

Abstract
Carboxylic acids are widely available and generally inexpensive from abundant biomass feedstocks, and they are suitable and generic coupling partners in synthetic chemistry. Reported herein is an electroreductive coupling of stable and versatile carboxylic acids with (hetero)arenes using protons as the hydrogen source. The application of an earth-abundant titanium catalyst has significantly improved the deoxygenative reduction process. Preliminary mechanistic studies provide insights into the deoxygenative reduction of in-situ generated ketone pathway, and the intermediacy generation of ketyl radical and alkylidene titanocene. Without the necessity of pressurized hydrogen or stoichiometric hydride as reductants, this protocol enables highly selective and straightforward synthesis of various functionalized and structurally diverse alkylbenzenes under mild conditions. The utility of this reaction is further demonstrated through practical and valuable isotope incorporation from readily available deuterium source.
Similar content being viewed by others
Introduction
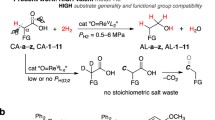
Direct alkylation of simple (hetero)arenes belongs to an ideal transformation to construct a carbon–carbon bond involving an aromatic moiety1. Conventional Friedel–Crafts alkylation, using an alkylating agent such as alkyl halide through an electrophilic aromatic substitution, has been acknowledged as one of the most fundamental methods (Fig. 1a)2. However, it suffers from the limitations of low chemo- and regioselectivities due to the undesired over-alkylation and carbocation rearrangement. Although a combination of Friedel–Crafts acylation3 and deoxygenative reduction4,5,6,7,8,9,10,11,12,13,14,15 can avoid these problems, the multistep manipulations and harsh reductive conditions still restrict their broad and practical applications. Carboxylic acid represents a class of cost-effective, widely available, and structurally diverse feedstock in synthetic organic chemistry16,17,18,19,20,21,22. The deoxygenative alkylation using carboxylic acids as electrophiles provides a rewarding direction to alleviate the reliance on classic halide chemistry23. Pioneered by Gribble, the possibility of direct deoxygenative arylation of trifluoroacetic acid was validated in 1985 (Fig. 1b)24. However, the harsh conditions involving borohydrides in great excess and limited scopes stimulated a surge of following research efforts. Later, Sakai described the deoxygenative functionalization of substituted benzoic acids, in which reductively generated silyl ethers by silane served as benzyl electrophiles in the presence of an indium catalyst25,26. Beller and his colleagues demonstrated a hydrogenative alkylation of substituted indoles using a wide range of carboxylic acids based on the Co/triphos system27. Despite these achievements, the use of sensitive hydride reductants or pressurized hydrogen can potentially result in serious safety issues, elaborate experimental setups as well as high costs, which limits a wide range of applications in synthetic chemistry.

a Traditional Friedel–Craft methods for alkylations of (hetero)arenes. b Reductive alkylations of (hetero)arenes with carboxylic acids using hydrides or hydrogen. c Electroreductive alkylations of (hetero)arenes with carboxylic acids using protons (this work).
Electrochemistry, in which electrons (e−) and protons (H+) can be used directly as sustainable and safe redox equivalent and hydrogen source, offers such an opportunity 28,29,30,31,32,33,34,35,36,37,38,39. To our knowledge, the application of carboxylic acids as coupling partners in decarboxylative or dehydroxylative functionalizations has been intensively explored in electrochemical synthesis40,41,42. In contrast to these elegant advances, electrochemical deoxygenative functionalization of carboxylic acids through complete deletion of the carboxylic oxygen remains limited, which holds intricate challenges in several aspects. The medium to low electrophilicity of a carboxyl group makes it often thermodynamically and kinetically inert43,44,45. In addition, the selectivity control of the carbonyl reduction remains challenging owing to the formation of alcohol through direct hydrogenation46,47,48,49 as well as methylene product through deoxygenative reduction50. Especially towards the desired methylene target constitutes a particular challenge since a higher negative reductive potential is required. Moreover, the carboxylic proton results in a hydrogen evolution reaction as the thermodynamic preference on the cathode competes with the desired carbonyl reduction process51,52,53. To date, a general electrosynthesis of alkylbenzenes via deoxygenative coupling of carboxylic acids has not been established, which is a significant but challenging goal in organic synthesis.
