
Abstract
γδ T cells play uniquely important roles in stress surveillance and immunity for infections and carcinogenesis. Human γδ T cells recognize and kill transformed cells independently of human leukocyte antigen (HLA) restriction, which is an essential feature of conventional αβ T cells. Vγ9Vδ2 γδ T cells, which prevail in the peripheral blood of healthy adults, are activated by microbial or endogenous tumor-derived pyrophosphates by a mechanism dependent on butyrophilin molecules. γδ T cells expressing other T cell receptor variable genes, notably Vδ1, are more abundant in mucosal tissue. In addition to the T cell receptor, γδ T cells usually express activating natural killer (NK) receptors, such as NKp30, NKp44, or NKG2D which binds to stress-inducible surface molecules that are absent on healthy cells but are frequently expressed on malignant cells. Therefore, γδ T cells are endowed with at least two independent recognition systems to sense tumor cells and to initiate anticancer effector mechanisms, including cytokine production and cytotoxicity. In view of their HLA-independent potent antitumor activity, there has been increasing interest in translating the unique potential of γδ T cells into innovative cellular cancer immunotherapies. Here, we discuss recent developments to enhance the efficacy of γδ T cell-based immunotherapy. This includes strategies for in vivo activation and tumor-targeting of γδ T cells, the optimization of in vitro expansion protocols, and the development of gene-modified γδ T cells. It is equally important to consider potential synergisms with other therapeutic strategies, notably checkpoint inhibitors, chemotherapy, or the (local) activation of innate immunity.
Similar content being viewed by others

Introduction
γδ T cells comprise a relatively small subset of T lymphocytes in the peripheral blood of adult individuals. While there is substantial interindividual variability, γδ T cells usually account for anywhere between 1 and 10% of CD3+ T cells in human blood,1 and there are age-dependent alterations in the proportion and T cell receptor (TCR) repertoire of γδ T cells in the blood.1,2 γδ T cells are more abundant at barrier sites such as the intestine; up to 20% of intraepithelial CD3+ T cells in the human colon express the γδ TCR.3 Interestingly, there are significant species-specific differences in the abundancy of γδ T cells. As an example, much higher numbers of γδ T cells are present in the blood of ruminants than in the blood of humans.4 In contrast to conventional T cells bearing an αβ TCR that recognizes antigen-derived peptides loaded onto MHC molecules (human leukocyte antigen [HLA] in humans), γδ T cells typically recognize their ligands independent of antigen processing and MHC/HLA restriction.5 The dominant population of γδ T cells in the blood of healthy adults expresses a TCR composed of the variable (V) gene Vγ9 paired with Vδ2. Such Vγ9Vδ2T cells (for simplicity referred to as Vδ2 in the following sections) account for anywhere from 50 to more than 95% of peripheral blood γδ T cells, with remarkable donor-dependent variability.6,7 The second most frequent γδ T cell subset in blood expresses the variable Vδ1 chain, which can be paired with any of the six expressed Vγ genes. Importantly, such Vδ1T cells (and other non-Vδ2 γδ T cells, mostly Vδ3) are more abundant in the intestinal mucosa,8 in line with differential ligand recognition of peripheral blood and mucosal γδ T cells.5,9 γδ T cells are considered to have their niche at the crossroad of innate and adaptive immunity.9 They share features of the adaptive immune system, with their expression of clonally rearranged TCR genes, but at the same time are similar to innate immune cells, with the lack of need for antigen processing to activate their effector functions. Therefore, γδ T cells rapidly respond to TCR triggering. Moreover, γδ T cells frequently coexpress functional receptors of innate immune cells, such as activating natural killer (NK) receptors such as NKG2D, NKp30, and/or NKp44, which directly trigger cytotoxic activity,10,11,12,13 in addition to certain Toll-like receptors (TLRs), which can provide costimulatory signals.14,15 At the level of effector activity, γδ T cells share many functions with αβ T cells. Activated γδ T cells have the capacity to be potent killers that can lyse a broad variety of solid tumor and leukemia/lymphoma cells and produce an array of cytokines.16,17,18,19 Depending on the local micromilieu, γδ T cells can differentiate into Th1-, Th2-, Th9- or Th17-like cells and produce prototypical cytokines such as interferon-γ (IFNγ) and interleukin (IL)-4/-10, IL-9, or IL-17.20,21,22,23
Ligand recognition by human γδ T cells
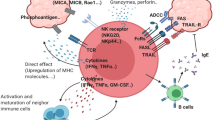
Although γδ T cells were discovered in the mid-1980s, the nature of the antigens recognized by the γδ TCR has largely remained a mystery that we continue to dissect. Today, some antigens with relevance for immune surveillance by specific subsets of γδ T cells have been well characterized. The most conspicuous ligands for human Vδ2T cells are small pyrophosphate molecules, which are intermediates of the cholesterol synthesis pathway in microbes and eukaryotic cells. Many bacteria and some parasites use the so-called non-mevalonate (or Rohmer) pathway of isoprenoid biosynthesis, of which (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMBPP) is an intermediate that exclusively activates Vδ2T cells at picomolar to nanomolar concentrations.24,25 The synthetic molecule bromohydrin pyrophosphate (BrHPP) is similarly active at nanomolar concentrations.26 Homologous pyrophosphate molecules, specifically isopentenyl pyrophosphate (IPP), are also generated in the mevalonate pathway of cholesterol synthesis in eukaryotic cells. However, much higher concentrations (in the micromolar range) of IPP are required to activate the same population of Vδ2T cells. While normal cells do not accumulate sufficient IPP to activate γδ T cells, many transformed cells have a dysregulated mevalonate pathway, leading to increased IPP accumulation and consequent γδ T cell activation.27,28,29,30 Importantly, the activation of human Vδ2T cells by such phosphoantigens is crucially dependent on transmembrane butyrophilin (BTN) molecules. The landmark study by Harly and coworkers has identified BTN3A1 and its intracellular B30.2 signaling domain as being indispensable for the activation of Vδ2T cells by phosphoantigens.31 Subsequently, detailed molecular studies have shown that pyrophosphates directly bind to the intracellular B30.2 domains and trigger inside-out signaling to activate Vδ2T cells.32,33 This process is modulated by the GTPase Rho in tumor cells, which is recruited to the B30.2 domain, thereby inducing changes in the cytoskeleton, as well as conformational changes in BTN3A1.34 However, very recent studies from three independent laboratories indicate that another member of the BTN family is also required. Using different experimental approaches, it was found that BTN2A1 collaborates with BTN3A1 in sensitizing pAg-exposed cells to recognition by human γδ T cells. While BTN2A1 directly binds to the TCR via germline-encoded regions of the Vγ9 chain, it is suggested that, following pAg binding to the B30.2 domain, the BTN2A1–BTN3A1 complex engages additional regions of the TCR. These exciting new results also indicate that there might be the recruitment of an additional as yet unidentified CDR3 ligand upon complex formation of BTN2A1, BTN3A1, and pAgs.35,36 Recent results from J. Kuball’s group have added greater complexity to the molecular mechanisms involved in BTN-dependent tumor recognition by Vδ2T cells. According to these studies, the binding of TCRγ regions between CDR2 and CDR3 to BTN2A1 is followed by the binding of the TCRδ CDR3 to an as yet unidentified ligand. This process is pAg independent. Full activation of the γδ TCR requires pAg- and RhoB-dependent recruitment of BTN3A1 (together with BTN2A1) to the immunological synapse.37 In any case, it is clear that pAg accumulation in transformed cells is critically important for BTN-dependent activation of tumor-reactive Vγ9Vδ2T cells. Interestingly, BTN-dependent selective activation of Vγ9Vδ2T cells can also be achieved with agonistic anti-BTN3A1 (CD277) monoclonal antibodies (mAbs). Agonistic antibodies (e.g., clone 20.1) mimic pAg-dependent activation, whereas antagonistic mAbs (e.g., clone 103.2) inhibit the activation of Vγ9Vδ2T cells.25,31 Importantly, recent insights revealed that the agonistic activity of mAb 20.1 also depended on cell surface-expressed BTN2A1.35 The currently discussed model38 of Vγδ2 T cell activation and the role of BTN molecules and pAgs are illustrated in Fig. 1.

Role of BTN2A1 and BTN3A1 in the activation of human Vγ9Vδ2 γδ T cells. The butyrophilin members BTN2A1 and BTN3A1 are loosely associated on the surface of target cells. a In the homeostatic “resting” state, the intracellular B30.2 signaling domain does not associate with endogenous (tumor-derived IPP) or exogenous (microbe-derived HMBPP) phosphoantigens (pAgs). However, BTN2A1 binds to germ-line-encoded regions of the Vγ9 chain in the homeostatic state. There is also evidence that the CDR3 region of the TCR δ chain interacts with another currently unidentified ligand.37 b In infected cells and tumor cells, exogenous (HMBPP) or endogenous (IPP) pAgs bind to the B30.2 domain and thereby induces a conformational change in the BTN2A1–BTN3A1 complex, resulting in TCR-dependent activation of Vγ9Vδ2 T cells. This step may involve other as yet unidentified CDR3 ligands.36,37 c Agonistic anti-BTN3A antibodies such as clone 20.1 mimic the activity of pAgs by inducing a conformational change in the BTN molecules, leading to γδ T cell activation. The depicted model is based on refs. 35,36,37,38
There is no doubt that the respective roles of pyrophosphate antigens and BTN2A1/BTN3A1 are the best understood mechanisms of activation requirements of a specific population of human γδ T cells. Additional reported ligands for the Vγ9Vδ2 subset include the complex of cell surface F1-ATPase and apolipoprotein A-I39 and the ectopically expressed DNA mismatch repair protein human MutS homologue 2 (hMSH2).40,41 The knowledge of specific ligands for other γδ T cell subsets is less advanced, but there are several interesting findings that have been reported that are worth noting. Early studies indicated that non-Vδ2 (mainly Vδ1 and Vδ3) γδ T cells were expanded in the blood of kidney transplant recipients who experienced cytomegalovirus (CMV) infection after transplantation.42 Subsequently, it was shown that such CMV-reactive non-Vδ2 γδ T cells also recognized various intestinal epithelial tumor cells.43 The endothelial protein C receptor (EPCR) expressed on CMV-infected endothelial cells, which is also aberrantly expressed on epithelial tumor cells, has been identified as a specific ligand for human Vγδ5 γδ T cells.44 Along the same line, the intracytoplasmic phospholipid binding protein Annexin A2 (Anx A2) translocates to the cell surface following exposure to oxidative stress. Anx A2 expressed on the surface of tumor cells is reported to be a ligand for certain Vδ2-negative, specifically Vγ8Vδ3, γδ T cells.45 Moreover, human intestinal Vγ4 γδ T cells coexpressing Vδ1 or Vδ3 were recently shown to recognize butyrophilin-like proteins BTNL-3 and BTNL-8 in a TCR-dependent manner on intestinal epithelial cells.46 In these instances, BTNL responsiveness is mediated by germline-encoded motifs within the Vγ4 chain.47,48 In addition, human Vδ1-expressing γδ T cells can recognize microbial and self-lipids bound to the nonclassical MHC protein CD1d.49 While it is to be expected that more specific ligands for various γδ T cell subsets will be identified in the future, the emerging overall picture clearly indicates that the TCR of γδ T cells is constantly being scrutinized for signs of “stress” on normal cells and those undergoing malignant transformation, thereby assigning γδ T cells an important place in local immune surveillance.50,51 This conclusion is well supported by a pivotal study demonstrating an increased susceptibility to tumor development in γδ T cell deficient mice.52
In addition to TCRs, γδ T cells usually express other activating receptors. As already mentioned, most human γδ T cells also carry the NKG2D receptor on the cell surface, which recognizes stress-inducible MHC class-I-related molecules frequently expressed on transformed cells but absent on normal cells. The NKG2D receptor also contributes to immune surveillance, as illustrated by increased tumor incidence in NKG2D-deficient mice.53 Most solid tumors as well as leukemias express at least one of the eight NKG2D ligands (MHC class I-related chain A/B [MICA/B], UL16-binding proteins [ULBP1–6]),54 and the cytotoxic effector function of γδ T cells can be triggered through NKG2D/NKG2D ligand interactions independent of TCR signaling.16,55 Notably, however, NKG2D ligands are not uniformly expressed on malignant cells. It appears that leukemia stem cells may lack NKG2D ligand expression,56 and this absence of expression can render them less susceptible to γδ T cell recognition. As an exhibit of validating redundancy, it has been found that the NKG2D ligand MICA can also be recognized by the Vδ1 TCR,57 and direct binding of MICA to Vδ1 has been demonstrated.58 Moreover, other NK receptors, such as NKp30, NKp44, and DNAM-1 (CD226), can also be expressed at varying levels on γδ T cells and contribute to tumor cell recognition and killing.12 Recently, NKp46 was shown to be specifically expressed on gut-resident intraepithelial human Vδ1 T cells endowed with potent antitumor activity.59 Taken together, γδ T cells are uniquely equipped with two independent recognition pathways to sense stressed and transformed cells, i.e., TCRs as well as activating NK receptors.12,16 A schematic overview of the major receptor–ligand interactions involved in the activation of human Vδ2 and non-Vδ2 γδ T cells is shown in Fig. 2.

