

Abstract
Mitochondrial RNA (mtRNA) in the cytosol can trigger the innate immune sensor MDA5, and autoinflammatory disease due to type I IFN. Here, we show that a dominant negative mutation in the gene encoding the mitochondrial exonuclease REXO2 may cause interferonopathy by triggering the MDA5 pathway. A patient characterized by this heterozygous de novo mutation (p.T132A) presented with persistent skin rash featuring hyperkeratosis, parakeratosis and acanthosis, with infiltration of lymphocytes and eosinophils around small blood vessels. In addition, circulating IgE levels and inflammatory cytokines, including IFNα, are found consistently elevated. Transcriptional analysis highlights a type I IFN gene signature in PBMC. Mechanistically, REXO2 (T132A) lacks the ability to cleave RNA and inhibits the activity of wild-type REXO2. This leads to an accumulation of mitochondrial dsRNA in the cytosol, which is recognized by MDA5, leading to the associated type I IFN gene signature. These results demonstrate that in the absence of appropriate regulation by REXO2, aberrant cellular nucleic acids may accumulate and continuously trigger innate sensors, resulting in an inborn error of immunity.
Similar content being viewed by others


Introduction
Innate immune cells express intracellular sensors involved in the surveillance of pathogens, which trigger a fast antimicrobial response. Nucleic acids, such as DNA or RNA found in viruses, are particularly potent triggers for antiviral type I IFN expression1. Intracellular RNA species are recognized by RIG-I-like receptors (RLR), RIG-I (retinoic acid-inducible gene I) and MDA5 (melanoma differentiation associated protein 5), as well as Protein Kinase R (PKR). Both MDA5 and RIG-I signal via the adaptor protein mitochondrial antiviral signaling protein (MAVS)2. Gain of function mutations in RIG-I and MDA5 can cause chronic upregulation of type I IFN signaling and disease such as Aicardi-Goutières syndrome (AGS)3,4,5, while mutations in PKR are associated with early onset isolated generalized dystonia, although an IFN signature was not established6. Most type I interferonopathy-related disease genes discovered so far actually encode molecules involved in nucleic acid processing, emphasizing the difficulty of self/non-self-discrimination7,8. In particular, it turns out that mitochondrial nucleic acids are potent immunostimulators, and frequently the target of regulatory pathways that prevent adverse inflammatory events9.
Mitochondria are organelles with their own genome. MtRNA go through stages of maturation and modification for proper mtDNA-encoded protein production. Mitochondrial RNA granules (MRG) regulate post-transcriptional processing of mtRNAs, ensuring full maturation before protein synthesis10,11. Polynucleotide phosphorylase (PNPase) and ATP-dependent RNA helicase SUPV3L1 (SUV3) form a degradosome in MRGs12. SUV3 is an NTP-dependent helicase that participates in the breakdown of properly processed RNA molecules, as well as the degradation of the damaged mtRNAs13,14,15. PNPase is the protein partner working with SUV3 and it can perform both 3′-to-5′ phosphorolysis and 5′-to-3′ RNA polymerization16,17. Depleting PNPase/SUV3 results in accumulation of antisense mtRNAs and cytosolic leakage of dsRNA derived from bidirectional mtDNA transcription, triggering IFN through a BAX/BAK-dependent mechanism18. Also, dysfunction of the PNPase/SUV3 leads to the formation of R-loops, which disrupt the maintenance of mtDNA19. Mutations in the gene that encodes PNPase (PNPT1) can cause severe hypotonia and movement abnormalities with severe but non-progressive encephalopathy accompanied by elevated levels of lactate in plasma and cerebrospinal fluid20. Although it has been shown that these mutations disrupt PNPase’s ability to homo-trimerize, which interferes with the protein’s ability to import RNA, the impact on exoribonuclease activity is unknown. Also, patients exhibit a type-I IFN signature, however, the contribution of this to disease pathogenesis is not yet established.
RNA exonuclease REXO2 is another protein that plays an important role in mtRNA degradation. In Escherichia coli, the Orn protein belonging to the 3’-to-5’ exonucleases of the DEDD family cleaves nanoRNAs and is the only significant exonuclease for mRNA degradation21,22,23. REXO2, also known as small fragment nuclease (Sfn), is believed to be the human counterpart of the Orn gene22,24. REXO2 is a homotetrameric 3′-to-5′ exonuclease located in the mitochondria, which degrades oligonucleotides in the matrix and is active on 5-nucleotide (nt) RNA substrates24,25. The degradosome may break down RNA into small oligo-ribonucleotides, which could then be further processed by REXO2 for later stages of decay25. Szewczyk et al. found that loss of REXO2 activity in mitochondria causes accumulation of non-coding mtRNA species, including short linear ncH2 RNA and longer structured tRNA-like molecules. REXO2-controlled RNAs like ncH2 can indirectly affect the mitochondrial degradosome, causing an increase in mitochondrial dsRNA accumulation26. Upon REXO2 inactivation, the steady-state levels of mtRNA species increase, even though they are not direct REXO2 substrates. The authors also analyzed the OriL transcript, a mitochondrial short RNA functioning as a primer for mtDNA replication27,28. Szewczyk et al. observed that REXO2 degraded synthetic ncH2, but only partially degraded the OriL primer. This was because of the difference in secondary structure between the two substrates. The authors predicted that ncH2 had a linear structure, while OriL had a stem-loop structure with single-stranded tails. The pattern of degradation showed that REXO2 only degraded the 3’ single-stranded tail of OriL26.
REXO2 can degrade unstructured RNAs with a preference for 2-nt substrates26,29, but it also has sequence preference and can efficiently degrade even 25-nt-long substrates, depending on substrate sequence and enzyme concentration26. However, degradation of longer substrates is distributive, and the third nucleotide from the 3’ end of RNA determines REXO2’s degradation efficiency26.
REXO2 depletion may increase ncH2, antisense RNA, and dsRNA levels in mitochondria also by activating mtDNA transcription. Prokaryote studies suggest this possibility. Oligoribonuclease depletion in Pseudomonas aeruginosa accumulated nanoRNAs, primers that initiate transcription and cause gene expression changes30. Nicholls et al. reported that nanoRNAs can stimulate mtDNA transcription in vitro29. The authors reported that REXO2 knockout in mice resulted in early embryonic death, thus demonstrating the essential function of REXO229. However, the impact of conditional heart and skeletal muscle-specific knockout on mtRNA levels was insignificant, raising questions about REXO2’s role in mtRNA biology in mouse29.
REXO2 has been linked to various diseases, including a family with four pheochromocytoma (PCC) affected patients across two generations who had a possible new pathogenic variant (c.531-1 G > T (NM 015523.3)) in the REXO2 gene that affects splicing and was inherited with the disease phenotype31. It was also reported that REXO2 expression is correlated with the worse prognosis of lower grade glioma (LGG) with higher immune infiltration32. Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNP) in REXO2 associated with diseases of the colon including inflammatory bowel diseases (IBD)33 and ulcerative colitis (UC)34.
In contrast to those previous associations, here we show that a functionally validated variant in REXO2 causes an interferonopathy associated with inflammatory skin disease. This de novo variant acts in a dominant negative way to prevent REXO2 cleavage of RNA, which instead accumulates in the cytosol to trigger MDA5. These findings highlight the critical role of REXO2 in maintaining immune homeostasis, its impact on human pathology, and potential therapeutic avenues for related inflammatory conditions.
Results
Identification of a de novo heterozygous variant in REXO2
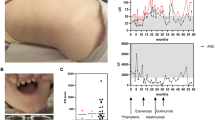
A female infant of Chinese ancestry, born to healthy non-consanguineous parents, presented at 2 years of age with whole-body rash (Fig. 1A). Laboratory tests showed a high level of immunoglobulin E (IgE) and IgG (Fig. 1B), episodes of elevated IgA (Supplementary Fig. 1B), episodes of elevated C-reactive protein (CRP) (Fig. 1B). Immunology workup showed episodes of elevated neutrophils, eosinophils, basophils (Fig. 1C and Supplementary Fig. 1C), as well as high level of B cells, T cells, Helper T cells and episodes of elevated white blood cell (WBC) counts (Supplementary Fig. 1A). Histopathologic examination of a skin biopsy revealed focal hyperkeratosis, parakeratosis and acanthosis with elongated rete ridges, focal liquefaction and degeneration of the basal layers of epidermis, vascular proliferation in the superficial dermis, infiltration of lymphocytes and eosinophils around small blood vessels in the dermis (Fig. 1D). She had recurrent infections (frequent and severe pneumonia). Neurologically and developmentally, the patient is normal. Currently, she continues to have skin symptoms with recurrent infections. Analysis of whole-exome sequencing of DNA samples from this family did not identify any known monogenic inborn errors of immunity (Fig. 1E). However, a monoallelic de novo variant in the REXO2 gene was nominated as a candidate on the basis of the gene’s known biological function, high degree of conservation and predicted pathogenicity scores (de novo c.394 A > G, NM_015523; p.T132A:(7Exons):exon4:missense; Fig. 1E, Supplementary Table 1). We identified several variants in known inborn errors of immunity (IEI) genes, but none of them was classified as pathogenic or likely pathogenic, and no other de novo mutation was found to be related with IEI (Supplementary Table 2).

