

Abstract
Protein degradation is critical to maintaining cellular homeostasis, and perturbation of the ubiquitin proteasome system leads to the accumulation of protein aggregates. These aggregates are either directed towards autophagy for destruction or sequestered into an inclusion, termed the aggresome, at the centrosome. Utilizing high-resolution quantitative analysis, here, we define aggresome assembly at the centrosome in human cells. Centriolar satellites are proteinaceous granules implicated in the trafficking of proteins to the centrosome. During aggresome assembly, satellites were required for the growth of the aggresomal structure from an initial ring of phosphorylated HSP27 deposited around the centrioles. The seeding of this phosphorylated HSP27 ring depended on the centrosomal proteins CP110, CEP97 and CEP290. Owing to limiting amounts of CP110, senescent cells, which are characterized by the accumulation of protein aggregates, were defective in aggresome formation. Furthermore, satellites and CP110–CEP97–CEP290 were required for the aggregation of mutant huntingtin. Together, these data reveal roles for CP110–CEP97–CEP290 and satellites in the control of cellular proteostasis and the aggregation of disease-relevant proteins.
Similar content being viewed by others


Main
The centrosome consists of a pair of centrioles surrounded by pericentriolar material (PCM) and serves as the main microtubule organizing centre (MTOC) of the cell1. It also provides a platform for localized protein degradation via the ubiquitin proteasome system (UPS). However, the contribution of centrosomal components to proteostasis remains unclear.
If the capacity of the UPS is exceeded, proteins accumulate into an inclusion termed the aggresome2. Aggresomes are membrane-free, juxta-nuclear, cytoplasmic inclusions that concentrate misfolded proteins, ubiquitin, molecular chaperones and proteasomes at the centrosome and the MTOC3. The organization of potentially toxic proteins into a single location is protective while also facilitating their clearance by autophagy4,5,6. Aggresome assembly is an active process that requires microtubules, HDAC6 and dynein7,8,9. Some centrosomal proteins can accumulate into aggresomes2,10,11,12 along with PCM1, which is a key component of centriolar satellites13. Satellites are proteinaceous granules associated with the centrosome that contribute to its assembly and function. Satellites also require microtubules and dynein for their association with centrosomes14,15. Growing evidence implicates satellites as mediators of protein stability via autophagy and the UPS16,17,18,19, and changes in the cellular proteome of satellite-deficient cells highlight satellites as regulators of global proteostasis20. However, the role of satellites in aggresome formation has yet to be explored.
Centrosome biology itself is tightly regulated by proteasomal degradation, and stringent regulation of centriole numbers is achieved through control of the abundance of centriole duplication factors21,22,23,24. Prolonged proteasome inhibition leads to centriole amplification25 and the appearance of elongated centrioles26. A number of proteins regulate centriole length, including CP110, which together with CEP97 forms a cap at the centriole distal end that restricts centriole elongation27,28,29. CP110 levels are regulated via the antagonistic actions of the SCFCyclinF ubiquitin ligase complex30 and the deubiquitinating enzyme USP33 (ref. 31). CP110 also interacts with the satellite protein CEP290 (ref. 32), and both CP110 and CEP97 are found in the satellite proteome and interactome33,34. Whether CP110 function extends beyond the control of centriole elongation is unknown.
Senescent cells cease dividing and undergo distinctive phenotypic changes35. Proteasomal activity decreases during senescence, which can result in the accumulation of protein aggregates that are associated with late-onset pathologies36,37,38,39. Neurodegenerative disorders such as Huntington’s disease (HD) are characterized by inclusions that contain misfolded proteins, with expanded polyglutamine (polyQ) tracts in the HD protein huntingtin (HTT) associated with its aggregation40. HTT interacts with PCM1 through HAP1, and expression of HTT-polyQ causes PCM1 to aggregate at the centrosome41. However, the role of satellites in HTT-polyQ inclusion formation remains to be determined.
Here, we describe the association of centriolar and PCM proteins with the aggresome. Centriolar satellites also accumulated into this structure, and aggresome assembly was reduced in cells depleted of the satellite proteins AZI1 (also known as CEP131), CCDC14, KIAA0753 and PIBF1 (also known as CEP90). In these cells, protein aggregates were directed towards autophagy rather than the aggresome. However, in cells devoid of satellites (via depletion of PCM1), inhibition of autophagy failed to rescue aggresome formation, which places satellites at the intersection of the aggresome and autophagy pathways. Furthermore, a module consisting of CP110, CEP97 and CEP290 operates early in the aggresome pathway, and aggresome formation was defective in senescent cells due to limiting levels of CP110. Finally, satellites and the CP110–CEP97–CEP290 module function in the formation of HTT-polyQ inclusions.
Results
Centrosomal proteins localize to the aggresome
Treatment of RPE-1 cells with proteasome inhibitors (MG132 and bortezomib) led to the collection of ubiquitinated proteins (Ub+) around the centrosome (Fig. 1a,b). The accumulation of aggresome markers, including phosphorylated HSP27 (pHSP27)42, HDAC6 (ref. 9), p62 (ref. 43), HSP70 (ref. 44), HAP1 (ref. 45) and dynein8, confirmed aggresome assembly (Extended Data Fig. 1a). More than 75% of cells formed an aggresome (Fig. 1c), which was universal to additional cell lines tested (Extended Data Fig. 1b,c).

a, RPE-1 cells treated with DMSO or MG132 were stained for CEP97, ubiquitinated proteins (Ub+) and DNA (4,6-diamidino-2-phenylindole (DAPI)). b, RPE-1 cells treated with bortezomib (BZ) were stained for CEP135, Ub+, PCM1 and DNA (DAPI). c, The percentage of cells that formed an aggresome in untreated cells and in cells treated with DMSO, MG132 or BZ as revealed by Ub+ staining. Data displayed as the mean ± s.d., n = 3 independent experiments. ****P < 0.0001 or not significant (NS) by two-tailed unpaired Student’s t-test. d, RPE-1 cells treated with DMSO or MG132 were stained for PCM1 and the indicated protein. e, A-375 cells treated with MG132 for 5 or 16 h were stained as indicated. f, RPE-1 cells treated with DMSO or MG132 were stained for PCM1, pHSP27 and the indicated protein. g, Super-resolution images of aggresomes in MG132-treated RPE-1 cells stained as indicated. h, Colocalization between the indicated protein pairs from individual z-planes of super-resolution images of RPE-1 cells treated with MG132 displayed using Pearson’s correlation co-efficient. Boxes represent the median, upper and lower quartiles, whiskers represent 1.5× the interquartile range, with individual values from two independent experiments superimposed. Scale bars, 1 μm (g), 2 μm (d,f; insets of a,b,e) or 10 μm (a,b,e). Numerical data and P values are provided as source data.
Given that CEP97 and PCM1 are recruited to the aggresome (Fig. 1a,b), and that γ-tubulin, PCNT and ninein accumulate during proteasome inhibition13, we surveyed the localization of centrosomal proteins following MG132 treatment (Fig. 1d). The centriole markers CP110 and CETN2 associated with the aggresome; however, CEP135, glutamylated tubulin (GT335) and SAS6 remained restricted to the centrioles. The PCM markers CEP215, γ-tubulin, HAUS6 and PCNT localized to the aggresome, whereas CEP192 and NEDD1 stayed limited to the PCM.
Proteolysis regulates centriole number46, and centriole amplification occurs in U-2 OS cells following prolonged proteasome inhibition25. Centriolar staining typically reveals two or four foci that correspond to two or four centrioles depending on the cell cycle phase. CEP135, SAS6 and GT335 staining revealed normal centriole complement during MG132 treatment (Extended Data Fig. 1d,e), which suggests that the extra foci of CETN2, CP110 and CEP97 did not correspond to supernumerary centrioles. This result was confirmed by transmission electron microscopy (TEM; n = 50; Fig. 5c and Extended Data Fig. 1f).
Overnight treatment with MG132 was not tolerated by RPE-1 cells; however, larger accumulations of pHSP27 and p62 occurred at the centrosome in A-375 cells (Fig. 1e). CP110 and PCM1 were limited to an area within the aggresome similar in size to the 5-h inclusions, which suggests that this structure forms first and may potentiate the further accumulation of protein aggregates.
In addition to PCM1, other satellite proteins localized to the aggresome (Fig. 1f and Extended Data Fig. 1g). Super-resolution imaging and colocalization analysis revealed that protein aggregates (pHSP27, Ub+ and p62), satellite proteins (PCM1 and PIBF1) and CP110 occupy overlapping domains within the aggresome (Fig. 1g,h and Extended Data Fig. 1h,i). However, the centrioles themselves (CEP135) were mostly devoid of protein aggregates.
Satellites accumulate in response to proteasome inhibition
To examine the aggresome pathway, we established image analysis pipelines to measure aggresome size (pHSP27) and satellite distribution (PCM1; Extended Data Fig. 2a and Methods). Knockdown of HSP27 demonstrated the specificity of the pHSP27 signal (Extended Data Fig. 2b–d) and blocked aggresome assembly, as seen by Ub+ and CP110 staining, which was rescued following the expression of short interfering RNA (siRNA)-resistant HSP27 (Extended Data Fig. 2b–e). Notably, satellites accumulated in the pericentrosomal region during proteasome inhibition, even when HSP27 was depleted (Extended Data Fig. 2f).
Next, we examined approaches reported to disrupt aggresome formation. Active protein translation is required for aggresome assembly47,48, and pretreatment with cycloheximide to inhibit translation before MG132 addition completely blocked pHSP27 and Ub+ recruitment to the aggresome and satellite accumulation (Fig. 2a–d). Aggresome formation also requires HDAC6 activity9, so we analysed the effects of the HDAC6 inhibitors ACY-1215 and ACY-738 (ref. 49). Increased levels of acetylated tubulin confirmed HDAC6 inhibition (Extended Data Fig. 2g), and we observed a moderate, but significant, reduction in aggresome size rather than a complete block to aggresome assembly as reported for HDAC6 depletion9, with pHSP27 and Ub+ restricted to the area immediately around the centrioles (Fig. 2e–g). The accumulation of satellites was largely unaffected (Fig. 2h).

a, RPE-1 cells treated with DMSO, MG132, cycloheximide (CHX), or CHX plus MG132 were stained as indicated. b, The area occupied by pHSP27 in cells treated as in a. n = 493 (DMSO), 630 (MG132), 459 (CHX) and 437 (CHX + MG132) aggresomes examined over 2 independent experiments. ****P < 0.0001. c, Cells treated with MG132 with or without pretreatment with CHX were stained as indicated. d, Intensity maps of PCM1 distribution relative to the centrosome in cells treated as in a. The percentage of PCM1 signal residing in the defined inner region is indicated. AU, arbitrary units. e, RPE-1 cells treated with DMSO or MG132 were treated concurrently with ACY-1215 or ACY-738 and stained as indicated. f, The area occupied by pHSP27 in cells treated as in e. n = 586 (DMSO), 566 (MG132), 470 (ACY-1215), 504 (ACY-1215 + MG132), 470 (ACY-738) and 395 (ACY-738 + MG132) aggresomes examined over 2 independent experiments. ****P < 0.0001. g, RPE-1 cells treated with MG132 with or without ACY-1215 or ACY-738 were stained as indicated. h, PCM1 distribution relative to the centrosome in cells treated as in e. i, RPE-1 cells were pretreated with nocodazole (Noc) or taxol followed by DMSO or MG132 and stained as indicated. j, The area occupied by pHSP27 in cells treated as in i. n = 554 (DMSO), 499 (MG132), 413 (Noc), 422 (Noc + MG132), 510 (taxol) and 485 (taxol + MG132) aggresomes examined over 2 independent experiments. ****P < 0.0001. k, PCM1 distribution relative to the centrosome in cells treated as in i. l, RPE-1 cells treated with MG132 and nocodazole were stained as indicated. For b, f and j, boxes represent the median, upper and lower quartiles, whiskers represent 1.5× the interquartile range, with individual values superimposed. Data were compared using Kruskal–Wallis analysis of variance (ANOVA) test, and post-hoc Dunn multiple comparison test was performed to calculate P values. Scale bars, 2 μm (a,c,e,g,i; inset of l) or 10 μm (l). Numerical data and P values are provided as source data. AU, arbitrary units.