Based on our persistent interest in electrohydrogenations54, we describe herein a mild and efficient electrochemical deoxygenative functionalizations of carboxylic acids mediated by an earth-abundant titanium catalyst (Fig. 1c). This method allows for the direct alkylations of (hetero)arenes to produce various functionalized and structurally diverse alkylbenzenes using protons as the hydrogen source. Specifically, a significant advantage of this electrochemical reaction is the highly practical and valuable chemo-divergent isotope incorporation into the alkylbenzenes from a readily available deuterium source.
Results
Optimization reaction conditions
In light of these challenges and opportunities, our studies were initiated by screening of reaction parameters for the electrochemical deoxygenative coupling of propionic acid 1 with n-butoxybenzene 2 (Table 1). The model reaction was carried out in an undivided cell equipped with a platinum cathode and a zinc anode in trifluoroacetic acid and acetonitrile mixture. Inspired by previous specific activity for deoxygenative couplings55,56,57,58,59, the earth-abundant titanium complex (Cp2TiCl2) was used as a precatalyst. Under the optimal condition, the desired n-propylation product 3 was obtained in an excellent 98% yield (Table 1, entry 1) with high regioselectivity (>50:1) at para-position of 2. The application of other commercially available Ti and Zr complexes such as Cp*2TiCl2, CpTiCl3, Cp*TiCl3, and Cp2ZrCl2 could afford the desired product 3 but in moderate yield (entries 2–5). In addition, the corresponding acylating side-product 3a was observed in 11–25% yields with these catalysts. The reactions did not proceed when DMAc or DMSO were employed as the solvent (entry 6). The choice of electrode material significantly impacts the efficiency of the electrochemical transformations. Other electrodes, such as Ni, graphite plate as cathodes, or Fe, Al as anodes, led to low or no catalytic activity in the model reaction (entries 7−10). As a comparison, under the pressure of hydrogen gas instead of electrolysis, the reaction did not provide any product (entry 11). In the absence of Cp2TiCl2 as a deoxygenating reagent, the transformation proceeded with decreased efficiency (entry 12). Moreover, using trifluoroacetic acid (TFA) as a co-solvent provided sufficient proton source and acidic condition for this transformation (entry 13). Notably, triflic anhydride (Tf2O) is essential to activate carboxylic acids and generate highly potent triflate electrophiles in this reaction (entry 14). The control experiment demonstrated that electricity is necessary for the deoxygenation process (entry 15).
Substrate scope
After establishing the optimized reaction conditions, the generality of this electrochemical alkylation was studied (Fig. 2). First, we tested various substituted and functionalized aliphatic and aromatic carboxylic acids to produce the corresponding alkylbenzenes. In addition to propionic acid 1, various short and longer-chain aliphatic carboxylic acids performed well, forming the corresponding products 4−10 in moderate to good yields (49−88%). Specifically, carboxylic acids with the chlorine, ester, olefin, and 1,3-benzodioxole functionalities underwent this transformation smoothly, and their successful conversion to substituted arenes 11−14 expands the scope of this reaction. The application of trifluorobutyric acid as a substrate led to the corresponding alkylating product 15, albeit with decreased yield and regioselectivity (12:1). Besides primary carboxylic acids, secondary carboxylic acids also proved compatible under this electrochemical condition. A wide variety of structurally diverse long-chain and cyclic acids performed well, and synthetic, useful yields of 16−24 were obtained. Gratifyingly, pharmaceutical molecules such as Ibuprofen and Naproxen were found to be suitable substrates to afford the corresponding alkylbenzenes 25 and 26 in moderate yields. Moreover, the use of pivalic acid as a substrate led to an 11% yield of the desired product 27, which is likely due to the challenging deoxygenative process caused by the high steric hindrance of the substrate. To our delight, beyond aliphatic carboxylic acids as substrates, the present system could be effectively applied to aromatic acids affording the corresponding diarylmethanes 28–31 in 53–78% yields. Notably, unless otherwise mentioned, the deoxygenative alkylations were highly selectively performed at the para-position of n-butoxybenzene or anisole with more than 50:1 regioselectivity.