A Skin rash observed in patient. B Laboratory tests for patient showed elevated IgE (left), episodes of elevated C-reactive protein CRP (right) and elevated IgG (middle). The gray areas indicate healthy donor control ranges. C Laboratory tests for patient showed episodes of elevated neutrophils (left), episodes of elevated basophils (right) and episodes of elevated eosinophils (middle). The gray areas indicate healthy donor control ranges. D Histological analysis showing active skin inflammation including focal hyperkeratosis and parakeratosis and increased cellular infiltration in the dermis (clinical data, n = 1) Scale bar 50 μm. E Pedigree and REXO2 variant in patient. Variant identification and Sanger sequencing validation of whole-exome sequencing (WES) data of heterozygous variant for patient and parents. F Location of T132 in the structure of REXO2 dimer (PDB:6STY) protomer 1 (surface representation in cyan), protomer 2 (ribbon representation in green), with bound nucleotide (pink). The mutation T132A would remove a H-bond from H161 and could shift Y164 away from the 3’ nucleotide where it is required for REXO2 exonuclease activity. Source data are provided as a Source data file.
Structural analysis of the REXO2 dimer with bound RNA (PDB:6STY) shows that T132 is present on the loop following helix 3 directly above C-terminally positioned H161 on helix 4, and therefore not directly involved in dimerization or proximal to active site residues (Fig. 1F). However, the loss of H bond between T132 and H161 could result in modest structural change to the loop following helix 4, where Y164 is part of an aromatic clamp on the bound 3’ nucleotides. The T132A mutation would potentially move Y164 away from the ligand which would most likely inactivate REXO2, as demonstrated for the Y164A mutation previously26. Taking these findings together, we suspected that the patient is manifesting an autosomal dominant syndrome of immune dysregulation and skin inflammation, caused by a monoallelic variant in REXO2.
REXO2 (T132A) patient inflammatory gene expression
To determine the inflammatory phenotype of the patient, we first examined the expression of interferon stimulated genes (ISG) in resting peripheral blood mononuclear cells (PBMC) from the patient. In contrast to healthy control, PBMCs from the patient showed spontaneous induction of IFNA4, IFNB1, IFNAR2, ISG15, ISG20, ISG20L2, ISG54, ISG56, and TNF (mRNAs) (Fig. 2A). Using Epstein-Barr Virus (EBV)-transformed B cell lines from controls (healthy control, father and mother) or the patient (REXO2-T132A), we confirmed that IFNA4, IFNB1, ISG15, ISG20, ISG54, and ISG56 were again upregulated for the patient (Supplementary Fig. 2A). ELISA analysis of the patient’s PBMCs showed significantly increased levels of IFNα, IFNβ and TNFα production compared to healthy controls, but not IL-6 and IL-1β (Fig. 2B). We then made similar findings in the patient’s serum and confirmed elevation of IFNα and IFNβ, but not IL-1β using ELISA analysis (Fig. 2C). Taken together, these data provide evidence that REXO2 (T132A) can predispose to increased IFN-stimulated gene expression in PBMCs and in immortalized B cells, with elevated IFNα, IFNβ, and TNFα production in PBMCs and serum.

A Expression of IFNA4 (P = 0.0007), IFNB1 (P = 0.0067), IFNAR2 (P = 0.0033), ISG15 (P = 0.0289), ISG20 (P = 0.0008), ISG20L2 (P = 0.0186), ISG54 (P < 0.0001), ISG56 (P = 0.0004), TNF (P = 0.0015), IL6 (P = 0.3358), and CCL5 (P = 0.0770) was measured by qPCR in peripheral blood mononuclear cell (PBMC) of healthy control and patient. Data are mean ± SD, pooled from 3 independent experiments. The p values were calculated using unpaired t test between healthy control and patient. Two-tailed. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. B Quantification of IFNα (P < 0.0001), IFNβ (P < 0.0001), TNFα (healthy control vs patient P = 0.0080, father vs patient P = 0.0359, mother vs patient P = 0.3454), IL-6 (healthy control vs patient P = 0.0004, father vs patient P = 0.0004, mother vs patient P > 0.9999) and IL-1β (healthy control vs patient P = 0.8122, father vs patient P = 0.8122, mother vs patient P = 0.2415) by ELISA in PBMC supernatant of healthy controls (healthy control, father and mother) and patient. Data are mean ± SD, pooled from 3 independent experiments. The p values were calculated using two-way ANOVA to patient. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. Adjusted p value. C Quantification of IFNα (P < 0.0001), IFNβ (P < 0.0001) and IL-1β (healthy control vs patient P = 0.1151, father vs patient P = 0.1151, mother vs patient P = 0.5878) by ELISA in the serum of healthy controls (healthy control, father and mother) and patient. Data are mean ± SD, pooled from 3 independent experiments (IL-1β) and 6 independent experiments (IFNα and IFNβ). The p values were calculated using two-way ANOVA to patient. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. Adjusted p value. Source data are provided as a Source data file.
To further determine whether the inflammatory phenotype of the patient is indeed due to the innate activation of IFN-I, we conducted additional experiments using patient’s PBMCs treated with Ruxolitinib (a JAK inhibitor) (Supplementary Fig. 2B). JAK inhibition ameliorates the type I IFN signature of REXO2 (T132A) patient cells.
REXO2 (T132A) variant spontaneously induces innate activation of IFN-I
To uncover molecular mechanisms underlying inflammation triggered by REXO2 (T132A), we first applied stable expression of empty vector (Ctrl), REXO2 (WT), or REXO2 (T132A) in a human monocytic cell line (THP-1), which confirmed upregulation of the type I IFN pathway in vitro (Fig. 3A–D). Specifically, REXO2 (T132A) spontaneously increased IFNA4, IFNAR2, IFNAR1, IFNB1, ISG15, ISG20, ISG20L2, CCL5, TNF, ISG54, ISG56, and IL6 (mRNAs) expression when compared with control (Fig. 3A). Moreover, global analysis revealed a significant IFN regulated gene signature35,36,37 due to REXO2 (T132A) (Fig. 3B). We also confirmed activation of the type I IFN pathway using western blotting, as the REXO2 (T132A) showed significantly increased phosphorylation of IRF3 (pIRF3) and TBK1 (pTBK1) compared to control and REXO2 (WT), in THP-1 stable cell lines (Fig. 3C). Finally, we verified increased production of IFNβ by REXO2 (T132A) cells compared to control using ELISA analysis (Fig. 3D). These results confirm that REXO2 (T132A) can intrinsically upregulate the type I IFN pathway for a model system in vitro, as seen in patient cells.

A Expression of IFNA4 (Ctrl vs WT P = 0.0019, Ctrl vs T132A P < 0.0001), IFNAR2 (Ctrl vs WT P = 0.0009, Ctrl vs T132A P < 0.0001), IFNAR1 (Ctrl vs WT P = 0.1308, Ctrl vs T132A P < 0.0001), IFNB1 (Ctrl vs WT P < 0.0001, Ctrl vs T132A P < 0.0001), ISG15 (Ctrl vs WT P = 0.0805, Ctrl vs T132A P < 0.0001), ISG20 (Ctrl vs WT P = 0.0036, Ctrl vs T132A P < 0.0001), ISG20L2 (Ctrl vs WT P = 0.0528, Ctrl vs T132A P < 0.0001), CCL5 (Ctrl vs WT P = 0.1788, Ctrl vs T132A P < 0.0001), TNF (Ctrl vs WT P = 0.1027, Ctrl vs T132A P < 0.0001), ISG54 (Ctrl vs WT P = 0.0003, Ctrl vs T132A P < 0.0001), ISG56 (Ctrl vs WT P = 0.0007, Ctrl vs T132A P < 0.0001) and IL6 (Ctrl vs WT P = 0.0286, Ctrl vs T132A P = 0.0002) was measured by qPCR analysis of inflammatory signaling pathways related to type I IFN in vector alone (Ctrl), REXO2-FLAG (WT) and REXO2-FLAG (T132A) THP-1 stable cell lines. Data are mean ± SD, pooled from 6 independent experiments. The p values were calculated using two-way ANOVA to Ctrl. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. Adjusted p value. B Heat map showing IFN regulated genes in REXO2 (T132A) compared with REXO2 (WT) THP-1 stable cell lines (n = 3). C Phosphorylated IRF3 (pIRF3) and total IRF3, phosphorylated TBK1 (pTBK1) and total TBK1 expression in vector alone (Ctrl), REXO2 (WT) and REXO2 (T132A) THP-1 stable cell lines by western blot (representative blots are shown of three independent repeats). D Quantification of IFNβ (Ctrl vs WT P < 0.0001, Ctrl vs T132A P < 0.0001) by ELISA in vector alone (Ctrl), REXO2 (WT) and REXO2 (T132A) THP-1 stable cell lines. Data are mean ± SD, pooled from 9 independent experiments. The p values were calculated using two-way ANOVA to Ctrl. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. Adjusted p value. Source data are provided as a Source data file.