Finally, as aggresome formation depends on the instability of microtubule dynamics50, we used nocodazole and taxol to disrupt microtubules. These compounds led to satellite dispersion throughout the cytoplasm15,51 (Extended Data Fig. 2h). Treatment of cells with MG132 led to reduced accumulation of pHSP27 and restricted recruitment of satellites (Fig. 2i–k). Ub+ in these cells formed multiple cytoplasmic aggregates that colocalized with PCM1 (Fig. 2l and Extended Data Fig. 2i), which places satellites with these proteins before their recruitment to the aggresome. In cells that lacked centrioles through disruption of STIL, Ub+ similarly colocalized with cytoplasmic PCM1 foci (Extended Data Fig. 2j). However, Ub+ aggregates in ACY-1215-treated cells did not contain PCM1 (Extended Data Fig. 2k), which suggests that this association requires HDAC6.
Together, these results demonstrate the robustness of our analysis for quantitative morphometric measurements of aggresomes and for distinguishing between complete and partial blocks to aggresome assembly. Furthermore, satellites accumulate around the centrosome in response to proteasomal inhibition, downstream of the requirement for active protein translation in aggresome biogenesis.
Centriolar satellites are required for aggresome formation
To test the requirement for satellites in aggresome assembly, we used CRISPR–Cas9 to disrupt AZI1, CCDC14, KIAA0753 and PIBF1 expression. Sequence analysis confirmed gene disruption (Supplementary Table 1), and immunoblotting and immunofluorescence (IF) microscopy confirmed protein loss (Extended Data Fig. 3a,b). In each knockout (KO) line, PCM1-positive satellites persisted, whereas AZI1, CCDC14, KIAA0753 and PIBF1 were restricted to the centrioles in a previously generated PCM1 KO line33 (Extended Data Fig. 3b). This result indicated satellite loss and is in line with previous studies17,20,52. Aggresome assembly was reduced to similar extents in each KO line despite PCM1-positive satellites accumulating in the pericentrosomal region of AZI1, CCDC14, KIAA0753 and PIBF1 KO cells (Fig. 3a–c and Extended Data Fig. 3c). Conversely, aggresome formation was not impeded in cells depleted of CETN2 (ref. 53), FOP54 or OFD1 (this study; Extended Data Fig. 3e–g and Supplementary Table 1), despite these proteins accumulating within the aggresome (Fig. 1d,f).

a, WT cells and ΔAZI1, ΔCCDC14, ΔKIAA0753, ΔPCM1 and ΔPIBF1 RPE-1 cells were treated with MG132 and stained as indicated. b, The area occupied by pHSP27 in cells treated as in a. n = 518 (WT-DMSO), 705 (WT-MG132), 498 (ΔAZI1), 560 (ΔCCDC14), 468 (ΔKIAA0753), 682 (ΔPCM1) and 550 (ΔPIBF1) aggresomes examined over 2 independent experiments. ****P < 0.0001. c, Intensity maps of PCM1 distribution relative to the centrosome in cells treated as treated in a. The percentage PCM1 signal residing in the defined inner region is indicated. d, Super-resolution images of pHSP27, CEP135 and PCM1 staining in WT and ΔPCM1 cells treated with MG132. e, WT and ΔPCM1 cells treated with MG132 were stained for CP110 and the indicated protein. f, WT and ΔPCM1 cells were treated with control (GL2), KIAA0753 or PIBF1 siRNAs (siGL2, siKIAA0753 and siPIBF1, respectively) as indicated for 48 h, then treated with MG132. Cells were stained for CEP135, pHSP27 and PCM1. g, The area occupied by pHSP27 in cells treated as in f. n = 511 (WT-siGL2), 493 (WT-siKIAA0753), 324 (WT-siPIBF1), 519 (ΔPCM1-siGL2), 452 (ΔPCM1-siKIAA0753) and 291 (ΔPCM1-siPIBF1) aggresomes examined over 2 independent experiments. ****P < 0.0001. h, Intensity maps of PCM1 distribution relative to the centrosome in cells treated as in f. The percentage PCM1 signal residing in the defined ‘inner’ region is indicated. i, Ub+ and CEP135 staining in WT and PCM1 KO cells treated as in f. For b and g, boxes represent the median, upper and lower quartiles, whiskers represent 1.5× the interquartile range, with individual values superimposed. Data were compared using Kruskal–Wallis ANOVA test, and post-hoc Dunn multiple comparison test was performed to calculate P values. Scale bars, 1 μm (d) or 2 μm (a,e,f,i). Numerical data and P values are provided as source data. AU, arbitrary units.
pHSP27 was restricted to a ring around each centriole in PCM1 KO cells, which corresponded to an early stage of aggresome biogenesis compared with wild-type (WT) cells at different points in aggresome assembly (Fig. 3d). Ub+ was similarly restricted (Extended Data Fig. 3d), and other aggresome markers failed to accumulate (Fig. 3e).
Despite lacking satellites, PCM1 KO cells retained a centrosomal pool of AZI1, CCDC14, KIAA0753 and PIBF1 (Extended Data Fig. 3b). As each KO cell line had similarly impaired aggresome formation to the PCM1 KO cell line, this suggests that the satellite, rather than centrosomal, pool of these proteins is involved in aggresome assembly. To test this, we depleted KIAA0753 and PIBF1 by siRNA in WT and PCM1 KO cells. Knockdown was confirmed by IF microscopy and immunoblotting (Extended Data Fig. 3h,i). In untreated cells, satellite distribution in control cells and in KIAA0753-depleted cells was comparable, but PIBF1 depletion reduced satellite abundance (Extended Data Fig. 3j,k). In WT cells, KIAA0753 and PIBF1 knockdown reduced aggresome size, with the greatest effect observed for PIBF1 depletion (Fig. 3f–h). The accumulation of Ub+ was also reduced (Fig. 3i). In PCM1 KO cells, KIAA0753 and PIBF1 knockdown further modestly reduced aggresome size, with PIBF1 again having the greatest effect (Fig. 3f,g); however, this was not significantly different to WT cells. This result suggests that the centrosomal pools of these proteins have a minor role in aggresome formation.
Centriolar satellites link aggresome assembly and autophagy
Given that they have different effects on satellites, it was surprising that AZI1, CCDC14, KIAA0753 and PIBF1 KO affected aggresome assembly to the same degree as PCM1 KO. To examine the consequence of impeded aggresome formation in these cell lines, we performed clonogenic survival assays following treatment with MG132. PCM1 KO cells displayed decreased fitness compared with the other KO cell lines (Extended Data Fig. 4a,b). However, there was no evidence of increased baseline proteotoxic stress in the PCM1 KO cells (Extended Data Fig. 4c).
The UPS and autophagy are interconnected, with disruption of one causing upregulation of the other55,56. Aggresome size is a function of the production, transport and clearance of protein aggregates, and satellites can regulate autophagy16; therefore we examined autophagy in PCM1 KO cells. Detection of LC3-II by immunoblotting showed that there was a stronger induction of autophagy in the PCM1 KO cells than in WT cells (Fig. 4a). Notably, activation of autophagy in WT cells reduced aggresome size to below levels observed in PCM1 KO cells treated with MG132 (Fig. 4a–c). It is probable that pre-activation of autophagy before the addition of MG132 accounts for this stronger effect. Inhibition of autophagy however, only moderately rescued aggresome size in the PCM1 KO cells, with aggresomes failing to reach the size seen in MG132-treated WT cells. The appearance of cytoplasmic pHSP27 aggregates in PCM1 KO cells suggested that aggregate production was not impeded (Fig. 4c), which was supported by the presence of HSP70, p62 and HSP27 in the insoluble fraction of PCM1 KO cells (Fig. 4d).

a, WT and ΔPCM1 cells treated and probed as indicated. KU, KU-0063794. b, pHSP27 area in WT and ΔPCM1 cells treated as indicated. n = 360 (WT-DMSO), 368 (WT-MG132), 323 (WT-MG132 + CQ), 386 (WT-MG132 + KU), 360 (ΔPCM1-DMSO), 375 (ΔPCM1-MG132), 366 (ΔPCM1-MG132 + CQ) and 418 (ΔPCM1-MG132 + KU) aggresomes examined over 2 independent experiments. ****P < 0.0001. c, Cells treated as in b were stained as indicated. d, Fractions from WT and ΔPCM1 cells treated and probed as indicated. e, pHSP27 area in siRNA-transfected WT, ΔKIAA0753 and ΔPIBF1 cells treated as indicated. Number of aggresomes examined over 2 experiments: WT: n = 522 (siGL2-DMSO), 525 (siGL2-MG132), 433 (siGL2-MG132 + CQ), 466 (siPCM1-DMSO), 422 (siPCM1-MG132) and 460 (siPCM1-MG132 + CQ); ΔKIAA0753: n = 509 (siGL2-DMSO), 563 (siGL2-MG132), 503 (siGL2-MG132 + CQ), 474 (siPCM1-DMSO), 442 (siPCM1-MG132) and 380 (siPCM1-MG132 + CQ); ΔPIBF1: n = 680 (siGL2-DMSO), 608 (siGL2-MG132), 478 (siGL2-MG132/CQ), 497 (siPCM1-DMSO), 531 (siPCM1-MG132) and 427 (siPCM1-MG132/CQ). ****P < 0.0001. f, siRNA-transfected WT cells and ΔPCM1, ΔKIAA0753 and ΔPIBF1 cells were treated and stained as indicated. g, WT and ΔPCM1 cells were treated and stained as indicated. h, pHSP27 area in WT and ΔPCMI cells treated as indicated. Number of aggresomes examined over 2 independent experiments: WT: n = 423 (0 h), 387 (1 h), 428 (2 h), 470 (3 h), 470 (4 h), 497 (5 h), 392 (8 h) and 287 (10 h); ΔPCM1: n = 524 (0 h), 581 (1 h), 580 (2 h), 598 (3 h), 640 (4 h), 635 (5 h), 593 (8 h) and 394 (10 h). ****P < 0.0001, **P < 0.01. i, siRNA-transfected A-375 cells were treated and stained as indicated. j, Quantitation of aggresome formation in A-375 cells transfected with control or PCM1 siRNAs, then treated with MG132 as indicated. Data displayed as the mean ± s.d., n = 3 independent experiments. ****P < 0.0001 by two-tailed unpaired Student’s t-test. For b, e and h, boxes represent the median, upper and lower quartiles, whiskers represent 1.5× the interquartile range, with individual values superimposed. Data compared using Kruskal–Wallis ANOVA test and post-hoc Dunn multiple comparison test. Scale bars, 10 μm (c,f,g,i) or 2 μm (insets of c,f,g,i). Unprocessed immunoblots, numerical data and P values are provided as source data.
The other KO cell lines also displayed stronger autophagy induction than WT cells (Extended Data Fig. 4d). However, inhibition of autophagy rescued aggresome size and led to the accumulation of CP110 and p62 at the centrosome in these cells (Extended Data Fig. 4e,f). Knockdown of PCM1 demonstrated that this rescue depended on satellites (Fig. 4e,f and Extended Data Fig. 4g).