Major receptor–ligand interactions between Vδ2/non-Vδ2 γδ T cells and tumor cells/antigen-presenting cells. Left side: The best characterized ligands for the human Vγ9Vδ2 TCR are phosphoantigens (pAgs), which are recognized in a BTN2A1-/BTN3A1-dependent manner. Other ligands for this TCR include the ectopically expressed DNA repair protein human MutS homologue 2 (hMSH2) and ectopically expressed F1-ATPase in conjunction with apolipoprotein A-I. Tumor antigen–TCR crosslinking bispecific T cell engagers (BiTEs) also activate Vδ2 T cells via the TCR. Therapeutically used antibodies against tumor (associated) antigens can activate γδ T cells via CD16/FcγRIII-dependent ADCC. Right side: Ligands for non-Vδ2 γδ T cells. Some ligands for specific subsets of human non-Vδ2 γδ T cells have been identified: endothelial protein C receptor (EPCR) on CMV-infected and intestinal tumor cells (Vγ4Vδ5), butyrophilin-like molecules BTNL3/BTNL8 on intestinal epithelia (Vγ4/Vδ1 or Vδ3), Annexin A2 (Anx A2) (Vγ8Vδ3), MICA (Vδ1), and lipids bound to CD1d (Vδ1). MICA/B and ULBP molecules are ligands for the activating NKG2D receptor expressed on both Vδ2 and non-Vδ γδ T cells. In addition to NKG2D, other NK receptors (NKp30, Nkp44, NKp46) can be expressed as well
Tumor-infiltrating γδ T cells: friends or foes?
Many studies with in vitro activated cells isolated from peripheral blood have demonstrated potent and HLA-independent activity of γδ T cells against various solid tumors and leukemia/lymphoma cells. γδ T cells can also infiltrate tumors where they exert protumorigenic activities or contribute to tumor regression. What information can then be gathered from the analysis of tumor-associated γδ T cells, and how does this correlate with prognostic significance? There are three ways of approaching this question: (i) transcriptional analysis of bulk tumor transcriptomes in large cohorts of patients; (ii) immunohistological characterization of tumor-infiltrating γδ T cells in the context of the in situ tumor micromilieu; and (iii) phenotypic and functional studies of tumor-infiltrating γδ T cells.
Analyzing transcriptomes from 18,000 tumor samples across 39 different cancer types using the CIBERSORT algorithm,60 Gentles et al. identified the abundance of γδ T cells as the single most favorable prognostic parameter out of 22 distinct leukocyte subsets.61 Figure 3c from their paper is frequently presented to support the notion that γδ T cells are critical for optimal tumor defense. Technical limitations of this approach were later noted, as it did not appropriately differentiate between γδ T cells and other T cell subsets and NK cells.62 Tosolini and coworkers improved the computational CIBERSORT identification of tumor-infiltrating Vγ9Vδ2T cells by the deconvolution of cancer microarray data sets using machine-learning methods, revealing more variability with respect to interindividual variation and the respective cancer type. Overall, the abundance of Vγ9Vδ2 tumor-infiltrating T cells in this study was associated with a favorable outcome in colorectal carcinoma, prostate carcinoma, chronic lymphocytic leukemia (CLL) and acute myeloblastic leukemia (AML).62 Several other tools have been developed to monitor T cell subset abundance from RNAseq and microarray expression data in cancer patients. ImmuCellAI has been recently introduced for estimating 18 T cell subsets, including γδ T cells. The method has been validated with flow cytometry results and was shown to allow predictions for immunotherapy responses.63 As an example, this algorithm revealed that γδ T cell infiltration was significantly greater in responders than in nonresponders in 58 melanoma samples from a clinical trial with anti-PD1 checkpoint inhibitor therapy.63 On the other hand, it was observed in a recent CIBERSORT-based analysis that the abundance of γδ T cells is associated with poor prognosis in pancreatic adenocarcinoma. Together with M0 macrophages and naïve CD4 T cells, γδ T cells contributed to an immune score that was superior to the classic TNM staging.64

How to enhance the efficacy of γδ T cell immunotherapy in vitro and in vivo. a Genetic engineering is used to transduce chimeric antigen receptors (CARs) into γδ T cells, to transfect αβ T cells with selected high affinity γδ TCRs (TEGs), or to render γδ T cells drug-resistant (drug-resistant immunotherapy, DRI). In vitro expansion of γδ T cells can be optimized by selected cytokines (e.g., IL-15, TGF-β), specific medium supplements (e.g., vitamin C), or the selection of novel activators (ZOL derivatives, novel pAgs). b Multiple strategies are available to activate γδ T cells in vivo, to target γδ T cells to tumor antigens, to target tumor-intrinsic suppressive pathways, or to increase the local inflammatory milieu
In addition to whole-tissue transcriptomic analysis and phenotypic/functional characterization of isolated tumor-infiltrating lymphocytes (TILs), immunohistochemistry can provide important insights into the localization of γδ T cells within tumors and the surrounding tissue. Such investigations (usually combined with functional analysis of isolated TILs) have been performed in several types of cancer, including melanoma, pancreatic adenocarcinoma, glioblastoma multiforme, colorectal carcinoma, hepatocellular carcinoma, breast cancer, and others.65,66,67,68,69,70 In some studies, γδ T cells comprised up to 20% of CD3+ TILs.68 Due to the limited availability of antibodies suitable for immunohistological analysis of paraffin-embedded tissue or frozen sections, most studies have focused on the detection of all γδ T cells using a pan-TCRγδ mAb. In several instances, it was observed that γδ T cells are localized in the periphery of the tumor or in peritumoral tissue, suggesting that strategies to enhance tumor infiltration might improve the antitumor activity of γδ T cells.65,68,69 A very recent study by Chabab and colleagues established a standardized protocol to analyze γδ T cells in tumor tissue microarrays using the pan-TCRγδ mAb clone H-41. They applied this approach to quantify γδ TILs in breast, colorectal, pancreatic and ovarian cancer, demonstrating the variability of γδ T cell infiltration in different tumor entities.71 Further optimization of staining protocols and antibody clone selection, which includes suitable mAbs directed against γδ T cell subsets, will help to improve immunohistological studies in the future.72 We anticipate that interest in γδ T cell subset analysis within tumors in situ will be invigorated with innovative technologies such as fully automated high-content imaging and quantitative whole-slide imaging analysis.73,74
Phenotypic and functional characterization of γδ T cells within TILs freshly isolated from various tumors has been extensively performed. A comparison between γδ T cells within the TILs and peripheral blood of the same patient frequently revealed an altered γδ T cell subset distribution across different tumor entities, with higher proportions of Vδ1T cells being present in the tumor than in the blood.68,75,76,77 Of particular interest is the functional analysis of γδ TILs and their TCR repertoire in comparison to blood to determine whether there is preferential recruitment of clonal γδ T cells to the tumor site. In different tumors, γδ TILs frequently produce IFN-γ,68,77 and no major difference was observed in this respect between Vδ1 and Vδ2 TILs in an ovarian cancer study.77 Furthermore, most, if not all, Vδ1 and Vδ2 TILs produced granzyme A and B, in line with their cytotoxic capacity.77 In several experimental settings, IL-17-producing γδ T cells have been implicated in tumorigenesis and metastasis formation through the recruitment of tumor-promoting macrophages and neutrophils78,79,80 or the induction of angiogenesis in response to human papilloma virus (HPV)-16 oncoprotein expression.81 In a recent study using conventional and germ-free mice, it was observed that commensal microbiota present in the lung can indirectly stimulate IL-17 production in lung-resident γδ T cells, thereby promoting inflammation and lung cancer development.82 There are multiple pathways through which IL-17 can promote tumorigenesis, including effects on angiogenesis, endothelial cell permeability, or the upregulation of adhesion molecules.83 It should be noted that the capacity for IL-17 production is much lower for human γδ T cells than for murine γδ T cells. Nevertheless, IL-17-producing cells among γδ TILs (usually of the Vδ1 subgroup) have been identified in several different tumors, and tumor-promoting activity has been suggested.84,85,86 However, IL-17 production is not a general feature of human γδ TILs. While both Vδ1 and Vδ2 TILs in ovarian cancer produced IFN-γ and granzyme A/B, very little, if any, IL-17 was detected.77 Moreover, there is controversy among published studies in the same tumor entity. Wu et al. detected high proportions of IL-17-producing cells among γδ TILs in colorectal cancer,84 whereas Meraviglia et al. reported only low numbers in a different cohort of colorectal cancer patients.68 Very few IL-17-expressing γδ T cells among breast-cancer-infiltrating γδ TILs were reported in a recent study by Janssen et al.87 Again, we expect that better conclusions can be drawn in future studies using automated high-content imaging of tumor tissue to more precisely define and quantify immune cell composition within the tumor and peritumoral tissue. As yet, limited information is available on the TCR repertoire of γδ TILs in human tumors. Based on antibody staining with available mAbs to identify expressed TCR Vγ chains,72 an increase in non-Vδ2 γδ T cells coexpressing Vγ2/3/4 among ascites and TIL γδ T cells compared to levels in peripheral blood was observed in ovarian cancer patients.77 In glioblastoma multiforme, unique TCR clonotypes were identified by next-generation sequencing in intratumoral Vγ9Vδ2 γδ T cells compared to peripheral blood, suggesting specific recruitment of selected γδ T cells to the tumor site.67 Furthermore, a distinct TCR repertoire was identified in gut-resident Vδ1 γδ T cells that expressed NKp46 and had high cytotoxic potential against colorectal cancer.59 The γδ TCR repertoire has been extensively studied during normal development and in infectious diseases.88 Some studies have also noted different γδ TCR repertoires in the blood of cancer patients than in healthy controls.89 However, to better understand the significance of clonal γδ T cell recruitment to the tumor site, more equivalent studies in various tumors and lymphomas are needed.