Reaction condition as shown in Table 1. a60 °C, 5 h. bTf2O (0.675 mmol), TFA (3.0 mL), MeCN (1.0 mL), 50 °C, 6 h. cTf2O (0.675 mmol), 60 °C, 6 h. dTf2O (0.3 mmol), r.t., 6 h. eTFA (1.0 mL), MeCN (2.0 mL), r.t., 6 h. f10 mol% Cp*2TiCl2 and diphenyl thiourea instead of Cp2TiCl2 and Tf2O, 90 °C.
Next, we examined the substrate scope by employing structurally diverse (hetero)arenes. Aryl ethers with different substitutions such as methyl, phenyl, and allyl groups were well compatible with this methodology and gave the desired products 32–36 in 36−88% yields and excellent regioselectivities (>50:1). Moreover, a range of disubstituted arenes proved to be good coupling partners, providing the corresponding alkylarenes 37–43 in moderate to good yields (38−65%). Notably, moderate regioselectivity of 2.2:1 was observed for product 40 when 3-methylanisole was applied as a substrate. Additionally, a scale-up reaction with 1,2-dimethoxybenzene as a substrate gave a 65% yield of the corresponding product 42. Benzenes bearing trisubstitutions such as 1,3,5-trimethoxybenzene 44 and 1-bromo-3,5-dimethoxybenzene 45 were also found to be suitable substrates and underwent these alkylations smoothly. Similarly, 1-methoxynaphthalene provided a moderate yield of the desired product 46. The Gemfibrozil derivative, a pharmaceutical used to reduce cholesterol and triglycerides, also furnished the desired products 47 in 34% yields. Interestingly, heteroaromatics such as substituted furan 48, benzofuran 49, and benzothiophene 50 participated in this transformation, highlighting the broad substrate scope of this protocol. Moreover, the deoxygenative coupling of various indoles with trifluoroacetic acid also performed efficiently and gave the corresponding trifluoroethylating products 51−55 in 41−86% yields without the necessity of triflic anhydride. In addition to intermolecular couplings, the use of 2-phenoxybenzoic acid as a substrate led to xanthene 56 in 31% yield through an intramolecular reductive cyclization.
Deuterium applications
Organic molecules labeled with hydrogen isotopes are highly desirable in analytical science and pharmaceutical chemistry60. The replacement of hydrogen by deuterium (D) at the benzylic position often gives rise to improved metabolic stability and bioactivity in drug discovery. Here the present electrocatalytic deoxygenative reduction provides a practical and cost-effective approach to benzylic deuterium labeling (Fig. 3a). The application of deuterated acetic acids with different arenes furnished valuable products 57–62 in 23–78% yields with 90−96% deuterium incorporations. Notably, in some cases, the aromatic C–H position was also partly deuterated at the ortho-position of ether functionality, as supported by natural population analysis (Supplementary Fig. 12). Remarkably, this catalytic system also allows for a chemo-divergent synthesis of different deuterium products starting from 2-ethylbenzofuran (Fig. 3b). The selectivity control was achieved by using different acetic acids (AcOH, d-AcOH and d4-AcOH) and trifluoroacetic acids (TFA and d-TFA) as deuterium sources. The use of non-deuterated AcOH and TFA under optimal conditions led to a 56% yield of ethylative product 63, whereas the CD3-labled product 64 is obtained in 38% yield with d4-AcOH. In addition, the application of d-TFA with two different d-AcOH and d4-AcOH resulted in a d2-methylene (–CD2–) product 65 and a fully deuterated ethyl (CD3CD2–) product 66 with excellent deuterium incorporation. These results also indicate that the hydrogen source for deoxygenative reduction mainly comes from trifluoroacetic acid.

a Deuterium applications. b Chemodivergent synthesis of deuterium products.
