

Abstract
Designing active and stable electrocatalysts with economic efficiency for acidic oxygen evolution reaction is essential for developing proton exchange membrane water electrolyzers. Herein, we report on a cobalt oxide incorporated with iridium single atoms (Ir-Co3O4), prepared by a mechanochemical approach. Operando X-ray absorption spectroscopy reveals that Ir atoms are partially oxidized to active Ir>4+ during the reaction, meanwhile Ir and Co atoms with their bridged electrophilic O ligands acting as active sites, are jointly responsible for the enhanced performance. Theoretical calculations further disclose the isolated Ir atoms can effectively boost the electronic conductivity and optimize the energy barrier. As a result, Ir-Co3O4 exhibits significantly higher mass activity and turnover frequency than those of benchmark IrO2 in acidic conditions. Moreover, the catalyst preparation can be easily scaled up to gram-level per batch. The present approach highlights the concept of constructing single noble metal atoms incorporated cost-effective metal oxides catalysts for practical applications.
Similar content being viewed by others


Introduction
Centuries of industrialization and human activities have consumed enormous amounts of fossil fuels, which has inevitably led to the current energy crisis and global warming1. Hydrogen (H2) is considered a promising alternative energy carrier of the future owing to its high energy density and potential for carbon-free emission2. Among many H2 production methods, water electrolysis has received much attention as a feasible technology for practical applications. Nevertheless, due to the sluggish kinetics of the complex four-electron transfer process, the oxygen evolution reaction (OER) at the anode side of the water electrolyzer demonstrates high overpotentials, which severely limit the overall operation efficiency and impede the large commercialization of water electrolyzers3,4,5,6. Therefore, tremendous effort has been devoted to the rational design of robust electrocatalysts which improve the OER activity and energy conversion efficiency.
Over the past few decades, the noble metal oxides based on iridium (Ir) and ruthenium (Ru) are considered the state-of-the-art OER electrocatalysts. However, their widespread applications are limited by the cost and scarcity of Ir and Ru7,8,9,10. As a result, the first row 3d transition metal oxides and their derivatives, with merits of high abundance and low cost have received much attention as alternative OER candidates11,12,13,14,15. Among all 3d transition metal oxides, cobalt-based oxides such as spinel Co3O4 have been widely used to activate OER activity, as demonstrated by both experimental data and theoretical calculations16,17,18,19. Nevertheless, Co3O4 still suffers from an overpotential of more than 400 mV for the OER at a current density of 10 mA cm−2, which is far from meeting the requirements for practical applications. Moreover, Co3O4 is unstable under the harsh OER conditions at the high oxidizing potential in corrosive media such as an acidic electrolyte20,21,22. It is well known that the 4d/5d noble elements possess a large d-electronic wave-function spatial extent, generating versatile electronic structures via the interaction between 3d and 4d/5d orbitals, which is beneficial for enhancing OER activity. One available method that has been frequently used is incorporating bulk amounts of noble metals with Co or its derivatives to obtain hybrid 3d/4d or 3d/5d nanomaterials as OER electrocatalysts23,24,25. In this regard, Li et al. reported that the Sr2CoIrO6-δ, which benefits from a synergy between Co and Ir active sites, exhibits a low overpotential toward OER26. Pi and co-workers also demonstrated that IrCo bimetallic nanoclusters can be employed as efficient OER electrocatalysts27. In addition, Shah et al. developed a catalyst with single Co atoms doped on RuO2 sphere, yielding remarkable catalytic performances for water splitting28. Notwithstanding these efforts to modify Co-based nanomaterials as alternative OER catalysts, it remains difficult to tip the balance between low cost and high performance. Recently, the strategy of constructing highly dispersed single-sites catalysts has attracted significantly attentions for a range of electrocatalytic reactions29. Downsizing the materials into isolated atoms is beneficial for reducing the amount of bulk metal, which can greatly maximize the atom-utilization efficiency, leading to remarkable catalytic mass activity30,31,32. Additionally, the unique electronic structure of uniformly dispersed active sites usually results in a strong interaction with the host material, effectively adjusting the electrochemical microenvironment over catalysts and potentially boosting the electrocatalytic activity33,34,35. Motivated by the abovementioned guidelines, the integration of spinel Co-based oxide with noble metal single atoms as the prospective OER electrocatalyst, possesses advantages of minimized noble metal usage and optimized catalytic performances. Nevertheless, the large lattice discrepancy between the diverse atoms and strong binding energy between the same atoms, are great challenges to disperse homogeneous noble metal single sites in the transition metal oxide host36. And the production of traditional methods to prepare the single-atom materials such as impregnation are usually limited to milligram level, it is extremely urgent to explore a mass-production method to meet the requirements for practical applications.
In this work, we deploy a facile and economical strategy to prepare atomically dispersed Ir atoms doped in spinel Co3O4 (Ir-Co3O4) via a mechanochemical method. By introducing a trace amount of Ir (~1.05 at%), Ir-Co3O4 displays a remarkable OER overpotential of 236 mV in an acid medium at the current density of 10 mA cm−2, which is significantly lower than that of as-prepared Co3O4 (412 mV). Significantly, both the normalized mass activity and TOF of Ir-Co3O4 can reach almost two order magnitudes higher than those of commercial IrO2 at an overpotential of 300 mV, respectively. Meanwhile, the stability can also be extended much longer after incorporating Ir single atoms within the lattice of Co3O4. Using operando X-ray absorption near edge structure (XANES) at the Ir-L3 and Co-K edges, it is observed that the Ir species are partially oxidized to OER-active Ir>4+, and the elevated valence states with increased voltage also indicate that both high-valence Ir and Co atoms with their bridged electrophilic O ligands serve as active sites, which are jointly responsible for charge transfer in electrochemical reactions. In addition, density functional theory (DFT) calculations confirm the improved electronic conductivity and facile energy barrier contributed by the incorporated Ir single atoms, resulting in a high OER catalytic activity. Moreover, the preparation of Ir-Co3O4 can be easily scaled up to achieve gram-level per batch with negligible activity loss, demonstrating this strategy as an economical approach to prepare advanced electrocatalysts in a larger scale for practical applications.
Results
Structural characterizations of Ir-Co3O4
Ir-Co3O4 was synthesized based on the mechanochemical method, in which a solid solution of NaCl and metal precursors were formed by ball milling and then calcination (see Methods for details). The morphology of Ir-Co3O4 was initially characterized by scanning electron microscopy (SEM). As shown in Fig. 1a, Ir-Co3O4 reveals nanosized and flake morphology. Typical transmission electron microscopy (TEM) images show that Ir-Co3O4 is structured as flexible ultrathin nanosheets with thickness of about 3 nm as indicated by atomic force microscopy (AFM) images. The ultrathin 2D structure of Ir-Co3O4 can potentially maximize the exposed active sites on the surface and increase the contact area with the electrolyte, leading to the enhanced catalytic performance (Fig. 1b, c & Supplementary Fig. 1)37. Detailed structural analysis by high-resolution TEM (HR-TEM) confirms that Ir-Co3O4 has lattice spacings of 2.48 Å and 3.02 Å, which can be ascribed to the Co3O4 (311) and (220) facets, respectively. Furthermore, due to the introduction of NaCl as the hard template in preparation, pores are generated and can be observed in the lattice structure of Ir-Co3O4. In general, those mesopores often result in more exposed active atoms on the edge, which are favorable for catalytic activity (Fig. 1d & Supplementary Fig. 2)38. Meanwhile, pure Co3O4 synthesized by the same method exhibits no evident differences in morphology as compared with Ir-Co3O4 (Supplementary Fig. 3). The crystal structures of Ir-Co3O4 and as-synthesized Co3O4 were characterized by X-ray diffraction (XRD) analysis, revealing similar patterns of face-centered cubic symmetry (space group: Fd-3 m) of the spinel structure. Rietveld refinements were further performed on those XRD patterns: the refined lattice parameters are a = 8.0972(5) Å, b = 8.0972(5) Å, c = 8.0972(5) Å, and V = 530.89(6) Å3 for Ir-Co3O4, as compared to a = 8.0754(3) Å, b = 8.0754(3) Å, c = 8.0754(3) Å, and V = 526.61(3) Å3 for as-prepared Co3O4 (Supplementary Figs. 4, 5 & Supplementary Table 1, 2). The expanded crystal cells of Ir-Co3O4 may be attributed to Ir atoms with a larger radius incorporated into the lattice of Co3O4. Subsequent composition evaluation of Ir-Co3O4 by inductively coupled plasma-optical emission spectrometry (ICP-OES) and SEM energy-dispersive spectroscopy (SEM-EDS) confirms that Ir has an atomic ratio of 1.05% (Supplementary Fig. 6). In addition, the possible Fe impurity in Ir-Co3O4 derived from the stainless-steel ball milling device was also determined to be 0.0667 wt% by ICP-OES, which can be ignored compared to the contents of other elements.