To exclude the possibility that the C-terminal tag attached to REXO2 was interfering in our assays, we expressed untagged REXO2 and assessed the IFN-I signature through qPCR analysis. In this analysis, the overexpression of untagged REXO2 (T132A) was consistent with the tagged version tested previously (Supplementary Fig. 3A). Untagged WT REXO2 triggered fewer inflammatory pathways than the tagged version, however, some minimal reactivity is still present which could represent misfolding or other issues associated with overexpression of WT REXO2 which destabilize it in a way similar to the patient mutation.
Additionally, we knocked out REXO2 in THP1 cells to observe inflammatory gene and ISG expression. Similar to the effect of REXO2 mutant overexpression, cells lacking REXO2 display increased IFNA4, IFNB1, IFNAR1, IFNAR2, CCL5, IL6, TNF, ISG15, ISG20, ISG20L2, ISG54, and ISG56 (mRNAs) expression when compared with control (Supplementary Fig. 3H).
Our structural analysis had indicated that T132A may impact its activity via an aromatic clamp from Y164 via H161 (Fig. 1C). To test this, we generated the REXO2 Y164A, which is known to be inactive26, and found that it promotes inflammation similar to the patient mutation T132A, although not as strongly (Supplementary Fig. 3С). On the other hand, we noted a variant present in healthy humans (H161T) and tested this as a negative control, which does not potentiate inflammatory responses (Supplementary Fig. 3С).
To differentiate the roles of REXO2’s mitochondrial and nonmitochondrial functions in preventing sterile inflammation, we conducted experiments involving WT REXO2 and the T132A variant without the mitochondrial localization sequence26. The REXO2 T132A construct that accumulates in the cytoplasm of cells, but not mitochondria, did not trigger inflammatory responses (Supplementary Fig. 3E, F). This confirms that inflammatory signaling from REXO2 (T132A) is due to its activity within mitochondria.
Inflammatory signaling from REXO2 (T132A) is dependent on MDA5/MAVS
To identify an innate immune sensor for the REXO2 (T132A) associated IFN I signature, we first genetically ablated RNA sensors upstream of IFN-I signaling. This revealed that IFIH1 (also known as MDA5), but not DDX58 (also known as RIG-I), or EIF2AK2 (also known as PKR), was responsible for the increased cytokine and ISGs due to overexpression of REXO2 (T132A) in THP-1 cells (Fig. 4A). We also confirmed this by activation of signaling molecules via western blot, where deletion of MDA5 returned type I IFN activation to baseline (Fig. 4B, Supplementary Fig. 4A). Finally, we verified that deletion of MAVS, which is downstream of MDA5, also returned type I IFN activation to baseline (Supplementary Fig. 4B, C). These results indicate that MDA5 as an important immune sensor regulating REXO2 (T132A) mediated inflammation.

A Expression of IFNB1 (NC Ctrl vs NC WT P = 0.0361, NC Ctrl vs NC T132A P = 0.0003, RIG-I sgRNA Ctrl vs RIG-I sgRNA WT P = 0.0021, RIG-I sgRNA Ctrl vs RIG-I sgRNA T132A P = 0.0003, PKR sgRNA Ctrl vs PKR sgRNA WT P = 0.0028, PKR sgRNA Ctrl vs PKR sgRNA T132A P < 0.0001), TNF (NC Ctrl vs NC T132A P < 0.0001, RIG-I sgRNA Ctrl vs RIG-I sgRNA T132A P < 0.0001, PKR sgRNA Ctrl vs PKR sgRNA T132A P < 0.0001), CCL5 (NC Ctrl vs NC T132A P = 0.0025, RIG-I sgRNA Ctrl vs RIG-I sgRNA T132A P < 0.0001, PKR sgRNA Ctrl vs PKR sgRNA T132A P = 0.0010), ISG15 (NC Ctrl vs NC WT P = 0.1752, NC Ctrl vs NC T132A P = 0.0045, RIG-I sgRNA Ctrl vs RIG-I sgRNA T132A P < 0.0001, PKR sgRNA Ctrl vs PKR sgRNA T132A P = 0.0032), ISG20 (NC Ctrl vs NC WT P = 0.0053, NC Ctrl vs NC T132A P = 0.0005, RIG-I sgRNA Ctrl vs RIG-I sgRNA WT P < 0.0001, RIG-I sgRNA Ctrl vs RIG-I sgRNA T132A P < 0.0001, PKR sgRNA Ctrl vs PKR sgRNA WT P = 0.0003, PKR sgRNA Ctrl vs PKR sgRNA T132A P < 0.0001), ISG20L2 (NC Ctrl vs NC WT P = 0.0039, NC Ctrl vs NC T132A P = 0.0004, RIG-I sgRNA Ctrl vs RIG-I sgRNA WT P < 0.0001, RIG-I sgRNA Ctrl vs RIG-I sgRNA T132A P < 0.0001, PKR sgRNA Ctrl vs PKR sgRNA WT P = 0.0154, PKR sgRNA Ctrl vs PKR sgRNA T132A P = 0.0276), ISG54 (NC Ctrl vs NC T132A P = 0.0037, RIG-I sgRNA Ctrl vs RIG-I sgRNA T132A P < 0.0001, PKR sgRNA Ctrl and PKR sgRNA T132A P = 0.0014), ISG56 (NC Ctrl vs NC WT P = 0.0502, NC Ctrl vs NC T132A P = 0.0001, RIG-I sgRNA Ctrl vs RIG-I sgRNA WT P < 0.0001, RIG-I sgRNA Ctrl vs RIG-I sgRNA T132A P < 0.0001, PKR sgRNA Ctrl vs PKR sgRNA WT P = 0.0006, PKR sgRNA Ctrl vs PKR sgRNA T132A P < 0.0001) and IL6 (NC Ctrl vs NC T132A P < 0.0001, RIG-I sgRNA Ctrl vs RIG-I sgRNA T132A P = 0.0004, PKR sgRNA Ctrl vs PKR sgRNA T132A P = 0.0002) was measured by qPCR analysis of inflammatory signaling pathways related to type I IFN in vector alone (Ctrl), REXO2 (WT) and REXO2 (T132A) THP-1 stable cell lines which were also transduced with a sgRNA vector (NC), MDA5 sgRNA, RIG-I sgRNA or PKR sgRNA. Data are mean ± SD, pooled from 3 independent experiments. The p values were calculated using one-way ANOVA to Ctrl. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. Adjusted p value. B Western blot of phosphorylated IRF3 (pIRF3), total IRF3, phosphorylated TBK1 (pTBK1) and total TBK1 in vector alone (Ctrl), REXO2-FLAG (WT), and REXO2-FLAG (T132A) THP-1 stable cell lines which were also transduced with a sgRNA vector (WT) or MDA5 sgRNA (representative blots are shown of three independent repeats). Source data are provided as a Source data file.
REXO2 (T132A) causes mtRNA release into the cytoplasm via BAK/BAX
Given that MDA5 is the innate immune sensor triggered downstream of REXO2 (T132A) we investigated if this was due to the presence of mtRNA in the cytosol. For this analysis, cytosolic extracts were prepared and qPCR primers specific for mitochondrial (MT-ND1 and MT-ND2) or nuclear (L1ORF1 and POLG1) RNA employed. Only mtRNA was significantly increased in the cytosol of REXO2 (T132A) THP-1 cells at baseline (Fig. 5A). To ascribe a direct contribution of mtRNA to IFN-I expression we made cells deficient in mtDNA using 2’,3’ dideoxycytidine (ddC). ddC inhibits the mtDNA polymerase POLG, causing depletion of mtDNA, leading to a secondary depletion of mtRNA38. mtRNA depletion blunted the increase in MT-ND1 and MT-ND2, but not L1ORF1 and POLG1 transcription in REXO2 (T132A) THP-1 cells (Fig. 5A). To validate the reduction in steady-state levels of mitochondrial DNA and mitochondrial RNA after ddC treatment, we analyzed total cell lysates from THP-1 cells (Supplementary Fig. 5A).