To determine whether aggresomes can grow larger in PCM1 KO cells, we conducted a time course of MG132 treatment. In WT cells, pHSP27 began to accumulate around the centrosome at 2 h and continued to increase in size until 8–10 h (Fig. 4g,h). By contrast, in PCM1 KOs, pHSP27 reached its maximum size by 3–5 h. Depletion of PCM1 similarly affected aggresome formation in A-375 cells treated with MG132 overnight (Fig. 4i,j), with pHSP27 and p62 failing to assemble into a single inclusion despite aggregates accumulating in these cells. Together, these data support the requirement for centriolar satellites in aggresome formation and that protein aggregates are directed towards autophagy in their absence.
The CP110 module is required for aggresome formation
Despite localizing to the aggresome, CETN2 was not required for aggresome formation (Extended Data Fig. 3g). To further assess the contribution of centriolar proteins to aggresome formation, we analysed CP110, CEP97 and CEP290. CP110 is stabilized by USP33 and degraded by the UPS via cyclin F (CCNF), with proteasome inhibition stabilizing both CP110 (ref. 31) and CEP97 levels (Extended Data Fig. 5a). Knockdown of CP110, CEP97, CEP290 and USP33 reduced aggresome assembly (Fig. 5a,b and Extended Data Fig. 5b). Depletion of the CP110 interactor TALPID3 (ref. 57), which also localizes to the aggresome, similarly reduced aggresome size (Extended Data Fig. 5c–e). CP110 depletion had the greatest effect, causing mis-localization of pHSP27 to actin filaments (Extended Data Fig. 5f), and RNA interference (RNAi)-resistant CP110 rescued aggresome formation in CP110-depleted cells (Extended Data Fig. 5g–j). Conversely, MG132 treatment of CEP290-transfected cells resulted in multiple inclusions that contained pHSP27, HSP70, p62, Ub+ and PCM1 throughout the cytoplasm (Extended Data Fig. 5k,l). Although CP110 and CEP97 did not localize to these inclusions, their accumulation at the centrosome was blocked.

a, siRNA-transfected cells were treated and stained as indicated. b, Quantitation of pHSP27 area in cells treated as in a. n = DMSO: 705 (siGL2), 376 (siCP110), 372 (siCEP97), 691 (siCEP290), 579 (siCCNF), 472 (siUSP33); MG132: 517 (siGL2), 455 (siCP110), 396 (siCEP97), 449 (siCEP290), 475 (siCCNF) and 555 (siUSP33) aggresomes examined over 2 independent experiments. ****P < 0.0001. c, Cells treated as in a were stained as indicated. d, PCM1 distribution relative to the centrosome in cells treated as in a. The percentage of PCM1 in the inner region is indicated. e, siRNA-transfected WT and ΔPCM1 cells were treated and stained as indicated. f, pHSP27 area in cells treated as in e, with data from the same experiment displayed with different y axis ranges to ease comparison between knockdowns. Number of aggresomes examined over 2 independent experiments: WT: n = 408 (siGL2), 491 (siCP110), 518 (siCEP97) and 578 (siCEP290); ΔPCM1: n = 518 (siGL2), 510 (siCP110), 494 (siCEP97) and 699 (siCEP290). ****P < 0.0001. g, Cells treated as in e were stained as indicated. h, siRNA-transfected cells were treated and stained as indicated. i, pHSP27 area in siRNA-transfected WT cells treated with MG132. Number of aggresomes examined over 2 independent experiments: siGL2: n = 524 (0 h), 664 (3 h), 533 (5 h), 510 (8 h) and 519 (10 h); siCP110: n = 413 (0 h), 229 (3 h), 312 (5 h), 430 (8 h) and 550 (10 h). ****P < 0.0001, **P < 0.01, *P < 0.05. j, A-375 cells transfected with siGL2 or siCP110 were treated and stained as indicated. k, Aggresome formation in A-375 cells transfected and treated as indicated. Data displayed as the mean ± s.d., n = 3 independent experiments. ****P < 0.0001, **P < 0.01, *P < 0.05 by two-tailed unpaired Student’s t-test. For b, f and i, boxes represent the median, upper and lower quartiles, whiskers represent 1.5× the interquartile range, with individual values superimposed. Data were compared using Kruskal–Wallis ANOVA test and post-hoc Dunn multiple comparison test. Scale bars, 2 μm (a,c,e,g; insets of h,j) or 10 μm (h,j). Numerical data and P values are provided as source data. AU, arbitrary units.
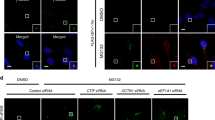
Satellite accumulation was reduced in cells depleted in CP110, CEP97 and CEP290 (Fig. 5c,d). In PCM1 KO cells, depletion of CP110, CEP97 or CEP290 had an additive effect on aggresome assembly (Fig. 5e,f), which suggests that satellites are required for a fraction of the recruitment of pHSP27 to the centrioles in the absence of CP110, CEP97 or CEP290. Moreover, CP110, CEP97 and CEP290 are responsible for the accumulation of a proportion of pHSP27 in the absence of satellites. The recruitment of Ub+ was restricted accordingly in these cells (Fig. 5g).
Fractionation of CP110-depleted cells revealed p62, HSP70 and pHSP27 in the insoluble fraction (Extended Data Fig. 5m), which demonstrates that protein aggregation occurs in these cells. However, autophagy in CP110-depleted cells was not more strongly induced (Extended Data Fig. 5n), and autophagy inhibition failed to rescue pHSP27 and p62 accumulation (Extended Data Fig. 5o,p). Time course analysis revealed that aggresomes do not grow in size beyond 5 h in CP110-depleted cells (Fig. 5h,i), and pHSP27 remained restricted to the centrioles even after overnight treatment in A-375 cells depleted of CP110, CEP97 or CEP290 (Fig. 5j,k). Together, these data assign a function to the CP110–CEP97–CEP290 module in aggresome formation, upstream of satellites.
CP110 levels limit aggresome formation in senescent cells
Protein aggregates accumulate in senescent cells due to decreased proteasomal activity37,39. We therefore wondered whether senescent cells utilize the aggresome pathway. Somatic cells in culture undergo replicative senescence after a finite number of divisions58. To obtain senescent cells, we grew low-passage primary HFF-1 cells for at least 140 days. Senescence-associated β-galactosidase activity59 and increased p53 and p21 levels60 confirmed the induction of senescencence (Extended Data Fig. 6a,b). Senescent cells were unable to form aggresomes, which correlated with cells staining negative for the proliferation marker Ki67 (Fig. 6a,b). Aggregates of Ub+ were observed throughout the cytoplasm (Extended Data Fig. 6c).

a, Cycling and senescent HFF-1 cells were treated with MG132 and stained as indicated. The number of passages (p) and total number of days in culture (d) are indicated. b, Quantitation of aggresome formation in Ki67 positive/negative cycling and senescent HFF-1 cells. c, Extracts from cycling and senescent HFF-1 cells treated with or without MG132 were probed as indicated. α-tubulin was used as the loading control. d, Cycling HFF-1 cells were treated with control (GL2), CP110 or CEP290 siRNAs, treated with MG132 and stained as indicated. e, Quantitation of aggresome formation in cycling HFF-1 cells treated as in d. f, Senescent HFF-1 cells were transfected with GFP–CP110 and treated with MG132. Cells were stained as indicated. g, Senescent HFF-1 cells were transfected with GFP–CP110, GFP–CP110-R586A,L588A or GFP–CP110-Δ67-82 and treated with MG132. Cells were stained for pHSP27. h, Quantification of senescent HFF-1 cells treated as in g that formed an aggresome. For b, e and h, data are displayed as the mean ± s.d., n = 3 independent experiments, and P values were calculated by two-tailed unpaired Student’s t-test; ****P 0.0001, ***P 0.001. Scale bars, 2 μm (insets of a,d,f,g) or 10 μm (a,d,f,g). Unprocessed immunoblots, numerical data and P values are provided as source data.
Senescent cells had decreased levels of many satellite proteins, although AZI1 and PCM1 levels were maintained (Fig. 6c), and disruption of PCM1 had no effect on the progression of IMR-90 cells towards senescence (Extended Data Fig. 6d–f). As CP110 levels were reduced, in line with a previous study61, we questioned whether this limited aggresome assembly in senescent cells. First, depeletion of CP110 and CEP290 in cycling HFF-1 cells recapitulated our earlier findings (Fig. 6d,e). Conversely, CP110 overexpression rescued aggresome assembly in senescent cells (Extended Data Fig. 6g,h). A possible explanation for this finding is that we observed the formation of green fluorescent protein (GFP)–CP110 aggregates that associated with pHSP27 rather than true aggresomes. However, pHSP27, p62, HSP70 and Ub+ (Fig. 6f), along with CEP97, CEP290 and PCM1 (Extended Data Fig. 6i), localization to these structures supported that they are aggresomes. Furthermore, a CP110 construct lacking the CEP290-binding domain (Δ67-82)32 failed to rescue aggresome formation, whereas mutation of the CCNF-binding domain (R586A,L588A)30 had little effect (Fig. 6g,h). This suggests that CP110 promotes aggresome formation through its interaction with CEP290. Together, these results demonstrate that senescent cells have a reduced capacity to form aggresomes due to reduced CP110 levels.
Satellites and the CP110 module drive HTT inclusion assembly
Enrichment of HTT into inclusions is a pathological feature of HD62. As HTT interacts with PCM1 through HAP1 (ref. 41), we asked whether satellites, or the CP110–CEP97–CEP290 module, are required for a HTT fusion protein containing 97 polyQ repeats (GFP–HTT97Q) to form inclusions. In a proportion of WT cells, GFP–HTT97Q formed a single large inclusion to which pHSP27 and Ub+ localized (Extended Data Fig. 7a,b). Measurement of inclusion size in PCM1 KO cells revealed that they were restricted to 4.35 ± 3.91 μm2 compared with 14.43 ± 3.93 μm2 in WT cells (Fig. 7a–c). However, polyQ-97 still accumulated in the insoluble fraction of PCM1 KO cells (Fig. 7d), which suggests that insoluble aggregates form in these cells. polyQ-25 was used to demonstrate the failure of a non-aggregating polyQ expansion to accumulate in the insoluble fraction (Extended Data Fig. 7b). IF microscopy using overexposed conditions revealed the presence of multiple small aggregates in PCM1 KO cells, thereby demonstrating aggregate formation in these cells, but that they failed to accumulate into a single inclusion. Similar results were obtained for cells depleted of CP110, CEP97 or CEP290 (Fig. 7f–j).

a, Images of WT and ΔPCM1 cells transfected with GFP–HTT97Q for 24 h. Mean aggregate size ± s.d. is indicated. b, Example GFP–HTT97Q aggregates in WT and ΔPCM1 cells. c, GFP–HTT97Q aggregate size in WT and ΔPCM1 cells. n = 75 cells for each condition over 2 independent experiments. ****P < 0.0001. d, Immunoblot of soluble (Sol.) and insoluble (Insol.) fractions from WT and ΔPCM1 cells transfected with GFP–HTT97Q. e, GFP–HTT97Q transfected WT and ΔPCM1 cells were imaged with overexposed conditions. f, Cells treated with siRNA then transfected with GFP–HTT97Q. Mean aggregate size ± s.d. is indicated. g, GFP–HTT97Q aggregates in cells treated with siGL2 or siCP110. h, GFP–HTT97Q aggregate size in WT cells treated with the indicated siRNAs. n = 57 (siGL2), 62 (siCP110), 68 (siCEP97) and 85 (siCEP290) over 2 independent experiments. ****P < 0.0001. i, Immunoblot of fractions from WT cells treated with siGL2 or siCP110, then transfected with GFP–HTT97Q. j, GFP–HTT97Q transfected siGL2 or siCP110 cells were imaged with overexposed conditions. k, Proteasome inhibition causes protein aggregates to accumulate in the cytoplasm. These aggregates are either directed towards autophagy or sequestered to the aggresome. Without satellites, autophagy is upregulated and becomes the predominant pathway. The earliest event in aggresome formation at the centrosome is the accumulation of pHSP27 on the centrioles and the recruitment of proteins to the aggresome. pHSP27 expansion requires CP110, CEP97 and CEP290, satellites and HDAC6. l, Senescent cells have a limited capacity to form aggresomes due to reduced levels of CP110. m, Assembly of GFP–HTT97Q aggregates into single inclusions requires the CP110 module, satellites, microtubules and a functional centrosome. For c and h, bars represent median and interquartile range, with individual values superimposed. P values were calculated by two-tailed unpaired Student’s t-test. Scale bars, 2 μm (insets of b,g) or 10 μm (a,e,f,j). Unprocessed immunoblots, numerical data and P values are provided as source data.