Vδ1 and Vδ2 γδ T cells exerting potent antitumor cytotoxic activity can be readily expanded from peripheral blood and ascites,18,90,91,92,93,94,95,96,97,98,99,100 despite the potential protumorigenic activity of γδ T cells residing in situ within the tumor. The mechanisms by which intratumoral γδ T cells might in fact promote tumorigenesis and metastasis formation have been recently reviewed in several excellent articles.83,101,102 Depending on the cancer type, such mechanisms might include various effects, such as the recruitment of myeloid-derived suppressor cells (MDSCs) by IL-17-secreting γδ T cells84 or the suppression of αβ T cell responses by tumor-infiltrating γδ T cells mediated by PD-L1-dependent mechanisms,103 TLR8-dependent pathways104 or CD73.105 However, again, conflicting observations about the role of specific γδ T cell subsets in a given tumor type have been reported. Vδ1 TILs in breast cancer were shown to suppress αβ T cells,104 whereas a recent study actually identified innate-like Vδ1 TILs as being associated with remission in triple-negative breast cancer.106 We conclude that the protumorigenic potential of γδ T cells must be well taken into account; however, there is a substantial body of evidence supporting the notion that γδ T cells are exceptional candidates for cellular immunotherapy.
γδ T cells: regulating and being regulated
γδ T cells to be applied for cancer immunotherapy should not exert suppressive activity. To ensure this, it is important to understand which signals can impose regulatory functions on γδ T cells. Transforming growth factor-β (TGF-β) is well known to induce a regulatory phenotype for CD4 T cells (iTregs).107 Casetti et al. first showed that TGF-β can induce FOXP3, the master transcription factor of Treg, and regulatory activity in human Vδ2T cells.108 The suppressive activity of Tregs critically depends on the demethylation of Treg-specific demethylated regions (TSDRs) in the FOXP3 gene.109,110 Vitamin C (Vit C) is a well-characterized cofactor for the activation of ten-eleven translocation (Tet) enzymes that mediate DNA hydroxymethylation, including the hypomethylation of FOXP3 TSDRs.111 We recently studied the effect of Vit C on TGF-β-induced regulatory activity and FOXP3 expression in human Vδ2T cells. We observed a strong enhancement of FOXP3 expression and regulatory activity of purified γδ T cells stimulated with phosphoantigen and TGF-β in the presence of Vit C. More importantly, strong hypomethylation of FOXP3 TSDRs was observed only in the presence of Vit C, suggesting that TGF-β frequently expressed in the tumor microenvironment might prime local γδ T cells for suppressive activity if additional epigenetically active signals are present.112 In some circumstances, however, it appears that γδ T cells can also downregulate αβ T cell responses independent of FOXP3 expression. Upon activation, γδ T cells transiently upregulate various inhibitory receptors and costimulatory molecules, including PD-1, PD-L1, CTLA4, and CD80/CD86.113 In our studies, we observed that activated Vδ2 γδ T cells inhibited the proliferative response of CD4 αβ T cells in a CD86/CTLA4-dependent manner, as suggested by antibody blocking studies.113 An alternative mechanism implies upregulated cell surface expression of inhibitory PD-L1 on γδ T cells, which may then lead to the inhibition of αβ T cell activation. This has been shown for tumor-infiltrating non-Vγ9 γδ T cells in pancreatic adenocarcinoma.103 In a recent study, Schilbach and coworkers performed detailed studies to characterize the PD-L1-dependent suppressive activity of human Vδ2 γδ T cells. It was observed that the suppressive activity of activated Vδ2T cells on autologous αβ T cells was dependent on the signal strength of the TCR stimulation and enforced by IL-15, but it was independent of TGF-β (in line with the independence of FOXP3).114 The molecular mechanism of how γδ T cells suppress their neighbors is not precisely known. Conceivably, suppression might, in some instances, result from direct killing of αβ T cells by activated γδ T cells. Taken together, however, it is obvious that γδ T cells can acquire suppressive activity through a variety of different mechanisms. This would certainly be an unwanted effect in the context of cancer immunotherapy. Given the very limited success of clinical trials with unmodified γδ T cells or γδ T cell transfer/in vivo activation without additional “costimulatory” strategies (see below), the potential role of a suppressive function of γδ T cells in vivo requires careful consideration when attempting to harness their significant anticancer potential for cancer immunotherapy.
The activity of γδ T cells is also subject to regulation by the cellular context and the tumor micromilieu. γδ T cells are susceptible to inhibition by Treg cells115, which might be relevant in the context of cancer, as suggested in a study with hepatocellular carcinoma patients.116 Furthermore, the activity of γδ T cells is also regulated by multifaceted interactions with neutrophils.117 Neutrophils can inhibit the activation of γδ T cells, which has been mainly ascribed to neutrophil-derived reactive oxygen species (ROS).118 We also observed that neutrophils exposed to zoledronic acid (ZOL) inhibited the activation of Vδ2T cells, which was revealed when comparing the activation of γδ T cells present in Ficoll-Hypaque isolated PBMCs to total leukocytes following red blood cell lysis.119 Within PBMCs, monocytes incorporate aminobisphosphonates, such as ZOL, and generate the phosphoantigen IPP, which then activates Vδ2T cells.120 While neutrophils also take up ZOL, they fail to produce IPP25 but rather inhibit the activation of the γδ T cells. Neutrophil-derived ROS were also identified as a major inhibitory mechanism in our study; however, based on the effect of specific inhibitors, we also found arginase and serine proteases contributing to neutrophil-mediated γδ T cell suppression.119 Further studies identified elastase as the inhibitory serine protease of neutrophils.121 Interestingly, using phosphoantigen HMBPP rather than ZOL (used in our studies), Towstyka et al. observed that neutrophils and elastase actually costimulated IFN-γ production in anti-CD3 activated γδ T cells rather than exerting inhibitory effects.122 This apparent discrepancy illustrates the complexity of the relationship between neutrophils and γδ T cells, as different modes of activation of γδ T cells were applied: the indirect activation with ZOL used by us119 induced a neutrophil burst, which was not the case using direct stimulation of γδ T cells with pAg HMBPP121 or BrHPP (used by us as a control119). Further analyzing the regulatory interactions between neutrophils, tumor cells and γδ T cells in vitro, we also observed contrasting effects of neutrophils, depending on situational factors and the activation status of cells. While neutrophils present in leukocyte preparations inhibited the tumor killing capacity of γδ T cells within leukocytes following activation with ZOL, isolated neutrophils actually enhanced the killing capacity of short-term expanded γδ T cells by increasing their release of cytotoxic mediators.123 Seemingly opposing effects of reciprocal interactions between neutrophils and γδ T cells in the tumor microenvironment have also been observed in in vivo models. Tumor-associated neutrophils strongly inhibited IL-17 production by γδ T cells via the induction of oxidative stress, thereby exerting antitumoral activity in the tumor microenvironment.124 On the other hand, IL-17-producing γδ T cells were found to expand neutrophils in a granulocyte colony-stimulating factor (G-CSF)-dependent manner in a breast cancer model, which then actually suppressed CD8 T cell responses, thereby promoting metastasis formation.80
The tumor microenvironment can be rich in multiple factors that negatively impact T cells, including γδ T cells. Tumor cells and suppressive cells such as MDSCs frequently express ligands for inhibitory checkpoint receptors; for instance, PD-L1 and γδ T cells can express such receptors to varying degrees.125 Moreover, tumor cells themselves, tumor-associated macrophages, MDSCs and other cells within the microenvironment can produce a range of inhibitory molecules, including (but not limited to) TGF-β, IL-4, galectins, and indoleamine-2,3-dioxygenase (IDO), all of which may inhibit intratumoral γδ T cells from attacking the tumor.126,127,128,129,130 Arginase-I, an enzyme that suppresses both Vδ2 T cell cytotoxicity and IFN-γ production, can be produced by both tumor cells and MDSCs.131 Targeting such inhibitory pathways is an important aspect for improving the efficacy of T cell-based immunotherapies.