a SEM, (b) TEM, (c) AFM and (d) HR-TEM images of Ir-Co3O4. Inset in (c) is the height profile of Ir-Co3O4 along the dashed line. e AC HAADF-STEM image and the corresponding FFT image (inset) of Ir-Co3O4. f The enlarged area in (e), with the Ir single atoms marked in circles, and (g) intensity profiles along the dashed rectangles. h 3D atom-overlapping Gaussian function fitting mapping of the selected area in (e). i AC HAADF-STEM image and the corresponding elemental mappings of Ir-Co3O4.
To determine the distribution of Ir species at the atomic scale, aberration corrected high angle annular dark field-scanning transmission electron microscopy (AC HAADF-STEM) images were collected on Ir-Co3O4. The corresponding fast Fourier transform (FFT) image identifies the exposure of (33\(\mathop{1}\limits^{-}\)), (311) and (02\(\mathop{2}\limits^{-}\)) planes of Co3O4 along the [\(\mathop{2}\limits^{-}\)33] zone axis. Interestingly, high-magnification images demonstrate the presence of monodispersed Ir single atoms (bright dots marked by red circles in Fig. 1e, f). On account of the higher Z number, the Ir atom brightness is more intense than neighboring Co atoms39. The dispersion of isolated Ir atoms was further verified by the profiles along the dashed rectangles in the AC HAADF-STEM image, in which the different atomic intensities can be clearly distinguished (Fig. 1g). Additionally, the 3D atom-overlapping Gaussian function fitting mapping of the selected area in Fig. 1e is employed to observe the atomic distribution. As shown in Fig. 1h, the highest peaks representing Ir atoms are evenly dispersed, providing further evidence of isolated atoms. Elemental mapping images also show a homogeneous dispersion of Ir, Co, and O atoms throughout the entire Ir-Co3O4 sample (Fig. 1i & Supplementary Fig. 7).
Electronic and atomic structure analysis of Ir-Co3O4
To understand the electronic structure of Ir-Co3O4, we carried out X-ray photoelectron spectroscopy (XPS). All XPS spectra were calibrated with C 1s using a binding energy of 284.8 eV. The survey XPS spectra of Ir-Co3O4 demonstrate the existence of Ir, Co and O elements on the surface (Supplementary Fig. 8). As shown in Fig. 2a, a typical IrO2 XPS shows one sets of doublets at 62.5 and 65.4 eV, which can be attributed to Ir4+. For Ir-Co3O4, the Ir 4f spectrum can be deconvoluted into two sets of doublets centered at 61.8 / 63.7 eV and 62.4 / 65.3 eV, which can be attributed to Ir4+ and Ir3+, respectively40,41,42. Accordingly, the Ir species on the surface of Ir-Co3O4 is oxidized with chemical states between +3 and +4. The peak located at 60.6 eV is produced by Co 3p. The introduction of Ir affects the electronic structure of Co3O4; as revealed in Fig. 2b, the main peaks of Co 2p3/2 of both Ir-Co3O4 and as-synthesized Co3O4 present signals that can be identified as Co2+ and Co3+, respectively. Nevertheless, the Co2+ over Co3+ ratio is increased by 8.33 % after Ir species are introduced (the ratios of Co2+: Co3+ in Co3O4 and Ir-Co3O4 are 0.48: 1 and 0.52: 1, respectively), suggesting a decreased valence state of Co on the surface of Ir-Co3O4. Moreover, since the incorporated Ir possesses stronger covalency, the O 1s XPS spectrum of Ir-Co3O4 reveals a more positive binding energy and larger lattice oxygen M-O concentration than that of Co3O4, which is induced by the incorporation of Ir atoms (Fig. 2c)43.

a Ir 4f XPS spectra of Ir-Co3O4 and IrO2. b Co 2p XPS spectra and (c) O 1 s XPS spectra of Ir-Co3O4 and Co3O4. d XANES spectra of Ir-L3 edge on Ir-Co3O4, Ir foil and IrO2. d The normalized Ir-L3 edge XANES spectra of Ir-Co3O4, Ir foil and IrO2. e Ir-L3 edge EXAFS spectra and (f) corresponding wavelet-transformed k3-weighted EXAFS spectra of Ir-Co3O4, Ir foil and IrO2. g The normalized Co-K edge XANES spectra and (h) Co-K edge XANES spectra of Ir-Co3O4 and Co3O4.
X-ray absorption spectroscopy (XAS) was conducted with the aim of distinguishing the electronic structure and the local atomic environment of Ir-Co3O4. The energy position of the white line of X-ray absorption near edge structure (XANES) at the 5d transition metals is highly sensitive to their valence states44,45,46,47,48. And it is well known that an increase of the valence state of the 5d metal ion by one results in a shift of the L2,3 XAS spectra by one or more eV toward higher energies44. Figure 2d shows the Ir L3 XANES of Ir-Co3O4 together with Ir foil and IrO2 for comparison. One can observe that the white line of Ir-Co3O4 shifts by 1.6 eV to a higher energy relative to Ir foil, but is located extremely close to that of IrO2, demonstrating that the Ir chemical state is close to +4, which is distinct from those in nanoparticles or supported single atoms. In addition, the white line intensity is in principle proportional to the d orbital density of states that are affected by the presence of multiple oxidation states and various electron configuration. The higher integrated white line intensity of Ir-Co3O4 indicates that it has more empty d orbital states (d holes) than IrO2, which may lead to enhanced electrocatalytic performances. Importantly, the information derived from Ir L3-edge extended X-ray absorption fine structure (EXAFS) of Ir-Co3O4 shows two distinct peaks in R space, in which the first peak can be clearly attributed to Ir-O bonds (Fig. 2e). The second bond cannot be distinguished directly due to the similar lengths of Ir-Ir bond (2.707 Å) and Ir-Co bond (2.91 Å) (discussed below). It was further recognized by the wavelet transform (WT)-EXAFS analysis, which exhibits two characteristic regions: a first shell domain for Ir-O scattering with a local maximum at R = 1.58 Å and k = 6.62 Å−1 and a second shell domain at R = 2.61 Å and k = 8.02 Å−1. Although the second shell domain in Ir-Co3O4 has a similar R value on that of Ir-Ir scattering in Ir foil (R = 2.65 Å and k = 10.04 Å−1), their k values are quite different, implying the existence of Ir-Co rather than Ir-Ir in the second shell scattering (Fig. 2f). Furthermore, a model-based EXAFS fit of Ir-Co3O4, IrO2 and Ir foil were performed and the results indicate that no Ir-Ir scattering can be discovered and that the second scattering can be attributed to Ir-Co with a distinct bond distance, which represents Ir-O-Co in the crystal structure (Supplementary Fig. 9 & Supplementary Table 3). Therefore, based upon of advanced electronic microscopy and spectroscopic techniques, we conclude that the Ir species exist as single atoms and are homogeneously dispersed in the Co3O4 host.