HTT can bind to microtubules and localize to the centrosome63, and microtubule depolymerization disrupts inclusion formation64,65. In line with this, we observed multiple small polyQ-97 aggregates in cells treated with nocodazole (Extended Data Fig. 7c,d). By contrast, in cells lacking functional centrosomes, multiple larger inclusions formed (Extended Data Fig. 7e,f). Taken together, these results suggest that satellites and the CP110 module participate in the concentration of polyQ-97 aggregates to the centrosome to facilitate the formation of a large single inclusion.
Finally, identification of CP110 proximity interactors during proteasome inhibition revealed an enrichment in pathways associated with protein aggregation diseases, including HD, and the proteasome (Extended Data Fig. 7g–k and Supplementary Table 2). This result therefore supports a role for CP110 in the regulation of proteostasis and aggregation in diseases.
Discussion
Here, we described the requirement for centriolar satellites and the CP110–CEP97–CEP290 module in aggresome formation (Fig. 7k). In the absence of protein translation, aggresomes were completely blocked. The initial recruitment of pHSP27 to the centrioles seeds aggresome assembly, which expands further depending on the CP110–CEP97–CEP290 module and satellites.
The formation of a ring of pHSP27 around the centrioles suggests this is an early event that lays the foundation for the assembly of aggresomal particles into a single structure (Fig. 7k). This implies that aggregate recruitment is regulated and that aggresome assembly is controlled to enable an ordered structure to form. Previous TEM studies revealed multiple particles loosely associating with each other within aggresomes2,7,66, and the images produced in this current study support the notion that aggresomes have higher-order structure. Aggregated proteins are sticky due to exposed hydrophobic residues. Order within the aggresome would bury these residues, which prevents further aggregation and interaction with other cellular components66,67.
The exact role that CP110 plays in aggresome assembly remains to be determined. One possibility is that during proteasomal inhibition, components of the CP110 module aggregate and their transport to the centrosome potentiates aggresome formation. Alternatively, as centriolar proteins, they could form a site on the centriole that facilitates aggresome seeding. Indeed, TALPID3 forms ring-like structures on the centriole57, and depletion of this CP110 interactor reduced aggresome formation. Notably, CEP120, which also interacts with TALPID3 (ref. 68), localized to two ring structures in the centre of the aggresome (Extended Data Fig. 1h), which provides support for the idea that a ring of centriolar proteins resides at the core of aggresome assembly. Finally, as CP110, CEP97 and CEP290 are reported in satellites33,34, they may act as adaptors between satellites and aggresomal cargo. Exactly how CP110 functions to target or anchor aggresomal particles to the centrosome will be an important focus of future studies.
Satellites regulate protein stability via the UPS and autophagy16,17, and these pathways regulate satellite composition and turnover17,69,70,71,72,73, which therefore places satellites at the intersection of these two pathways. The satellite protein KO lines had increased autophagy activity in response to proteasome inhibition, which accounted for the reduction in aggresome size in these cells (presumably because protein aggregates are directed towards autophagy rather than the aggresome). However, depletion of PCM1, which in turn reduces the level of satellites, prevented rescue of aggresome assembly in cells with inhibited autophagy, thereby providing strong evidence for the involvement of satellites in aggresome formation (Fig. 7k).
Sequestration of proteins at the MTOC allows proteins to be transported from throughout the cytoplasm to a central location. Both aggresome assembly and satellite movement depends on dynein-dependent transport along microtubules7,8,14,15, so it therefore fits that aggregates and satellites traffic to the aggresome together. Indeed, placement of satellites at the aggresome centre suggests that their disruption would affect aggresome formation by interfering with the homing of aggregate-loaded dynein motors. A key function of satellites is the recruitment of proteins to the centrosome via dynein–dynactin14, with several satellite proteins interacting directly with dynein–dynactin. Future studies will focus on identifying the interactions between protein aggregates, satellites and the aggresomal machinery during aggresome assembly.
The localization of satellites around the pHSP27 ring that assembles early during aggresome formation (Fig. 3d) supports their association with the aggresome as it grows (Fig. 7k). PCM1 KO cells assemble a pHSP27 ring, but its expansion is impeded, which suggests that satellites are required for aggresome growth. The degree of expansion that occurred in the absence of satellites depended on the status of autophagic activity. Although slightly more pHSP27 was recruited when autophagy was inhibited in PCM1 KO cells, it was not completely rescued, which suggests that some aggresome expansion can occur in the absence of satellites and autophagy. Conversely, autophagy activation before proteasomal inhibition caused less pHSP27 to assemble. This therefore places satellites at the nexus between centrosomes and proteostasis networks. Strikingly, satellite accumulation did not depend on interactions with aggresomal cargo, as they still accumulated in the pericentrosomal region when HDAC6 was inhibited or HSP27 was depleted.
Ageing is the biggest risk factor associated with neurodegenerative disorders74, and protein aggregation, due to decreased proteasomal activity75, is a common feature of aged cells76. Senescent cells were unable to form aggresomes due to reduced amounts of CP110 (Fig. 7l). CP110 lacking the CEP290 binding domain could not rescue aggresome formation; therefore, these proteins function together to promote aggresome assembly. Satellites and the CP110–CEP97–CEP290 module also facilitated HTT-polyQ inclusion formation (Fig. 7m). The role of proteinaceous inclusions to disease pathology remains unclear. Primary cilia are crucial to neuron function77, and cilium structure and function are altered in HD78. Expression of HTT-polyQ caused centrosomal accumulation of PCM1 and altered cilium structure41, which suggests a link between protein aggregation and cilium function in neurodegeneration. Given the emergent connection between satellites and aggregate processing, and the requirement for satellites in cilia formation and function17,20, future studies that elucidate the role of satellites in these processes will provide valuable insight into the pathophysiology of neurodegenerative diseases. Indeed, the identification of proteins associated with aggregation-prone diseases as CP110 interactors raises the tantalizing possibility that targeting CP110 might be a potential therapeutic avenue for such diseases.
Methods
Cell culture and drug treatments
All cell lines were cultured in a 5% CO2 humidified atmosphere at 37 °C. hTERT RPE-1 (female, human epithelial cells immortalized with hTERT; CRL-4000), A-375 (female, human malignant melanoma epithelial; CRL-1619), BJ-5ta (male, human fibroblasts immortalized with hTERT; CRL-4001), HFF-1 (male, human primary fibroblasts; SCRC-1041), HeLa (female, human adenocarcinoma epithelial; CRM-CCL-2) and U-2 OS (female, human osteosarcoma epithelial; HTB-96) cells from American Type Culture Collection (ATCC) were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% v/v fetal bovine serum (FBS). IMR-90 (female, human diploid fibroblasts; CCL-186) were cultured in Eagle’s minimum essential medium (EMEM, ATCC) supplemented with 10% v/v FBS. To inhibit the proteasome, cells were treated with 10 μM MG132 (Millipore-Sigma) or 1 μM bortezomib (Santa Cruz Biotechnology) for 5 h. To disrupt microtubules, cells were treated with nocodazole (Millipore-Sigma) at 10 μM or taxol (paclitaxel; Millipore-Sigma) at 5 μM for 2 h before the addition of MG132. Cycloheximide (Enzo Life Sciences) was used to inhibit protein translation at 5 μg ml–1 for 30 min before the addition of MG132. ACY-1215 and ACY-738 (Selleck Chemicals) at 50 μM each were used to inhibit HDAC6. To activate autophagy, cells were treated with the mTOR inhibitor KU-0063794 (Selleck Chemicals) at 10 μM for 2 h before the addition of MG132. To inhibit autophagy, cells were treated with 40 μM chloroquine (Millipore-Sigma) for 2 h before MG132 addition. As control, cells were treated with vehicle (dimethylsulfoxide (DMSO)) alone. To verify senescent populations, senescence associated β-galactosidase activity was assayed using a senescence detection kit (Abcam) according to the manufacturer’s instructions.
Lentiviral methods
To produce lentivirus, 4 × 106 HEK293T (female, human kidney; ATCC, ACS-4500) cells were seeded in a T-75 flask and transfected with the relevant lentiviral transfer vector, 3 μg psPAX2 and 2 μg pCMV-VSV-G using Lipofectamine 3000 (Invitrogen). After 24 h, the growth medium was replaced with fresh medium containing 30% FBS, and the virus supernatant collected after a further 48 h, centrifuged to remove cells and stored at −80 °C. To generate cell lines, 200,000 cells were seeded with varying amounts of virus supernatant and 4 μg ml–1 polybrene. Medium was changed after 24 h and cells selected with 3 μg ml–1 puromycin (IMR-90) or 600 μg ml–1 G418 (RPE-1) until uninfected control cells were dead. Samples with approximately 30% survival or less compared with unselected cells were used to enrich for cells with a single integration event.
Generation of KO cell lines
To generate hTERT RPE-1 AZI1, CCDC14, KIAA0753, PIBF1 and OFD1 KO cell lines, guide RNA targeting the respective gene (Supplementary Table 1) was selected and transcribed in vitro before being transfected into WT hTERT RPE-1 cells constitutively expressing Cas9 using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. After 5 days, the transfected cells were diluted to isolate clones. Gene disruption was confirmed by PCR amplification of genomic DNA with the primers detailed in Supplementary Table 1 and sequence analysis using inference of CRISPR edits (Synthego; Supplementary Table 1). Loss of signal of the respective protein was demonstrated by immunoblotting and IF microscopy. To disrupt PCM1 in IMR-90 cells, we used lentivirus-mediated CRISPR gene disruption. PCM1 guide RNA (Supplementary Table 1) was cloned into pLentiCRISPRv2 plasmid33,79, and KOs were generated using the lentiviral methods described above. FAM83G (guide sequence: agcccgggtcacgccgcggt; genomic forward primer: aacaataaagaggccctttgagacgg; genomic reverse primer: ctcatcaggtctttcccgcagatt) was targeted as a control80. The U-2 OS STIL KO81, RPE-1 CETN2 KO53 and RPE-1 FOP KO54 have previously been published.
Clonogenic survival assays
For RPE-1 cells, 250 cells per well were seeded in 6-well dishes. The next day, cells were treated with MG132 for 5 h before drug washout with three changes of medium. After 12 days, plates were rinsed once with PBS, then fixed and stained with 0.5% w/v crystal violet in 20% v/v methanol for 20 min. Plates were washed extensively with water, dried and scanned. Images were segmented using the Trainable Weka Segmentation tool in ImageJ. A new model was built for each replicate. The resulting segmentation image was thresholded and used as a mask to overlay the original image that was inverted and background subtracted using a 50-pixel rolling circle. The colony intensity per well was then measured within the masked region. Data in Extended Data Fig. 4b are presented as the mean and standard deviation of four replicates from two independent experiments.