Clinical studies with γδ T cells
Following the original observation by Kunzmann et al. of increased numbers of γδ T cells in the blood of patients with multiple myeloma treated with aminobisphosphonates for increased bone resorption,132 a number of small clinical studies have been performed to investigate the safety and efficacy of γδ T cell therapy in cancer patients. Two different approaches have been explored: (i) the in vivo activation of γδ T cells with aminobisphosphonates (usually ZOL) or (in rare instances) a phosphoantigen (BrHPP) plus low-dose IL-2 and (ii) the adoptive transfer of autologous or (rarely so far) allogeneic γδ T cells following in vitro expansion (again with ZOL or phosphoantigens). Such studies have been performed in various cancer diseases, including renal cell carcinoma, lung cancer, hepatocellular carcinoma, breast cancer, prostate cancer, and multiple myeloma. The general conclusions from those studies are as follows: (i) ZOL plus low-dose IL-2 application induces transient γδ T cell activation in vivo; (ii) adoptive transfer of expanded γδ T cells is safe, with usually only low levels of adverse events being observed; and (iii) even though clinical responses were recorded in most studies (ranging from partial remission and stable disease to complete remissions in exceptional cases), there is still—not surprisingly—room for substantial improvement. Several recent reviews have extensively documented past studies with in vivo activation or with adoptive transfer of γδ T cells, and the reader is referred to these publications for further information.133,134,135,136 In view of the HLA independence of γδ T cells, the application of allogeneic γδ T cells obtained from healthy donors could be considered. Haploidentical transplantation of hematopoietic stem cell (HSC) preparations depleted of αβ T cells and CD19+ B cells (thus containing NK cells and γδ T cells) for treatment of acute leukemia is now an established procedure,137,138 and haploidentical γδ T cells obtained by the depletion of CD4 and CD8 T cells from PBMCs have been infused into patients with advanced hematological malignancies.139 In this setting, γδ T cells are thought to contribute to the anti-leukemia effect.140 The limited experience thus far with adoptive transfer of allogeneic γδ T cells expanded from healthy donors in vitro has also shown a good safety profile in a case report in a patient with a solid tumor.141 While it needs to be considered that some γδ T cells might also display allo-reactivity,140 the application of γδ T cells freshly isolated or expanded from healthy donors rather than the patient’s own autologous γδ T cells might be a reasonable strategy for future application (see below). Some of the currently ongoing trials are mentioned in Table 1, while others are described in the literature.136,142
How to improve the in vitro expansion and effector activity of γδ T cells for adoptive transfer
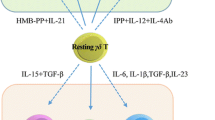
Both Vδ1 and Vδ2 γδ T cells are in clinical development for adoptive cell therapy. Since mice do not express γδ TCRs homologous to human Vδ2 (i.e., mouse γδ T cells are not activated by phosphoantigens), immunodeficient or humanized mice transplanted with human tumors and γδ T cells are frequently used as a preclinical in vivo model.143 While both subsets can kill a broad range of malignant cells and show efficacy in xenograft models,18,97,98,144,145,146,147,148 they display different patterns of NK receptor and accessory molecule expression,82 and they also display related yet distinct cytotoxic hallmarks as revealed by recent single-cell RNAseq studies.149 The expansion of Vδ1 T cells with a specific protocol involving a two-step process with selected cytokines including IL-15 in the second step has been defined as DOT (“delta one T cells”).97 Other protocols for expanding highly cytolytic γδ T cells mainly of the Vδ1 variety utilized mitogen phytohemagglutinin (PHA) plus IL-7 stimulation150 or artificial antigen-presenting cells (APCs) expressing costimulatory molecules and CMV-pp65 antigens.144 Furthermore, polyclonal γδ T cells expressing various TCR VγVδ elements and broad cytotoxic activity against various tumor cells have also been generated in the presence of CD137L-expressing artificial APC and IL-2 plus IL-21.98 The most widely used protocol for selectively expanding Vδ2T cells relies on ZOL stimulation of PBMCs in the presence of IL-2,100 but similarly efficient Vδ2T cell activation in vitro can be achieved with synthetic pAgs such as BrHPP20 and HMBPP.24,25 After expansion for two weeks with continuous supply of IL-2 and careful control of the growth pattern, cell cultures starting from PBMCs (with 2–4% γδ T cells) can easily and reproducibly be expanded to contain 95% Vδ2T cells.100 Such short-term expanded Vδ2 T cell lines display strong cytotoxic activity against some tumor targets, but they show limited activity against others.77,151 Therefore, various optimization strategies have been investigated. ZOL has a rather narrow concentration range and is toxic at high concentrations.25 However, pulsing PBMCs with high concentrations (100 μM) of ZOL for four hours with subsequent washing steps was found to result in more efficient Vδ2 T cell expansion.148 Moreover, the use of bisphosphonate prodrugs rather than ZOL152 and the use of artificial APCs in addition to ZOL153 were also found to enhance the proliferative expansion and functional activity of Vδ2 γδ T cells. It should be mentioned that cellular cross-talk can significantly modulate the efficacy of Vδ2 T cell expansion and overall antitumor activity. In patients with multiple myeloma, ZOL-treated dendritic cells were superior to monocytes in expanding Vδ2T cells. In the additional presence of peptides of an HLA-A2-restricted tumor-associated antigen (survivin), this coculture system also amplified survivin-specific CD8 αβ T cells.154 Cytokines are also a critical component of in vitro γδ T cell expansion protocols. In this regard, common γ-chain family cytokines are pivotal for supporting proliferative expansion and cytotoxic effector function.155 Among those cytokines, IL-15 is particularly active in promoting cellular expansion and cytotoxic effector function.148,155,156 IL-15 has also been shown to upregulate the expression of CD56,157 which is known to be expressed on γδ T cells with potent cytotoxic activity.51,158 We also observed the upregulation of CD56 on Vδ2T cells by IL-15.159 Cytokines other than common γ-chain cytokines also modulate the cytotoxic potential of γδ T cells. Interestingly, we recently observed that TGF-β significantly increased the cytotoxic activity of isolated γδ T cells that were activated in vitro with pAgs in the presence of IL-2 and/or IL-15. TGF-β is usually considered an immunosuppressive cytokine, and TGF-β inhibits γδ T cell expansion if PBMCs are stimulated with ZOL. Mechanistically, we found that TGF-β strongly upregulated CD103 (the αE chain of the αEβ7 integrin), which is a receptor for E-cadherin frequently expressed on epithelial tumor cells. CD103-positive Vδ2T cells form prolonged synapses with E-cadherin-expressing tumor cells, and anti-CD103 antibodies reduced the killing capacity of TGF-β-expanded Vδ2T cells.159 The superior antitumor activity of tumor-specific CD8 αβ T cells expressing CD103 has been previously demonstrated.160,161 CD103 is a marker for resident memory cells, and CD103-positive TILs were associated with increased survival in high-grade serous ovarian cancer.162 CD103-expressing Vδ2T cells might migrate more efficiently into E-cadherin-positive tumor tissue. Therefore, it could be considered to induce CD103 expression on γδ T cells before adoptive transfer into patients with E-cadherin-expressing tumors. TGF-β-treated Vδ2T cells also potently produce IL-9, which might be an added benefit for adoptive transfer, given that IL-9 has multiple antitumor activities.163
Other strategies to improve proliferative expansion and effector functions of γδ T cells target metabolic pathways. In a recent study, it was reported that systemic β-adrenergic receptor activation, which was accomplished by dynamic physical exercise, mobilized γδ T cells to the blood and significantly augmented their subsequent in vitro expansion capacity and cytotoxic antitumor activity.164 A placebo-controlled crossover study applying adrenergic receptor inhibitors indicated that effects on the γδ T cell compartment were mediated by the β2—rather than the β1 adrenergic receptor.164
T cell activation and differentiation are also modified by vitamins. Vitamin C (Vit C, L-ascorbic acid), an essential vitamin, plays an important role in remodeling the epigenome165 and impacts T cell activation at multiple levels.166 The mechanistic basis of its action implies an interplay between antioxidant potential and (epi)genetic regulation of gene expression. We have investigated the effects of Vit C and the more stable phospho-modified L-ascorbic acid 2-phosphate (pVC) on the in vitro activation of Vδ2T cells. Proliferation and cytokine induction were significantly increased, and pVC strongly increased the proliferative expansion of short-term expanded Vδ2T cells following restimulation with pAg BrHPP, a condition that typically induces massive activation-induced cell death. Further studies showed that pVC reduced intracellular ROS levels and increased cell cycle progression and Ki-67 expression in surviving γδ T cells, thereby promoting the expansion of surviving cells rather than preventing cell death.167 Vδ2T cells expanded in the presence of pVC also displayed stronger cytotoxicity against tumor cells in vitro and were more active upon transfer into immunodeficient mice transplanted with a human lung tumor cell line (Yu et al., unpublished results). To conclude, the effect of Vit C on γδ T cell plasticity depends on the overall environmental signals.166,167 As discussed above, Vit C actually conveys a regulatory phenotype and induces FOXP3 hypomethylation in the additional presence of TGF-β.112 However, in the absence of TGF-β during the expansion phase, Vit C substantially enhances effector functions desired in the context of cancer immunotherapy. This may also include the potent production of IL-13,167 which is known to contribute to antitumor immunity.168 Therefore, we suggest including Vit C/pVC in in vitro γδ T cell expansion protocols for adoptive immunotherapy.
Despite the many strategies briefly summarized here to enhance the in vitro expansion and functionality of expanded γδ T cells, the question remains whether the expanded γδ T cells are sufficiently effective to induce a clinically important response in patients. This issue is mainly related to clonal heterogeneity even among a defined cell population such as Vγ9Vδ2T cells.37 Therefore, it is important to consider how γδ T cells can be engineered for optimal functionality.
Design your desired γδ T cells
In recent years, genetic engineering of αβ T cells has been widely explored as a tool to improve cancer immunotherapy.169 Chimeric antigen receptor-modified T (CAR-T) cells that express CAR molecules that target surface antigens on tumor cells have revolutionized the treatment of B-cell malignancies but have yet to achieve the same level of success for solid tumors.170 γδ T cells are interesting recipient cells for CAR constructs as the transfection should result in effector cells with two-fold antitumor activity, e.g., (i) through the endogenous γδ TCRs and (ii) through the CAR specificity.171 In fact, CAR-transduced Vδ2T cells showed enhanced cytotoxicity towards relevant tumor target cells.172 Activated Vδ2T cells can act as APCs and cross-present tumor-derived peptides to CD8 αβ T cells upon the killing of tumor cells.173 Importantly, the ability for cross-presentation of tumor antigens to αβ T cells was preserved in CAR-transduced Vδ2T cells.172 γδ T cells can also be transfected with tumor antigen-specific αβ TCRs, such as HLA-A2-restricted melanoma-related gp100-specific αβ TCRs, again resulting in effector cells with dual antitumor specificity.174 An alternative and different approach is to transduce the αβ T cells of cancer patients with high-affinity Vγ9Vδ2 TCRs, termed T cells engineered with defined γδ TCRs (TEGs).175 This is based on the fact that not all Vγ9Vδ2 TCRs display equally high affinity for pAgs and, thus, tumor cell recognition. An added advantage of the strategy is that CD8, as well as CD4 αβ T cells are transduced with the selected Vγ9Vδ2 TCRs, thereby enabling CD4 T cells to exert helper functions such as the induction of dendritic cell maturation.135 TEGs expressing a high-affinity Vγ9Vδ2TCR have been manufactured under GMP conditions, and a clinical trial exploring safety and tolerability has been initiated (https://www.trialregister.nl/trial/6357).176 Alternatively, NK cells might be suitable recipient cells for the transduction of selected high-affinity γδ TCRs. Such an approach has been recently reported for the generation of anti-CD19 CAR-expressing NK cells, which mediated clinical responses in patients with relapsed or refractory CD19-expressing malignancies.177 The combination of intrinsic NK cell properties with a high-affinity antitumor-directed γδ TCR might reveal significant synergistic potential.
Genetic engineering is also used to render γδ T cells resistant to chemotherapeutic drugs used to treat cancer patients. Glioblastoma multiforme is a cancer in which local instillation of γδ T cells following surgery is considered a promising immunotherapeutic approach and has been demonstrated to be effective in a preclinical model.147 Local administration of γδ T cells appears feasible from a safety perspective, as allogeneic γδ T cells do not injure normal brain tissue.178 Lamb and colleagues designed a lentiviral vector expression system encoding a DNA repair enzyme. The transduction of γδ T cells conferred resistance to temozolomide, the widely used standard chemotherapeutic drug to treat glioblastoma patients.179 Temozolomide resistance did not alter the expansion capacity and cytotoxic activity of γδ T cells, suggesting that drug-resistant γδ T cells might be applicable in a clinical setting. A phase I study investigating the safety and tolerability of drug-resistant γδ T cell infusion is currently ongoing (https://clinicaltrials.gov/ct2/show/NCT04165941).