Unlike the XANES spectra at the 5d-L3 edge, where the strong white line represents an unoccupied 5d state, the dominate peak at the 3d-K edge represents an unoccupied 4p state, whose energy position and shape are determined primarily by the crystal structure. In such a case, the valence state of 3d elements can be confirmed by the energy position of the leading edge (at a normalized absorption position of 0.8)48,49,50. The sensitivity of the absorption edge to the valence state of 3d elements originates from a relatively large Ucd (1s core hole and 3d Coulomb interaction) as compared with Udp (3d and 4p Coulomb interactions). As shown in Fig. 2g, the normalized Co K-edge XANES of Ir-Co3O4 clearly displays a slightly decreased valence state of the Co species induced by Ir incorporation. The corresponding Co K-edge EXAFS of Co3O4 and Ir-Co3O4 reveal three distinct peaks, identified as Co-O, octahedral Co-Co and tetrahedral Co-Co bonds, respectively51,52. However, a lower intensity of the Co-O and Co-Co bonds can be found in Ir-Co3O4 in comparison with the Co3O4, indicating higher atomic chaos caused by introducing Ir atoms (Fig. 2h). The WT-EXAFS analysis on Co K-edge is consistent with the EXAFS results (Supplementary Fig. 10).
Electrochemical performance of Ir-Co3O4
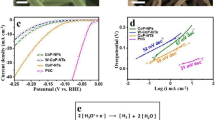
The OER performances of Ir-Co3O4 and as-prepared Co3O4 in 0.5 M H2SO4 were investigated on glassy carbon electrode (GCE) together with the commercial IrO2 and commercial Co3O4 (denoted as IrO2 and C-Co3O4, respectively) for comparison (Supplementary Fig. 11). All measured potentials in this study were calibrated to the reversible hydrogen electrode (RHE) scale (Supplementary Fig. 12). As shown in Supplementary Fig. 13, Ir-Co3O4 exhibits a negatively shifted Co redox peak (III/IV) in the cyclic voltammetry (CV) curves compared to the Co3O4, indicating the isolated Ir sites can expedite the proton-electron transfer-mediated activation of Co to OER-active Co4+53. As expected, Ir-Co3O4 exhibits the best OER performances among all studied samples. In terms of reaching the current density of 10 mA cm−2, Ir-Co3O4 requires an overpotential (ƞ) of 236 mV, which is lower than that of Co3O4 (412 mV) and C-Co3O4 (511 mV). Notably, the low overpotential of Ir-Co3O4 even suppresses that of benchmark IrO2 (298 mV) with the same amounts of catalysts loading on electrode, as shown in Fig. 3a. Likewise, Ir-Co3O4 displays the lowest onset potential among these catalysts, showing that it has the fastest OER activation rate (Supplementary Fig. 14). The LSV curves of those catalysts without iR compensation are also displayed in Supplementary Fig. 15. In addition, the outstanding OER activity of Ir-Co3O4 is also reflected by the smallest Tafel slope of 52.6 mV dec−1 compared to that of IrO2 (75.8 mV dec−1), Co3O4 (109.8 mV dec−1) and C-Co3O4 (131.3 mV dec−1), indicating that the OER kinetic of Ir-Co3O4 is significantly accelerated (Fig. 3b). Corresponding graphs that plot the Tafel slope as a function of the overpotential on these catalysts are depicted in Supplementary Fig. 16, from which the boundary between kinetically-limited and mass transport limited currents can be clear distinguished. An obvious tendency of the boundary is followed as: Ir-Co3O4 < IrO2 < Co3O4 < C-Co3O4. The evaluation of an electrocatalyst system is generally assessed by two key factors: specific activity and turnover frequency (TOF). The electrochemical active surface area (ECSA) was firstly estimated from the double-layer capacitance (Cdl) measurements as shown in Supplementary Fig. 1754. By normalizing the OER current to ECSA at ƞ = 300 mV, Ir-Co3O4 shows the highest specific activity (0.098 mA cm−2ECSA), almost 2.8 and 9.8 times higher than those of IrO2 (0.035 mA cm−2ECSA) and Co3O4 (0.01 mA cm−2ECSA). Moreover, Ir-Co3O4 delivers a high intrinsic activity of TOF value normalized to Ir sites per geometric area of 1.665 s−1, which is nearly 70-fold higher than that of IrO2 (0.0237 s−1) (Fig. 3c). Also, Ir-Co3O4 shows a higher TOF value than both Co3O4 and C-Co3O4 when normalized to Co sites per geometric area. In addition, ECSA-normalized TOF values further indicate the better intrinsic activity of Ir-Co3O4 (Supplementary Fig. 18). BET-normalized specific activities were calculated to exclude the morphology effects on activity assessment, in which Ir-Co3O4 also outperforms that of Co3O4 (Supplementary Fig. 19). The above results confirm the incorporated Ir single atoms can efficiently boost the catalytic performances of Co spinel oxide beyond that of the noble IrO2. A synopsis of previous literatures in Fig. 3d, highlights the merits of Ir-Co3O4, including low overpotential and Tafel slope are shown to be superior to most reported Ir-based OER electrocatalysts (Supplementary Table 4). In addition to the consideration of catalytic performance, cost-effectiveness is also of great significance for catalysts for practical applications. Thereafter the OER polarization currents of Ir-Co3O4 and commercial IrO2 were normalized by the loading mass of Ir at ƞ = 300 mV. In addition, the mass activities were calculated from the LSV on carbon paper electrodes with higher electrode surface areas (1 cm2) (Supplementary Fig. 20). As revealed in Fig. 3e, the mass activity of Ir-Co3O4 is 3343.37 A g−1Ir, which is 51.16 times higher than that of IrO2 (65.35 A g−1Ir) at the overpotential of 300 mV. The superb mass activity derived from each Ir atoms indicates the remarkable economic efficiency of Ir-Co3O4 towards OER. Electrochemical impedance spectroscopy (EIS) measurements of these four samples were tested under the same condition, as shown in Fig. 3f. In comparison with the charge transfer resistance (Rct) among IrO2 (4.18 Ω), Co3O4 (20.55 Ω) and C-Co3O4 (115.5 Ω), Ir-Co3O4 displays the smallest Rct of 2.37 Ω by establishing the equivalent circuit and fitting the Nyquist plots, implying that it has the lowest charge transfer resistance and the fastest charge transfer rate for the OER reaction (Supplementary Fig. 21 & Supplementary Table 5)55.

a Polarization curves of Ir-Co3O4, IrO2, Co3O4 and C-Co3O4 in 0.5 M H2SO4 at a scanning rate of 5 mV s−1. b Tafel plots derived from the polarization curves in (a). c Corresponding specific activities of Ir-Co3O4, IrO2, Co3O4 and C-Co3O4; TOF values of Ir-Co3O4 and IrO2 at the overpotential of 300 mV. d Comparison of overpotentials and Tafel plots in reported Ir-based OER electrocatalysts. e Comparison of the mass activities normalized to Ir mass on Ir-Co3O4 and IrO2. f EIS Nyquist plots and fitting curves of samples recorded at the 1.43 V vs. RHE. g Chronopotentiometric measurements of Ir-Co3O4, IrO2, Co3O4 and C-Co3O4 at 10 mA cm−2, carbon paper was used as the catalyst support. h Dissolved Ir (left-y axis) and Co (right-y axis) ion concentrations measured for Ir-Co3O4, IrO2, Co3O4 and C-Co3O4 in the electrolyte by ICP-OES. i Generated O2 amount and corresponding Faradaic efficiency over a range of overpotentials on Ir-Co3O4. Note: error bars represent the standard deviation of three independent measurements.