RNA-mediated interference
All siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Details of the siRNA oligonucleotides utilized in this study are provided in Supplementary Table 3. hTERT RPE-1 cells were transfected with 20 nM (final concentration) of the respective siRNA for 48 or 72 h, as indicated. Effective knockdown was confirmed by immunoblotting and/or IF microscopy. The GFP–CP110 expression construct has been previously published61, and the HSP27 entry clone was a gift from B. Piette and M. Taipale82. RNAi-resistant mutants of CP110 and HSP27, and the CP110-Δ67-82 and CP110-R586A,L588A mutants, were generated using site-directed mutagenesis (primers detailed in Supplementary Table 3). To rescue the CP110 RNAi phenotype, cells were transfected with 1 μg of DNA using Lipofectamine 3000 (Invitrogen) 24 h after siRNA transfection. After a further 24 h, cells were treated with MG132 for 5 h. To rescue the HSP27 RNAi phenotype, the siRNA-resistant HSP27 complementary DNA was cloned into a pInducer20, a tetracycline-inducible lentiviral vector83, using Gateway technology (Invitrogen). siRNA infection was performed as detailed above for 24 h, before expression of siRNA-resistant HSP27 was induced by the addition of 0.5 μg ml–1 tetracycline to the culture medium. Cells were treated with MG132 24 h after induction and collected for analysis.
Transient transfections
Transfections were performed using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. In brief, 1 μg plasmid DNA was complexed with Lipofectamine 3000 in serum-free Opti-MEM and added to cells at 70–80% confluency. After 24 h of transfection, cells were treated with MG132 for 5 h before analysis by IF microscopy. The HTT-polyQ constructs (25Q and 97Q) were a gift from G. Hesketh, who modified them from ref. 84 to use in human cells, and the FLAG–CEP290 construct has been previously published33.
IF microscopy
Details of the primary and secondary antibodies used for IF microscopy are provided in Supplementary Table 3. For IF microscopy, cells were fixed with ice-cold methanol for 10 min at −20 °C, with the exception of the cells presented in Fig. 6a and Extended Data Fig. 5f, which were fixed with 4% paraformaldehyde for 5 min at room temperature before permeabilization with methanol for 2 min. All cells were blocked with 2% BSA in PBS for 10 min, then incubated with primary antibody in blocking solution for 50 min at room temperature. After washing with PBS, cells were incubated with fluorophore-conjugated secondary antibodies (Supplementary Table 3) and 4,6-diamidino-2-phenylindole (0.1 μg ml–1) in blocking solution for 50 min at room temperature. After washing with PBS, coverslips were mounted onto glass slides using ProLong Gold antifade (Molecular Probes). Cells were imaged using a DeltaVision Elite high-resolution imaging system equipped with a sCMOS 2,048 × 2,048 pixel camera (GE Healthcare). Z-stacks (0.2-μm step) were collected using a ×60, 1.42 NA plan apochromat oil-immersion objective (Olympus) and deconvolved using softWoRx (v.6.0, GE Healthcare). Images are shown as maximum intensity projections. The images presented in Figs. 1g and 3d and Extended Data Fig. 1h were captured using a Nikon CSU-W1 SoRa spinning disk super-resolution system equipped with a LUN-F XL laser unit and a Hamamatsu FusionBT sCMOS camera. Z-stacks (0.125-μm step) were collected using a ×60, 1.4 NA plan apochromat λ oil-immersion objective (Nikon) and deconvolved in NIS-Elements (Nikon). Images are presented as maximum intensity projections. All images were processed for publication using Adobe Photoshop 2020 and figures prepared using Adobe Illustrator 2020.
Colocalization analysis from super-resolution images
Super-resolution SoRa images were used for colocalization analysis. Each image was cropped using a 7.5 × 7.5 μm area centred around the aggresome. The Pearson’s correlation co-efficient for each image pair was determined using the JACoP plug-in for ImageJ85. At least ten images were used for each pair of antibody stainings.
Automated quantitative image analysis
Images were analysed using CellProfiler86 (v.3.0; https://cellprofiler.org) and R (https://www.r-project.org). We chose pHSP27 staining as the marker for measurement of aggresome size due to its low background in control-treated cells and distinct signal around the centrosome in cells treated with MG132. Knockdown of HSP27 demonstrated the specificity of this signal. For pHSP27 analysis, centrosomes were identified using the CEP135 signal. Foci closer than 8 pixels were merged, and all centrosomes identified were shrunk to a single pixel. The aggresome area was found using the centrosome coordinates as seed points and expanding outwards on the pHSP27 channel using the Watershed algorithm. The lower quartile of each pHSP27 image was used as a background subtraction value before the total pHSP27 intensity in the aggresome area was determined. The results from two independent experiments, each using at least 291 cells, were pooled and plotted (source data are provided). Box-and-whisker plots show the median, upper and lower quartiles (boxes), and 1.5× the interquartile range (whiskers), with individual data points plotted. Data distribution was assumed to be normal, but this was not formally tested. The data were analysed using a Kruskal–Wallis analysis of variance (ANOVA) to determine whether at least one distribution was statistically different from the others. A post-hoc Dunn test was then performed to assess pairwise differences and to calculate P values. To measure centriolar satellites, we used PCM1 staining, as per convention of the field. For PCM1 analysis, the same images were analysed and centrosomes were identified as above using the CEP135 signal (or CETN2 in the case of Extended Data Fig. 3j). The centrosome points were expanded 13 pixels to define the ‘inner PCM’ area that approximates the average size of an aggresome. The inner region was further expanded 100 pixels to create an annulus defining the ‘outer PCM’ area that generally encompassed the entire cell and were non-overlapping between cells. The inner satellites were segmented using an adaptive Otsu method and the outer satellites segmented with a global Otsu algorithm. The (x,y) position for every satellite was calculated relative to the cell centrosome position. We assumed that the cells were rotationally similar and overlaid all cells in each condition centred on the centrosome. The PCM1 signal was background subtracted using the lower quartile of each image and further subtracted based on PCM1 KO images to correct for spurious objects detected in the absence of PCM1. The centroid of each satellite was weighted by the total corrected PCM1 intensity of the object and visualized using a hexbin plot normalized to the total number of cells and replicates. On the inner satellite plots, the percentage of total PCM1 intensity within this pericentrosomal region is displayed.
Soluble/insoluble fractionation
The following protocol was adapted from that used by Runwal et al.87. Cells grown in 60-mm dishes and treated as indicated were washed twice with PBS then scraped into 360 μl RIPA (50 mM Tris-HCl pH 8, 1 mM EDTA, 150 mM NaCl, 0.25% sodium-deoxycholate, 0.1% SDS and 1% NP40) supplemented with complete mini protease inhibitor cocktail (Sigma) and 1:100 phosphatase inhibitor cocktail 3 (Sigma). The lysates were then passed through 20 gauge needles 8 times, incubated on ice for 15 min and then centrifuged at 16,000g for 15 min at 4 °C. The supernatant was collected as the soluble fraction, to which 200 μl of 2× SDS–PAGE buffer was added before boiling at 95 °C for 5 min. The pellet was washed once with 250 μl RIPA and centrifuged again for 5 min at 4 °C. The supernatant was discarded and the pellet resuspended in 72 μl RIPA containing 2 M urea (Sigma). An equal volume of 2× SDS–PAGE buffer was added, and the insoluble fractions boiled at 95 °C for 5 min. All samples were stored at −80 °C before being re-boiled for 5 min and resolved by SDS–PAGE and subjected to immunoblotting as detailed below.
Immunoblotting
Details of the primary and secondary antibodies used for immunoblotting are provided in Supplementary Table 3. For immunoblotting, total cell lysates were collected in 2× SDS–PAGE buffer and treated with benzonase nuclease (Millipore-Sigma). Proteins were separated on SDS–PAGE gels and transferred to polyvinylidenedifluoride membrane (Amersham Hybond P, Cytiva). Membranes were blocked for 30 min with 5% w/v milk powder (BioShop) in TBS, then incubated with primary antibodies in 5% w/v milk powder-TBST (TBS, 0.1% Tween-20) at 4 °C overnight. Following washing in TBST, blots were incubated with IRDye-conjugated secondary antibodies (LI-COR) in the dark for 1 h at room temperature. Blots were then washed three times in TBST and once in TBS, before imaging on a LI-COR Odyssey CLx Infrared Imager. Unprocessed blots are presented as source data.
TEM
For thin-section TEM, hTERT RPE-1 cells were grown in 10-cm dishes and treated with DMSO or MG132 for 5 h. Cells were then pelleted and washed twice with PBS, before primary fixation in 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer overnight at 4 °C, and secondary fixation in 2% osmium tetroxide. Samples were dehydrated through an ethanol gradient, followed by propylene oxide and embedded in EMbed 812 resin (Electron Microscopy Sciences). Ultra-thin sections were cut on a RMC MT6000 ultramicrotome and stained with 2% uranyl acetate in 70% methanol and aqueous lead citrate. Sections were viewed on a FEI Tecnai 20 transmission electron microscope.
Generation of Flp-In T-REx 293 cells for miniTurbo proximity labelling
Proximity labelling was carried out utilizing miniTurbo. Full-length CP110 cDNA was cloned into a pcDNA5-FRT/TO-miniTurbo-FLAG destination vector via the Gateway cloning system. Using the Flp-In system, HEK293 cells stably expressing miniTurbo-FLAG (control) and miniTurbo-FLAG–CP110 were generated. After selection with 200 μg ml–1 hygromycin B, 5 × 150-cm2 plates of subconfluent (~70%) cells per biological replicate were incubated in complete medium supplemented with 1 μg ml–1 tetracycline (BioShop) to induce expression of the miniTurbo construct. The next day, cells were treated with DMSO or MG132 as indicated for 5 h, with 50 μM biotin (BioShop) added for the final hour. Cells were scraped into PBS and pelleted at 500g for 5 min at 4 °C. Cell pellets were washed twice with PBS and then snap-frozen.
Sample preparation for mass spectrometry
Each cell pellet was resuspended in 10 ml of lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% SDS, 1:500 protease inhibitor cocktail (Sigma-Aldrich) and 1:1,000 benzonase nuclease (Novagen)), incubated on an end-over-end rotator at 4 °C for 1 h, briefly sonicated to disrupt any visible aggregates and then centrifuged at 45,000g for 30 min at 4 °C. The supernatant was transferred to a fresh 15-ml conical tube. A total of 30 μl of packed, pre-equilibrated streptavidin sepharose beads (GE Healthcare) was added and the mixture was incubated for 3 h at 4 °C with end-over-end rotation. Beads were pelleted by centrifugation at 820g for 2 min and transferred with 1 ml of lysis buffer to a fresh Eppendorf tube. Beads were washed once with 1 ml lysis buffer and twice with 1 ml 50 mM ammonium bicarbonate (pH 8.3). Beads were transferred in ammonium bicarbonate to a fresh centrifuge tube and washed twice more with 1 ml ammonium bicarbonate buffer. Tryptic digestion was performed by incubating the beads with 1 μg MS-grade TPCK trypsin (Promega) dissolved in 200 μl 50 mM ammonium bicarbonate (pH 8.3) overnight at 37 °C. The following morning, 0.5 μg MS-grade TPCK trypsin was added, and beads were incubated for an additional 2 h at 37 °C. Beads were pelleted by centrifugation at 2,000g for 2 min, and the supernatant transferred to a fresh Eppendorf tube. Beads were washed twice with 150 μl of 50 mM ammonium bicarbonate, and these washes were pooled with the first eluate. The sample was lyophilized and resuspended in buffer A (0.1% formic acid). One-fifth of the sample was analysed per mass spectometry (MS) run.