How to activate and target γδ T cells in vivo
As summarized in recent reviews, ZOL in combination with low-dose IL-2 has been used in an attempt to activate (and possibly expand) tumor-reactive Vδ2T cells in vivo.133,134,135,136 While the treatment regimens varied in these studies, an increase in circulating γδ T cells was usually observed within one week.180,181,182 In addition, ZOL treatment substantially shifted the phenotype of Vδ2T cells to effector memory (TEM) cells.183,184 However, the expansion of γδ T cells in the peripheral blood was not sustained. In fact, repeated application of ZOL infusions resulted in a progressive decline in γδ T cells,180,184 an observation that we also made in patients with osteoporosis newly prescribed i.v. bisphosphonate therapy.185 Haploidentical HSC transplantation of αβ T cell/CD19 B-cell-depleted cells is now an established therapeutic procedure for certain blood malignancies. γδ T cells comprise the major CD3+ T cell population during reconstitution early after transplantation. In addition to their potential anti-leukemia effect, γδ T cells are also important for the control of Epstein-Barr virus (EBV) reactivation and related lymphoproliferative diseases and for the containment of CMV reactivation after HSC transplantation. In fact, the inflammatory milieu might be important for early γδ T cell control.186 Patients undergoing haploidentical HSC transplantation are routinely treated with immunosuppressive drugs such as mycophenolate mofetil (MMF) to prevent graft-versus-host disease (GvHD). Early reduction of MMF was associated with improved Vδ2 T cell recovery and decreased EBV reactivation.187 ZOL has also been used for in vivo activation of γδ T cells after αβ T cell/CD19 B-cell-depleted haploidentical HSC transplantation in children with acute leukemia. Following repeated ZOL administration, the γδ T cell numbers declined more than in the control group; however, γδ T cells differentiated into TEM cells and were more cytotoxic in the ZOL-treated cohort. Despite decreasing γδ T cell counts, the incidence of GvHD and transplant-related mortality were lower in patients receiving ≥3 ZOL infusions.188,189 These studies suggest that in vivo activation of γδ T cells with ZOL is a useful treatment option after HSC transplantation with αβ T cell/CD19 B-cell-depleted stem cell preparations. However, even though objective responses were observed in some patients (see the abovementioned reviews), continuous therapy with ZOL and low-dose IL-2 is not an efficient option for γδ T cell immunotherapy of patients with solid cancers (also in view of the effect of low-dose IL-2 in Treg activation190) but might be useful as a transient procedure together with other strategies. In vivo activation and expansion of Vδ2T cells has also been achieved with i.v. infusion of pAg BrHPP (Phosphostim) together with s.c. low-dose IL-2.191 Originally developed as a γδ T cell immunotherapy, this strategy was abandoned as it failed in further clinical trials.
As mentioned earlier, agonistic mAbs directed against BTN3A1/CD277, such as clone 20.1, are very potent and selective activators of Vδ2T cells.25,31 BTN3A1 is expressed on tumor cells,192 and sensitizing tumor cells with an anti-BTN3A1 mAb drastically increases sensitivity to γδ T cell killing.31 Therefore, a novel strategy for in vivo activation of tumor-reactive Vδ2 T cells is the therapeutic application of a humanized anti-BTN3A1 mAb. In fact, a phase I/IIa clinical trial with such an antibody (ICT01) has just started (https://www.clinicaltrials.gov/ct2/show/NCT04243499). However, given that BTN3A1 is also widely expressed on normal cells, a concern is that normal cells—and not just transformed cells—could also be sensed and killed after binding of an anti-BTN3A1 mAb. The difference might be related to much more efficient BTN3A1 clustering on tumor cells than on normal cells,192,193 but potential adverse effects will need to be closely monitored in the ongoing trial. Based on the wide distribution of the target molecule, the risk of inducing autoimmunity is a potential concern.
Another strategy to specifically target γδ T cells to cancer cells in vivo is bispecific antibody constructs that cross-link the TCRs on γδ T cells with tumor surface antigens. Using Her2-neu as a model antigen, it was shown that a bispecific Her2-Vγ9 antibody construct (designed in a “tribody” format with two anti-Her2 single-chain variable fragments [scFvs] linked to the Fab fragment of an anti-Vγ9 mAb)194,195 efficiently triggered the killing of Her2-expressing tumor cells by short-term expanded Vγ9Vδ2 T cell lines.194,195 Such antibody constructs also efficiently triggered the cytotoxic activity of γδ TILs against autologous cancer cells.77 There are multiple strategies for the rational design of even smaller molecules, such as single-domain nanobodies with low immunogenicity that may increase the likelihood of accumulating around the actual tumor site for the activation of tumor-resident γδ T cells.196,197 In preclinical proof-of-principle studies, the therapeutic efficacy of such anti-EGFR-Vγ9 nanobodies has been demonstrated,198 and bispecific γδ T cell engagers are in development for targeting γδ T cells in cancer patients. Lava Therapeutics’ first bispecific γδ T cell engager (see Table 1) against a novel target is entering clinical trials with first patients with a hematological indication anticipated for the end of 2020 (P. Parren, personal communication).
Taken together, there are various strategies available for the in vivo activation and targeting of γδ T cells as opposed to the adoptive transfer of in vitro expanded γδ T cells. Clinical studies will reveal the respective advantages and disadvantages. Overall, however, we believe that γδ T cell-targeted immunotherapy will need to be combined with other approaches to optimally enhance efficacy. Some possible strategies are discussed in the following section.
Combination matters: how to improve γδ T cell therapy
Successful T cell immunotherapy of cancer requires the optimization of several key issues: (i) tumor-targeted effector activity of T cells; (ii) the sensitivity of cancer cells to T cell attack; (iii) the infiltration of T cells into the tumor tissue (particularly important in the case of “cold” tumors199); and (iv) overriding the immunosuppressive tumor micromilieu. In principle, this applies both to in vivo activation and to adoptive transfer of γδ T cells, as discussed above.
Antibodies and checkpoint inhibitors
A substantial proportion of γδ T cells express the low-affinity Fc receptor for IgG (FcγRIII; CD16), and CD16-dependent antibody-dependent cellular cytotoxicity (ADCC) can be mediated by Vδ2T cells. Therefore, the combination of clinically used therapeutic antibodies with adoptive Vδ2 T cell transfer might enhance the efficacy.200,201 Further, γδ T cell activity is also regulated by checkpoint receptors. Vδ2T cells transiently upregulate PD-1 expression upon activation.113,202,203 PD-1 blockade with antibodies such as pembrolizumab does not significantly modulate the killing capacity202,203 but enhances IFN-γ production in ZOL-activated Vδ2T cells.203 Depending on the status of PD-1 expression on γδ T cells, combination with pembrolizumab might be envisaged for γδ T cell therapy, similar to what has recently been shown for allogeneic NK cell therapy.204 Another example of an inhibitory receptor is NKG2A, which can be expressed together with CD94 on γδ T cells.205 NKG2A has recently been identified as a novel checkpoint inhibitor, and the humanized anti-NKG2A mAb monalizumab unleashes antitumor immunity mediated by CD8 T cells and NK cells.206,207 While the role of γδ T cells has not been specifically addressed in these studies, it will be interesting to investigate the possible effects of monalizumab on the activation and effector function of NKG2A-expressing γδ T cells.
Chemotherapy and epigenetic drugs
Several studies have demonstrated that standard chemotherapeutic drugs or kinase inhibitors frequently increase the cancer cell susceptibility to γδ T cell killing, for instance, for colon cancer,208 glioblastoma,209 ovarian cancer,210 or chronic lymphocytic leukemia.211 Thus, relevant preclinical model systems suggest synergistic effects of combined chemotherapy and adoptive transfer of allogeneic Vδ2T cells.210 A large number of substances have been shown to epigenetically modify gene expression at the level of DNA methylation and histone modification, and several drugs, including the histone deacetylase inhibitor valproic acid (VPA) and the DNA demethylating agent decitabine, are clinically used.212 VPA was shown to synergize with ZOL in enhancing γδ T cell cytotoxicity at the level of pAg production213 but also affected the interaction between γδ T cells and tumor cells at the level of the NKG2D receptor/ligand axis.214 By contrast, decitabine increased the sensitivity of osteosarcoma cells to Vδ2 T cell killing by upregulating the cell surface expression of NKG2D ligands.215 Overall, epigenetic drugs offer interesting perspectives for combination with γδ T cell immunotherapies.216
Accessibility of solid tumors
Adoptively transferred or endogenous γδ T cells need to infiltrate into the tumor to exert their antitumor activity. It has been demonstrated that local low-dose gamma irradiation can cause the normalization of aberrant vasculature and efficient recruitment of tumor-specific T cells,217 and this may also apply to enhancing γδ T cell migration into the tumor. The remodeling of the extracellular matrix by an inhibitor of hyaluronan synthesis has been shown to enhance γδ T cell cytotoxicity against pancreatic adenocarcinoma cells in vitro and to promote the infiltration of γδ T cells into tumor tissue, thereby suppressing tumor growth in xenografted mice.218 Another strategy to enhance antitumor immunity is the activation of innate immunity by ligands for Toll-like receptors (TLRs) or cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING). The modulation of tumorigenesis by TLR/STING ligands is complex and context-dependent. In fact, it can also promote tumor development under certain conditions.219 Similarly, the modulation of γδ T cell activation by specific innate receptors, such as TLR8, also depends on the cellular context and functional outcome parameters. We recently observed strong and monocyte-dependent costimulation of IFN-γ production in Vδ2T cells by TLR8 ligands, whereas proliferative expansion of Vδ2T cells in response to pAgs was simultaneously inhibited.220 Among the many translational perspectives, TLR/STING ligands are considered adjuvants for cancer vaccines.219,221 The intratumoral application of TLR222,223 and STING ligands224 may additionally be used to increase the inflammatory condition of the tumor in situ, thereby allowing more efficient migration of T cells, including γδ T cells, into the tumor microenvironment.
Reversal of the immunosuppressive tumor microenvironment
Tumor cells, tumor-associated macrophages, MDSCs, and other tumor stromal cells can work together to potently suppress intratumoral immune responses. Tumor-intrinsic mechanisms that have been identified to impair γδ T cell attack include the release of large amounts of prostaglandin E2 by tumor cells with strong expression of cyclooxygenase-2 (COX-2),151 the activity of indoleamine-2,3-dioxygenase (IDO) and its metabolite kynurenine,225 the release of galectin-3,226 and the hypoxic tumor microenvironment.227 Inhibitors for the respective pathways can enhance tumor killing by Vδ2T cells in vitro, and it seems reasonable to propose that these strategies can also work in vivo, given the availability of approved drugs such as COX2 inhibitors. In addition to galectin-3, other tumor-expressed galectins, such as galectin-9, also suppress T cell activation.228 The galectin-9 receptor, namely, T cell immunoglobulin domain and mucin domain 3 (Tim-3), on Vδ2T cells has recently been shown to suppress their killing capacity by reducing perforin and granzyme B expression.229 A human anti-galectin-9 antibody has been developed for clinical application,230 and its combined use with γδ T cell immunotherapy may be synergistic. Another important aspect is the hostile metabolic environment for T cells within the tumor, which includes hypoxia, glucose depletion, and lactate accumulation. There exist multiple strategies to optimize T cell metabolism to improve cellular immunotherapy,231 and it will be important to consider these for harnessing the full potential of γδ T cell immunotherapy.