Mechanistic investigations
To gain insights into this electroreductive coupling, a series of mechanistic experiments were performed (Fig. 4). Firstly, the interactions between the carboxylic acid, arene, and triflic anhydride were monitored by 13C NMR. As shown in Fig. 4a, a significant chemical shift from 181.1 to 189.6 ppm was observed after the addition of Tf2O to the propionic acid 1 solution, indicating the rapid formation of a highly electrophilic triflate intermediate. Subsequently, the addition of n-butoxybenzene 2 resulted in the observation of a chemical shift downfield to 212.7 ppm, suggesting a ketone intermediate generation. Next, the catalytic reactions of possible intermediates were performed under standard conditions (Fig. 4b). The model hydrogenative coupling of acetic acid with anisole gave the desired 4-ethylanisole 67 in 51% yield. Under the standard conditions, deoxygenative reduction of 4-methoxyacetophenone 68 led to 86% yield, confirming that the in-situ generated ketone by Friedel–Crafts acylation is the key intermediate. This ketone intermediate then participates in the subsequent electrochemical hydrogenolysis12,13,14. We also investigated the potential intermediacy of an alcohol which could be produced by ketone hydrogenation and corresponding alkene generated by dehydration of the alcohol61. However, the application of 1-(4-methoxyphenyl)ethanol 69 and 4-methoxystyrene 70 did not afford the desired ethylating product, indicating the proposed alcohol and alkene are not intermediates in this transformation.

a NMR experiment for the interactions. b Reactions of possible intermediates. c Radical capture experiments. d Observation of alkylidiene titanocene intermediates. e Kinetic isotope effect experiments. f Performances of TFA concentration-dependent product yield and cyclic voltammetry (CV) studies.
Next, more experiments were conducted to provide an understanding of the reaction mechanism. We firstly performed radical capture experiments to explore the reaction patterns of carboxylic acids (Fig. 4c). The addition of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) led to a significant decreasing yield of the desired product 3 and the adduct of TEMPO with a ketyl radical was observed by high-resolution mass. Interestingly, the use of 2,2,3,3-tetramethylcyclopropanecarboxylic acid as a substrate generated a six-membered bicycle 71 in 45% yield through a sequence of cyclopropane ring opening and intramolecular cyclization. These results indicate a ketyl radical species formed via single-electron reduction of a ketone could be related62. In addition, high-resolution mass analysis of the hydrogenative coupling of benzoic acid with anisole suggested the involvement of a titanium carbene intermediate 72 in this transformation63, and a tetrasubstituted alkene 73 by McMurry reaction was also observed as a minor product (Fig. 4d)64,65,66. Moreover, the kinetic isotope effect experiments were performed using deuterated and non-deuterated reagents (Fig. 4e). The KH/KD value was found to be 1.71, revealing a primary kinetic isotope effect for the hydrogen transfer process.
In this reaction, trifluoroacetic acid (TFA) is not only used as the proton source for deoxygenative electroreduction, but also provides suitable acidic conditions to achieve the desired transformation (Fig. 4f). We found that the concentration of TFA significantly impacts the reaction efficiency. In the absence of TFA, the model reaction proceeded with low activity, whereas increasing TFA concentration led to obvious higher yields of desired product 3. Additionally, cyclic voltammetry (CV) experiments were investigated to acquire a further understanding of the interaction of TFA with Cp2TiCl2 and ketone intermediate. The CV profile of Cp2TiCl2 at 100 mV/s showed reversible Ti redox couples (black line)67,68. The addition of TFA to the titanium complex solution led to an obvious positive shift in the reduction potential (red line). On the other hand, adding TFA to a ketone intermediate 68 solution also resulted in the observation of heightened current response and a more positive peak shift of the reduction potential (blue line and green line). Moreover, the anodic wave corresponding to oxidation of 68 is not observed on the return scan. Such observations are indicative of a more feasible reduction of titanium catalyst and an irreversible ketone electroreduction to ketyl radical in the presence of TFA.
Based on these results, a possible mechanism for this electroreductive coupling is proposed in Fig. 5. Initial electrophilic activation of a carboxylic acid with triflic anhydride leads to the formation of a highly electrophilic triflate, which is prone to attack by a (hetero)arene to furnish a ketone A. This intermediate is reduced through a single electron transfer process to afford a ketyl radical B which has been confirmed by radical trapping experiments (Fig. 4c). Subsequently, reductive deoxygenation in B takes place and generates a carbene C69. There are two pathways for the subsequent transformation. In the presence of a titanium catalyst (path 1), the carbene C is trapped by the Cp2Ti(II) species, which is generated from the reduction of Cp2TiX2 (X = Cl or OTFA) on the cathode, to provide an alkylidene titanocene D. This key intermediate was observed by the high-resolution mass (Fig. 4d). Protonation of D yields the desired alkylating product and regenerates Cp2TiX2. Alternatively, this electroreductive coupling also provided the desired product in moderate yield (67%) in the absence of a titanium catalyst. Therefore, a direct electrochemical hydrogenolysis (path 2) through stepwise protons and electrons transfer on the cathode might also deliver the corresponding hydrocarbons70.