MS analysis
MS/MS was done as previously described88. In brief, high-performance liquid chromatography was conducted using a 2 cm pre-column (Acclaim PepMap, 50 mm × 100 μm inner diameter) and 50 cm analytical column (Acclaim PepMap, 500 mm × 75 μm diameter, C18, 2 μm, 100 Å; Thermo Fisher Scientific), running a 120-min reversed-phase buffer gradient at 225 nl min–1 on a Proxeon EASY-nLC 1000 pump in-line with a Thermo Q Exactive HF Quadrupole-Orbitrap mass spectrometer. A parent ion scan was performed using a resolving power of 60,000, then up to the 20 most intense peaks were selected for MS/MS (minimum ion count of 1,000 for activation), using higher energy collision-induced dissociation (HCD) fragmentation. Dynamic exclusion was activated such that MS/MS of the same m/z (within a range of 10 ppm; exclusion list size of 500) detected twice within 5 s was excluded from analysis for 15 s. For protein identification, Thermo .RAW files were converted to the .mzXML format using ProteoWizard89, then searched using X!Tandem90 and Comet91 against the human Human RefSeq Version 45 database (containing 36,113 entries). Search parameters specified a parent ion mass tolerance of 10 ppm and a MS/MS fragment ion tolerance of 0.4 Da, with up to 2 missed cleavages allowed for trypsin. Variable modifications of +16 at methionine and tryptophan residues, +32 at methionine and tryptophan residues, +42 at the amino terminus and +1 at asparagine and glutamine residues were allowed.
Protein identification and functional enrichment analysis
Both SAINT express (v.3.6.1)92 and MiST93 analyses were performed with proteins detected with an iProphet Probability of ≥0.9 and number of unique peptides ≥2. High-confidence interactors were defined when SAINT’s BFDR value was <0.01 and MiST score was >0.75. Functional enrichment analysis was performed with the high-confidence interactors using g:Profiler with the KEGG Pathway database94,95. Terms with adjusted enrichment P < 0.05 are presented.
Statistics and reproducibility
All experiments were independently repeated at least twice, with similar results obtained. No statistical method was used to predetermine the sample size. No data were excluded from the analyses. For statistical analysis, data distribution was assumed to be normal, but this was not formally tested; individual data points are presented on all plots to show data distribution. Samples were not randomized to specific treatment groups because samples depended on specific genetic cell lines or knockdown conditions; therefore, randomization was not appropriate. Data collection and analysis were not performed blind to the conditions of the experiments. Source data and exact sample sizes are provided as source data files for each figure. Statistical analyses were performed using two-tailed unpaired Student’s t-tests and significance was assumed by P < 0.05. P values are defined as follows: ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, NS, not significant. Statistical analysis of aggresome size was performed using Kruskal–Wallis ANOVA with post-hoc Dunn test as detailed in the ‘Automated quantitative image analysis’ section.
Data presented in Figs. 1c, 4j, 5k and 6b,e,h and Extended Data Figs. 1c,e, 4b and 5j were collected over three independent experiments. Data presented in Figs. 1h, 2b,d,f,h,j,k, 3b,c,g,h, 4b,d,h, 5b,d,f,i and 7c,h and Extended Data Figs. 1i, 2d,f, 3c,g,k, 4e, 5e,o and 6f,h were collected over two independent experiments. Immunoblots presented in Figs. 4a,d, 6c and 7d,i and Extended Data Figs. 2c,g, 3a,f,i, 4c,d,g, 5a–c,h,m,n, 6b and 7,b,d,f,h are representative of at least two independent experiments. Micrographs presented in Figs. 1a,b,d–f, 2a,c,e,g,i,l, 3a,d–f,i, 4c,f,g,i, 5a,c,e,g,h,j, 6a,d,f,g and 7e,j and Extended Data Figs. 1a,b,d,g, 2b,e,h–k, 3b,d,e,h,k, 4f, 5d,f,g,i,k,l,p, 6c,d,i and 7a,c,e,g are representative of at least three independent experiments. Micrographs presented in Figs. 1g and 7a,b,f,g and Extended Data Figs. 1f,g and, 6g are representative of two independent experiments.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All mass spectrometry data regarding CP110 proximity interactors have been deposited in the MassIVE (https://massive.ucsd.edu) repository under accession MSV000088076. The previously published KEGG Pathway database95 (https://www.genome.jp/kegg/pathway.html) that was re-analysed here was accessed via the web-based toolset gProfiler94 (https://biit.cs.ut.ee/gprofiler/gost, version e104_eg51_p15_3922dba, updated on 2021-05-07). All other data supporting the findings of this study are available from the corresponding authors on reasonable request. Source data are provided with this paper.
Code availability
The analysis pipelines for measurement of aggresomes and centriolar satellites were generated using CellProfiler86 (https://cellprofiler.org). The pipelines are available on Zenodo (https://www.zenodo.org/record/5834143; https://doi.org/10.5281/zenodo.5834143) or from the corresponding authors on reasonable request.
References
Joukov, V. & De Nicolo, A. The centrosome and the primary cilium: the yin and yang of a hybrid organelle. Cells 8, 701 (2019).
Johnston, J. A., Ward, C. L. & Kopito, R. R. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898 (1998).
Johnston, H. E. & Samant, R. S. Alternative systems for misfolded protein clearance: life beyond the proteasome. FEBS J. 288, 4464–4487 (2021).
Fortun, J., Dunn, W. A., Joy, S., Li, J. & Notterpek, L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 23, 10672–10680 (2003).
Hao, R. et al. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 51, 819–828 (2013).
Wong, E. S. P. et al. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum. Mol. Genet. 17, 2570–2582 (2008).
García-Mata, R., Bebök, Z., Sorscher, E. J. & Sztul, E. S. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 146, 1239–1254 (1999).
Johnston, J. A., Illing, M. E. & Kopito, R. R. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil. Cytoskeleton 53, 26–38 (2002).
Kawaguchi, Y. et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 (2003).
Chiba, Y. et al. Immunohistochemical localization of aggresomal proteins in glial cytoplasmic inclusions in multiple system atrophy. Neuropathol. Appl. Neurobiol. 38, 559–571 (2012).
McNaught, K. S. P., Shashidharan, P., Perl, D. P., Jenner, P. & Olanow, C. W. Aggresome-related biogenesis of Lewy bodies. Eur. J. Neurosci. 16, 2136–2148 (2002).
Szebenyi, G. et al. Hook2 contributes to aggresome formation. BMC Cell Biol. 8, 19–11 (2007).
Didier, C., Merdes, A., Gairin, J.-E. & Jabrane-Ferrat, N. Inhibition of proteasome activity impairs centrosome-dependent microtubule nucleation and organization. Mol. Biol. Cell 19, 1220–1229 (2008).
Dammermann, A. & Merdes, A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 159, 255–266 (2002).
Kubo, A., Sasaki, H., Yuba-Kubo, A., Tsukita, S. & Shiina, N. Centriolar satellites: molecular characterization, ATP-dependent movement toward centrioles and possible involvement in ciliogenesis. J. Cell Biol. 147, 969–980 (1999).
Joachim, J. et al. Centriolar satellites control GABARAP ubiquitination and GABARAP-mediated autophagy. Curr. Biol. 27, 2123–2136.e7 (2017).
Wang, L., Lee, K., Malonis, R., Sanchez, I. & Dynlacht, B. D. Tethering of an E3 ligase by PCM1 regulates the abundance of centrosomal KIAA0586/Talpid3 and promotes ciliogenesis. eLife 5, e12950 (2016).
Gerdes, J. M. et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 39, 1350–1360 (2007).
Liu, Y. P. et al. Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators. J. Clin. Invest. 124, 2059–2070 (2014).
Odabasi, E., Gul, S., Kavakli, I. H. & Firat-Karalar, E. N. Centriolar satellites are required for efficient ciliogenesis and ciliary content regulation. EMBO Rep. 20, e47723 (2019).
Arquint, C., Sonnen, K. F., Stierhof, Y. D. & Nigg, E. A. Cell-cycle-regulated expression of STIL controls centriole number in human cells. J. Cell Sci. 125, 1342–1352 (2012).
Korzeniewski, N. et al. Cullin 1 functions as a centrosomal suppressor of centriole multiplication by regulating polo-like kinase 4 protein levels. Cancer Res. 69, 6668–6675 (2009).
Rogers, G. C., Rusan, N. M., Roberts, D. M., Peifer, M. & Rogers, S. L. The SCFSlimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J. Cell Biol. 184, 225–239 (2009).
Strnad, P. et al. Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev. Cell 13, 203–213 (2007).
Duensing, A. et al. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene 26, 6280–6288 (2007).
Korzeniewski, N., Cuevas, R., Duensing, A. & Duensing, S. Daughter centriole elongation is controlled by proteolysis. Mol. Biol. Cell 21, 3942–3951 (2010).
Kohlmaier, G. et al. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr. Biol. 19, 1012–1018 (2009).
Schmidt, T. I. et al. Control of centriole length by CPAP and CP110. Curr. Biol. 19, 1005–1011 (2009).
Spektor, A., Tsang, W. Y., Khoo, D. & Dynlacht, B. D. Cep97 and CP110 suppress a cilia assembly program. Cell 130, 678–690 (2007).
D’Angiolella, V. et al. SCFCyclin F controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 466, 138–142 (2010).
Li, J. et al. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature 495, 255–259 (2013).
Tsang, W. Y. et al. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev. Cell 15, 187–197 (2008).
Gheiratmand, L. et al. Spatial and proteomic profiling reveals centrosome-independent features of centriolar satellites. EMBO J. 38, e101109 (2019).
Quarantotti, V. et al. Centriolar satellites are acentriolar assemblies of centrosomal proteins. EMBO J. 38, e101082 (2019).
van Deursen, J. M. The role of senescent cells in ageing. Nature 509, 439–446 (2014).
Cuanalo-Contreras, K., Mukherjee, A. & Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 1638083 (2013).
Fernández-Cruz, I. & Reynaud, E. Proteasome subunits involved in neurodegenerative diseases. Arch. Med. Res 52, 1–14 (2020).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Saez, I. & Vilchez, D. The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genomics 15, 38–51 (2014).
Adegbuyiro, A., Sedighi, F., Pilkington, A. W. IV, Groover, S. & Legleiter, J. Proteins containing expanded polyglutamine tracts and neurodegenerative disease. Biochemistry 56, 1199–1217 (2017).
Keryer, G. et al. Ciliogenesis is regulated by a huntingtin–HAP1–PCM1 pathway and is altered in Huntington disease. J. Clin. Invest. 121, 4372–4382 (2011).
Ito, H. et al. Inhibition of proteasomes induces accumulation, phosphorylation, and recruitment of HSP27 and αβ-crystallin to aggresomes. J. Biochem. 131, 593–603 (2002).
Zhou, L. et al. p62/Sequestosome 1 regulates aggresome formation of pathogenic Ataxin-3 with expanded polyglutamine. Int. J. Mol. Sci. 15, 14997–15010 (2014).
Wigley, W. C. et al. Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol. 145, 481–490 (1999).
Fujinaga, R. et al. Microtubule-dependent formation of the stigmoid body as a cytoplasmic inclusion distinct from pathological aggresomes. Histochem. Cell Biol. 132, 305–318 (2009).
Nigg, E. A. & Holland, A. J. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat. Rev. Mol. Cell Biol. 19, 297–312 (2018).
Meriin, A. B., Zaarur, N. & Sherman, M. Y. Association of translation factor eEF1A with defective ribosomal products generates a signal for aggresome formation. J. Cell Sci. 125, 2665–2674 (2012).
Qin, S., Jiang, C. & Gao, J. Transcriptional factor Nrf2 is essential for aggresome formation during proteasome inhibition. Biomed. Rep. 11, 241–252 (2019).
Wang, X.-X., Wan, R.-Z. & Liu, Z.-P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 143, 1406–1418 (2018).
Bauer, N. G. & Richter-Landsberg, C. The dynamic instability of microtubules is required for aggresome formation in oligodendroglial cells after proteolytic stress. J. Mol. Neurosci. 29, 153–168 (2006).