A summary of current strategies to enhance cellular expansion/effector activity in vitro and the clinical efficacy of γδ T cell therapy in vivo is illustrated in Fig. 3.
Future directions
As discussed in this article, there are multiple fronts for future development and optimization to bring to fruition the promise of γδ T cells into clinically effective cellular therapeutics. Here, we highlight just a few of the many noteworthy advances.
Preclinical evaluation
The only relevant animal model to evaluate the in vivo activity of human Vδ2 γδ T cells is nonhuman primates, which also harbor Vγ9Vδ2T cells. Given the lack of readily available appropriate tumor models and the exorbitant costs, it is not feasible to properly evaluate antitumor activity of Vδ2T cells in nonhuman primates. Conventional mice harbor neither homologous γδ TCRs nor BTN2A/3A-homologous butyrophilins; therefore, these cannot be used to address the functionality of Vδ2 γδ T cells. As a result, immunodeficient mice or different types of humanized mice are routinely used to test for antitumor activity of human γδ T cells, but again, this approach does not allow the extrapolation of predictions as to safety and efficacy when applied to humans.143,146 We suggest that more efforts should be devoted to the development of advanced in vitro models, such as three-dimensional spheroid cultures, which allow better predictions than regular two-dimensional tumor γδ T cell cocultures.232
Off-the-shelf γδ T cell products for immunotherapy
As discussed, there is good evidence that the application of allogeneic rather than autologous γδ T cells might be feasible. This opens up the perspective that γδ T cells from healthy blood donors can be manufactured under GMP conditions and stored until required for adoptive immunotherapy of cancer patients. Several companies are pursuing this strategy, as summarized in Table 1.
Biomarkers for tumor susceptibility
Not all transformed cells are equally susceptible to γδ T cell killing. Whenever possible, patients who are being considered for γδ T cell immunotherapy should be selected on the basis of predictive biomarkers. In the case of acute myeloid leukemia (AML), the expression of the NKG2D ligand ULBP1 on AML blasts may be a predictive biomarker for efficacy.233 Conversely, off-the-shelf produced GMP-expanded γδ T cell products generated from different blood donors might not be equally effective against various tumor entities. Pretesting against a panel of tumor targets could help select the most suitable product for each patient.
Coactivation of innate immunity
Above, we discussed the role of TLR/STING activation of innate immunity to support the antitumor activity of γδ T cells. There are multiple additional pathways in the innate immune system that need to be investigated for their potential ability to optimally harness and support the effector functions of tumor-reactive γδ T cells.234 An exciting new perspective arises from recent evidence that organ-specific activation of innate immunity can be triggered by inactivated microbes that are endogenous within specific organ sites. Based on this concept, microbial preparations, called site-specific immunomodulators (SSIs), derived from Klebsiella variicola, Escherichia coli, and Staphylococcus aureus, have been developed and shown in mouse models to have organ-specific effects on the lung, intestine or skin and can stimulate organ-specific antitumor responses.235 SSIs can also enhance the efficacy of adoptively transferred antitumor T cells by supporting the infiltration of T cells into the tumor microenvironment and increasing tumor immunogenicity.236 Therefore, it is anticipated that SSIs would provide important additional activation of tumor-reactive γδ T cells in an organ-specific manner, a concept that we believe should be explored in the future.
High-dose Vit C
The results from our studies have proven that γδ T cell activation and cytotoxicity in vitro can be enhanced by Vit C. These results raise the question of whether Vit C could also enhance γδ T cell activity in vivo. Recently, it was shown that high-dose Vit C increased the efficacy of cancer immunotherapy in various mouse models by enhancing the cytotoxic activity of CD8 T cells and by cooperating with immune checkpoint inhibition.237 In fact, high-dose i.v. application of Vit C in cancer patients is used in some centers, and studies have shown a very good safety profile of high-dose Vit C therapy.238 Therefore, high-dose i.v. Vit C therapy might also be considered when thinking of ways to enhance the efficacy of γδ T cell therapy.
Concluding remarks
Even though there is now fierce competition for determining which cells should be invested in and taken forward for cancer immunotherapy, we believe that the unique properties of γδ T cells put them at the forefront. Support for this idea is evident in the recent burst of interest from small and not-so-small biotech companies exploring the immunotherapeutic potential of γδ T cells, as summarized in Table 1. After 35 years of research to understand the peculiarities of γδ T cells, it is rewarding to witness the current multiple activities to bring these cells into clinical application to treat cancer patients.
Change history
01 September 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Parker, C. M. et al. Evidence for extrathymic changes in the T cell receptor gamma/delta repertoire. J. Exp. Med. 117, 1597–1612 (1990).
Vasudev, A. et al. γ/δ T cell subsets in human aging using the classical α/β T cell model. J. Leukoc. Biol. 96, 647–655 (2014).
Mayassi, T. & Jabri, B. Human intraepithelial lymphocytes. Mucosal Immunol. 11, 1281–1289 (2018).
Guerra-Maupome, M., Slate, J. R. & McGill, J. L. Gamma delta T cell function in ruminants. Vet. Clin. North Am. Food Anim. Pr. 35, 453–469 (2019).
Vantourout, P. & Hayday, A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat. Rev. Immunol. 13, 88–100 (2013).
Hinz, T. et al. Identification of the complete expressed human TCR V gamma repertoire by flow cytometry. Int. Immunol. 9, 1065–1072 (1997).
Wesch, D., Hinz, T. & Kabelitz, D. Analysis of the TCR Vgamma repertoire in healthy donors and HIV-1-infected individuals. Int. Immunol. 10, 1067–1075 (1998).
Deusch, K. et al. A major fraction of human intraepithelial lymphocytes simultaneously expresses the gamma/delta T cell receptor, the CD8 accessory molecule and preferentially uses the V delta 1 gene segment. Eur. J. Immunol. 21, 1053–1059 (1991).
Kalyan, S. & Kabelitz, D. Defining the nature of human γδ T cells: a biographical sketch of the highly empathetic. Cell. Mol. Immunol. 10, 21–29 (2013).
Groh, V. et al. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc. Natl Acad. Sci. USA 96, 6879–6884 (1999).
Hudspeth., K., Silva-Santos, B. & Mavilio, D. Natural cytotoxicity receptors: broader expression patterns and functions in innate and adaptive immune cells. Front. Immunol. 4, 69 (2013).
Correia, D. V., Lopes, A. & Silva-Santos, B. Tumor cell recognition by γδ T lymphocytes: T-cell receptor vs. NK-Cell receptors. Oncoimmunology 2, e22892 (2013).
Simões, A. E., Di Lorenzo, B. & Silva-Santos, B. Molecular Determinants of Target Cell Recognition by Human γδ T Cells. Front. Immunol. 9, 929 (2018).
Pietschmann, K. et al. Toll-like receptor expression and function in subsets of human gammadelta T lymphocytes. Scand. J. Immunol. 70, 245–255 (2009).
Wesch, D., Peters, C., Oberg, H. H., Pietschmann, K. & Kabelitz, D. Modulation of gammadelta T cell responses by TLR ligands. Cell. Mol. Life Sci. 68, 2357–2370 (2011).
Wrobel, P. et al. Lysis of a broad range of epithelial tumour cells by human gamma delta T cells: involvement of NKG2D ligands and T-cell receptor- versus NKG2D-dependent recognition. Scand. J. Immunol. 66, 320–328 (2007).
Kunzmann, V. & Wilhelm, M. Anti-lymphoma effect of gammadelta T cells. Leuk. Lymphoma 46, 671–680 (2005).
Di Lorenzo, B. et al. Broad cytotoxic targeting of acute myeloid leukemia by polyclonal delta one T cells. Cancer Immunol. Res. 7, 552–558 (2019).
Kabelitz, D. & He, W. The multifunctionality of human Vgamma9Vdelta2 gammadelta T cells: clonal plasticity or distinct subsets? Scand. J. Immunol. 76, 213–222 (2013).
Wesch, D., Glatzel, A. & Kabelitz, D. Differentiation of resting human peripheral blood gamma delta T cells toward Th1- or Th2-phenotype. Cell. Immunol. 212, 110–117 (2001).
Ness-Schwickerath, K. J., Jin, C. & Morita, C. T. Cytokine requirements for the differentiation and expansion of IL-17A- and IL-22-producing human Vgamma2Vdelta2 T cells. J. Immunol. 184, 7268–7280 (2010).
Caccamo, N. et al. Differentiation, phenotype, and function of interleukin-17-producing human Vγ9Vδ2 T cells. Blood 118, 129–138 (2011). (2011).
Peters, C., Häsler, R., Wesch, D. & Kabelitz, D. Human Vdelta2 T cells are a major source of interleukin-9. Proc. Natl Acad. Sci. USA 113, 12520–12525 (2016).
Hintz, M. et al. Identification of (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate as a major activator for human gammadelta T cells in Escherichia coli. FEBS Lett. 509, 317–322 (2001).
Nerdal, P. T. et al. Butyrophilin 3A/CD277-dependent activation of human gammadelta T cells: accessory cell capacity of distinct leukocyte populations. J. Immunol. 197, 3059–3068 (2016).
Espinosa, E. et al. Chemical synthesis and biological activity of bromohydrin pyrophosphate, a potent stimulator of human gamma delta T cells. J. Biol. Chem. 276, 18337–18344 (2001).
Freed-Pastor, W. A. et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258 (2012).
Gruenbacher, G. et al. Stress-related and homeostatic cytokines regulate Vγ9Vδ2 T-cell surveillance of mevalonate metabolism. Oncoimmunology 3, e953410 (2014).
Gruenbacher, G. & Thurnher, M. Mevalonate Metabolism in Immuno-Oncology. Front. Immunol. 8, 1714 (2017).
Gober, H. J. et al. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 197, 163–168 (2003).
Harly, C. et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset. Blood 120, 2269–2279 (2012).
Sandstrom, A. et al. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vγ9Vδ2 T cells. Immunity 40, 490–500 (2014).
Yang, Y. et al. A structural change in butyrophilin upon phosphoantigen binding underlies phosphoantigen-mediated Vγ9Vδ2 T cell activation. Immunity 50, 1043–1053 (2019).
Sebestyen, Z. et al. RhoB mediates phosphoantigen recognition by Vγ9Vδ2 T cell receptor. Cell Rep. 15, 1973–1985 (2016).
Rigau, M. et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science. 367, eaay5516 (2020).
Karunakaran, M. M. et al. Butyrophilin-2A1 directly binds germline-encoded regions of the Vγ9Vδ2 TCR and is essential for phosphoantigen sensing. Immunity 52, 487–498 (2020).
Vyborova, A. et al. γ9δ2T cell diversity and the receptor interface with tumor cells. J. Clin. Invest. 132489. https://doi.org/10.1172/JCI132489 (2020).
Herrmann, T., Fichtner, A. S. & Karunakaran, M. M. The molecular basis of recognition of phosphoantigens by human Vγ9Vδ2 T cells. Cells 9, 1433 (2020).
Scotet, E. et al. Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity 22, 71–80 (2005).
