3 ml of ice-cold polyamine buffer (15 mM Tris-HCl, 2 mM EDTA, 0.5 mM (ethylenebis(oxonitrilo))tetra-acetate (EGTA), 80 mM KCl, 3 mM dithiothreitol, 0.25% Triton X-100, 0.2 mM spermine tetrahydrochloride and 0.5 mM spermidine trihydrochloride, pH 7.7) and incubated for 15 min on ice. Mitotic chromosomes were released by vortexing (30 s, maximum speed) and syringing through a 22.5-gauge needle. Chromosome suspensions were centrifuged for 2 min at 200g and RT. The supernatant containing mitotic chromosomes was filtered through a 20-μm mesh CellTrics filter (Sysmex). Mitotic chromosomes were stained with 5 μg ml−1 of Hoechst 33258 (Sigma-Aldrich), 50 μg ml−1 of chromomycin A3 (Sigma-Aldrich) and 10 mM MgSO4, overnight at 4 °C. Sodium citrate (10 mM final) and sodium sulfite (25 mM final) were added to chromosome suspensions 1 h before FACS sorting. Chromosomes were analyzed and sorted using a Becton Dickinson Influx equipped with spatially separated lasers using BD FACS software. As previously described36, Hoechst 33258 was excited using a 355-nm air-cooled laser (Spectra Physics Vanguard) with a power output of 350 mW, and fluorescence was collected using a 400-nm long-pass filter in combination with a 500-nm short-pass filter. Chromomycin A3 was excited using a water-cooled, 460-nm laser (Coherent Genesis) with a power output of 500 mW. Chromomycin A3 fluorescence was collected using a 500-nm long-pass filter in combination with a 600-nm short-pass filter. As previously described36, forward scatter was measured using a Coherent Sapphire 488-nm laser with a power output of 200 mW and this was used as the trigger signal for data collection. Chromosomes were sorted using a 70-μm nozzle tip, a drop drive frequency of ~96 kHz and a sheath pressure of 65 p.s.i (448 kPa).
Immunofluorescence on flow-sorted chromosomes
Flow-sorted chromosomes (105) were cytospun (Cytospin3, Shandon) on to poly(l-lysine) slides at 1,200 r.p.m. for 10 min, as previously described36. Samples were incubated with blocking buffer (5% normal goat serum in 10 mM Hepes, 2 mM MgCl2, 100 mM KCl and 5 mM EGTA) for 30 min at RT, and incubated overnight at 4 °C with primary antibodies to Cenpa (catalog no. 2048S, Cell Signaling), H3K9me3 (catalog no. 07-523 or 07-442, Millipore), H3K27me3 (catalog no. ab6002, Abcam or catalog no. 07-449, Millipore), Esrrb (catalog no. PP-H6705-00, Perseus Proteomics) and Sox2 (catalog no. ab97959, Abcam). All antibodies were diluted 1:200 in blocking buffer. Chromosomes were incubated with appropriate secondary antibodies (anti-mouse Alexa 488 (catalog no. A11001, Invitrogen), anti-rabbit Alexa 488 (catalog no. A11008, Invitrogen) or anti-mouse Alexa 568 (catalog no. A11031, Invitrogen)) for 1 h at RT. All secondary antibodies were diluted 1:400 in blocking buffer. Stained chromosomes were mounted in DAPI-containing Vectashield (Vector Laboratories). Wide-field epifluorescence microscopy was performed on an Olympus IX70 inverted microscope using a UPlanApo ×100/1.35 oil objective lens and Micro-Manager software.
Immunofluorescence on ESCs
ESCs were cultured on gelatin-coated glass coverslips, fixed with 1% formaldehyde 10 min at RT and crosslinked with 2 mM disuccinimidyl glutarate (DSG, Sigma-Aldrich) for 50 min at RT, as previously described32. Cells were permeabilized with 0.1% Triton X-100 for 15 min at RT and blocked with 1% bovine serum albumin and 5% goat serum. Cells were incubated with primary antibodies Esrrb or H3S10ph (catalog no. ab5176, Abcam), 1:200, at 4 °C overnight. After three phosphate-buffered saline (PBS) washes, cells were labeled with appropriate secondary antibodies and mounted in DAPI-containing Vectashield. Images were acquired with an Olympus IX70 inverted microscope using a UPlanApo ×100/1.35 oil objective lens and Micro-Manager software.