Staples, C. J. et al. The centriolar satellite protein Cep131 is important for genome stability. J. Cell Sci. 125, 4770–4779 (2012).
Hoang-Minh, L. B. et al. PCM1 depletion inhibits glioblastoma cell ciliogenesis and increases cell death and sensitivity to temozolomide. Transl. Oncol. 9, 392–402 (2016).
Prosser, S. L. & Morrison, C. G. Centrin2 regulates CP110 removal in primary cilium formation. J. Cell Biol. 208, 693–701 (2015).
Mojarad, B. A. et al. CEP19 cooperates with FOP and CEP350 to drive early steps in the ciliogenesis programme. Open Biol. 7, 170114 (2017).
Kocaturk, N. M. & Gozuacik, D. Crosstalk between mammalian autophagy and the ubiquitin–proteasome system. Front Cell Dev. Biol. 6, 712–727 (2018).
Wang, X. J. et al. A novel crosstalk between two major protein degradation systems. Autophagy 9, 1500–1508 (2014).
Kobayashi, T., Kim, S., Lin, Y.-C., Inoue, T. & Dynlacht, B. D. The CP110-interacting proteins Talpid3 and Cep290 play overlapping and distinct roles in cilia assembly. J. Cell Biol. 204, 215–229 (2014).
Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell. Res. 37, 614–636 (1965).
Dimri, G. P. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA 92, 9363–9367 (1995).
Rufini, A., Tucci, P., Celardo, I. & Melino, G. Senescence and aging: the critical roles of p53. Oncogene 32, 5129–5143 (2019).
Breslin, L., Prosser, S. L., Cuffe, S. & Morrison, C. G. Ciliary abnormalities in senescent human fibroblasts impair proliferative capacity. Cell Cycle 13, 2773–2779 (2014).
Olzmann, J. A., Li, L. & Chin, L. S. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr. Med. Chem. 15, 47–60 (2008).
Hoffner, G., Kahlem, P. & Djian, P. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with β-tubulin: relevance to Huntington’s disease. J. Cell Sci. 115, 941–948 (2002).
Muchowski, P. J., Ning, K., D’Souza-Schorey, C. & Fields, S. Requirement of an intact microtubule cytoskeleton for aggregation and inclusion body formation by a mutant huntingtin fragment. Proc. Natl Acad. Sci. USA 99, 727–732 (2002).
Kaminosono, S. et al. Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability. J. Neurosci. 28, 8747–8755 (2008).
Garcia-Mata, R., Gao, Y.-S. & Sztul, E. Hassles with taking out the garbage: aggravating aggresomes. Traffic 3, 388–396 (2002).
Drummond, D. A. How infidelity creates a sticky situation. Mol. Cell 48, 663–664 (2012).
Tsai, J.-J., Hsu, W.-B., Liu, J.-H., Chang, C.-W. & Tang, T. K. CEP120 interacts with C2CD3 and Talpid3 and is required for centriole appendage assembly and ciliogenesis. Sci. Rep. 9, 6037 (2019).
Douanne, T. et al. CYLD regulates centriolar satellites proteostasis by counteracting the E3 ligase MIB1. Cell Rep. 27, 1657–1665.e4 (2019).
Han, K.-J. et al. Deubiquitylase USP9X maintains centriolar satellite integrity by stabilizing pericentriolar material 1 protein. J. Cell Sci. 132, jcs221663 (2019).
Li, X. et al. USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat. Commun. 8, 14866 (2017).
Wang, Q. et al. The X-linked deubiquitinase USP9X is an integral component of centrosome. J. Biol. Chem. 292, 12874–12884 (2017).
Holdgaard, S. G. et al. Selective autophagy maintains centrosome integrity and accurate mitosis by turnover of centriolar satellites. Nat. Commun. 10, 4176 (2019).
Hou, Y. et al. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581 (2019).
Keller, J. N., Hanni, K. B. & Markesbery, W. R. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech. Ageing Dev. 113, 61–70 (2000).
Santra, M., Dill, K. A. & de Graff, A. M. R. Proteostasis collapse is a driver of cell aging and death. Proc. Natl Acad. Sci. USA 116, 22173–22178 (2019).
Lee, J. H. & Gleeson, J. G. The role of primary cilia in neuronal function. Neurobiol. Dis. 38, 167–172 (2010).
Kaliszewski, M., Knott, A. B. & Bossy-Wetzel, E. Primary cilia and autophagic dysfunction in Huntington’s disease. Cell Death Differ. 22, 1413–1424 (2015).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
Steinhart, Z. et al. Genome-wide CRISPR screens reveal a Wnt–FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 23, 60–68 (2017).
Liu, Y. et al. Direct binding of CEP85 to STIL ensures robust PLK4 activation and efficient centriole assembly. Nat. Commun. 9, 1731–15 (2018).
Taipale, M. et al. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 158, 434–448 (2014).
Meerbrey, K. L. et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl Acad. Sci. USA 108, 3665–3670 (2011).
Krobitsch, S. & Lindquist, S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl Acad. Sci. USA 97, 1589–1594 (2000).
Bolte, S. & Cordelières, F. P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 (2006).
Carpenter, AnneE. et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 (2006).
Runwal, G. et al. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 9, 10147–14 (2019).
Hua, R. et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 216, 367–377 (2017).
Kessner, D., Chambers, M., Burke, R., Agus, D. & Mallick, P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 (2008).
Craig, R. & Beavis, R. C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 (2004).
Eng, J. K., Jahan, T. A. & Hoopmann, M. R. Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 (2013).
Teo, G. et al. SAINTexpress: improvements and additional features in significance analysis of INTeractome software. J. Proteom. 100, 37–43 (2014).
Jäger, S. et al. Global landscape of HIV–human protein complexes. Nature 481, 365–370 (2011).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists. Nucleic Acids Res. 47, W191–W198 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Acknowledgements
We thank members of the Pelletier Lab for their scientific feedback during the project. We are grateful to G. Hesketh for sharing the GFP–HTT-polyQ constructs and B. Piette and M. Taipale for the HSP27 entry clone; T. (C.) Li and J. Brumell, and G. (J.) Mastromarco and H. (K.) Lee for sharing reagents and providing expert guidance and technical advice; R. Buijs for help generating U-2 OS STIL KO cells; and S. Cheung and K. Lane for proofreading the manuscript. S.L.P. was funded by a European Union Horizon 2020 Marie Skłodowska-Curie Global Fellowship (number 702601). This work was funded by CIHR Foundation (FDN number 167279) and Krembil Foundation grants to L.P. L.P is a Tier 1 Canada Research Chair in Centrosome Biogenesis and Function. The Network Biology Collaborative Centre at the LTRI is supported by the Canada Foundation for Innovation, the Ontario Government, and Genome Canada and Ontario Genomics (OGI-139).
Author information
Authors and Affiliations
Contributions
Conceptualization: S.L.P., C.G.M. and L.P. Methodology: S.L.P., J.T. and B.R. Formal analysis: S.L.P., J.T. and J.K. Investigation: S.L.P. Resources: J.T. and L.G. Writing (original draft): S.L.P. and L.P. Writing (review and editing): S.L.P, J.T., C.G.M. and L.P. Visualization: S.L.P., J.T. and J.K. Supervision: C.G.M. and L.P. Funding acquisition: S.L.P. and L.P.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks Michael Sherman, Tso-Pang Yao, and the other, anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Centrosomal proteins localize to the aggresome upon proteasome inhibition.
(a) RPE-1 cells treated with DMSO or MG132 were stained for CP110 and the indicated protein. Scale bar 2 μm. (b) The indicated cell lines were treated with MG132 and stained for pHSP27, CEP135 and PCM1. DNA was stained with DAPI. Scale bar 10 μm, inset 2 μm. (c) Quantification of the percentage cells forming an aggresome upon DMSO, MG132 and BZ treatment, as indicated. Data displayed as mean ± s.d., n = 3 independent experiments. ****p < 0.0001; ***p < 0.001. (d) RPE-1 cells treated with MG132 were stained for PCM1 and the indicated protein. Scale bar 2 μm. (e) Histogram showing the percentage of cells with greater than 4 foci of CEP135, SAS6, GT335, CETN2, CP110 and CEP97 following MG132 treatment. Data displayed as mean ± s.d., n = 3 independent experiments. ****p < 0.0001; ***p < 0.001; ns, not significant. p values: 0.218071 (CEP135), 0.058423 (SAS6), 0.160943 (GT335), 0.000219 (CETN2), 0.000055 (CP110), and 0.000150 (CEP97). (f) TEM images of centrioles from RPE-1 cells treated with DMSO or MG132. Scale bar 500 nm. (g) RPE-1 cells treated with DMSO or MG132 for 5 hours were stained for PCM1, pHSP27 and the indicated protein. Scale bar 2 μm. (h) Super resolution images of aggresomes in MG132 treated RPE-1 cells stained as indicated. Scale bar 1 μm. (i) Co-localization between the indicated protein pairs from individual z-planes of super resolution images of RPE-1 cells treated with MG132 displayed using Pearson correlation co-efficient. Boxes represent the median, upper and lower quartiles, whiskers represent 1.5x the interquartile range, with individual values from two independent experiments superimposed. For c,e: p determined by two-tailed unpaired Student’s t-test. Numerical data and p values are provided as source data.
Extended Data Fig. 2 High-resolution quantitative analysis confirms a requirement for protein translation, HDAC6 and microtubules in aggresome formation.
(a) Schematic representation of the automated aggresome quantification and satellite mapping pipeline utilized in this study (see Methods). (b) RPE-1 cells were transfected with iRNAs and treated and stained as indicated. Scale bar 2 μm. (c) Immunoblots of extracts from MG132 treated RPE-1 cells expressing an inducible siRNA resistant HSP27-FLAG construct following treatment with control (GL2) or HSP27 siRNAs. Blots were probed as indicated. (d) Box-and-whisker plot showing the area occupied by pHSP27 in RPE-1 pInducer siRNA resistant HSP27-FLAG cells treated as indicated. Boxes represent the median, upper and lower quartiles, whiskers represent 1.5x the interquartile range, with individual values superimposed. n = siGL2: 482 (un-induced DMSO), 449 (un-induced MG132), 358 (induced DMSO), 386 (induced MG132); siHSP27 437 (un-induced DMSO), 360 (un-induced MG132), 399 (induced DMSO) and 333 (induced MG132) aggresomes examined over 2 independent experiments. Data were compared using a Kruskal-Wallis ANOVA test and a post-hoc Dunn multiple comparison test performed to calculate p-values. ****p < 0.0001. (e) Cells transfected with siRNAs were treated and stained as indicated. Scale bar 2 μm. (f) Intensity maps of PCM1 distribution relative to the centrosome in cells treated as indicated. The percentage PCM1 signal residing in the ‘inner’ region is indicated. Arb, arbitrary units. (g) Immunoblot of extracts from cells treated and probed as indicated. (h) RPE-1 cells were treated and stained as indicated. Scale bar 10 μm, inset 2 μm. (i) RPE-1 cells treated with MG132 and taxol were stained as indicated. Scale bar 10 μm, inset 2 μm. (j) STIL KO cells were treated with MG132 then stained for PCM1 and Ub+. Scale bar 10 μm, inset 2 μm. (k) RPE-1 cells treated with MG132 and the HDAC6 inhibitor ACY-1215 were stained as indicated. Scale bar 10 μm, inset 2 μm. Unprocessed immunoblots, numerical data and p values are provided as source data.
Extended Data Fig. 3 Centriolar satellites are required for aggresome formation.