Proposed mechanism.
Discussion
In summary, we have developed a selective and efficient electrochemical deoxygenative alkylation of (hetero)arenes using stable and versatile carboxylic acids. The reaction is significantly improved by an earth-abundant titanium catalyst as the deoxygenative reagent. Mechanistic investigations revealed that the in-situ generated ketone, identified as the key intermediate, is involved in the subsequent electrochemical deoxygenative reduction. The present method exhibits a broad substrate scope with good functional group compatibility and is amenable to valuable isotope incorporation from readily available protons. Given the broad availability and diversity of carboxylic acids and (hetero)arenes, we anticipate this protocol will find potential utility and facilitate further explorations in synthetic chemistry.
Methods
Representative procedure for the synthesis of compound 3
A dried 10 mL glass tube equipped with a magnetic stirring bar was added Cp2TiCl2 (0.03 mmol), n-butoxybenzene 2 (0.3 mmol), propionic acid 1 (1.5 mmol), TBABF4 (0.6 mmol), Tf2O (0.6 mmol), CF3COOH (2.0 mL) and MeCN (2.0 mL). The reactor was equipped with a Zn electrode (2 cm × 1.5 cm × 0.05 cm) as the anode and a Pt electrode (2 cm × 1 cm × 0.02 cm) as the cathode. The reaction was bubbled with N2 for 5 min. Then the mixture was electrolyzed under a constant current of 30 mA for 3.5 h at 70 °C. After the reaction was completed, the reaction solvent was diluted with 40 mL ethyl acetate and washed with sat. NaHCO3 (aq.) solution three times, dried over Na2SO4 and organic layers were combined and concentrated in vacuo. The resulting residue was purified by silica gel flash chromatography to give the product 3 in a 93% isolated yield.
Data availability
The data reported in this paper are available within the article and its Supplementary Information files. All data are also available from the corresponding author upon request.
References
Evano, G. & Theunissen, C. Beyond Friedel and Crafts: directed alkylation of C−H bonds in arenes. Angew. Chem. Int. Ed. 58, 7202–7236 (2019).
Roberts, R. M. & Khalaf, A. A. Friedel–Crafts Alkylation Chemistry: A Century of Discovery (Marcel Dekker, 1984).
Sartori, G. & Maggi, R. Advances in Friedel–Crafts Acylation Reactions: Catalytic and Green Processes (CRC Press, 2009).
Kishner, N. J. Russ. Phys. Chem. Soc. 43, 582 (1911).
Wolff, L. Methode zum ersatz des sauerstoffatoms der ketone und aldehyde durch wasserstoff. Justus Liebigs Ann. Chem. 394, 86 (1912).
Huang, M. A simple modification of the Wolff–Kishner reduction. J. Am. Chem. Soc. 68, 2487–2488 (1946).
Clemmensen, E. Reduktion von ketonen und aldehyden zu den entsprechenden kohlenwasserstoffen unter anwendung von amalgamiertem zink und salzsäure. Ber 46, 1837 (1913).
Wang, S. & König, B. Catalytic generation of carbanions through carbonyl umpolung. Angew. Chem. Int. Ed. 60, 21624–21634 (2021).
Dai, X.-J., Li, C.-C. & Li, C.-J. Carbonyl umpolung as an organometallic reagent surrogate. Chem. Soc. Rev. 50, 10733–10742 (2021).
Li, J., He, L.-F., Liu, X., Cheng, X. & Li, G.-G. Electrochemical hydrogenation with gaseous ammonia. Angew. Chem. Int. Ed. 58, 1759–1763 (2019).
Ou, W. et al. Room-temperature palladium-catalyzed deuterogenolysis of carbon oxygen bonds towards deuterated pharmaceuticals. Angew. Chem. Int. Ed. 60, 6357–6361 (2021).