Chromosome size and histone modification quantification
Chromosome images acquired on the Olympus IX70 wide-field microscope were analyzed in Fiji82. Chromosome and centromere size measurements were performed as previously described36 by estimating whole chromosome (total DAPI) and centromere (DAPI high) areas. H3K27me3 and H3K9me3 maximum intensity projections of z-planes were quantified for both centromere and whole chromosome areas of each individual chromosome.
Metaphase spreads
Metaphase spreads were prepared using the following method, as previously described36. Metaphase-arrested cells were resuspended in hypotonic solution (40 mM KCl, 0.5 mM EDTA, 20 mM Hepes, pH 7.4) for 25 min at 37 °C. Nuclei were pelleted (8 min at 500g) and supernatant was removed, leaving a small drop for resuspension. Next, a 3:1 mixture of MeOH and glacial acetic acid fixative was added to the top of the tube and tubes were stored at −20 °C. The next day nuclei were pelleted (8 min at 500g) and washed in fresh 3:1 MeOH:glacial acetic acid 3×. To prepare metaphase spreads, nuclei were pelleted and resuspended in a small volume of fixative (to a pale-gray solution). A 20-μl drop of 45% acetic acid in water was pipetted on to a glass Twinfrost microscope slide and 23 μl of spread mixture was dropped on to it, tilting the slide to spread the nuclei. Slides were air dried at RT. Mouse chromosome X and 19 painting probes (Metasystems Probes) were used together with mouse γSat probes (DNA a gift from N. Dillon) labeled with FluoroRed (Amersham Life Science) by nick translation, to detect chromosome X or 19 with pericentromeric DNA. Metaphase chromosome painting was performed according to the Metasystems Probes protocol and samples were mounted in Vectashield containing DAPI. A Leica SP5 II confocal microscope was used for imaging using LAS-AF software.
ATAC-seq
ATAC-seq83 was performed in duplicate on asynchronous and mitotic WT and Suv39h dn ESCs and on purified mitotic chromosomes, with the method details as previously described36. Nuclei were obtained from 50,000 asynchronous ESCs according to the Omni-ATAC-seq protocol84 by resuspending in 50 μl of ATAC-resuspension buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Igepal CA-630, 0.1% Tween-20 and 0.01% digitonin) and incubating on ice for 3 min. After adding 1 ml of ATAC-resuspension buffer without Igepal and digitonin, nuclei were pelleted at 500g (10 min at 4 °C). To perform ATAC-seq, nuclei, mitotic cells (50,000) or purified chromosomes (2 × 106 pelleted at 10,000g for 10 min at 4 °C) were resuspended in 50 μl of transposase mixture (25 μl of Illumina TD buffer, 22.5 μl of H2O, and 2.5 μl of Illumina TDE1 transposase) and incubated at 37 °C for 30 min, shaking at 1,000 r.p.m. DNA was purified and amplified with seven PCR cycles (NEBNext High Fidelity master mix; primers in Supplementary Table 1)83. Libraries were purified twice with Ampure XP beads (Beckman Coulter), including size selection with 0.5× beads. Libraries were assessed by Qubit, Bioanalyzer and KAPA Library Quantification (Roche) before Illumina NextSeq500 sequencing (75 bp, paired end).
Data were processed using RTA (v.2.11.3, default settings) and reads were demultiplexed with bcl2fastq2 (v.2.20.0; allowing 0 mismatches). Reads were trimmed with Trim Galore! (trim_galore_v0.4.4; --trim-n --paired; www.bioinformatics.babraham.ac.uk/projects/trim_galore) and aligned to University of California Santa Cruz (UCSC) mm10 using bowtie2 (bowtie2/2.2.9; -p8 -t -very-sensitive -X 2000)85. Aligned bam files were sorted with Samtools (v.1.2)86 and technical replicate sequencing runs were merged. Duplicate reads were marked with Picardtools MarkDuplicates (v.1.90; http://broadinstitute.github.io/picard). The bamQC function from R/Bioconductor package ATACseqQC87 was used to keep properly mapped paired-end reads, and remove duplicates and mitochondrial alignments. Chromosome-wide accessibility profiles and accessibility trend plots were generated from the resulting bam files in Seqmonk (v.1.48.0; www.bioinformatics.babraham.ac.uk/projects/seqmonk) as previously described36. Accordingly, chromosome-wide accessibility profiles show transposase insertion frequency (5′-read ends, offset +4 bp/−5 bp) in each 25-kb window, relative to the genome-wide average. Accessibility trend plots show average relative distribution of insertion sites (extended ±25 bp) across 2-kb windows centered on Esrrb peak summits (taken from ref. 32 and converted to mm10 using the UCSC LiftOver tool). Bam files were further processed with deepTools2 (ref.88) by using the alignmentSieve function to shift and extract nucleosome-free fragments (<100 bp). Peaks were called from nucleosome-free fragments with model-based analysis for ChIP-seq 2 (MACS2: -f BAMPE)89. Peaks overlapping ENCODE blacklist v.2 regions90 were removed. For downstream analysis, we separately defined consensus peaks for cells and purified chromosomes. For each, we defined nonredundant peaks using the reduce function from R/Bioconductor package GenomicRanges91 and only kept peaks appearing in at least two samples. Peak-based read counts were obtained with the featureCounts function from R/Bioconductor package Rsubread92,93 and normalized using the calcNormFactors function (method = ‘TMM’) in R/Bioconductor package edgeR (v.3.28.1)94,95. Differential accessibility analysis was performed with R/Bioconductor package limma (v.3.42.2)96, after applying the voom function97 to estimate the mean-variance relationship within the count-based datasets.