A Expression of MT-ND1 (UT Ctrl vs UT T132A P < 0.0001), MT-ND2 (UT Ctrl vs UT T132A P = 0.0005), L1ORF1, and POLG1 was measured by qPCR in vector alone (Ctrl), REXO2 (WT), and REXO2 (T132A) THP-1 stable cell lines with or without ddC treatment. Data are mean ± SD, pooled from 3 independent experiments. The p values were calculated using one-way ANOVA to Ctrl. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. Adjusted p value. B Confocal microscopy showing the cellular localization of REXO2 in HeLa cells (P = 0.0080). The plot shows Pearson’s correlation coefficients for the colocalization analysis between REXO2-mCherry and mitochondria is shown on the right (n = 8 cells). Scale bar 7.5 μm. Data are mean ± SD. The p values were calculated using unpaired t test. Two-tailed. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. C Confocal microscopy showing the colocalization of REXO2 and dsRNA in HeLa cells (P < 0.0001). The plot shows Pearson’s correlation coefficients for the colocalization analysis between REXO2-mCherry and dsRNA is shown on the right (n = 9 cells). Scale bar 7.5 μm. Data are mean ± SD. The p values were calculated using unpaired t test. Two-tailed. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. D Confocal microscopy showing the cellular localization of dsRNA in HeLa cells (P = 0.0097). The plot shows Pearson’s correlation coefficients for the colocalization analysis between mitochondria and dsRNA is shown on the right (n = 8 cells). Scale bar 10 μm. Data are mean ± SD. The p values were calculated using unpaired t test. Two-tailed. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. Source data are provided as a Source data file.
To confirm these results we also performed confocal imaging. This demonstrates that REXO2 colocalized with mitochondria although somewhat less for REXO2 (T132A) (Fig. 5B and Supplementary Fig. 5B). This imaging analysis also showed that REXO2 (T132A) had a substantially increased colocalization with dsRNA (Fig. 5C and Supplementary Fig. 5C). Finally, we observed decreased colocalization of dsRNA with mitochondria due to REXO2 (T132A) compared to WT (Fig. 5D and Supplementary Fig. 5D), which suggested that the mutation leads to mtRNA accumulation outside mitochondria, as we had found by subcellular fractionation and qPCR.
As an orthogonal and translational approach, we also tested the role of mtRNA using patient B cells. Specifically, healthy control and patient B-cell lines were treated with 2’,3’ dideoxycytidine (ddC) as before, to deplete mtDNA, and thus also mtRNA. In agreement with the previous data, depletion of mtRNA using ddC ameliorates the type I IFN signature of REXO2 (T132A) patient cells (Supplementary Fig. 5E).
We next determined whether mutant REXO2 induces mtRNA release through the mitochondrial permeability transition pore (mPTP) or VDAC1-pores using chemical inhibitors, cyclosporine A (CsA)39,40, and VBIT-441, respectively. While the administration of CsA and VBIT-4 had no effect, Ruxolitinib, a JAK inhibitor used as positive control, returned type I IFN activation of REXO2 (T132A) in THP-1 cells to baseline (Supplementary Fig. 6A). We then conducted qPCR experiments (IFN-I signature) using cells treated with BAX/BAK sgRNA to investigate their potential role in the cytosolic leakage of dsRNA-induced innate immune response. We found that REXO2 (T132A) drives type I IFN production dependent on BAK/BAX (Supplementary Fig. 6B, C). This establishes that these molecules are indeed required for the inflammatory response observed.
Dominant negative effect of REXO2 (T132A) preventing RNA cleavage
The degradation of dsRNA has typically been evaluated through the analysis of substrate residues using gel electrophoresis and spectrometer absorbance at OD26042,43. Alternatively, a continuous fluorescence intensity measurement method was developed for Dicer cleavage assays, which allows for more precise quantitative analysis44. In this study, we modified that method by selecting a suitable fluorophore and quencher pair, and developed a fluorescence-based assay for quantifying RNA degradation activity using the 3’ to 5’ Exonuclease Activity Assay Kit (Fluorometric) from Abcam.
For the fluorescence assay, we used the fluorescent conjugated OriL transcript (a mitochondrial short RNA functioning as a primer for mtDNA replication)27,28, which was degraded by REXO2 in the study by Szewczyk et al.26. The OriL primer is a dsRNA, formed a stem-loop with single-stranded tails at both ends. The 5′ end was labeled with fluorophore and 3′ end was labeled with quencher (Supplementary Fig. 7A). No fluorescence is emitted if the substrate remains intact. When RNA nuclease is added to the reaction tube, the OriL primer will be gradually degraded, leading to separation of the fluorophore group and the quencher group, and thus the emission of the corresponding fluorescence signal.
We measured the cleavage of this OriL RNA substrate by THP-1 cell extracts expressing REXO2 (T132A). When overexpressing REXO2 (WT), these cell extracts contained readily detectable RNA cleavage activity (Fig. 6A, B, Supplementary Fig. 7B). The results show that this is lacking for the REXO2 mutation T132A. Moreover, we observed that REXO2 (T132A) could inhibit RNA cleavage by REXO2 (WT) in a titratable fashion (Fig. 6C, D, Supplementary Fig. 7C), thus acts as a dominant negative mutation.

A Cell-free protein extracts were assayed for RNA cleavage activity with a dsRNA probe, with a stem-loop and with single-stranded tails at both ends in vector alone (Ctrl), REXO2 (WT), and REXO2 (T132A) THP-1 stable cell lines. Data are mean ± SD, pooled from 3 independent experiments. The p values were calculated using one-way ANOVA to Ctrl. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. B Maximum enzyme velocity (Vmax) (Ctrl vs WT P < 0.0001, Ctrl vs T132A P = 0.9994) from data in panel (A). Data are mean ± SD, pooled from 3 independent experiments. The p values were calculated using one-way ANOVA to Ctrl. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. Adjusted p value. C RNA cleavage activity using a dsRNA probe, with a stem-loop and single-stranded tails at both ends in 6 titration combinations with REXO2 (WT) and REXO2 (T132A) THP-1 stable cell extracts (the volume of all combinations was equal, by adding different amount of vector alone cell extract). Data are mean ± SD, pooled from 3 independent experiments. The p values were calculated using one-way ANOVA to WT. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. D Maximum enzyme velocity (Vmax) (WT 50% vs WT 50% + T132A 10% P = 0.9999, WT 50% vs WT 50% + T132A 20% P = 0.9445, WT 50% vs WT 50% + T132A 30% P = 0.0146, WT 50% vs WT 50% + T132A 40% P < 0.0001, WT 50% vs WT 50% + T132A 50% P < 0.0001) from data in panel (C). Data are mean ± SD, pooled from 3 independent experiments. The p values were calculated using one-way ANOVA to WT. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS not significant. Adjusted p value. Source data are provided as a Source data file.
We then conducted experiments with recombinant REXO2 to confirm its specific responsibility for the observed activity in THP-1 total cell lysates. Our results revealed clear RNA cleavage activity with recombinant REXO2 (WT) (Supplementary Fig. 7D–F, H). This provides further evidence that the conclusions drawn from our ribonuclease activity assay in THP-1 cell extracts are indeed due to REXO2, and purified REXO2 p.T132A is catalytically inactive (Supplementary Fig. 7D–F, I).
Finally, in order for REXO2 (T132A) to function in a dominant negative fashion, it would need to interact with the WT REXO2. Therefore, we examined the interaction between WT REXO2 and T132A mutant using immunoprecipitation. This experiment verified the binding of both WT-WT and WT-T132A pairs (Supplementary Fig. 7G). Overall, the accumulated data support the hypothesis that REXO2 (T132A) acts as a dominant negative mutation.
Discussion
Our study describes an interferonopathy caused by the mutation of REXO2, a mitochondrial exonuclease that plays a critical role in regulating mitochondrial RNA. We found that the heterozygous de novo mutation (p.T132A) impairs REXO2’s ability to cleave RNA, and that this can also prevent the action of WT REXO2, by acting in a dominant negative fashion. Szewczyk et al.26 emphasized the role of dimerization in the formation of the active REXO2 molecule, which is consistent with previous research29. A mutation that removes the protein function, but retains the dimerization domain could cause a dominant negative phenotype, because some fraction of protein dimers would be missing one of the functional domains. Consistent with this, we observed that the T132A mutant inhibited the activity of the wild-type REXO2 in a titratable manner in THP-1 cell extracts.
Our study also delves into the distinction between the RNA degradation mechanisms of mutant REXO2 and WT REXO2, which is important for proper mtRNA processing and maturation. Specifically, we observed accumulation of mtRNA in REXO2 mutant overexpressing cells, and this led to activation of the dsRNA sensor MDA5, despite REXO2 acting as an exonuclease for ssRNA. This is consistent with the findings of Szewczyk et al.26, who demonstrated accumulation of dsRNA in REXO2-deficient cells. In that paper, it was found that the degradosome, a dynamic protein structure that performs a regulatory function for post-transcriptional processing of mtRNA, was less functional in REXO2-deficient cells. This could occur because REXO2 substrates accumulate, and overwhelm/inhibit the PNPase and SUPV3L1 degradosome machinery26. Notably, the cleavage assay in THP-1 cell extracts in this study was conducted using cell lysates which contain not only REXO2 but also other components of the degradosome complex, including PNPase and SUPV3L1. Given that the substrate tested was a double-stranded hair-pin with single-stranded ends, the measured activity may reflect REXO2 and the entire degradosome complex.