(a) CRISPR/Cas9 mediated gene disruption was used to generate AZI1, CCDC14, KIAA0753, PCM1 and PIBF1 knockout (KO) RPE-1 cell lines. Extracts from WT and KO cells were probed as indicated. (b) Loss of protein in AZI1, CCDC14, KIAA0753, PCM1 and PIBF1 KO cells was also confirmed by IF microscopy. Scale bar 2 μm. (c) Intensity maps of PCM1 distribution relative to the centrosome in WT and KO cells treated as indicated for 5 hours. Arb, arbitrary units. (d) Ub+ and CEP135 staining in WT and satellite protein KO cells treated with MG132. Scale bar 2 μm. (e) WT and OFD1 KO cells were stained for OFD1, PCM1 and CETN2. Scale bar 10 μm, inset 2 μm. (f) Immunoblot of extracts from WT and OFD1 KO cells probed as indicated. (g) Box-and-whisker plot showing the area occupied by pHSP27 in the indicated cell lines treated with MG132. Boxes represent the median, upper and lower quartiles, whiskers represent 1.5x the interquartile range, with individual values superimposed. n = WT: 400 (DMSO), 414 (MG132); ΔCETN2: 411 (DMSO), 434 (MG132); ΔFOP: 456 (DMSO), 352 (MG132); ΔOFD1: 422 (DMSO), 377 (MG132); ΔPCM1: 519 (DMSO) and 429 (MG132) aggresomes examined over 2 independent experiments. Data were compared using a Kruskal-Wallis ANOVA test and a post-hoc Dunn multiple comparison test performed to calculate p-values. ****p < 0.0001; n.s. not significant. (h) WT cells were transfected with siRNAs and stained as indicated. Scale bar 2 μm. (i) Immunoblot of extracts from WT and PCM1 KO cell lines transfected with siRNAs. All samples were run on the same gels; however, an intervening lane between the WT and PCM1 KO cell lines was excised, thereby placing non-adjacent lanes next to each other in the panel. Numbers at the bottom indicate the mean band intensity expressed as a percentage of signal of the corresponding control for the indicated proteins. (j) WT and PCM1 KO cells treated with control, KIAA0753 or PIBF1 siRNAs were stained as indicated. Scale bar 2 μm. (k) Intensity maps of PCM1 distribution relative to the centrosome in WT and PCM1 KO cells treated and stained as in j. Arb, arbitrary units. Unprocessed immunoblots, numerical data and p values are provided as source data.
Extended Data Fig. 4 Centriolar satellites direct protein aggregates to the aggresome in the absence of autophagy.
(a) WT, AZI1, CCDC14, KIAA0753, PCM1 and PIBF1 KO cells were seeded for clonogenic assays and treated with MG132 for 5 hours. The drug was then extensively washed out and the cells left to grow for 12 days. (b) Colony density was quantified and growth normalized to WT. Mean ± s.d. is shown, n = 4 from two independent experiments. *p < 0.05 by two-tailed unpaired Student’s t-test. (c) Immunoblots of extracts from WT and PCM1 KO cells treated with DMSO or MG132 probed as indicated. α-tubulin was used as a loading control. (d) Immunoblots of extracts from WT, AZI1, CCDC14, KIAA0753, PCM1 and PIBF1 KO cells treated with DMSO or MG132 and probed as indicated. The fold change in the amount of LC3-II present in MG132 treated cells as compared to the DMSO treated counterpart is indicated. GAPDH was used as a loading control. (e) Box-and-whisker plot showing the area occupied by pHSP27 in WT, AZI1, CCDC14, KIAA0753 and PIBF1 KO cells treated with DMSO, MG132 or MG132 plus chloroquine (CQ). Boxes represent the median, upper and lower quartiles, whiskers represent 1.5x the interquartile range, with individual values superimposed. n = WT: 470 (DMSO), 529 (MG132), 513 (MG132/CQ); ΔAZI1: 484 (DMSO), 458 (MG132), 524 (MG132/CQ); ΔCCDC14: 387 (DMSO), 739 (MG132), 404 (MG132/CQ); ΔKIAA0753: 381 (DMSO), 426 (MG132), 411 (MG132/CQ); ΔPIBF1: 470 (DMSO), 529 (MG132) and 513 (MG132/CQ) aggresomes examined over 2 independent experiments. Data were compared using a Kruskal-Wallis ANOVA test and a post-hoc Dunn multiple comparison test performed to calculate p-values. ****p < 0.0001. (f) WT, PCM1, AZI1, CCDC14, KIAA0753 and PIBF1 cells were treated with MG132 or MG132 plus CQ and stained for CP110 and p62. Scale bar 10 μm, 2 μm. (g) Immunoblot of extracts from WT, KIAA0753 and PIBF1 KO cells transfected with siRNAs against PCM1. α-tubulin was used as a loading control. Unprocessed immunoblots, numerical data and p values are provided as source data.
Extended Data Fig. 5 A CP110-CEP97-CEP290 module is required for aggresome formation.
(a) MG132 treated extracts were probed as indicated. (b) Extracts from siRNA transfected cells were probed as indicated. (c) Extracts from control or Talpid3 siRNAs treated cells were probed as indicated. (d) Cells treated with DMSO or MG132 were stained as indicated. Scale bar 10 μm, inset 2 μm. (e) Plot of pHSP27 area in cells transfected with siRNAs and treated with MG132. Boxes represent the median, upper and lower quartiles, whiskers represent 1.5x the interquartile range, with individual values superimposed. n = siGL2: 459 (DMSO), 463 (MG132); siTalpid3: 372 (DMSO) and 388 (MG132) aggresomes examined over 2 independent experiments. Data were compared using a Kruskal-Wallis ANOVA test and a post-hoc Dunn multiple comparison test performed to calculate p-values. ****p < 0.0001. (f) RPE-1 cells were treated and stained as indicated. Scale bar 10 μm. (g) RPE-1 cells transfected with GFP-CP110 were treated and stained as indicated. Scale bar 10 μm, inset 2 μm. (h) Extracts from RPE-1 cells treated with CP110 siRNAs and transiently transfected with RNAi-resistant GFP-CP110 were probed as indicated. (i) Cells transfected as in h were treated with MG132 and stained as indicated. Scale bar 2 μm. (j) Percentage cells treated as in i that formed an aggresome. Data displayed as mean ± s.d., n = 3 independent experiments. ****p < 0.0001 by two-tailed unpaired Student’s t-test. (k, l) Cells transfected with FLAG-CEP290 were treated and stained as indicated. Scale bars 10 μm, inset 2 μm. (m) Fractions from cells transfected with siRNAs and treated as indicated (CQ, chloroquine). (n) Extracts from cells transfected with siRNAs were treated and probed as indicated. The fold change in LC3-II levels in MG132 treated cells is shown as compared to their control treated counterparts. (o) Plot showing pHSP27 area in WT cells transfected siRNAs and treated with MG132 or MG132 and CQ. Boxes represent the median, upper and lower quartiles, whiskers represent 1.5x the interquartile range, with individual values superimposed. n = siGL2: 766 (DMSO), 719 (MG132), 741 (MG132/CQ); siCP110: 810 (DMSO), 577 (MG132) and 659 (MG132/CQ) aggresomes examined across 2 independent experiments. Data were compared using a Kruskal-Wallis ANOVA test and a post-hoc Dunn multiple comparison test performed to calculate p-values. ****p < 0.0001. (p) siRNA transfected cells were treated with MG132 or MG132 plus CQ and stained as indicated. Scale bar 10 μm, inset 2 μm. Unprocessed immunoblots, numerical data and p values are provided as source data.
Extended Data Fig. 6 Senescent cells have a reduced capacity to form aggresomes.
(a) Bright-field image of cycling and senescent HFF-1 cells that were subjected to a senescence associated β-galactosidase (SA-β-gal) assay. The percentage of cells displaying SA-β-gal activity is indicated. Scale bar 10 μm. (b) Extracts from cycling and senescent HFF-1 cells treated with or without MG132 were probed for CP110, CEP290, Ki67, p53, phospho-p38, p38, p21, pHSP27 and total HSP27, as indicated. α-tubulin was used as a loading control. (c) Cycling and senescent HFF-1 cells were treated with DMSO or MG132, then stained for Ub+ and CP110. DNA was stained with DAPI. Scale bar 10 μm. (d) crFAM83 and crPCM1 IMR-90 cells were stained for PCM1. Scale bar 10 μm. (e) Bright-field image of crFAM83G and crPCM1 IMR-90 cells that were subjected to a senescence associated β-galactosidase (SA-β-gal) assay. Scale bar 20 μm (f) Quantification of the percentage of senescent cells as determined by SA-β-gal activity in crFAM83 and crPCM1 IMR-90 cells, n = 2 biological replicates. (g) Senescent HFF-1 cells were transfected with GFP-CP110 for 48 hours before treatment with MG132. Cells were stained as indicated. Scale bar 10 μm, inset 2 μm. (h) Histogram of the percentage of senescent HFF-1 cells transfected with GFP or GFP-CP110 that formed an aggresome. Data displayed as mean and range. n = 2 independent experiments. (i) Senescent HFF-1 cells were transfected with GFP-CP110, treated with MG132 and stained for CEP97, CEP290 and PCM1. Scale bar 10 μm, inset 2 μm. Unprocessed immunoblots and numerical data are provided as source data.
Extended Data Fig. 7 HTT-polyQ inclusion formation requires microtubules and a functional centrosome.
(a) RPE-1 cells were transiently transfected with GFP-HTT97Q and stained for pHSP27 or Ub+. DNA was stained with DAPI. Scale bar 10 μm. (b) Immunoblot of soluble and insoluble fractions from WT and PCM1 KO cells transfected with GFP-HTT25Q. GAPDH and Histone H3 were used controls for the soluble and insoluble fractions, respectively. (c) Cells were transfected with GFP-HTT97Q for 5 hours, then treated with DMSO or nocodazole for 5 hours before being fixed and stained for GFP and α-tubulin. Scale bar 10 μm. (d) Immunoblot of soluble and insoluble fractions from cells prepared as in c. GAPDH and Histone H3 were used controls for the soluble and insoluble fractions, respectively. (e) WT and STIL KO U-2 OS cells were transiently transfected with GFP-HTT97Q then stained for GFP, CEP135 and α-tubulin. Scale bar 10 μm. (f) Immunoblot of soluble and insoluble fractions from cells prepared as in e. GAPDH and Histone H3 were used controls for the soluble and insoluble fractions, respectively. (g) HEK293T-FLAG-miniTurbo-CP110 cells were treated and stained as indicated. Scale bar 10 μm, 2 μm. (h) Immunoblot of HEK293T-FLAG-miniTurbo-CP110 cells treated and probed as indicated. α-tubulin was used as a loading control. (i) The number of preys from DMSO- or MG132-treated groups. Preys were defined as detailed in the Methods. (j) Functional enrichment analysis was performed with the preys from i. using g:Profiler and the KEGG database. (k) Spectral counts from the genes which are implicated in Parkinson’s disease, proteasome function or Huntington’s disease are shown. Genes which are related to the respective pathways are marked in green. Unprocessed immunoblots are provided as source data. For i,j,k: the raw mass spectrometry data and analysis giving rise to these panels is available in Supplementary Table 2.
Supplementary information
Supplementary Tables
File contains a workbook with multiple tabs assigned to Supplementary Tables 1, 2 and 3.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 6
Unprocessed western blots.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 7
Unprocessed western blots.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 2
Unprocessed western blots.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 3
Unprocessed western blots.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 7
Unprocessed western blots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prosser, S.L., Tkach, J., Gheiratmand, L. et al. Aggresome assembly at the centrosome is driven by CP110–CEP97–CEP290 and centriolar satellites. Nat Cell Biol 24, 483–496 (2022). https://doi.org/10.1038/s41556-022-00869-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41556-022-00869-0
- Springer Nature Limited