Proteomics
Proteomics was performed on mitotic lysate pellets and mitotic chromosomes from WT and Suv39h dn MEFs and ESCs, Suv39h dn ESCs treated with GSK343 or Hesperadin and Suv39h1-EGFP ESCs, using a method previously described36. Samples were digested with trypsin by an in-Stage Tip digestion protocol98 using commercially available iST Kits (Preomics) according to the manufacturer’s recommendations. Protein digests were analyzed by LC-MS/MS using a data-dependent acquisition method with a 50-cm EasySpray column at a flow rate of 250 nl min−1 coupled to a Q-Exactive HF-X mass spectrometer, as described previously36. Experiments were carried out in biological triplicate and digests were analyzed by LC-MS/MS as technical duplicates.
Raw data were analyzed using MaxQuant99,100 with the in-built LFQ algorithm101. Statistical analysis and data visualization were performed using Perseus software102. Analysis details are provided as previously described36, with minor modifications. After processing raw proteomic data in MaxQuant, the proteinGroups.txt file was imported into Perseus (v.1.6.2.2 or v.1.6.7.0) with the respective LFQ intensities as the main columns. We filtered on categorical columns to remove reverse decoy hits, potential contaminants and protein groups that were ‘only identified by site’. GO annotations for taxonomy Mus musculus (mainAnnot.mus_musculus.txt) were downloaded from http://annotations.perseus-framework.org. GO annotations for molecular function and cellular compartment were imported by annotating columns based on majority protein IDs. Groups of technical replicate injections and biological replicates of the two conditions (‘mitotic lysate pellet’ and ‘sorted chromosome pellet’) were defined in categorical annotation rows. Missing values were replaced with ‘NaN’ (Quality→Convert to NaN), technical duplicates were averaged (annot. rows→average groups→mean value; min. one valid value) and data were log(transformed) (Basic→Transform→log2(x)). Principal component analysis of proteomic datasets was performed after the filtering steps. Volcano plots were generated based on LFQ intensities with the following settings: test, Student’s t-test; side, both; number of randomizations, 250; preserve grouping in randomizations, <None>; permutation-based FDR, 0.05; and s0, 0.1. Hierarchical clustering analysis was carried out after filtering rows based on a minimum of two valid values in at least one group, z-scoring of values in rows and a two-sample Student’s t-test of the conditions using the following settings: test, Student’s t-test; s0, 0; side, both; and permutation-based FDR, 0.05. After filtering rows retaining Student’s t-test significant hits only, hierarchical clustering analysis was generated with the following settings for both the rows tree and the columns tree: distance, Euclidean; linkage, average; constraint, none; preprocess with k-means selected (number of clusters, 300; maximal number of iterations, 10; number of restarts, 1).
Majority protein IDs were extracted from the list of enriched hits from the volcano plots (listed in Supplementary Data 1 and 2) and were used to generate Venn diagrams and in GO analysis (http://geneontology.org; Fisher’s exact test with FDR correction).
Comparison of transcript abundance with proteomics
Raw poly(A) RNA-seq data from WT and Suv39h dn ESCs were obtained from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA; accession no. GSE57092)27. Reads were aligned to the UCSC mm10 genome using the STAR aligner (v.2.7.7a)103 based on the Ensembl genome annotation (v.2.7.7)104. Gene-based read counts were obtained with STAR and normalized by calculating transcripts per million. Differential expression analysis was performed using DESeq2 (v.1.30.1)105 to generate log2(fold-changes). Gene symbols were obtained for transcripts using Ensembl BioMart (https://www.ensembl.org/biomart/martview) and used to match transcriptomics and proteomics data. Ambiguous gene symbols were removed from the proteomics data. In total, 5,664 genes were mapped between proteomics and transcriptomics datasets for correlation analyses.