Mo, C., Dai, Y., Kang, N., Cui, L. & He, W. Ectopic expression of human MutS homologue 2 on renal carcinoma cells is induced by oxidative stress with interleukin-18 promotion via p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) signaling pathways. J. Biol. Chem. 287, 19242–19254 (2012).
Dai, Y., Chen, H., Mo, C., Cui, L. & He, W. Ectopically expressed human tumor biomarker MutS homologue 2 is a novel endogenous ligand that is recognized by human γδ T cells to induce innate anti-tumor/virus immunity. J. Biol. Chem. 287, 16812–16819 (2012).
Déchanet, J. et al. Implication of gammadelta T cells in the human immune response to cytomegalovirus. J. Clin. Invest. 103, 1437–1449 (1999).
Halary, F. et al. Shared reactivity of Vδ2neg γδ T cells against cytomegalovirus-infected cells and tumor intestinal epithelial cells. J. Exp. Med. 201, 1567–1578 (2005).
Willcox, C. R. et al. Cytomegalovirus and tumor stress surveillance by binding of a human γδ T cell antigen receptor to endothelial protein C receptor. Nat. Immunol. 13, 872–879 (2012).
Marlin, R. et al. Sensing of cell stress by human γδ TCR-dependent recognition of annexin A2. Proc. Natl Acad. Sci. USA 114, 3163–3168 (2017).
Barros, Di. Marco et al. A. epithelia use butyrophilin-like molecules to shape organ-specific γδ T cell compartments. Cell 167, 203–218 (2016).
Melandri, D. et al. The γδTCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nat. Immunol. 19, 1352–1365 (2018).
Willcox, C. R. et al. Butyrophilin-like 3 directly binds a human Vγ4+ T cell receptor using a modality distinct from clonally-restricted antigen. Immunity 51, 813–825 (2019).
Adams, E. J., Gu, S. & Luoma, A. M. Human gamma delta T cells: evolution and ligand recognition. Cell. Immunol. 296, 31–40 (2015).
Hayday, A. C. γδ T cell update: adaptate orchestrators of immune surveillance. J. Immunol. 203, 311–320 (2019).
Nussbaumer, O. & Thurnher, M. Functional phenotypes of human Vγ9Vδ2 T cells in lymphoid stress surveillance. Cells 9, 772 (2020).
Girardi, M. et al. Regulation of cutaneous malignancy by gammadelta T cells. Science 294, 605–609 (2001).
Guerra, N. et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28, 571–580 (2008).
Liu, H. et al. Role of NKG2D and its ligands in cancer immunotherapy. Am. J. Cancer Res. 9, 2064–2078 (2019).
Rincon-Orozco, B. et al. Activation of V gamma 9V delta 2 T cells by NKG2D. J. Immunol. 175, 2144–2151 (2005).
Paczulla, A. M. et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion [published correction appears in Nature. Nature 572, 254–259 (2019).
Wu, J., Groh, V. & Spies, T. T cell antigen receptor engagement and specificity in the recognition of stress-inducible MHC class I-related chains by human epithelial gamma delta T cells. J. Immunol. 169, 1236–1240 (2002).
Xu, B. et al. Crystal structure of a gammadelta T-cell receptor specific for the human MHC class I homolog MICA. Proc. Natl Acad. Sci. USA 108, 2414–2419 (2011).
Mikulak, J. et al. NKp46-expressing human gut-resident intraepithelial Vδ1 T cell subpopulation exhibits high antitumor activity against colorectal cancer. JCI Insight 4, e125884 (2019).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Gentles, A. J. et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 21, 938–945 (2015).
Tosolini, M. et al. Assessment of tumor-infiltrating TCRVγ9Vδ2 γδ lymphocyte abundance by deconvolution of human cancers microarrays. Oncoimmunology 6, e1284723 (2017).
Miao, Y. R. et al. ImmuCellAI: a unique method for comprehensive t-cell subsets abundance prediction and its application in cancer. Immunother. Adv. Sci. 7, 1902880 (2020).
Xu, C. et al. The landscape of immune cell infiltration and its clinical implications of pancreatic ductal adenocarcinoma. J. Adv. Res. 24, 139–148 (2020).
Cordova, A. et al. Characterization of human γδ T lymphocytes infiltrating primary malignant melanomas. PLoS ONE 7, e49878 (2012).
Oberg, H. H. et al. Monitoring and functional characterization of the lymphocytic compartment in pancreatic ductal adenocarcinoma patients. Pancreatology 16, 1069–1079 (2016).
Lee, M. et al. Preferential infiltration of unique Vγ9Jγ2-Vδ2 T cells into glioblastoma multiforme. Front. Immunol. 10, 555 (2019).
Meraviglia, S. et al. Distinctive features of tumor-infiltrating gammadelta T lymphocytes in human colorectal cancer. Oncoimmunology 6, e1347742 (2017).
Cai, X. Y. et al. Low counts of γδ T cells in peritumoral liver tissue are related to more frequent recurrence in patients with hepatocellular carcinoma after curative resection. Asian Pac. J. Cancer Prev. 15, 775–780 (2014).
Allaoui, R. et al. Infiltration of γδ T cells, IL-17+ T cells and FoxP3+ T cells in human breast cancer. Cancer Biomark. 20, 395–409 (2017).
Chabab, G. et al. Diversity of tumor-infiltrating γδ T cell abundance in solid cancers. Cells 9, E1537 (2020).
Beucke, N. et al. Pitfalls in the characterization of circulating and tissue-resident human γδ T cells. J. Leukoc. Biol. 107, 1097–1105 (2020).
Reiß, S. et al. Characterization and classification of glioblastoma multiforme using the novel multiparametric cyclic immunofluorescence analysis system MACSima. Cancer Res. 79(13 Suppl), Abstract nr 245 (2019).
Yoo, S. Y. et al. Whole-slide image analysis reveals quantitative landscape of tumor-immune microenvironment in colorectal cancers. Clin. Cancer Res. 26, 870–881 (2020).
Rong, L. et al. Analysis of tumor-infiltrating gamma delta T cells in rectal cancer. World J. Gastroenterol. 22, 3573–3580 (2016).
Vella, M. et al. Characterization of human infiltrating and circulating gamma-delta T cells in prostate cancer. Investig. Clin. Urol. 60, 91–98 (2019).
Oberg, H. H. et al. Bispecific antibodies enhance tumor-infiltrating T cell cytotoxicity against autologous HER-2-expressing high-grade ovarian tumors. J. Leukoc. Biol. 107, 1081–1095 (2020).
Wakita, D. et al. Tumor-infiltrating IL-17-producing gammadelta T cells support the progression of tumor by promoting angiogenesis. Eur. J. Immunol. 40, 1927–1937 (2010).
Rei, M. et al. Murine CD27(-) Vγ6(+) γδ T cells producing IL-17A promote ovarian cancer growth via mobilization of protumor small peritoneal macrophages. Proc. Natl Acad. Sci. USA 111, E3562–E3570 (2014).
Coffelt, S. B. et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348 (2015).
Van Hede, D. et al. Human papillomavirus oncoproteins induce a reorganization of epithelial-associated γδ T cells promoting tumor formation. Proc. Natl Acad. Sci. USA 114, E9056–E9065 (2017).
Jin, C. et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell 176, 998–1013 (2019).
Silva-Santos, B., Mensurado, S. & Coffelt, S. B. γδ T cells: pleiotropic immune effectors with therapeutic potential in cancer. Nat. Rev. Cancer 19, 392–404 (2019).
Wu, P. et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 40, 785–800 (2014).
Patil, R. S. et al. IL17 producing γδT cells induce angiogenesis and are associated with poor survival in gallbladder cancer patients. Int. J. Cancer 139, 869–881 (2016).
Lo Presti, E. et al. Squamous cell tumors recruit γδ T cells producing either IL17 or IFNγ depending on the tumor stage. Cancer Immunol. Res. 5, 397–407 (2017).
Janssen, A. et al. γδ T-cell receptors derived from breast cancer-infiltrating T lymphocytes mediate antitumor reactivity. Cancer Immunol. Res. 8, 530–543 (2020).
Fichtner, A. S., Ravens, S. & Prinz, I. Human γδ TCR repertoires in health and disease. Cells 9, E800 (2020).
Chen, H. et al. Profiling the pattern of the human T-cell receptor γδ complementary determinant region 3 repertoire in patients with lung carcinoma via high-throughput sequencing analysis. Cell. Mol. Immunol. 16, 250–259 (2019).
Kobayashi, H. et al. Safety profile and anti-tumor effects of adoptive immunotherapy using gamma-delta T cells against advanced renal cell carcinoma: a pilot study. Cancer Immunol. Immunother. 56, 469–476 (2007).
Kobayashi, H., Tanaka, Y., Shimmura, H., Minato, N. & Tanabe, K. Complete remission of lung metastasis following adoptive immunotherapy using activated autologous γδ T-cells in a patient with renal cell carcinoma. Anticancer Res. 30, 575–579 (2010).
Kakimi, K., Matsushita, H., Murakawa, T. & Nakajima, J. γδ T cell therapy for the treatment of non-small cell lung cancer. Transl. Lung Cancer Res. 3, 23–33 (2014).
Correia, D. V. et al. Differentiation of human peripheral blood Vδ1+ T cells expressing the natural cytotoxicity receptor NKp30 for recognition of lymphoid leukemia cells. Blood 118, 992–1001 (2011).
Siegers, G. M. et al. Human Vδ1 γδ T cells expanded from peripheral blood exhibit specific cytotoxicity against B-cell chronic lymphocytic leukemia-derived cells. Cytotherapy 13, 753–764 (2011).
Knight, A., Mackinnon, S. & Lowdell, M. W. Human Vdelta1 gamma-delta T cells exert potent specific cytotoxicity against primary multiple myeloma cells. Cytotherapy 14, 1110–1118 (2012).
Knight, A. et al. CMV-independent lysis of glioblastoma by ex vivo expanded/activated Vδ1+ γδ T cells. PLoS ONE 8, e68729 (2013).
Almeida, A. R. et al. Delta one T cells for immunotherapy of chronic lymphocytic leukemia: clinical-grade expansion/differentiation and preclinical proof of concept. Clin. Cancer Res. 22, 5795–5804 (2016).
Deniger, D. C. et al. Activating and propagating polyclonal gamma delta T cells with broad specificity for malignancies. Clin. Cancer Res. 20, 5708–5719 (2014).
Abe, Y. et al. Possible application of ascites-infiltrating gamma-delta T cells for adoptive immunotherapy. Anticancer Res. 38, 4327–4331 (2018).
Peters, C., Kouakanou, L., Oberg, H. H., Wesch, D. & Kabelitz, D. In vitro expansion of Vγ9Vδ2 T cells for immunotherapy. Methods Enzymol. 631, 223–237 (2020).
Chitadze, G., Oberg, H. H., Wesch, D. & Kabelitz, D. The ambiguous role of γδ T lymphocytes in antitumor immunity. Trends Immunol. 38, 668–678 (2017).
Fleming, C., Morrissey, S., Cai, Y. & Yan, J. γδ T cells: unexpected regulators of cancer development and progression. Trends Cancer 3, 561–570 (2017).