In addition to the activity, durability is another crucial criterion for the electrocatalytic performance of catalysts towards OER under operating conditions. As plotted in Fig. 3g, Ir-Co3O4 shows long-term stability with a current density of 10 mA cm−2 for almost 30 h, which is superior to that of IrO2, Co3O4 and C-Co3O4 catalysts. Specifically, the commercial IrO2 and as-synthesized Co3O4 can only maintain the current densities for nearly 15 h and 9 h, respectively, before they deactivate. CV scanning experiments further highlight that Ir-Co3O4 exhibits a negligible increase of overpotential after 3000 cycles, whereas both Co3O4 and C-Co3O4 lost most of their activities (Supplementary Fig. 22). Both chronopotentiometric and CV scanning measurements demonstrate that the isolated Ir single sites enable activating the spinel Co oxide more durable in harsh OER environment. Further ICP-OES investigations show that leaching occurs preferentially at the Co species in Ir-Co3O4, but the dissolution effect of hosting Co3O4 is alleviated by dispersed Ir single sites during OER reaction. In contrast, IrO2, Co3O4 and C-Co3O4 suffer from severe Ir and Co ions leaching after the OER durability test (Fig. 3h & Supplementary Fig. 23)56. The enhanced stability of Ir-Co3O4 is induced by the doped effect of Ir single atoms, which is the main reason for the observed long-term stability in the acidic medium. In addition, the morphology characterizations by means of SEM and TEM reveal that Ir-Co3O4 preserves the original ultrathin nanosheets with slight fractures after the chronopotentiometry at 10 mA cm−2. No obvious structural changes can be found from the XRD pattern and HR-TEM images. Elemental mapping images show that Ir, Co and O elements are still homogeneously distributed over the entire Ir-Co3O4 sample after OER (Supplementary Fig. 24). However, both the Ir 4f and Co 2p XPS spectra of post-OER Ir-Co3O4 show increased valence states of Ir and Co, which is common for OER catalysts under oxidative voltages. The O 1s XPS binding energy of post-OER Ir-Co3O4 shifts to a more positive position, further confirming the increase of valence state on overall catalyst after the stability test (Supplementary Fig. 25). The stabilities of Ir-Co3O4 and IrO2 were further tested at a higher current density of 50 mA cm−2. As shown in Supplementary Fig. 26, Ir-Co3O4 maintains the long-term stability up to 6 h, while IrO2 loses its activity after only 2 hours, further indicating the activity retention of Ir-Co3O4 even at a high current density. On the other hand, complex OER is always accompanied with other side reactions such as catalyst/support oxidation, material dissolution and capacitive current effects, which may lead to the overestimation of actual activity and thus it is essential to estimate the associated OER Faradaic efficiency (FE) of the catalyst57. Gas chromatography (GC) was used to continuously monitor the generated O2 amount and calculate the FE on Ir-Co3O4 over a wide range of overpotentials (Supplementary Fig. 27). As depicted in Fig. 3i, the corresponding O2 production increases with the increasing OER overpotentials; the calculated FE for OER remains at almost 100% at small overpotentials but drops slightly to 97% for large overpotentials. The relatively high FE values signify that the majority of energy is applied to drive OER and almost no side reactions occur on Ir-Co3O4. Overall, the high electrode/intrinsic activity and electrochemical durability conjointly endow Ir-Co3O4 as a promising OER electrocatalyst candidate for practical applications in the acidic water electrolyzer, namely proton exchange membrane water electrolyzer. Going further, practical applications usually demand that the catalysts preparation process is contamination-free and non-toxic, as well as can easily realize gram-level preparation of catalysts in one batch58. Nevertheless, traditional synthesis methods for single-atom materials, including impregnation and solvothermal strategies usually require pollution-carrying organic solvent and their productions are seriously limited. For example, the similar atomic Ir-doped Co oxides have also been reported, but in which the multistep impregnation methods they employed can only prepare the electrocatalysts with milligram-level productions59,60. In contrast, the mechanochemical protocol we adopted for synthesizing Ir-Co3O4 is environmental-friendly, adding only NaCl and NaOH for preparation besides metal precursors; the production can be readily scaled up to 2.5 g per batch. Resulting SEM, TEM, HR-TEM images, XRD pattern and SEM-EDS spectrum reveal no obvious structural and compositional changes on the mass-produced Ir-Co3O4. Elemental mappings show that Ir, Co and O are homogeneously distributed over the Ir-Co3O4. In addition, this mass-produced sample can largely maintain its original OER activity, demonstrating the potential of Ir-Co3O4 for application in practical water electrolyzer (Supplementary Fig. 28).
Mechanistic insights into OER process
Since the doped Ir single atoms promote the electrocatalytic performances of Co spinel oxide, the operando XANES tests at both Ir-L3 edge and Co K-edge were carried out on Ir-Co3O4 to learn more about the active sites and examine the chemical states evolution during the OER process. In the measurements, the operando XANES data were sequential collected under the open circuit potential (OCP), one representative potential (1.6 V vs. RHE) and then back to OCP again. Corresponding schematic diagram of the custom-built electrochemical cell and the obtained current-time (i-t) curves under applied voltages used for collecting the operando XAS data were depicted in Supplementary Fig. 29. As shown in Fig. 4a, the Ir-L3 edge white line position at OCP shows a slight positive shift in energy as compared with that of fresh Ir-Co3O4, probably resulting from the delocalization of electron caused by adsorbed H2O in the electrolyte. Upon increasing the applied voltage to 1.6 V, the peak position of Ir-L3 shifts higher in energy by 0.6 eV, which suggests that isolated Ir atoms are evidently oxidized with an increased valence state during OER. In addition, the oxidized Ir atoms retain high chemical states even when the applied potential is reversed to OCP, indicating that the valence change is irreversible on Ir-Co3O4 (Fig. 4c). As depicted in Supplementary Fig. 30, through further comparison of the white line position with standard references, it can be seen that the Ir-L3 energy peak of Ir-Co3O4 at the anodic potential of 1.6 V is located between the standard spectra of the Ir4+ reference La2CoIrO6 and the Ir5+ reference Sr2CoIrO6, confirming it has a chemical state between +4 and +5. The average state of Ir single atoms is further estimated to be +4.46 according to the absorption energy-valence state standard curve26,48. In other words, there is a transition from Ir4+ state to Ir>4+ state in part of the Ir-Co3O4 catalysts during the electrochemical reactions. The phenomena discovered by operando XAS is definitely meaningful, as demonstrated by previous research showing that the Ir species with high valence state in combination with electrophilic O ligands act as the active sites of OER61,62. It is believed that the formed covalent Ir-O sits and electrophilic O ligands promote the O-O formation via a nucleophilic attack and thus support the enhanced OER performances. This suggests that the isolated Ir atoms are efficiently activated, endowing Ir-Co3O4 with enhanced activity in OER63,64,65,66.

a The Ir L3-edge XANES and (b) the Co K-edge XANES of Ir-Co3O4 measured in 0.5 M H2SO4 under pristine state, OCP, OER operating condition of 1.6 V and OCP after OER operation. Ir L3-edge XANES of Ir foil and IrO2, and Co K-edge XANES of Co3O4 are used as references. c The white line positions and the absorption edge positions of all measured XANES. d The top and side panels of charge density difference on Ir-Co3O4. e Comparison of the d-band center of Ir-Co3O4 and Co3O4. Corresponding Co 3d, O 2p and Ir 5d pDOS spectra of (f) Ir-Co3O4 and (g) Co3O4. h The Gibbs free energy diagrams of the four-electron OER process on the Ir sites and Co sites of these catalysts under the applied overpotentials of 1.23 V vs. RHE, respectively.
Subsequently, the valence change of Co species in Ir-Co3O4 was also probed by operando Co K-edge XANES (Fig. 4b). As shown in Fig. 4c, the Co-K absorption edge of Ir-Co3O4 at OCP is close to that of a Co3O4 reference, but it gradually shifts to a higher energy by 1.0 eV under an applied voltage of 1.6 V, indicating an increase in the Co valence state by 0.47 through comparison with the standard references of Co2+ reference (La2CoIrO6), Co3+ reference (EuCoO3) and Co4+ reference (BaCoO3) in Supplementary Fig. 3148. When the applied voltage was set back to OCP, the Co species in Ir-Co3O4 remain in the highly oxidized states. As a consequence, the continuously increased Ir and Co chemical states of Ir-Co3O4 under operating voltages demonstrates that the both the Ir single atoms and Co atoms are functionalized as the OER active sites in Ir-Co3O4, which are responsible for charge transfer in catalytic reactions together.