Live-cell imaging
Esrrb–tdTomato ESCs were plated in 0.1% gelatin-coated, 8-well Ibidi μ-Slides. On the next day, cells were treated with 100 nM chaetocin or DMSO in fresh ESC medium for 24 h. Cells were switched to fully supplemented phenol red-free medium, buffered with 20 mM Hepes, 1 h before imaging. SiR-DNA (1 µM, SpiroChrome) was added 30 min before imaging. Time-lapse images were acquired on an Olympus IX70 inverted wide-field microscope using a UPlanApo ×100/1.35 oil objective lens and an environmental chamber kept at 37 °C with a 5% CO2 supply. The z-stacks were collected every 120 s with a step size of 2 µm. Images were deconvolved with Huygens Professional (v.19.10, Scientific Volume Imaging, http://svi.nl), using the classic maximum likelihood estimation algorithm. Esrrb–tdTomato fluorescence signal was measured in Fiji82 on maximum intensity projections of z-planes for both interphase nuclei and metaphase chromosomes.
Probe labeling
Mouse Cot-1 (Invitrogen) or γSat DNA (a gift from N. Dillon), 1 μg, was labeled with Cy3-dUTP or Cy5-dUTP (Cytiva or Sigma-Aldrich) using a Prime-It Flour Fluorescence labeling kit (v.B, Agilent Technologies) according to the manufacturer’s instructions. Briefly, a DNA template was annealed with random 9-mer primers at 95 °C for 5 min, then the RNA extended using exonuclease-free Klenow polymerase for 30 min at 37 °C. The reaction was stopped and the probe stored at 4 °C.
RNA–FISH
Cells were grown on coverslips, fixed using 2.6% formaldehyde in PBS for 10 min, then permeabilized using 0.4% Triton X-100 in PBS for 5 min on ice. Cells were washed with PBS and dehydrated using an ethanol series (3 min in 70%, 80%, 95% and 100% EtOH). RNA probe, 2.5 μl, in 12 μl of hybridization buffer (1 ml 50% Denhardt’s solution, 200 μl of saline–sodium citrate (SSC) containing 20% dextran sulfate, 800 μl of formamide) was used for each coverslip of 22 × 22 mm2. Probe was denatured at 75 °C for 8 min, then placed on ice for 2 min. Cells were inverted on to the probe on a slide, sealed with rubber cement and placed in a humid chamber at 37 °C overnight. After hybridization, the rubber cement was removed and cells washed 3× for 3 min with 2× SSC containing 50% formamide at 42 °C followed by 3× 3-min washes with 2× SSC at 42 °C, and briefly rinsed in water before mounting in DAPI-containing Vectashield. Images were acquired with a Leica SP5 II confocal microscope using LAS-AF software.
ChIP
WT and Suv39h dn asynchronous and metaphase-arrested ESCs (5 × 106) were crosslinked in 1 ml of 2 mM DSG (Sigma-Aldrich) for 50 min at RT with occasional shaking. After PBS washes, cells were fixed in 1 ml of PBS, 1% methanol-free formaldehyde (Thermo Fisher Scientific) for 10 min at RT as previously described for Esrrb ChIP31,32. Cells were quenched with 1× glycine (Active Motif) and washed once with cold PBS containing protease inhibitor cocktail (Active Motif). Cell pellets were resuspended in 50 µl of lysis buffer containing 1% sodium dodecylsulfate, 10 mM EDTA and 50 mM Tris, pH 8.1, supplemented with 1 µl of protease inhibitor cocktail and incubated on ice for 15 min. Samples were diluted by the addition of 320 µl of ice-cold shearing buffer (Active Motif) as described in ref. 106, and sonicated in 1.5-ml tubes (Diagenode) using a Bioruptor Pico (Diagenode) for 7 cycles of 30 s on:30 s off at maximum power, in circulating ice-cold water. Sheared chromatin samples were centrifuged for 20 min at 18,000g and 4 °C, and supernatants were carefully transferred to fresh microcentrifuge tubes (Active Motif). Then, 50 μl of this sheared chromatin was used to assess DNA concentration and sonication efficiency (typically DNA between 200 and 800 bp) using ChIP-IT Express kit (Active motif) and ChIP clean & concentrator kit (Zymo research).