Dysfunctional degradosome-dependent mtRNA decay pathway leads to the accumulation of antisense mtRNAs and cytosolic leakage of dsRNA, triggering the innate immune response through a BAX/BAK-dependent mechanism18. We observed that the mtRNA release into the cytoplasm does not proceed via mPTP- or VDAC-dependent channels. Subsequent qPCR experiments, incorporating BAX/BAK sgRNA-treated cells, support the role of these molecules in the cytosolic leakage of dsRNA-induced innate immune response due to mutant REXO2. Whether there is a specific pathway leading to BAX/BAK pore formation downstream of defective mtRNA cleavage, or this represents a non-specific response to a loss of mitochondrial homeostasis, is currently under investigation.
In REXO2 mutant cells, we find that dsRNA is recognized by the innate immune sensor MDA5, leading to the associated type I interferonopathy. This is characterized by elevated levels of IgE, IFNα, and a type I IFN gene signature in peripheral blood mononuclear cells along with persistent skin rash, hyperkeratosis, parakeratosis, and acanthosis with infiltration of lymphocytes and eosinophils around small blood vessels. Dermatological involvement is a major feature of the type I interferonopathies, which include the allelic disorders familial chilblain lupus (FCL) and Aicardi-Goutières syndrome (AGS). X-linked reticulate pigmentary disorder (XLPDR), is also classified as an interferonopathy due to a mutation in POLA145. In AGS, cutaneous lesions are observed in 40% of cases and were first described by Bursztejn et al. in the context of mutations in MDA546. MDA5 expression is markedly increased in the skin of patients with dermatomyositis, chronic discoid lupus and lichen planus47, and MDA5 polymorphisms have been associated with an elevated risk of developing systemic lupus erythematosus, psoriasis and chronic periodontitis48. Our study highlights the link between skin manifestations in MDA5 and REXO2 genes and validates the interferon signature as a diagnostic tool in this context. Although neurological involvement is a prominent feature of many interferonopathies, our patient is neurologically and developmentally normal.
Overall, our study provides insights into the role of REXO2 in immune dysregulation and skin inflammation, and into the pathogenesis of interferonopathies. This highlights the critical role of REXO2 in regulating mitochondrial RNA processing, which is required to prevent adverse inflammatory events. The identification of a de novo heterozygous variant in REXO2 gene could help to better understand the genetic basis of this syndrome and could facilitate earlier diagnosis and treatment. Given that the molecular mechanism of disease is driven by MDA5, leading to type I IFN production, JAK inhibitors may be a suitable therapy for such patients moving forward.
Methods
Patient
Patient blood and other samples were collected at Guangzhou Women and Children’s Medical Centre, Guangzhou, China. No compensation was provided. The Medical Ethics Committee of Guangzhou Women and Children’s Medical Centre approved the study procedures (ID:2021083B00). The implementation was in line with the International Ethical Guidelines for Research Involving Human Subjects as stated in the Declaration of Helsinki. Informed written consent was obtained from the legal representatives of the patient. The patient’s baseline demographics were recorded, including clinical signs, duration of illness, family history and laboratory test results.
Patient sequencing
Whole exome sequencing and sanger sequencing were performed at the Beijing Genomic Institute (BGI). Predictions of variant pathogenicity were generated using dbNSFP v449.
Skin biopsy
Skin biopsy was performed at the Guangzhou Women and Children’s Medical Centre.
Isolation and culture of human peripheral blood mononuclear cells
PBMCs from healthy donors and patient were isolated by Ficoll density gradients and dispensed in 96-well plates at a density of 2–3 × 105 cells/well in 200 μl of media (RPMI 1640 (Gibco) medium supplemented with 10% FBS, 2 mM L-glutamine (Gibco) and 1% Penicillin-Streptomycin), and incubated at 37 °C in 5% CO2. Cell lysates and supernatants taken for RT-qPCR and ELISA analysis, respectively.
Antibodies and reagents
Rabbit monoclonal anti-phospho-IRF-3 (Ser386) (E7J8G) antibody (37829), rabbit monoclonal anti-IRF-3 (D83B9) (D614C) antibody (4302), rabbit monoclonal anti-phospho-TBK1/NAK (Ser172) (D52C2) antibody (5483), rabbit monoclonal anti-TBK1/NAK (D1B4) antibody (3504), mouse monoclonal anti-β-Actin (8H10D10) antibody (3700), anti-rabbit IgG, HRP-linked antibody (7074), anti-mouse IgG, HRP-linked antibody (7076), rabbit monoclonal anti-RIG-I (D33H10) antibody (4200), rabbit monoclonal anti-MDA5 (D74E4) antibody (5321), rabbit monoclonal anti-PKR (D7F7) antibody (12297), rabbit monoclonal Mitofusin-2 (D1E9) antibody (11925), rabbit monoclonal anti-BAK (D4E4) antibody (12105), rabbit monoclonal anti-BAX (D2E11) antibody (5023), mouse monoclonal anti-Myc-Tag (9B11) antibody (2276) and rabbit monoclonal anti-MAVS (D5A9E) antibody (24930) were purchased from Cell Signaling Technology. Mouse monoclonal anti-FLAG M2 (F3165) antibody was purchased from Sigma-Aldrich. Rabbit polyclonal anti-Tubulin beta (AF7011) antibody was purchased from Affinity Biosciences. Mouse monoclonal anti-TOM20 (sc-17764) antibody was purchased from Santa Cruz. Mouse monoclonal anti-dsRNA antibody J2 (RNT-SCI-10010200) was purchased from Jena Bioscience. Goat anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (A-11029) and Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 (A-11012) were purchased from Invitrogen. Rabbit polyclonal anti-REXO2 (ab206694) antibody was purchased from Abcam. The dilution of anti-β-actin, anti-rabbit IgG HRP-linked antibody, anti-mouse IgG HRP-linked antibody, anti-FLAG and anti-Tubulin beta was 1:5000, the dilution of anti-Myc-Tag was 1:2000. The other antibodies were used as 1:1000 dilution. 2’3’-Dideoxycytidine (ddC), cyclosporine A (CsA), VBIT-4 and Ruxolitinib were purchased from MedChemExpress.
Cell culture and transfection
HeLa, 293T, and THP-1 cells were obtained from American Type Culture Collection (ATCC). HeLa and 293T were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Corning) supplemented with 10% FBS, 2 mM L-glutamine, and 1% Penicillin-Streptomycin (Gibco). THP-1 cells were grown in RPMI 1640 (Gibco) medium supplemented with 10% FBS, 2 mM L-glutamine (Gibco), and 1% Penicillin-Streptomycin. All cells were cultured at 37 °C incubator with 5% CO2 at 100% humidity. Plasmids were transfected using lipofectamine 3000 reagent (Invitrogen). siRNAs (TsingKe Biological Technology) were transfected using lipofectamine RNAi max reagent (Invitrogen).
Cytosolic RNA isolation and quantification
Subcellular fractionation was performed as a previously published protocol50,51. Cells were washed with PBS and resuspended with permeabilization buffer (0.0045% digitonin/MELB buffer [20 mM Hepes-KOH (pH 7.5), 250 mM sucrose, 1 mM EDTA, 50 mM KCl, and 2.5 mM MgCl2] at 4 °C for 10 min under rotation. Cell lysates were centrifuged twice at 13,000 × g for 5 min at 4 °C to separate the cytosolic fraction from a pellet that contains the heavy membrane fraction. The heavy membrane pellet was discarded; supernatant was collected for cytosolic RNA isolation using RNA kit (EZ Bioscience). Quantitative PCR was performed using nuclear RNA primers (POLG1 and L1ORF1) and mtRNA primers (MT-ND1 and MT-ND2).
Mitochondrial RNA depletion
4 μM ddC were added to the culture mediums as described to achieve mtRNA ablation38.
Immunoprecipitation and immunoblotting assay
Cell lysates were extracted with low-salt lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 1.5 mM MgCl2, 10% glycerol, 1% Triton X-100), containing 5 mg/mL protease inhibitor cocktail (Roche). For immunoprecipitation, cell lysates were incubated overnight with anti-Flag M2 magnetic beads (Sigma). The beads were then washed 4 times with the low-salt lysis buffer before being resuspended with 3×SDS Loading Buffer (FD Biotechnology) and resolved by SDS–polyacrylamide gel electrophoresis (SDS-PAGE). Concentration of the protein was determined using the BCA protein assay kit (Thermo Fisher Scientific). Protein lysates (20 μg) with 5×SDS Loading Buffer (FD Biotechnology) were separated by SDS-PAGE. Proteins were transferred to PVDF membranes (Bio-Rad). After blocking with 5% skim milk, membranes were incubated with primary and secondary antibodies and detected through enhanced chemiluminescence (Millipore).