To further understand the OER mechanism and the synergistic function on Ir-Co3O4, a theoretical study was performed based on density functional theory (DFT) calculations. As shown in Fig. 4d, the charge density difference on Ir-Co3O4 indicates that the incorporation of Ir into Co3O4 can effectively regulate the charge redistribution, where the Ir atom tends to lose electrons and the delocalized electrons accumulate around adjacent Ir-O bonds. This result also confirms a strong electronic interaction between the Ir single site and the host Co3O467. In addition, as shown in Fig. 4e it can be obtained that the d-band center position of Ir-Co3O4 (−1.512 eV) is higher than that of Co3O4 (−1.834 eV). The positive shift of d-band center would stabilize the interaction between the catalyst surface and the adsorbates, which is in strong alignment with the calculated free energy in Fig. 4h68. The projected density of states (pDOS) suggests that the introduction of Ir causes new hybridized electronic states in Ir-Co3O4, leading to a widening of the total density of states (TDOS) near the Fermi level, whereas the TDOS of Co3O4 displays a band gap. The apparent difference demonstrates that the introduction of Ir single atoms endow the Co3O4 with metallic characters, leading to a better electronic conductivity than that of Co3O4 for faster OER kinetics, which is consistent with our EIS measurements. Furthermore, the synergistic function between Ir atom and Co3O4 was investigated by checking the Co 3d, O 2p and Ir 5d pDOS, in which it can be clearly observed that the Ir 5d orbit is substantially overlapped with Co 2p orbit in pDOS, suggesting the strong interaction and more covalent character between Co and Ir atoms (Fig. 4f, g)26. In addition, the Gibbs free energy changes of acidic OER intermediates (OH*, O*, OOH*) in the elementary steps under the applied potential of 1.23 and 0 V vs. RHE are displayed in Fig. 4h and Supplementary Fig. 32, respectively. The potential determining steps (PDS) of Ir sites were first calculated and results reveal that the limiting steps occur at the conversion from O* to OOH* on both Ir-Co3O4 and IrO2. Their corresponding PDS values are 0.288 eV and 0.378 eV, suggesting the lower energy barrier and better OER performance of Ir-Co3O4. Furthermore, the calculations on Co sites indicate that the PDS on Co3O4 is primarily derived from the transformation from H2O to OH* with an energy barrier of 1.31 eV. However, the PDS is changed as the formation of OOH* and the corresponding energy barrier is further reduced to 1.11 eV on Ir-Co3O4, emphasizing the promoting effect toward OER derived from Ir single sites29. It is believed that the electrophilic characters render these oxygen ligands caused by high-valence Ir susceptible to nucleophilic acid-base-type O-O bond formation at reduced kinetic barrier, leading to enhanced OER activity61,62. Therefore, both the operando XAS characterizations and DFT calculations conjointly demonstrate that the remarkable catalytic performance on Ir-Co3O4 is due to the synergistic cooperation between Ir and Co single sites with their bridged O ligands as well as the distinctly reduced energy barrier toward OER.
Discussion
In conclusion, the active Ir-Co3O4 OER catalyst was successfully synthesized via a facile and economical mechanochemical method. Both AC HAADF-STEM characterization and XAS technique confirm that isolated Ir single atoms are homogeneously dispersed in the Co3O4 host material. Consequently, OER measurement in acidic medium reveals that the overpotential of as-prepared Co3O4 (412 mV) sharply decreases to 236 mV by doping a trace amount of Ir atoms, and its long-term stability is prolonged. Notably, the normalized mass activity and TOF of Ir-Co3O4 are higher than those of commercial IrO2. By virtue of the operando XANES analysis, we recognize that the Ir single atoms are partially activated to higher valence under anodic voltages, simultaneously both Co and Ir atoms with their electrophilic O ligands served as active sites are synergistically responsible for charge transferring during OER process. DFT calculations further demonstrate the improved electronic conductivity and lower energy barrier towards OER by the electrophilic characters of oxygen ligands induced by high-valence Ir, boosting OER activity. Additionally, in contrast to most traditional synthesis methods for single-atom catalysts, which are limited to milligram-level production, the Ir-Co3O4 preparation can be easily scaled up to gram-level production with negligible activity loss. This study highlights the rational design of a promising, cost-effective and energy-efficient catalyst for boosting oxygen evolution in the acidic medium, and this prototype would potentially stimulate industrial interest in a larger scale for practical applications.
Methods
Materials
Iridium trichloride (IrCl3, solid, 99.9%) was purchased from Ark Pharm. Cobalt chloride hexahydrate (CoCl2.6H2O, solid, AR) was purchased from Aladdin. Sodium chloride (NaCl, solid, 99.8%) and sodium hydroxide (NaOH, solid, 97%) were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Cobalt oxide (Co3O4, solid, 99.7%) was purchased from Alfa Aesar. Iridium oxide (IrO2, solid, 99.9%) was purchased from Innochem. Isopropanol (C3H8O, liquid, ≥ 99.7%) and sulfuric acid (H2SO4, liquid, ≥ 96%) were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China).
Catalyst synthesis
The preparation of Ir-Co3O4 was based on mechanochemical method. In a typical synthesis, 475.9 mg CoCl2.6H2O (2 mmol) and 29.9 mg IrCl3 (0.1 mmol) were initially mixed in agate mortar for 5 min. Afterwards, 0.5 g NaCl was added into the evenly mixed precursors and ground for 10 min. A homogeneous powder was obtained and then put into a high-energy milling pot (Planetary Mono Mill, Pulverisette 6, Fritsch) with stainless steel balls for grinding for 1 h under a rotation speed of 500 rpm. After the first milling, 1 mL NaOH solution (5 mmol mL−1) was added into the above mixture and ball milling was continued for 1 h. Finally, the obtained slurry was collected from the milling pot and dried at 60 °C in an oven under air atmosphere, then it was calcined in air at 350 °C for 6 h with a heating rate of 5 °C min−1. After calcination, the sample was washed by water 3 times and dried overnight at 80 °C. The as-synthesized Co3O4 was prepared by a similar method as Ir-Co3O4, except that IrCl3 was not added as a precursor.
Scale-up synthesis
To realize the mass production of Ir-Co3O4, 2855.4 mg CoCl2.6H2O (12 mmol) and 179.4 mg IrCl3 (0.6 mmol) were initially mixed in agate mortar for 20 min. Afterwards, 3.0 g NaCl was added into the evenly mixed precursors and ground for 20 min. The homogeneous powder was obtained and then put into the high-energy milling pot with stainless steel balls and ground for 3 h at a rotation speed of 500 rpm. After the first milling, 6 mL NaOH solution (5 mmol L−1) was added into the above mixture and ball milling was continued for 3 h. Finally, the obtained slurry was collected from the milling pot and then calcined in air at 350 °C for 6 h at a heating rate of 5 °C s−1. After calcination, the sample was washed by water 3 times and dried overnight at 80 °C.
Characterization
Scanning electron microscopy (SEM) images were collected on a Zeiss Supra 55 at an acceleration voltage of 5 kV. Transmission electron microscopy (TEM) and High-resolution TEM (HR-TEM) images were determined by a JEOL 2100F instrument at a working voltage of 200 kV. The aberration-corrected high angle annular dark field-scanning TEM (AC HAADF-STEM) images and corresponding elemental mapping were collected on an aberration correction Hitachi 2700D microscope operated at 200 kV. The N2 absorption/desorption isotherms were tested on a Autosorb iQ Quantachrome device and the specific surface area with pore distribution were obtained by the Brunauer–Emmett-Teller (BET) analysis. The mass fraction of Ir in Ir-Co3O4 and the dissolved ion concentrations after stability tests were determined by ICP-OES on an Agilent ICP-OES 730. The amount of O2 generated over a range of overpotentials were recorded by Agilent 7890B equipped with a 5 Å molecular sieve column thermal conductivity detector; Ar was used as the carrier gas.