Immunoprecipitation was performed using the ChIP-IT Express kit according to the manufacturer’s recommendations and 1 μg of Esrrb mouse monoclonal antibody (catalog no. PP-H6705-00, Perseus Proteomics). Input and immunoprecipitated DNA fractions were purified using ChIP clean & concentrator kit. Real-time qPCR (BioRad, CFX96 system with CFX manager software) was performed in technical triplicate for each biological replicate, using SYBR Green PCR Master Mix (QIAGEN) and the primers listed in Supplementary Table 2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
ATAC-seq data generated in the present study have been deposited at the GEO SRA under accession no. GSE195767. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE107 partner repository with the dataset identifier PXD039521. Raw poly(A) RNA-seq data from WT and Suv39h dn ESCs, corresponding to accession no. GSE57092, were obtained from the National Center for Biotechnology Information SRA. UCSC mm10 genome and annotation files were retrieved from Illumina iGenome (http://igenomes.illumina.com.s3-website-us-east-1.amazonaws.com/Mus_musculus/UCSC/mm10/Mus_musculus_UCSC_mm10.tar.gz). The ENCODE mm10-blacklist.v2 used to filter peaks is available at https://github.com/Boyle-Lab/Blacklist. GO annotations for use in Perseus were downloaded from http://annotations.perseus-framework.org (mainAnnot.mus_musculus.txt). All other relevant data supporting the key findings of the present study are available within the article and its supplementary files or from the corresponding author upon reasonable request. Proteomics data are provided in Supplementary Data 1 and 2. Source data are provided with this paper.
References
Politz, J. C. R., Scalzo, D. & Groudine, M. The redundancy of the mammalian heterochromatic compartment. Curr. Opin. Genet. Dev. 37, 1–8 (2016).
Bizhanova, A. & Kaufman, P. D. Close to the edge: heterochromatin at the nucleolar and nuclear peripheries. Biochim. Biophys. Acta Gene Regul. Mech. 1864, 194666 (2021).
Arunkumar, G. & Melters, D. P. Centromeric transcription: a conserved Swiss-army knife. Genes 11, 911 (2020).
Saksouk, N., Simboeck, E. & Dejardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenet. Chromatin 8, 3 (2015).
Jones, K. W. Chromosomal and nuclear location of mouse satellite DNA in individual cells. Nature 225, 912–915 (1970).
Fransz, P., De Jong, J. H., Lysak, M., Castiglione, M. R. & Schubert, I. Interphase chromosomes in Arabidopsis are organized as well defined chromocenters from which euchromatin loops emanate. Proc. Natl Acad. Sci. USA 99, 14584–14589 (2002).
Probst, A. V., Santos, F., Reik, W., Almouzni, G. & Dean, W. Structural differences in centromeric heterochromatin are spatially reconciled on fertilisation in the mouse zygote. Chromosoma 116, 403–415 (2007).
Martin, C. et al. Genome restructuring in mouse embryos during reprogramming and early development. Dev. Biol. 292, 317–332 (2006).
Horz, W. & Altenburger, W. Nucleotide sequence of mouse satellite DNA. Nucleic Acids Res. 9, 683–696 (1981).
Vissel, B. & Choo, K. H. Mouse major (gamma) satellite DNA is highly conserved and organized into extremely long tandem arrays: implications for recombination between nonhomologous chromosomes. Genomics 5, 407–414 (1989).
Guenatri, M., Bailly, D., Maison, C. & Almouzni, G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 166, 493–505 (2004).
Rea, S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000).
Peters, A. H. et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323–337 (2001).
O’Carroll, D. et al. Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol. Cell Biol. 20, 9423–9433 (2000).
Tachibana, M., Sugimoto, K., Fukushima, T. & Shinkai, Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 276, 25309–25317 (2001).
Tachibana, M. et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev 19, 815–826 (2005).
Schultz, D. C., Ayyanathan, K., Negorev, D., Maul, G. G. & Rauscher, F. J. 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16, 919–932 (2002).
Yang, L. et al. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene 21, 148–152 (2002).
Yamane, K. et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125, 483–495 (2006).
Cloos, P. A. et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442, 307–311 (2006).
Shi, Y. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004).
Metzger, E. et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439 (2005).
Krishnan, S., Horowitz, S. & Trievel, R. C. Structure and function of histone H3 lysine 9 methyltransferases and demethylases. ChemBioChem 12, 254–263 (2011).