Sun, K.-H., Xu, Z.-M., Ramadoss, V., Tian, L.-F. & Wang, Y.-H. Electrochemical deoxygenative reduction of ketones. Chem. Commun. 58, 11155–11158 (2022).
Sun, Z.-H. et al. Electrochemical deoxygenative hydrogenation and deuteration of aldehydes/ketones by protic acids in water. Adv. Synth. Catal. 365, 476–481 (2023).
Bi, C. et al. Electrochemical reduction of diarylketones and aryl alkenes. ChemCatChem 15, e202300258 (2023).
Liu, B. & Liu, Q. A Pincer cobalt complex as catalyst with dual hydrogenation activities for hydrodeoxygenation of ketones with H2. Chin. J. Chem. 42, 3528–3532 (2023).
Xuan, J., Zhang, Z.-G. & Xiao, W.-J. Visible-light-induced decarboxylative functionalization of carboxylic acids and their derivatives. Angew. Chem. Int. Ed. 54, 15632–15641 (2015).
Wei, Y., Hu, P., Zhang, M. & Su, W. Metal-catalyzed decarboxylative C–H functionalization. Chem. Rev. 117, 8864–8907 (2017).
Moon, P. J. & Lundgren, R. J. Metal-catalyzed ionic decarboxylative cross-coupling reactions of C(sp3) acids: reaction development, mechanisms, and application. ACS Catal. 10, 1742–1753 (2020).
Leech, M. C. & Lam, K. Electrosynthesis using carboxylic acid derivatives: new tricks for old reactions. Acc. Chem. Res. 53, 121–134 (2020).
Varenikov, A., Shapiro, E. & Gandelman, M. Decarboxylative halogenation of organic compounds. Chem. Rev. 121, 412–484 (2021).
Zeng, Z., Feceu, A., Sivendran, N. & Gooßen, L. J. Decarboxylation-initiated intermolecular carbon–heteroatom bond formation. Adv. Synth. Catal. 363, 2678–2722 (2021).
Kitcatt, D. M., Nicolle, S. & Lee, A.-L. Direct decarboxylative Giese reactions. Chem. Soc. Rev. 51, 1415–1453 (2022).
Li, J.-B., Huang, C.-Y. & Li, C.-J. Deoxygenative functionalizations of aldehydes, ketones and carboxylic acids. Angew. Chem. Int. Ed. 61, e202112770 (2022).
Nutaitis, C. F. & Gribble, G. W. Reactions of sodium borohydride in acidic media; XV1. A convenient synthesis of 1,1,1-trifluoro-2,2-diarylethanes from arenes and sodium borohydride/trifluoroacetic acid. Synthesis 8, 756–758 (1985).
Sakai, N., Kawana, K., Ikeda, R., Nakaike, Y. & Konakahara, T. InBr3-catalyzed deoxygenation of carboxylic acids with a hydrosilane: reductive conversion of aliphatic or aromatic carboxylic acids to primary alcohols or diphenylmethanes. Eur. J. Org. Chem. 2011, 3178–3183 (2011).
Moriya, T., Takayama, K., Konakahara, T., Ogiwara, Y. & Sakai, N. Indium(III)-catalyzed reductive monoalkylation of electron-rich benzenes with aliphatic carboxylic acids leading to arylalkane derivatives. Eur. J. Org. Chem. 2015, 2277–2281 (2015).
Cabrero-Antonino, J. R., Adam, R., Junge, K. & Beller, M. Cobalt-catalysed reductive C–H alkylation of indoles using carboxylic acids and molecular hydrogen. Chem. Sci. 8, 6439–6450 (2017).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Beil, S. B., Pollok, D. & Waldvogel, S. R. Reproducibility in electroorganic synthesis—myths and misunderstandings. Angew. Chem. Int. Ed. 60, 14750–14759 (2021).
Novaes, L. F. T. et al. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 50, 7941–8002 (2021).
Zhu, C., Ang, N. W. J., Meyer, T. H., Qiu, Y. & Ackermann, L. Organic electrochemistry: molecular syntheses with potential. ACS Cent. Sci. 7, 415–431 (2021).
Yang, J., Qin, H., Yan, K., Cheng, X. & Wen, J. Advances in electrochemical hydrogenation since 2010. Adv. Synth. Catal. 363, 5407–5416 (2021).