Quantitative real-time PCR
Total RNA was isolated using the EZ-press RNA Purification Kit (EZ Bioscience) as per manufacturer’s instructions and reverse transcribed to cDNA using 4× Reverse Transcription Master Mix Kit (with gDNA Remover) (A0010GQ) (EZ Bioscience). Quantitative real-time PCR was performed using 2× Color SYBR Green qPCR Master Mix (ROX2 plus) (A0012-R2) (EZ Bioscience). Each sample was run in duplicate, and samples were normalized using the housekeeping gene.
qPCR primer sequences:
Human IFNA4 forward: ACCTGGTTCAACATGGAAATG
Human IFNA4 reverse: ACCAAGCTTCTTCACACTGCT
Human IFNAR1 forward: CAGTGGCTCCACGCCTTTTTA
Human IFNAR1 reverse: GCTTGTACGCGGAGAAGGTA
Human IFNAR2 forward: ACCACTCCATTGTACCAACTCA
Human IFNAR2 reverse: TGTGCTTCTCCACTCATCTGT
Human IFNB1 forward: GTCACTGTGCCTGGACCATAG
Human IFNB1 reverse: GTTTCGGAGGTAACCTGTAAGTC
Human ISG15 forward: TCCTGGTGAGGAATAACAAGGG
Human ISG15 reverse: GTCAGCCAGAACAGGTCGTC
Human ISG20 forward: TCTACGACACGTCCACTGACA
Human ISG20 reverse: CTGTTCTGGATGCTCTTGTGC
Human ISG20L2 forward: GAACTCCACCCAAGCTCATTC
Human ISG20L2 reverse: GCTACATCGAGCCAAGGAACT
Human ISG54 forward: AAGCACCTCAAAGGGCAAAAC
Human ISG54 reverse: TCGGCCCATGTGATAGTAGAC
Human ISG56 forward: TTGATGACGATGAAATGCCTGA
Human ISG56 reverse: CAGGTCACCAGACTCCTCAC
Human TNF forward: CCAGACCAAGGTCAACCTCC
Human TNF reverse: CAGACTCGGCAAAGTCGAGA
Human IL6 forward: AGAGGCACTGGCAGAAAACAAC
Human IL6 reverse: AGGCAAGTCTCCTCATTGAATCC
Human CCL5 forward: ATCCTCATTGCTACTGCCCTC
Human CCL5 reverse: GCCACTGGTGTAGAAATACTCC
Human MAVS forward: CAGGCCGAGCCTATCATCTG
Human MAVS reverse: GGGCTTTGAGCTAGTTGGCA
Human REXO2 forward: GAACTGTGCAGACGCTGGTAT
Human REXO2 reverse: CATCAAGTGCCCTATGAGAAGC
Human MT-ND1 forward: CTCTTCGTCTGATCCGTCCT
Human MT-ND1 reverse: TGAGGTTGCGGTCTGTTAGT
Human MT-ND2 forward: GTAGACAGTCCCACCCTCAC
Human MT-ND2 reverse: TTGATCCCGTTTCGTGCAAG
Human POLG1 forward: CTGCCATAAGGTCTGCAGGT
Human POLG1 reverse: CTCCTTTCCGTCAACAGCTC
Human L1ORF1 forward: AGAACGCCACAAAGATACTCCTCG
Human L1ORF1 reverse: CTCTCTTCTGGCTTGTAGGGTTTCTG
Human RNA18S forward: GTAACCCGTTGAACCCCATT
Human RNA18S reverse: CCATCCAATCGGTAGTAGCG
Enzyme-linked immunosorbent assay
All enzyme-linked immunosorbent assay (ELISA) experiments were carried out using serum of healthy controls and patient, and cell supernatants according to the manufacturer’s instructions. ELISA kits for human IFNα (CSB-E08636h), human IFNβ (CSB-E09889h), human IL-1β (CSB-E08053h), human IL-6 (CSB-E04638h), and human TNFα (CSB-E04740h) were from Cusabio.
Plasmids and lentiviruses
For overexpression studies, full-length human REXO2 (NM_015523) was inserted into backbone plasmid Ubi-MCS-3FLAG-SV40-Cherry-IRES-puromycin and CMV enhancer-MCS-3FLAG-sv40-puromycin. For human nonmitochondrial REXO2 (NM_015523 (Delta1-32aa)) was inserted into pGC-FU-CMV enhancer-mCherry-3FLAG-sv40-puromycin. For human untagged REXO2 (NM_015523) the full-length sequence was inserted into CMV-MCS-3FLAG-EF1a-mCherry-T2A-puromycin. Site-directed mutagenesis technology was used to generate T132A, H161T and Y164A mutations as required, according to manufacturers instructions (Stratagene).
For lentiviral constructs, virus was prepared by transfecting a combination of plasmids including REXO2 construct (20 μg), psPAX2 (15 μg), and pMD2.G (10 μg) into 293T cells with 60 μl Lipofectamine 2000 reagent (Invitrogen). At 3 days post transfection the viral supernatant was concentrated by ultracentrifugation. The resulting supernatant was collected and filtered through a 0.45 µm PES membrane filter, aliquoted into sterile 1 ml Eppendorf tubes, and stored at −80 °C.
Designing and synthesis of plasmids, and lentiviruses production were done at Genechem (Genechem, Shanghai, China).
For 293T cells and HeLa cells, plasmids were transfected using Lipofectamine 3000 reagent (Invitrogen) following the manufacturer’s instructions for 24 h.
For lentivirus transduction into THP-1 cells, concentrated lentiviruses (Titer: 3 × 107 TU/mL), along with HiTransG A reagent (Genechem) in complete medium, were added to the cells. After 16 h of incubation, the medium was replaced with fresh medium, and the plate was returned to the incubator. Antibiotic treatment was performed 72 h after infection.
To knockdown MDA5, RIG-I, PKR, REXO2, BAK and BAX, CRISPR-Cas9 technology was used to generate MDA5-, RIG-I-, PKR-, REXO2- and BAK-/BAX- THP-1 stable cell lines as described previously52. Briefly, MDA5 (NM_022168), RIG-I (NM_014314), PKR (NM_001135651), REXO2 (NM_015523.4) and BAX (NM_138761) sgRNA constructs were inserted in hU6-sgRNA-EF1a-Cas9-FLAG-CMV-EGFP-P2A-puromycin lentivirus vector. BAK (NM_001188) sgRNA constructs were inserted in hU6-sgRNA-CMV-NLS-cas13d-T2A-EGFP-sv40-neomycin lentivirus vector. Lentiviruses production was done at Genechem (Genechem, Shanghai, China). THP-1 cells were transduced by indicated lentiviruses to generate Cas9/EGFP/puromycin or Cas9/EGFP/neomycin resistance gene expressing cells. FACS sorting and antibiotic treatment were performed for positive cell selection during three weeks after infection.
Cells were treated with 2 µg/mL puromycin (Santa Cruz Biotechnology) for 3 weeks to generate MDA5-, RIG-I-, PKR-, REXO2- or overexpressed wild type and mutated, T132A REXO2 THP-1 stable cell line. Cells were treated with 2 µg/mL puromycin (Santa Cruz Biotechnology) and with 500 µg/mL geneticin (Thermo Fisher) for 3 weeks to generate BAK-/BAX-, or overexpressed wild type and mutated, T132A REXO2 THP-1 stable cell line. The pure cells expressing REXO2 construct and Cas9 construct were selected by three consecutive times FACS sorting. The knockdown efficiencies and stable overexpression were verified by Western blotting assay (Figs. 3C and 4B, Supplementary Figs. 4A, 3B, D, F, H and 6C).