Fadloun, A., Eid, A. & Torres-Padilla, M. E. Mechanisms and dynamics of heterochromatin formation during mammalian development: closed paths and open questions. Curr. Top. Dev. Biol. 104, 1–45 (2013).
Maison, C., Quivy, J. P., Probst, A. V. & Almouzni, G. Heterochromatin at mouse pericentromeres: a model for de novo heterochromatin formation and duplication during replication. Cold Spring Harb. Symp. Quant. Biol. 75, 155–165 (2010).
Lehnertz, B. et al. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13, 1192–1200 (2003).
Bulut-Karslioglu, A. et al. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell 55, 277–290 (2014).
Kadauke, S. et al. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell 150, 725–737 (2012).
Caravaca, J. M. et al. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 27, 251–260 (2013).
Deluz, C. et al. A role for mitotic bookmarking of SOX2 in pluripotency and differentiation. Genes Dev. 30, 2538–2550 (2016).
Festuccia, N. et al. Mitotic binding of Esrrb marks key regulatory regions of the pluripotency network. Nat. Cell Biol. 18, 1139–1148 (2016).
Festuccia, N. et al. Transcription factor activity and nucleosome organization in mitosis. Genome Res. 29, 250–260 (2019).
Teves, S. S. et al. A dynamic mode of mitotic bookmarking by transcription factors. eLife 5, e22280 (2016).
Higgins, J. M. & Prendergast, L. Mitotic mysteries: the case of HP1. Dev. Cell 36, 477–478 (2016).
Ginno, P. A., Burger, L., Seebacher, J., Iesmantavicius, V. & Schubeler, D. Cell cycle-resolved chromatin proteomics reveals the extent of mitotic preservation of the genomic regulatory landscape. Nat. Commun. 9, 4048 (2018).
Djeghloul, D. et al. Identifying proteins bound to native mitotic ESC chromosomes reveals chromatin repressors are important for compaction. Nat. Commun. 11, 4118 (2020).
Campos, E. I., Stafford, J. M. & Reinberg, D. Epigenetic inheritance: histone bookmarks across generations. Trends Cell Biol. 24, 664–674 (2014).
Chu, L. et al. SUV39H1 orchestrates temporal dynamics of centromeric methylation essential for faithful chromosome segregation in mitosis. J. Mol. Cell Biol. 4, 331–340 (2012).
Velazquez Camacho, O. et al. Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA-nucleosome association and RNA:DNA hybrid formation. eLife 6, e25293 (2017).
Akram, S. et al. LRIF1 interacts with HP1alpha to coordinate accurate chromosome segregation during mitosis. J. Mol. Cell Biol. 10, 527–538 (2018).
Montavon, T. et al. Complete loss of H3K9 methylation dissolves mouse heterochromatin organization. Nat. Commun. 12, 4359 (2021).
Peters, A. H. et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 12, 1577–1589 (2003).
Saksouk, N. et al. Redundant mechanisms to form silent chromatin at pericentromeric regions rely on BEND3 and DNA methylation. Mol. Cell 56, 580–594 (2014).
Puschendorf, M. et al. PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat. Genet. 40, 411–420 (2008).
Sivakumar, S., Daum, J. R. & Gorbsky, G. J. Live-cell fluorescence imaging for phenotypic analysis of mitosis. Methods Mol. Biol. 1170, 549–562 (2014).
Greiner, D., Bonaldi, T., Eskeland, R., Roemer, E. & Imhof, A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat. Chem. Biol. 1, 143–145 (2005).
Greiner, D., Bonaldi, T., Eskeland, R., Roemer, E. & Imhof, A. Reply to ‘Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases’. Nat. Chem. Biol. 9, 137 (2013).
Law, M. J. et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143, 367–378 (2010).
Gibbons, R. J. et al. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat. Genet. 24, 368–371 (2000).
Latos, P. A. et al. Fgf and Esrrb integrate epigenetic and transcriptional networks that regulate self-renewal of trophoblast stem cells. Nat. Commun. 6, 7776 (2015).
Healton, S. E. et al. H1 linker histones silence repetitive elements by promoting both histone H3K9 methylation and chromatin compaction. Proc. Natl Acad. Sci. USA 117, 14251–14258 (2020).
Fan, Y. et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212 (2005).
Fyodorov, D. V., Zhou, B. R., Skoultchi, A. I. & Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 19, 192–206 (2018).
Routh, A., Sandin, S. & Rhodes, D. Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proc. Natl Acad. Sci. USA 105, 8872–8877 (2008).
Gibbs, E. B. & Kriwacki, R. W. Linker histones as liquid-like glue for chromatin. Proc. Natl Acad. Sci. USA 115, 11868–11870 (2018).