Leech, M. C. & Lam, K. A practical guide to electrosynthesis. Nat. Rev. Chem. 6, 275–286 (2022).
Malapit, C. A. et al. Advances on the merger of electrochemistry and transition metal catalysis for organic synthesis. Chem. Rev. 122, 3180–3218 (2022).
Kaefer, N. & Leitner, W. Electrocatalysis with molecular transition-metal complexes for reductive organic synthesis. JACS Au 2, 1266–1289 (2022).
Cheng, X. et al. Recent applications of homogeneous catalysis in electrochemical organic synthesis. CCS Chem. 4, 1120–1152 (2022).
Li, Y., Wen, L. & Guo, W. A guide to organic electroreduction using sacrificial anodes. Chem. Soc. Rev. 52, 1168–1188 (2023).
Liu, Y., Li, P., Wang, Y. & Qiu, Y. Electroreductive cross-electrophile coupling (eXEC) reactions. Angew. Chem. Int. Ed. 62, e202306679 (2023).
Zeng, L., Wang, J., Wang, D., Yi, H. & Lei, A. Comprehensive comparisons between directing and alternating current electrolysis in organic synthesis. Angew. Chem. Int. Ed. 62, e202309620 (2023).
Ramadoss, V., Zheng, Y., Shao, X., Tian, L. & Wang, Y. Advances in electrochemical decarboxylative transformation reactions. Chem. Eur. J. 27, 3213–3228 (2021).
Chen, N., Ye, Z.-H. & Zhang, F.-Z. Recent progress on electrochemical synthesis involving carboxylic acids. Org. Biomol. Chem. 19, 5501–5520 (2021).
Li, L., Yao, Y. & Fu, N. Free carboxylic acids: the trend of radical decarboxylative functionalization. Eur. J. Org. Chem. 26, e202300166 (2023).
Dub, P. A. & Ikariya, T. Catalytic reductive transformations of carboxylic and carbonic acid derivatives using molecular hydrogen. ACS Catal. 2, 1718–1741 (2012).
Pritchard, J., Filonenko, G. A., van Putten, R., Hensen, E. J. M. & Pidko, E. A. Heterogeneous and homogeneous catalysis for the hydrogenation of carboxylic acid derivatives: history, advances and future directions. Chem. Soc. Rev. 44, 3808–3833 (2015).
Qu, R., Junge, K. & Beller, M. Hydrogenation of carboxylic acids, esters, and related compounds over heterogeneous catalysts: a step toward sustainable and carbon-neutral processes. Chem. Rev. 123, 1103–1165 (2023).
Brewster, T. P., Miller, A. J. M., Heinekey, D. M. & Goldberg, K. I. Hydrogenation of carboxylic acids catalyzed by half-sandwich complexes of iridium and rhodium. J. Am. Chem. Soc. 135, 16022–16025 (2013).
vom Stein, T. et al. Highly versatile catalytic hydrogenation of carboxylic and carbonic acid derivatives using a Ru–Triphos complex: molecular control over selectivity and substrate scope. J. Am. Chem. Soc. 136, 13217–13225 (2014).
Korstanje, T. J., van der Vlugt, J. I., Elsevier, C. J. & de Bruin, B. Hydrogenation of carboxylic acids with a homogeneous cobalt catalyst. Science 350, 298–302 (2015).
Cui, X., Li, Y., Topf, C., Junge, K. & Beller, M. Direct ruthenium-catalyzed hydrogenation of carboxylic acids to alcohols. Angew. Chem. Int. Ed. 54, 10596–10599 (2015).
Han, B., Ren, C., Jiang, M. & Wu, L. Titanium-catalyzed exhaustive reduction of oxo-chemicals. Angew. Chem. Int. Ed. 61, e202209232 (2022).
Luo, G.-G. et al. Recent progress in ligand-centered homogeneous electrocatalysts for hydrogen evolution reaction. Inorg. Chem. Front. 6, 343–354 (2019).
Tong, L., Duan, L., Zhou, A. & Thummel, R. P. First-row transition metal polypyridine complexes that catalyze proton to hydrogen reduction. Coord. Chem. Rev. 402, 213079 (2020).