sgRNA sequences:
IFIH1 (PCA11759): CGAATTCCCGAGTCCAACCA
IFIH1 (PCA11760): GCTTCTAGTTAGAGACGTCT
IFIH1 (PCA11761): CTGAATGTTCTTCGTCAAAC
DDX58 (PCA11756): GGGTCTTCCGGATATAATCC
DDX58 (PCA11757): GTCTGAATGTTTAATTAATC
DDX58 (PCA11758): TTTGAAATCCCAACTTTCAA
EIF2AK2 (PCA11762): TAATACATACCGTCAGAAGC
EIF2AK2 (PCA11763): TTATCCATGGGGAATTACAT
EIF2AK2 (PCA11764): AGCTGTTGAGATACTTAATA
BAX (PCA14716): TTTCTGACGGCAACTTCAAC
BAX (PCA14717): TCTGATCAGTTCCGGCACCT
BAX (PCA14718): TCCAGCCCAACAGCCGCTCC
BAK1 (PCA14719): TCCTGTTCCTGCTGATGGCGGTA
BAK1 (PCA14720): TCCTCTGTGTCCTGGGCTACCTG
BAK1 (PCA14721): CAGAGGTAAGGTGACCATCTCTG
REXO2 (PCA15963): CAGCATGTCAGATTGGTGTA
REXO2 (PCA15964): ATGCTGTCCAGCAACTCATC
REXO2 (PCA15965): ATCAGAGTCAGTTATCAGAC
Primer sequences:
nonmitochondrial WT REXO2 (ZF-KL-02979-P3): TCGGGCGGTGGTGGGTCGGGTGGCGGCGGATCTATGGTGAGCAAGGGCGAGGAG
nonmitochondrial WT REXO2 (ZF-KL-02979-P4): CCCGACCCACCACCGCCCGAGCCACCGCCACCACTCACGGTCTTCTCATTTTCC
nonmitochondrial T132A REXO2 (ZF-KL-02980-P3): TCGGGCGGTGGTGGGTCGGGTGGCGGCGGATCTATGGTGAGCAAGGGCGAGGAG
nonmitochondrial T132A REXO2 (ZF-KL-02980-P4): CCCGACCCACCACCGCCCGAGCCACCGCCACCACTCACGGTCTTCTCATTTTCC
untagged WT REXO2 (REXO2(93708-1)-p1): GATCTATTTCCGGTGAATTCCGCCACCATGCTAGGCGGCTCCCTGGG
untagged WT REXO2 (REXO2(93708-1)-p2): TCCTTGTAGTCCATGGATCCACTCACGGTCTTCTCATTTTCCC
untagged T132A REXO2 (REXO2(93707-1)-p1): GATCTATTTCCGGTGAATTCCGCCACCATGCTAGGCGGCTCCCTGGG
untagged T132A REXO2 (REXO2(93707-1)-p2): TCCTTGTAGTCCATGGATCCACTCACGGTCTTCTCATTTTCCC
H161T REXO2 (REXO2(97173-3)-p1): AGGTCGACTCTAGAGGATCCCGCCACCATGCTAGGCGGCTCCCTGGG
H161T REXO2 (REXO2(97173-3)-p2): TCCTTGTAGTCCATGGATCCCTTGTACAGCTCGTCCATGCCGC
Y164A REXO2 (REXO2(97174-11)-p1): AGGTCGACTCTAGAGGATCCCGCCACCATGCTAGGCGGCTCCCTGGG
Y164A REXO2 (REXO2(97174-11)-p2): TCCTTGTAGTCCATGGATCCCTTGTACAGCTCGTCCATGCCGC
siRNA
siRNA sequences:
MAVS: 5’-AUGUGGAUGUUGUAGAGAUUCTT-3’.
Fluorescence microscopy
Cells cultured on glass bottom culture dishes (Nest) were fixed with 4% paraformaldehyde for 15 min. After rinsed for 3 times, cells were blocked for 1 h with 5% fetal goat serum in PBS containing 0.3% Triton X-100, followed by overnight incubation with primary antibodies (diluted in 10% bull serum albumin) at 4 °C. Fluorescent labeled secondary antibodies were incubated for 2 h at room temperature. Nuclei were stained with DAPI (Invitrogen). Cells were observed through Carl Zeiss confocal microscopy (LSM 710) using 100× oil objectives.
Exonuclease assays
REXO2 exonuclease activity assays were carried out in Exonuclease assay buffer (3’ to 5’ Exonuclease Activity Assay Kit) (ab273269) (Abcam) containing dithiothreitol (DTT), according to the manufacturer’s instructions. OriLprimer substrate was incubated with vector alone (Ctrl), WT (REXO2-overexpressing) and T132A REXO2 cell-free protein extracts of THP-1 stable cell lines, and 6 titration combinations, and with recombinant proteins. Fluorescence was measured in kinetic mode for 50–90 min at 37 °C every 30 s.
Recombinant proteins were purchased from SinoBiological.
GGAAMAAGESMAQRMVWVDLEMTGLDIEKDQIIEMACLITDSDLNILAEGPNLIIKQPDELLDSMSDWCKEHHGKSGLTKAVKESTITLQQAEYEFLSFVRQQTPPGLCPLAGNSVHEDKKFLDKYMPQFMKHLHYRIIDVSTVKELCRRWYPEEYEFAPKKAASHRALDDISESIKELQFYRNNIFKKKIDEKKRKIIENGENEKTVS
GGAAMAAGESMAQRMVWVDLEMTGLDIEKDQIIEMACLITDSDLNILAEGPNLIIKQPDELLDSMSDWCKEHHGKSGLTKAVKESTITLQQAEYEFLSFVRQQAPPGLCPLAGNSVHEDKKFLDKYMPQFMKHLHYRIIDVSTVKELCRRWYPEEYEFAPKKAASHRALDDISESIKELQFYRNNIFKKKIDEKKRKIIENGENEKTVS
His-SUMO tag and 3C protease site are added at N-terminal of proteins.
Mitochondrial isolation
Mitochondria were obtained following the manufacturer’s protocol (Thermo Fisher). Briefly, 2 × 107 cells were collected and resuspended in 800 μl of mitochondria isolation reagent A. The mixture was vortexed and incubated on ice for 2 min, followed by the addition of 10 μl of mitochondria isolation reagent B. After sufficient vortexing and incubation, 800 μl of Reagent C was added, and the solution was centrifuged at 700 × g for 10 min. The supernatant was centrifuged at 12,000 × g for 15 min. Mitochondria sit in the pellet.
RNA-sequencing data processing
Total RNA was first extracted from cultured cells, and the concentration and RNA integrity were measured using NanoDrop 2000 (Thermo Fisher) and Agilent 2100, respectively. For RNA sequencing, mRNA was first enriched, fragmented and reversed-transcribed into cDNA, and then subjected to end-repairing and adapter ligation processes. The resulting mRNA library was paired-end sequenced (2 × 150 bp) with the sequencer DNBSEQ-T7 (BGI Genomics).
Adapters introduced in the sequencing reads during the library preparation were trimmed using the Cutadapt (ver. 1.18)53. For further read cleaning, the software fastp (ver. 0.23.2)54 was used and the following criteria were used to define low-quality reads: (1) more than 2 ambiguous bases (base N) in a read; (2) number of bases with quality score less than 15 exceeding 20% of the read length; If one of the above criteria is met, the read was considered as low-quality read and the pair of reads was removed.
The remaining high-quality reads were then aligned to human reference genome GRCh38 using STAR aligner (ver. 2.7.9a)55 to generate the binary alignment file (.bam), which was then passed to HTSeq (ver. 0.11.1)56 for gene expression quantification. The resulting gene expression matrix was then normalized using DESeq2 (ver. 1.34.0)57.
Data visualization was accomplished using the R software pheatmap (ver. 1.0.12).
Data representation and statistical analyses
Data presented are representative of at least three independent experiments and are expressed as mean ± s.d. and analyzed by t test between two groups or one- or two-way ANOVA. GraphPad Prism 7 (GraphPad Software) was used to generate all charts and statistical analyses. A P value < 0.05 was considered significant. Detailed descriptions of statistical tests are specified in the relevant figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq raw sequencing data generated in this study has been deposited under supervision and control of the Genome Sequence Archive of the Beijing Institute of Genomics (BIG), Chinese Academy of Sciences (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA004834). Please refer to GSA for detailed application guidance. All other data is available in the main text or the supplementary materials. All unique materials are readily available from the authors. Source data are provided with this paper.
References
Ablasser, A. & Hur, S. Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 21, 17–29 (2020).
Goubau, D., Deddouche, S. & Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 38, 855–869 (2013).
Rodero, M. P. & Crow, Y. J. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J. Exp. Med. 213, 2527–2538 (2016).
Rice, G. I. et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 46, 503–509 (2014).
Oda, H. et al. Aicardi-Goutieres syndrome is caused by IFIH1 mutations. Am. J. Hum. Genet 95, 121–125 (2014).
Kuipers, D. J. S. et al. EIF2AK2 missense variants associated with early onset generalized dystonia. Ann. Neurol. 89, 485–497 (2021).
Uggenti, C., Lepelley, A. & Crow, Y. J. Self-awareness: nucleic acid-driven inflammation and the type I interferonopathies. Annu. Rev. Immunol. 37, 247–267 (2019).
Davidson, S., Steiner, A., Harapas, C. R. & Masters, S. L. An update on autoinflammatory diseases: interferonopathies. Curr. Rheumatol. Rep. 20, 38 (2018).
Fu, Y., Tigano, M. & Sfeir, A. Safeguarding mitochondrial genomes in higher eukaryotes. Nat. Struct. Mol. Biol. 27, 687–695 (2020).
Mai, N., Chrzanowska-Lightowlers, Z. M. & Lightowlers, R. N. The process of mammalian mitochondrial protein synthesis. Cell Tissue Res. 367, 5–20 (2017).