Gibson, B. A. et al. Organization of chromatin by intrinsic and regulated phase separation. Cell 179, 470–484.e421 (2019).
Willcockson, M. A. et al. H1 histones control the epigenetic landscape by local chromatin compaction. Nature 589, 293–298 (2021).
Yusufova, N. et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 589, 299–305 (2021).
Arimura, Y., Shih, R. M., Froom, R. & Funabiki, H. Structural features of nucleosomes in interphase and metaphase chromosomes. Mol. Cell 81, 4377–4397.e12 (2021).
Hall, L. L., Byron, M., Pageau, G. & Lawrence, J. B. AURKB-mediated effects on chromatin regulate binding versus release of XIST RNA to the inactive chromosome. J. Cell Biol. 186, 491–507 (2009).
Hall, L. L. et al. Stable C0T-1 repeat RNA is abundant and is associated with euchromatic interphase chromosomes. Cell 156, 907–919 (2014).
Lu, J. & Gilbert, D. M. Proliferation-dependent and cell cycle regulated transcription of mouse pericentric heterochromatin. J. Cell Biol. 179, 411–421 (2007).
de la Barre, A. E. et al. Core histone N-termini play an essential role in mitotic chromosome condensation. EMBO J. 19, 379–391 (2000).
Johansen, K. M. & Johansen, J. Regulation of chromatin structure by histone H3S10 phosphorylation. Chromosome Res. 14, 393–404 (2006).
Williamson, I. et al. Anterior-posterior differences in HoxD chromatin topology in limb development. Development 139, 3157–3167 (2012).
Nicetto, D. & Zaret, K. S. Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr. Opin. Genet. Dev. 55, 1–10 (2019).
Janssen, A., Colmenares, S. U. & Karpen, G. H. Heterochromatin: guardian of the Ggenome. Annu. Rev. Cell Dev. Biol. 34, 265–288 (2018).
Nishioka, N. et al. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev. Cell 16, 398–410 (2009).
Russell, R. et al. A dynamic role of TBX3 in the pluripotency circuitry. Stem Cell Rep. 5, 1155–1170 (2015).
Gao, Z. et al. Determination of protein interactome of transcription factor Sox2 in embryonic stem cells engineered for inducible expression of four reprogramming factors. J. Biol. Chem. 287, 11384–11397 (2012).
Masui, S. et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat. Cell Biol. 9, 625–635 (2007).
Xie, L. et al. A dynamic interplay of enhancer elements regulates Klf4 expression in naive pluripotency. Genes Dev. 31, 1795–1808 (2017).
Weirich, S., Khella, M. S. & Jeltsch, A. Structure, activity and function of the Suv39h1 and Suv39h2 protein lysine methyltransferases. Life 11, 703 (2021).
Bradley, S. P., Kaminski, D. A., Peters, A. H., Jenuwein, T. & Stavnezer, J. The histone methyltransferase Suv39h1 increases class switch recombination specifically to IgA. J. Immunol. 177, 1179–1188 (2006).
Yaseen, I., Choudhury, M., Sritharan, M. & Khosla, S. Histone methyltransferase SUV39H1 participates in host defense by methylating mycobacterial histone-like protein HupB. EMBO J. 37, 183–200 (2018).
Piao, L., Suzuki, T., Dohmae, N., Nakamura, Y. & Hamamoto, R. SUV39H2 methylates and stabilizes LSD1 by inhibiting polyubiquitination in human cancer cells. Oncotarget 6, 16939–16950 (2015).
Sone, K. et al. Critical role of lysine 134 methylation on histone H2AX for gamma-H2AX production and DNA repair. Nat. Commun. 5, 5691 (2014).
Eckersley-Maslin, M. A. et al. Epigenetic priming by Dppa2 and 4 in pluripotency facilitates multi-lineage commitment. Nat. Struct. Mol. Biol. 27, 696–705 (2020).
Gretarsson, K. H. & Hackett, J. A. Dppa2 and Dppa4 counteract de novo methylation to establish a permissive epigenome for development. Nat. Struct. Mol. Biol. 27, 706–716 (2020).
Muller, I. et al. MPP8 is essential for sustaining self-renewal of ground-state pluripotent stem cells. Nat. Commun. 12, 3034 (2021).