The electronic structure was investigated by XPS on a Thermo Scientific Escalab 250Xi with an Al-Kα source. XAS of Co K-edge and Ir L3-edge were carried out at the 17 C beamline at the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. The operando XAS experiments of Co K-edge and Ir L3-edge were performed at the 17 C beamline at the NSRRC with a custom-built operando XAS instruments, corresponding data was collected by applying the constant voltage on the catalyst and the electrochemical bias was controlled as 1.6 V vs. RHE. All XAS experiments were carried out in ambient air under room temperature and analyzed using the standard program Demeter. For wavelet-transformed k3-weighted EXAFS, the χ(k) exported from Athena was imported into the Hama Fortran code designed by Harald Funke and Marina Chukalina. The parameters were listed as follows: R range, 0-6 Å, k range, 0-15 Å−1; k weight, 0; and Morlet function with kappaMorlet = 10, sigmaMorlet = 1 was used as the mother wavelet to provide the overall distribution.
The structure was analyzed by XRD with a Bruker D8 Advance powder diffractometer (operating at 40 kV, 40 mA) equipped with a Cu-Kα source (λ1 = 1.5405 Å, λ2 = 1.5443 Å) and fitted with a beryllium window at room temperature. Rietveld refinements for XRD data were carried out with the Full-Prof program, and the refined parameters were background parameters, line shift errors (zero shift), Caglioti coefficients (U, V and W), scale factor, lattice parameters, atomic position, atomic rate occupancy and isotropic atomic displacement parameters.
Electrochemical measurement
The OER activity measurements in O2-saturated 0.5 M H2SO4 were performed at room temperature on a CHI 760E electrochemistry workstation with a rotating disk electrode (RDE) configuration (Pine Research Instrumentation) via using a standard three-electrode electrochemical cell. A glassy carbon (GC) electrode (0.196 cm2), calomel electrode and graphite rod were used as the working, reference and counter electrode, respectively. Prior to each testing, the working electrode was ground and polished three times by fine aluminum oxide powder and washed with distilled water to remove the residual catalyst. The counter electrode was also cleaned by distilled water before being used. Before the electrocatalytic tests, samples (5 mg), carbon black (5 mg, Vulcan XC-72R) and 5 wt% Nafion solution (0.1 mL, Aldrich) were dispersed in absolute isopropanol (1.9 mL) via mild sonication to produce a homogeneous catalysts ink. Then, 20 μL of the catalyst ink was dropped onto the GC electrode with a diameter of 0.5 cm and dried under infrared light. The active materials that dropped on the electrode were 50 wt%. Metal mass loadings on the electrode for all samples were controlled at 0.255 mg cm−2. The Ir mass loadings for Ir-Co3O4 and IrO2 were calculated to be 0.018 mg cm−2 and 0.218 mg cm−2, respectively. The Co mass loadings for Ir-Co3O4, Co3O4 and C-Co3O4 were calculated to be 0.174 mg cm−2, 0.187 mg cm−2 and 0.187 mg cm−2, respectively. All OER potentials were calibrated with respect to RHE scale (Supplementary Fig. 12). The calibration was performed by using a Pt wire as the working electrode, the calomel electrode as the reference electrode and a graphite rod as the counter electrode in a H2-saturated electrolyte (0.5 M H2SO4). Linear sweep voltammetry (LSV) was carried out with a scan rate of 1 mV s−1 and the potential at which the current crossed zero was the calibration value. Cyclic voltammetry (CV) scans were initially performed at the scan rate of 100 mV s−1 between 1.1 and 1.5 V (vs. RHE) with no iR compensation for 50 cycles to activate and clean the catalyst surfaces. Subsequently, LSV with iR correction was conducted at a scan rate of 5 mV s−1 under room temperature to test the OER activity. The iR correction was automatic compensation by the used electrochemical workstation and was controlled as 95% rather than 100% to prevent the resonance and large data error. Tafel slopes were probed on Ir-Co3O4, IrO2, Co3O4 and C-Co3O4 within the potential range of 1.43–1.48 V vs. RHE, 1.48–1.56 V vs. RHE, 1.58–1.7 V vs. RHE and 1.69–1.81 V vs. RHE, respectively. And the Tafel slopes were determined by plotting the overpotential vs. the logarithm of current density (log |j|). Stability was evaluated using catalysts loaded carbon paper (1 cm2) with mass loadings of 0.255 mg cm−2 to carry out chronopotentiometry at a constant OER current density of 10 mA cm−2. Cyclic stability tests on Ir-Co3O4, IrO2, Co3O4 and C-Co3O4 were performed under room temperature within the potential range of 1.1–1.5 V vs. RHE, 1.16–1.56 V vs. RHE, 1.28–1.68 V vs. RHE and 1.38–1.78 V vs. RHE for 3000 cycles, respectively. All scan rates were controlled as 100 mV s−1. Electrochemical impedance spectroscopy was measured on the Bio-Logic SP-200 workstation in the frequency range of 0.01–100 kHz at the 1.43 V vs. RHE; a corresponding equivalent circuit and fitting resistance value were analyzed by EC-lab Software Analysis.
Calculation of the electrochemically surface area
The electrochemically active surface area (ECSA) for also catalysts was estimated from the electrochemical double-layer capacitance (Cdl) of the catalytic surface according to Eq. (1):
where Cdl was measured from the scan-rate-dependent CVs in the non-Faradaic region of 0.75–0.95 V vs. RHE in 0.5 M H2SO4 with scan rates of 20, 40, 60, 80 and 100 mV s−1. The units for Rf and S are cm2real cm−2geo and cm2geo, respectively. S stands for the real surface area of the smooth metal electrode, which was generally equal to the geometric area of the glassy carbon electrode (S = 0.196 cm2). The specific capacitance (Cs) for a flat surface was found to be in the range of 20-60 μF cm−2. In this work, a Cs value of 60 μF cm−2 was used to estimate the ECSA.
Calculation of the turnover frequency
The turnover frequency (TOF) of these catalysts was calculated using Eq. (2):
the O2 turnover per geometric area was obtained from the geometric current density for the LSV curves according to Eq. (3):
the number of the active sites per geometric area can be calculated from the results of the ICP-AES analysis. For example, the upper limit of Ir site density of Ir-Co3O4 is:
At the overpotential of 300 mV, the OER current density for Ir-Co3O4 is 58.84 mA cm−2. And the corresponding TOF value normalized to geometric area was calculated to be:
In addition, the O2 turnover per ECSA was obtained according to Eq. (4):
Thus the corresponding TOF value of Ir-Co3O4 normalized to ECSA polarization information was calculated to be:
The TOF per metal site is calculated based hypothesis that all Ir or all Co atoms are accessible to the electrolyte in all cases. Other catalysts were calculated according to the same procedure.
Calculation of OER Faradaic efficiency
The amount of produced O2 was tested for Ir-Co3O4 dropped carbon paper with a mass loading of 0.255 mg cm−2 as the working electrode to conduct OER in a sealed cell. The applied potentials on sample to generated O2 were 1.28 V, 1.33 V, 1.43 V, 1.53 V and 1.63 V vs. RHE, respectively. Then Ar was used as the carrier gas to purge the produced O2 into a gas chromatograph (Agilent 7890B) equipped with a 5 Å molecular sieve column thermal conductivity detector to generate the O2 signal peak. By fitting the relationship between a certain amount of O2 and the corresponding GC O2 signal area, a standard curve was established. By transforming the generated O2 peak area at different overpotentials into the standard curve, the generated O2 amounts on Ir-Co3O4 were calculated. The OER Faradaic efficiency (FE) of Ir-Co3O4 was obtained according to:
where e is the number of electrons transferred for generating O2, Q is the total charge, n is the amount of generated O2 (in moles) and F is the Faradaic constant.