Pearce, S. F. et al. Regulation of mammalian mitochondrial gene expression: recent advances. Trends Biochem. Sci. 42, 625–639 (2017).
Borowski, L. S., Dziembowski, A., Hejnowicz, M. S., Stepien, P. P. & Szczesny, R. J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 41, 1223–1240 (2013).
Minczuk, M., Lilpop, J., Boros, J. & Stepien, P. P. The 5’ region of the human hSUV3 gene encoding mitochondrial DNA and RNA helicase: promoter characterization and alternative pre-mRNA splicing. Biochim. Biophys. Acta 1729, 81–87 (2005).
Szczesny, R. J. et al. Down-regulation of human RNA/DNA helicase SUV3 induces apoptosis by a caspase- and AIF-dependent pathway. Biol. Cell 99, 323–332 (2007).
Kazak, L. et al. Alternative translation initiation augments the human mitochondrial proteome. Nucleic Acids Res. 41, 2354–2369 (2013).
Wang, D. D., Shu, Z., Lieser, S. A., Chen, P. L. & Lee, W. H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3’-to-5’ directionality. J. Biol. Chem. 284, 20812–20821 (2009).
Cameron, T. A., Matz, L. M. & De Lay, N. R. Polynucleotide phosphorylase: not merely an RNase but a pivotal post-transcriptional regulator. PLoS Genet 14, e1007654 (2018).
Dhir, A. et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242 (2018).
Silva, S., Camino, L. P. & Aguilera, A. Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc. Natl Acad. Sci. USA 115, 11024–11029 (2018).
Vedrenne, V. et al. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am. J. Hum. Genet. 91, 912–918 (2012).
Ghosh, S. & Deutscher, M. P. Oligoribonuclease is an essential component of the mRNA decay pathway. Proc. Natl Acad. Sci. USA 96, 4372–4377 (1999).
Mechold, U., Ogryzko, V. & Ngo, S. A. Danchin, Oligoribonuclease is a common downstream target of lithium-induced pAp accumulation in Escherichia coli and human cells. Nucleic Acids Res. 34, 2364–2373 (2006).
Chu, L. Y., Agrawal, S., Chen, Y. P., Yang, W. Z. & Yuan, H. S. Structural insights into nanoRNA degradation by human Rexo2. RNA 25, 737–746 (2019).
Nguyen, L. H., Erzberger, J. P., Root, J. & Wilson, D. M. 3rd, The human homolog of Escherichia coli Orn degrades small single-stranded RNA and DNA oligomers. J. Biol. Chem. 275, 25900–25906 (2000).
Bruni, F., Gramegna, P., Oliveira, J. M., Lightowlers, R. N. & Chrzanowska-Lightowlers, Z. M. REXO2 is an oligoribonuclease active in human mitochondria. PLoS ONE 8, e64670 (2013).
Szewczyk, M. et al. Human REXO2 controls short mitochondrial RNAs generated by mtRNA processing and decay machinery to prevent accumulation of double-stranded RNA. Nucleic Acids Res. 48, 5572–5590 (2020).
Mercer, T. R. et al. The human mitochondrial transcriptome. Cell 146, 645–658 (2011).
Wanrooij, S. et al. In vivo mutagenesis reveals that OriL is essential for mitochondrial DNA replication. EMBO Rep. 13, 1130–1137 (2012).
Nicholls, T. J. et al. Dinucleotide degradation by REXO2 maintains promoter specificity in mammalian mitochondria. Mol. Cell 76, 784–796 e786 (2019).
Goldman, S. R. et al. NanoRNAs prime transcription initiation in vivo. Mol. Cell 42, 817–825 (2011).
Laitman, Y., Tzur, S., Attai, R., Tirosh, A. & Friedman, E. Germline variant in REXO2 is a novel candidate gene in familial pheochromocytoma. Genet Res. 102, e3 (2020).
Wang, H., Wang, X., Xu, L., Zhang, J. & Cao, H. RUNX1 and REXO2 are associated with the heterogeneity and prognosis of IDH wild type lower grade glioma. Sci. Rep. 11, 11836 (2021).
Hulur, I. et al. Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci. BMC Genomics 16, 138 (2015).
Palmieri, O. et al. Genome-wide pathway analysis using gene expression data of colonic mucosa in patients with inflammatory Bowel disease. Inflamm. Bowel Dis. 21, 1260–1268 (2015).
Samarajiwa, S. A., Forster, S., Auchettl, K. & Hertzog, P. J. INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res. 37, D852–D857 (2009).
Metz, P., Reuter, A., Bender, S. & Bartenschlager, R. Interferon-stimulated genes and their role in controlling hepatitis C virus. J. Hepatol. 59, 1331–1341 (2013).
Schoggins, J. W. & Rice, C. M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1, 519–525 (2011).
Nelson, I., Hanna, M. G., Wood, N. W. & Harding, A. E. Depletion of mitochondrial DNA by ddC in untransformed human cell lines. Somat Cell Mol. Genet 23, 287–290 (1997).
Nicolli, A., Basso, E., Petronilli, V., Wenger, R. M. & Bernardi, P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, and cyclosporin A-sensitive channel. J. Biol. Chem. 271, 2185–2192 (1996).
Xian, H. et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 55, 1370–1385 e1378 (2022).
Kim, J. et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366, 1531–1536 (2019).
Arimatsu, Y. et al. Purification and properties of double-stranded RNA-degrading nuclease, dsRNase, from the digestive juice of the silkworm, Bombyx mori. J. Insect Biotechnol. Sericol. 76, 1_57–51_62 (2007).
Liu, J., Swevers, L., Iatrou, K., Huvenne, H. & Smagghe, G. Bombyx mori DNA/RNA non-specific nuclease: expression of isoforms in insect culture cells, subcellular localization and functional assays. J. Insect Physiol. 58, 1166–1176 (2012).
Podolska, K., Sedlak, D., Bartunek, P. & Svoboda, P. Fluorescence-based high-throughput screening of dicer cleavage activity. J. Biomol. Screen 19, 417–426 (2014).
Starokadomskyy, P. et al. DNA polymerase-alpha regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat. Immunol. 17, 495–504 (2016).
Bursztejn, A. C. et al. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutieres and Singleton-Merten syndromes. Br. J. Dermatol. 173, 1505–1513 (2015).
Zahn, S. et al. Enhanced skin expression of melanoma differentiation-associated gene 5 (MDA5) in dermatomyositis and related autoimmune diseases. J. Am. Acad. Dermatol. 64, 988–989 (2011).
Chen, G. et al. Genetic variants in IFIH1 play opposite roles in the pathogenesis of psoriasis and chronic periodontitis. Int. J. Immunogenet. 39, 137–143 (2012).
Liu, X., Li, C., Mou, C., Dong, Y. & Tu, Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 12, 103 (2020).
Yu, C. H. et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183, 636–649 e618 (2020).
Davidson, S. et al. Protein kinase R is an innate immune sensor of proteotoxic stress via accumulation of cytoplasmic IL-24. Sci. Immunol. 7, eabi6763 (2022).
Baker, P. J. & Masters, S. L. Generation of genetic knockouts in myeloid cell lines using a lentiviral CRISPR/Cas9 system. Methods Mol. Biol. 1714, 41–55 (2018).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Putri, G. H., Anders, S., Pyl, P. T., Pimanda, J. E. & Zanini, F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics 38, 2943–2945 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
We are grateful to all members of the Zhang and Masters laboratories for support. We would like to acknowledge Prof. Andrey V Zavialov, Prof. Jongdae Lee, Liya Shen and Fengyuan Yang for supplying reagents and technical advice or assistance. This study is supported by The National Natural Science Foundation of China (Grant number 32250410295 to E.I., Grant number 82125015 to Y.Z.), Guangzhou Women and Children’s Medical Center Fund.
Author information
Authors and Affiliations
Contributions
S.L.M. and Y.Z. conceived the project; E.I. conducted all experiments with assistance from M.A.; E.I. performed data analysis, drafted figures and wrote the first version of manuscript; K.H.-S. performed structural analysis; Z.L. assisted with bioinformatic analysis; L.L., X.L., and C.G. assisted with provision of human samples; M.L., Y.L., S.G., J.C., and W.Z. were involved in clinical management; C.G. assisted with immortalized B cells; X.C. was involved in WES sequencing analysis. S.L.M. supervised the project and wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
S.L.M. is a scientific advisor for Odyssey Therapeutics and NRG Therapeutics. Other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Satoshi Okada and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Idiiatullina, E., Al-Azab, M., Lin, M. et al. Heterozygous de novo dominant negative mutation of REXO2 results in interferonopathy. Nat Commun 15, 6685 (2024). https://doi.org/10.1038/s41467-024-50878-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50878-w
- Springer Nature Limited