Zhang, P. & Sun, L. Electrocatalytic hydrogenation and oxidation in aqueous conditions. Chin. J. Chem. 38, 996–1004 (2020).
Wang, T., He, F., Jiang, W. & Liu, J. Electrohydrogenation of nitriles with amines by cobalt catalysis. Angew. Chem. Int. Ed. 63, e202316140 (2024).
Beaumier, E. P., Pearce, A. J., See, X. Y. & Tonks, I. A. Modern applications of low-valent early transition metals in synthesis and catalysis. Nat. Rev. Chem. 3, 15–34 (2019).
Manßen, M. & Schafer, L. L. Titanium catalysis for the synthesis of fine chemicals – development and trends. Chem. Soc. Rev. 49, 6947–6994 (2020).
Pang, X. & Shu, X.-Z. Titanium: a unique metal for radical dehydroxylative functionalization of alcohols. Synlett 32, 1269–1274 (2021).
Hilche, T., Younas, S. L., Gansäuer, A. & Streuff, J. A guide to low-valent titanocene complexes as tunable single-electron transfer catalysts for applications in organic chemistry. ChemCatChem 14, e202200530 (2022).
Wu, X., Chang, Y. & Lin, S. Titanium radical redox catalysis: recent innovations in catalysts, reactions, and modes of activation. Chem 8, 1805–1821 (2022).
Di Martino, R. M. C., Maxwell, B. D. & Pirali, T. Deuterium in drug discovery: progress, opportunities and challenges. Nat. Rev. Drug Discov. 22, 562–584 (2023).
Pang, X. & Shu, X.-Z. Reductive deoxygenative functionalization of alcohols by first-row transition metal catalysis. Chin. J. Chem. 41, 1637–1652 (2023).
Peter, A., Agasti, S., Knowles, O., Pye, E. & Procter, D. J. Recent advances in the chemistry of ketyl radicals. Chem. Soc. Rev. 50, 5349–5365 (2021).
Hartley, R. C., Li, J., Main, C. A. & McKiernan, G. J. Titanium carbenoid reagents for converting carbonyl groups into alkenes. Tetrahedron 63, 4825–4864 (2007).
Mc Murry, J. E. & Fleming, M. P. New method for the reductive coupling of carbonyls to olefins. Synthesis of.beta.-carotene. J. Am. Chem. Soc. 96, 4708–4709 (1974).
McMurry, J. E. Carbonyl-coupling reactions using low-valent titanium. Chem. Rev. 89, 1513–1524 (1989).
Bongso, A., Roswanda, R. & Syah, Y. M. Recent advances of carbonyl olefination via McMurry coupling reaction. RSC Adv. 12, 15885–15909 (2022).
Liedtke, T., Spannring, P., Riccardi, L. & Gansäuer, A. Mechanism-based condition screening for sustainable catalysis in single-electron steps by cyclic voltammetry. Angew. Chem. Int. Ed. 57, 5006–5010 (2018).
Liedtke, T., Hilche, T., Klare, S. & Gansäuer, A. Condition screening for sustainable catalysis in single-electron steps by cyclic voltammetry: additives and solvents. ChemSusChem 12, 3166–3171 (2019).
Dang, Y. & Geise, H. J. Low-valent organotitanium species and their applications to organic synthesis. J. Organomet. Chem. 405, 1–39 (1991).
Yuan, X., Lee, K., Eisenberg, J. B., Schmidt, J. R. & Choi, K.-S. Selective deoxygenation of biomass-derived carbonyl compounds on Zn via electrochemical Clemmensen reduction. Nat. Catal. 7, 43–54 (2024).
Acknowledgements
The authors are grateful for the natural population analysis by Prof. Shuanglin Qu’s and financial support from the Natural Science Foundation of China (22301073), the Natural Science Foundation of Hunan Province (2021JJ40043, 2021RC3056), and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
B.W. and J.L. conceived and designed the project. B.W., X.H., and H.B. conducted the experiments. B.W. and J.L. wrote the manuscript. All authors contributed to analyzing the data and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yahui Zhang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, B., Huang, X., Bi, H. et al. Electroreductive alkylations of (hetero)arenes with carboxylic acids. Nat Commun 15, 4970 (2024). https://doi.org/10.1038/s41467-024-49355-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-49355-1
- Springer Nature Limited