Computational details
The geometries and energies were performed by the density functional theory (DFT) calculations with the Vienna Ab-initio Simulation Package (VASP)69,70. The interactions between ion cores and valence electrons were described by projector augmented wave (PAW) method71. The exchange correlation interaction was described with the generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) functional72. Dispersion correction (DFT-D3) was considered in all energy-related calculations. The wave functions at each k-point were expanded with a plane wave basis set with a kinetic cutoff energy of 400 eV. Geometries were optimized using a force-based conjugate-gradient method until the energy was converged to 1.0 × 10−7 eV/atom and the force to 0.02 eV/Å. The Brillouin zone was sampled with a 3 × 4 × 1 Monkhorst−Pack k-point mesh. The optimized (111) plane was adopted as the model for surface reaction pathways. Co3O4 (111) was constructed by cutting the bulk Co3O4 alone 111 directions. Ir doped Co3O4 was prepared by replacing one Co atom on the surface. During the optimization the up-layer atoms were allowed to relax and the atoms in the bottom layer were fixed to match the bulk structure. A vacuum layer of 15 Å was used along the c direction normal to the surface to avoid periodic interactions. The pDOS calculations were taken from the bare surfaces of studied samples and were calculated by hybrid functional HSE06 for the accuracy of electron distribution73. The d-band center was extracted from the average of the integral of d-band pDOS information. The Gibbs free energy was calculated based on the four-electron OER mechanism proposed by Nørskov16, corresponding formulas are listed as:
Both free energy profiles were calculated on the Ir and Co sites in samples, respectively. The applied overpotentials were set as 0 and 1.23 V vs. RHE, respectively. Free energy diagrams were estimated as ΔG = ΔE + ΔZPE - TΔS, where E, ZPE, T, and S are the total energy obtained from DFT calculations, the zero-point energy (ZPE), the temperature (298.15 K), the entropy obtained from vibrational frequency calculations, respectively74,75. In addition, the frequencies were set to be 50 cm−1.
Data availability
All relevant data are available from the corresponding authors on request. Source data are provided with this paper.
Change history
15 February 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41467-024-45791-1
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012).
Smalley, R. E. Future global energy prosperity: the terawatt challenge. MRS Bull. 30, 412–417 (2011).
Seitz, L. C. et al. A highly active and stable IrOX/SrIrO3 catalyst for the oxygen evolution reaction. Science 353, 1011–1014 (2016).
King, L. A. et al. A non-precious metal hydrogen catalyst in a commercial polymer electrolyte membrane electrolyser. Nat. Nanotechnol. 14, 1071–1074 (2019).
Koper, M. T. M. Theory of multiple proton-electron transfer reactions and its implications for electrocatalysis. Chem. Sci. 4, 2710–2723 (2013).
Reier, T., Nong, H. N., Teschner, D., Schlögl, R. & Strasser, P. Electrocatalytic oxygen evolution reaction in acidic environments – reaction mechanisms and catalysts. Adv. Energy Mater. 7, 1601275 (2017).
Shan, J. et al. Charge-redistribution-enhanced nanocrystalline Ru@IrOX electrocatalysts for oxygen evolution in acidic media. Chem 5, 445–459 (2019).
Zhang, Z. et al. Selectively anchoring single atoms on specific sites of supports for improved oxygen evolution. Nat. Commun. 13, 2473 (2022).
Meng, G. et al. Strain regulation to optimize the acidic water oxidation performance of atomic layer IrOX. Adv. Mater. 31, 1903616 (2019).
Wang, J. et al. Exceptionally active and stable RuO2 with interstitial carbon for water oxidation in acid. Chem 8, 1673–1687 (2022).
Xiao, Z. et al. Operando identification of the dynamic behavior of oxygen vacancy-rich Co3O4 for oxygen evolution reaction. J. Am. Chem. Soc. 142, 12087–12095 (2020).
Liu, Z. et al. Optimal geometrical configuration of cobalt cations in spinel oxides to promote oxygen evolution reactions. Angew. Chem. Int. Ed. Engl. 59, 4736–4742 (2020).
Wu, Y. et al. Electron density modulation of NiCo2S nanowires by nitrogen incorporation for highly efficient hydrogen evolution catalysis. Nat. Commun. 9, 1425 (2018).
Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212–222 (2021).
Man, I. C. et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159–1165 (2011).
Blasco-Ahicart, M., Soriano-López, J., Carbó, J. J., Poblet, J. M. & Galan-Mascaros, J. R. Polyoxometalate electrocatalysts based on Earth-abundant metals for efficient water oxidation in acidic media. Nat. Chem. 10, 24–30 (2017).
Yuan, X. et al. Controlled phase evolution from Co nanochains to CoO nanocubes and their application as OER catalysts. ACS Energy Lett. 2, 1208–1213 (2017).
Li, A. et al. Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid. Nat. Catal. 5, 109–118 (2022).
Jiao, F. & Frei, H. Nanostructured cobalt oxide clusters in mesoporous silica as efficient oxygen-evolving catalysts. Angew. Chem. Int. Ed. 48, 1841–1844 (2009).
Zhang, L. et al. Facet dependent oxygen evolution activity of spinel cobalt oxides. J. Electrochem. 28, 2108481 (2022).
Huynh, M., Ozel, T., Liu, C., Lau, E. C. & Nocera, D. G. Design of template-stabilized active and Earth-abundant oxygen evolution catalysts in acid. Chem. Sci. 8, 4779–4794 (2017).
Song, J. et al. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49, 2196–2214 (2020).
Zhang, B. et al. Homogeneously dispersed multimetal oxygen-evolving catalysts. Science 352, 333–337 (2016).
Zhang, J. et al. Single-atom Au/NiFe layered double hydroxide electrocatalyst: probing the origin of activity for oxygen evolution reaction. J. Am. Chem. Soc. 140, 3876–3879 (2018).
Li, L. et al. In situ/operando capturing unusual Ir6+ facilitating ultrafast electrocatalytic water oxidation. Adv. Funct. Mater. 31, 2104746 (2021).
Pi, Y., Shao, Q., Wang, P., Guo, J. & Huang, X. General formation of monodisperse IrM (M = Ni, Co, Fe) bimetallic nanoclusters as bifunctional electrocatalysts for acidic over water splitting. Adv. Funct. Mater. 27, 1700886 (2017).
Shah, K. et al. Cobalt single atom incorporated in ruthenium oxide sphere: a robust bifunctional electrocatalyst for HER and OER. Angew. Chem. Int. Ed. 61, e202114951 (2022).
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOX. Nat. Chem. 3, 634–641 (2011).
Zhang, J. et al. Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction. Nat. Catal. 1, 985–992 (2018).
Jiang, K. et al. Single platinum atoms embedded in nanoporous cobalt selenide as electrocatalyst for accelerating hydrogen evolution reaction. Nat. Commun. 10, 1743 (2019).
Jiang, K. et al. Isolated Ni single atoms in graphene nanosheets for high-performance CO2 reduction. Energy Environ. Sci. 11, 893–903 (2018).
Mao, J. et al. Accelerating water dissociation kinetics by isolating cobalt atoms into ruthenium lattice. Nat. Commun. 9, 4958 (2018).
Yan, Q.-Q. et al. Reversing the charge transfer between platinum and sulfur-doped carbon support for electrocatalytic hydrogen evolution. Nat. Commun. 10, 4977 (2019).
Luo, Z. et al. Chemically activating MoS2 via spontaneous atomic palladium interfacial doping towards efficient hydrogen evolution. Nat. Commun. 9, 2120 (2018).
Hai, X. et al. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nat. Nanotechnol. 17, 174–181 (2022).
Zhao, Z. et al. Boosting the oxygen evolution reaction using defect-rich ultra-thin ruthenium oxide nanosheets in acidic media. Energy Environ. Sci. 13, 5143–5151 (2020).