Gribble, S. M., Ng, B. L., Prigmore, E., Fitzgerald, T. & Carter, N. P. Array painting: a protocol for the rapid analysis of aberrant chromosomes using DNA microarrays. Nat. Protoc. 4, 1722–1736 (2009).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Ou, J. et al. ATACseqQC: a Bioconductor package for post-alignment quality assessment of ATAC-seq data. BMC Genomics 19, 169 (2018).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Amemiya, H. M., Kundaje, A. & Boyle, A. P. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 9, 9354 (2019).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N. & Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 (2014).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteom. 13, 2513–2526 (2014).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Djeghloul, D. et al. Age-associated decrease of the histone methyltransferase SUV39H1 in HSC perturbs heterochromatin and B lymphoid differentiation. Stem Cell Rep. 6, 970–984 (2016).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
We thank the London Institute of Medical Sciences (LMS)/National Institute for Health Research Imperial Biomedical Research Centre Flow Cytometry Facility, as well as the LMS microscopy facility and the LMS Genomics and LMS Bioinformatics facilities for support. We thank N. Festuccia and P. Navarro (Institut Pasteur, Paris) for providing the Esrrb–tdTomato ESC line. We thank N. Dillon (MRC LMS) for the mouse γSat DNA plasmids. Research in the laboratory of T. Jenuwein is supported by the Max Planck Society and by additional funds from the German Research Foundation within the CRC992 consortium ‘MEDEP’. The present study was funded by core support from the Medical Research Council UK to the LMS (nos. MC_U120027516, MC_UP_1605/12 and MC_UP_1605/11 to A.G.F and M.M.). D.D. was supported by a Wellcome Trust Institutional Strategic Support Fund Springboard award (no. WCMA_PSN102). A.D. is supported by a Kay Kendall Leukaemia Fund Junior Research Fellowship (no. KKL1334).
Author information
Authors and Affiliations
Contributions
A.G.F. and D.D. designed the study, wrote the manuscript and supervised the work. D.D. performed most of the experiments, conducted data analysis and designed the figures. A.D. conducted experiments, analyzed data and contributed to writing the manuscript. S.C., K.B. and N.V. conducted experiments. H.K. and A.M. conducted MS and helped with proteomic analysis. B.P. conducted all of the chromosome flow sorting. C.W. helped with live-cell imaging experiments and analysis. Y.W. and M.E.F. performed bioinformatic analysis. T.J., T.M. and M.M. contributed to the design of the study and provided scientific advice and support with experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks Michiel Vermeulen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Carolina Perdigoto, Beth Moorefield and Dimitris Typas were the primary editors on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary figs. 1–6, methods, tables 1–3, references and source data for Supplementary Fig. 4c.
Supplementary Data 1
Tab 1: Protein group file: proteomics on sorted mitotic chromosomes and mitotic lysate pellets of WT and Suv39h dn MEFs. Tab 2: Number of protein hits identified in WT and Suv39h dn MEF samples. Tab 3: List of proteins that are enriched on mitotic chromosome in both WT and Suv39h dn MEFs (enrichment maintained), list of proteins that are not enriched in Suv39h dn MEF samples compared with WT (enrichment lost) and list of proteins that are enriched in Suv39h dn MEFs but not in the WT (enrichment gained).
Supplementary Data 2
Tab 1: Protein group file: proteomics on sorted mitotic chromosomes and mitotic lysate pellets of WT and Suv39h dn ESCs. Tab 2: Number of protein hits identified in WT and Suv39h dn ESC samples. Tab 3: List of proteins that are enriched on mitotic chromosome in both WT and Suv39h dn ESCs (enrichment maintained), list of proteins that are not enriched in Suv39h dn ESC samples compared with WT (enrichment lost) and list of proteins that are enriched in Suv39h dn ESCs but not in the WT (enrichment gained).
Supplementary Data 3
Source data for supplementary figures.
Live-cell imaging of Esrrb–tdTomato mouse ESCs pretreated with DMSO cultured with SiR-DNA (red). Scale bar, 5 μm. Source data file for Fig. 4d.
Live-cell imaging of Esrrb–tdTomato mouse ESCs pretreated with 100 nM chaetocin cultured with SiR-DNA (red). Scale bar, 5 μm. Source data file for Fig. 4d.
Source data
Source Data
Raw values for plots in Figs. 1, 2, 4 and 5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Djeghloul, D., Dimond, A., Cheriyamkunnel, S. et al. Loss of H3K9 trimethylation alters chromosome compaction and transcription factor retention during mitosis. Nat Struct Mol Biol 30, 489–501 (2023). https://doi.org/10.1038/s41594-023-00943-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-023-00943-7
- Springer Nature America, Inc.