Cao, K. et al. Efficient direct H2O2 synthesis enabled by PdPb nanorings via inhibiting the O-O bond cleavage in O2 and H2O2. ACS Catal. 11, 1106–1118 (2021).
Zhang, Y. et al. Atomic iridium incorporated in cobalt hydroxide for efficient oxygen evolution catalysis in neutral electrolyte. Adv. Mater. 30, 1707522 (2018).
Pfeifer, V. et al. The electronic structure of iridium oxide electrodes active in water splitting. Phys. Chem. Chem. Phys. 18, 2292 (2016).
Pfeifer, V. et al. The electronic structure of iridium and its oxides. Surf. Interface Anal. 48, 261–273 (2016).
Pfeifer, V. et al. In situ observation of reactive oxygen species forming on oxygen-evolving iridium surfaces. Chem. Sci. 8, 2143–2149 (2017).
Li, P. et al. Boosting oxygen evolution of single-atomic ruthenium through electronic coupling with cobalt-iron layered double hydroxides. Nat. Commun. 10, 1711 (2019).
Baroudi, K. et al. Structure and properties of α-NaFeO2-type ternary sodium iridates. J. Solid. State Chem. 210, 195–205 (2014).
Deng, H. et al. Strong enhancement of spin ordering by A-site magnetic ions in the ferrimagnet CaCu3Fe2Os2O12. Phys. Rev. B 94, 024414 (2016).
Feng, H. et al. Ba2NiOsO6: A Dirac-Mott insulator with ferromagnetism near 100 K. Phys. Rev. B 94, 235158 (2016).
Feng, H. et al. Room-temperature ferrimagnetism of anti-site-disordered Ca2MnOsO6. Phys. Rev. Mater. 3, 124404 (2019).
Agrestini, S. et al. Nature of the magnetism of iridium in the double perovskite Sr2CoIrO6. Phys. Rev. B 100, 014443 (2019).
Guan, D. et al. Exceptionally robust face-sharing motifs enable efficient and durable water oxidation. Adv. Mater. 33, 2103392 (2021).
Chen, G. et al. Ultrahigh-performance tungsten-doped perovskites for the oxygen evolution reaction. J. Mater. Chem. A 6, 9854–9859 (2018).
Liu, H. et al. Insight into the role of metal–oxygen bond and O 2p hole in high-voltage cathode LiNixMn2–xO4. J. Phys. Chem. C. 121, 16079–16087 (2017).
Guan, D. et al. Utilizing ion leaching effects for achieving high oxygen-evolving performance on hybrid nanocomposite with self-optimized behaviors. Nat. Commun. 11, 3376 (2020).
Dai, Y. et al. Bridging the charge accumulation and high reaction order for high-rate oxygen evolution and long stable Zn-air batteries. Adv. Funct. Mater. 32, 2111989 (2022).
Guan, D. et al. Screening highly active perovskites for hydrogen-evolving reaction via unifying ionic electronegativity descriptor. Nat. Commun. 10, 3755 (2019).
Li, G. et al. The synergistic effect of Hf-O-Ru bonds and oxygen vacancies in Ru/HfO2 for enhanced hydrogen evolution. Nat. Commun. 13, 1270 (2022).
Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: toward boosted and sustained water oxidation catalysis. Joule 5, 2164–2176 (2021).
Zheng, Y.-R. et al. Monitoring oxygen production on mass-selected iridium–tantalum oxide electrocatalyst. Nat. Energy 7, 55–64 (2022).
Xiong, Y. et al. Gram-scale synthesis of high-loading single-atomic-site Fe catalysts for effective epoxidation of styrene. Adv. Mater. 32, 2000896 (2020).
Lu, Y. et al. Tuning the selective adsorption site of biomass on Co3O4 by Ir single atoms for electrosynthesis. Adv. Mater. 33, 2007056 (2021).
Shan, J. et al. Short-range ordered iridium single atoms integrated into cobalt oxide spinel structure for highly efficient electrocatalytic water oxidation. J. Am. Chem. Soc. 143, 5201–5211 (2021).
Nong, H. et al. Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 587, 408–413 (2020).
Nong, H. et al. A unique oxygen ligand environment facilitates water oxidation in hole-doped IrNiOX core-shell electrocatalysts. Nat. Catal. 1, 841–851 (2018).
Gao, J. et al. Breaking long-range order in iridium oxide by alkali ion for efficient water oxidation. J. Am. Chem. Soc. 141, 3014–3023 (2019).
Casalongue, H. G. S. et al. A. In situ observation of surface species on iridium oxide nanoparticles during the oxygen evolution reaction. Angew. Chem. Int. Ed. 53, 7169–7172 (2014).
Zhao, G., Li, P., Cheng, N., Dou, S. X. & Sun, W. An Ir-Ni(OH)2 heterostructured electrocatalyst for the oxygen evolution reaction: breaking the scaling relation, stabilizing iridium (V), and beyond. Adv. Mater. 32, 2000872 (2020).
Zheng, X. et al. Origin of enhanced water oxidation activity in an iridium single atom anchored on NiFe oxyhydroxide catalyst. Proc. Natl. Acad. Sci. U.S.A. 118, e2101817118 (2021).
Zhao, Y. et al. Modulating Pt-O-Pt atomic clusters with isolated cobalt atoms for enhanced hydrogen evolution catalysis. Nat. Commun. 13, 2430 (2022).
Seh, Z. W. et al. Combing theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998 (2017).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 118, 8207 (2003).
Rossmeisl, J., Logadottir, A. & Nørskov, J. K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 319, 178–184 (2005).
Bajdich, M., García-Mota, M., Vojvodic, A., Nørskov, J. K. & Bell, A. T. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 135, 13521–13530 (2013).
Acknowledgements
The work leading to these results has received funding from the National Natural Science Foundation of China (22179098) and the Fundamental Research Funds for the Central Universities to J.M. Z.H. acknowledges support from the Max Planck-POSTECH-Hsinchu Center for Complex Phase Materials. The calculations were supported by the Welch Foundation (F-1841) and the Texas Advanced Computing Center to G.H. M.K. and P.S. acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG) through Project "Iridium" (STR 596/11-1) and by the Federal Ministry of Education and Research (BMBF) through the Verbundprojekt "HyThroughGen" (FKZ 03Hy108D) in the framework of the Leitprojekt "H2Giga". T.K. acknowledges support by the Deutsche Forschungsgemeinschaft (DFG) through the German-French Project "REALcharge" (STR 596/13-1).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
J.M. conceived and supervised the research. Y.Z. and J.M. designed the experiments. Y.Z., T.K., M.K., J.C., S.H., Z.H., P.S. and J.M. performed the experiments and data analysis. J.W., Y.Z. and G.H. performed the theoretical computations and analyzed the theoretical results. Y.Z., T.K., J.C., S.H., Z.H., P.S. and J.M. participated in various aspects of the experiments and discussions. Y.Z., J.W., T.K., Z.H., P.S. and J.M. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Subrata Kundu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, Y., Wang, J., Koketsu, T. et al. Iridium single atoms incorporated in Co3O4 efficiently catalyze the oxygen evolution in acidic conditions. Nat Commun 13, 7754 (2022). https://doi.org/10.1038/s41467-022-35426-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-35426-8
- Springer Nature Limited
This article is cited by
-
Facilitating alkaline hydrogen evolution reaction on the hetero-interfaced Ru/RuO2 through Pt single atoms doping
Nature Communications (2024)
-
Co-catalytic metal–support interactions in single-atom electrocatalysts
Nature Reviews Materials (2024)
-
Modulation in the electronic structure of Ir-rich shell on AuIr solid solution as OER electrocatalyst for PEM electrolyzer
Journal of Applied Electrochemistry (2024)
-
Theoretical insights into efficient oxygen evolution reaction using non-noble metal single-atom catalysts on W2CO2 MXene
Rare Metals (2024)
-
Cation-π interactions regulate electrocatalytic water oxidation over iridium single atoms supported on conjugated polymers
Science China Chemistry (2024)