Daley, D. et al. γδ T cells support pancreatic oncogenesis by restraining αβ T cell activation. Cell 166, 1485–1499 (2016).
Peng, G. et al. Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity 27, 334–348 (2007).
Chabab, G. et al. Identification of a regulatory Vd1 gamma delta T cell population expressing CD73 in human breast cancer. J. Leukoc. Biol. 107, 1057–1067 (2020).
Wu, Y. et al. An innate-like Vδ1+ γδ T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci. Transl. Med. 11, 513 (2019).
Kanamori, M., Nakatsukasa, H., Okada, M., Lu, Q. & Yoshimura, A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol. 37, 803–811 (2016).
Casetti, R. et al. Cutting edge: TGF-beta1 and IL-15 Induce FOXP3+ gammadelta regulatory T cells in the presence of antigen stimulation. J. Immunol. 183, 3574–3577 (2009).
Yue, X. et al. Control of Foxp3 stability through modulation of TET activity. J. Exp. Med. 213, 377–397 (2016).
Someya, K. et al. Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and activation. Int. Immunol. 29, 365–375 (2017).
Sasidharan Nair, V., Song, M. H. & Oh, K. I. Vitamin C facilitates demethylation of the Foxp3 enhancer in a Tet-dependent manner. J. Immunol. 196, 2119–2131 (2016).
Kouakanou, L. et al. Vitamin C supports conversion of human γδ T cells into FOXP3-expressing regulatory cells by epigenetic regulation. Sci. Rep. 10, 6550 (2020).
Peters, C., Oberg, H. H., Kabelitz, D. & Wesch, D. Phenotype and regulation of immunosuppressive Vδ2-expressing γδ T cells. Cell. Mol. Life Sci. 71, 1943–1960 (2014).
Schilbach, K. et al. Suppressive activity of Vδ2+ γδ T cells on αβ T cells is licensed by TCR signaling and correlates with signal strength. Cancer Immunol. Immunother. 69, 593–610 (2020).
Kunzmann, V., Kimmel, B., Herrmann, T., Einsele, H. & Wilhelm, M. Inhibition of phosphoantigen-mediated gammadelta T-cell proliferation by CD4+ CD25+ FoxP3+ regulatory T cells. Immunology 126, 256–267 (2009).
Yi, Y. et al. The functional impairment of HCC-infiltrating γδ T cells, partially mediated by regulatory T cells in a TGFβ- and IL-10-dependent manner. J. Hepatol. 58, 977–983 (2013).
Kalyan, S. & Kabelitz, D. When neutrophils meet T cells: beginnings of a tumultuous relationship with underappreciated potential. Eur. J. Immunol. 44, 627–633 (2014).
Sabbione, F. et al. Neutrophils suppress γδ T-cell function. Eur. J. Immunol. 44, 819–830 (2014).
Kalyan, S., Chandrasekaran, V., Quabius, E. S., Lindhorst, T. K. & Kabelitz, D. Neutrophil uptake of nitrogen-bisphosphonates leads to the suppression of human peripheral blood γδ T cells. Cell. Mol. Life Sci. 71, 2335–2346 (2014).
Roelofs, A. J. et al. Peripheral blood monocytes are responsible for gammadelta T cell activation induced by zoledronic acid through accumulation of IPP/DMAPP. Br. J. Haematol. 144, 245–250 (2009).
Fazio, J., Kalyan, S., Wesch, D. & Kabelitz, D. Inhibition of human γδ T cell proliferation and effector functions by neutrophil serine proteases. Scand. J. Immunol. 80, 381–389 (2014).
Towstyka, N. Y. et al. Modulation of γδ T-cell activation by neutrophil elastase. Immunology 153, 225–237 (2018).
Oberg, H. H., Wesch, D., Kalyan, S. & Kabelitz, D. Regulatory interactions between neutrophils, tumor cells and T cells. Front. Immunol. 10, 1690 (2019).
Mensurado, S. et al. Tumor-associated neutrophils suppress pro-tumoral IL-17+ γδ T cells through induction of oxidative stress. PLoS Biol. 16, e2004990 (2018).
Belkina, A. C. et al. Multivariate computational analysis of gamma delta t cell inhibitory receptor signatures reveals the divergence of healthy and ART-suppressed HIV+ Aging. Front. Immunol. 9, 2783 (2018).
Munn, D. H. & Mellor, A. L. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 37, 193–207 (2016).
Chou, F. C., Chen, H. Y., Kuo, C. C. & Sytwu, H. K. Role of galectins in tumors and in clinical immunotherapy. Int. J. Mol. Sci. 19, 430 (2018).
Batlle, E. & Massagué, J. Transforming growth factor-β signaling in immunity and cancer. Immunity 50, 924–940 (2019).
Dysthe, M. & Parihar, R. Myeloid-derived suppressor cells in the tumor microenvironment. Adv. Exp. Med. Biol. 1224, 117–140 (2020).
Mao, Y. et al. A new effect of IL-4 on human γδ T cells: promoting regulatory Vδ1 T cells via IL-10 production and inhibiting function of Vδ2 T cells. Cell. Mol. Immunol. 13, 217–228 (2016).
Sacchi, A. et al. Myeloid-derived suppressor cells specifically suppress IFN-γ production and antitumor cytotoxic activity of Vδ2 T cells. Front. Immunol. 9, 1271 (2018).
Kunzmann, V., Bauer, E. & Wilhelm, M. Gamma/delta T-cell stimulation by pamidronate. N. Engl. J. Med. 340, 737–738 (1999).
Hoeres, T., Smetak, M., Pretscher, D. & Wilhelm, M. Improving the efficiency of Vγ9Vδ2 T-cell immunotherapy in cancer. Front. Immunol. 9, 800 (2018).
Nussbaumer, O. & Koslowski, M. The emerging role of γδ T cells in cancer immunotherapy. Immuno-Oncol. Technol. 1, 3–10 (2019).
Sebestyen, Z., Prinz, I., Déchanet-Merville, J., Silva-Santos, B. & Kuball, J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 19, 169–184 (2020).
Liu, Y. & Zhang, C. The role of human gamma delta T cells in anti-tumor immunity and their potential for cancer immunotherapy. Cells 9, 1206 (2020).
Schumm, M. et al. Depletion of T-cell receptor alpha/beta and CD19 positive cells from apheresis products with the CliniMACS device. Cytotherapy 15, 1253–1258 (2013).
Locatelli, F. et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130, 677–685 (2017).
Wilhelm, M. et al. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J. Transl. Med. 12, 45 (2014).
Kierkels, G. J. J. et al. Identification of a tumor-specific allo-HLA-restricted γδTCR. Blood Adv. 3, 2870–2882 (2019).
Alnaggar, M. et al. Allogenic Vγ9Vδ2 T cell as new potential immunotherapy drug for solid tumor: a case study for cholangiocarcinoma. J. Immunother. Cancer 7, 36 (2019).
Raverdeau, M., Cunningham, S. P., Harmon, C. & Lynch, L. γδ T cells in cancer: a small population of lymphocytes with big implications. Clin. Transl. Immunol. 8, e01080 (2019).
Joalland, N. & Scotet, E. Emerging challenges of preclinical models of anti-tumor immunotherapeutic strategies utilizing Vγ9Vδ2 T cells. Front. Immunol. 11, 992 (2020).
Polito, V. A. et al. Universal ready-to-use immunotherapeutic approach for the treatment of cancer: expanded and activated polyclonal γδ memory T cells. Front. Immunol. 10, 2717 (2019).
Kabelitz, D., Wesch, D., Pitters, E. & Zoller, M. Characterization of tumor reactivity of human V gamma 9V delta 2 gamma delta T cells in vitro and in SCID mice in vivo. J. Immunol. 173, 6767–6776 (2004).
Xiang, Z. et al. Targeted activation of human Vγ9Vδ2-T cells controls epstein-barr virus-induced B cell lymphoproliferative disease. Cancer Cell. 26, 565–576 (2014).
Jarry, U. et al. Stereotaxic administrations of allogeneic human Vγ9Vδ2 T cells efficiently control the development of human glioblastoma brain tumors. Oncoimmunology 5, e1168554 (2016).
Nada, M. H., Wang, H., Workalemahu, G., Tanaka, Y. & Morita, C. T. Enhancing adoptive cancer immunotherapy with Vγ2Vδ2 T cells through pulse zoledronate stimulation. J. Immunother. Cancer 5, 9 (2017).
Pizzolato, G. et al. Single-cell RNA sequencing unveils the shared and the distinct cytotoxic hallmarks of human TCRVδ1 and TCRVδ2 γδ T lymphocytes. Proc. Natl Acad. Sci. USA 116, 11906–11915 (2019).
Wu, D. et al. Ex vivo expanded human circulating Vδ1 γδT cells exhibit favorable therapeutic potential for colon cancer. Oncoimmunology 4, e992749 (2015).
Gonnermann, D. et al. Resistance of cyclooxygenase-2 expressing pancreatic ductal adenocarcinoma cells against γδ T cell cytotoxicity. Oncoimmunology 4, e988460 (2015).
Tanaka, Y. et al. Expansion of human γδ T cells for adoptive immunotherapy using a bisphosphonate prodrug. Cancer Sci. 109, 587–599 (2018). (2018).
Xiao, L. et al. Large-scale expansion of Vγ9Vδ2 T cells with engineered K562 feeder cells in G-Rex vessels and their use as chimeric antigen receptor-modified effector cells. Cytotherapy 20, 420–435 (2018).
Castella, B. et al. Immune modulation by zoledronic acid in human myeloma: an advantageous cross-talk between Vγ9Vδ2 T cells, αβ CD8+ T cells, regulatory T cells, and dendritic cells. J. Immunol. 187, 1578–1590 (2011).
Van Acker, H. H. et al. The role of the common gamma-chain family cytokines in γδ T cell-based anti-cancer immunotherapy. Cytokine Growth Factor Rev. 41, 54–64 (2018).
Van Acker, H. H. et al. Interleukin-15 enhances the proliferation, stimulatory phenotype, and antitumor effector functions of human gamma delta T cells. J. Hematol. Oncol. 9, 101 (2016). (2016).
Correia, M. P., Costa, A. V., Uhrberg, M., Cardoso, E. M. & Arosa, F. A. IL-15 induces CD8+ T cells to acquire functional NK receptors capable of modulating cytotoxicity and cytokine secretion. Immunobiology 216, 604–612 (2011).
Van Acker, H. H., Capsomidis, A., Smits, E. L. & Van Tendeloo, V. F. CD56 in the immune system: more than a marker for cytotoxicity? Front. Immunol. 8, 892 (2017).
Peters, C. et al. TGF-β enhances the cytotoxic activity of Vδ2 T cells. Oncoimmunology 8, e1522471 (2018).
Le Floc’h, A. et al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J. Exp. Med. 204, 559–570 (2007).
Franciszkiewicz, K. et al. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res. 73, 617–628 (2013).
Webb, J. R., Milne, K., Watson, P., Deleeuw, R. J. & Nelson, B. H. Tumor infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin. Cancer Res. 20, 434–444 (2014).
Zheng, N. & Lu, Y. Targeting the IL-9 pathway in cancer immunotherapy. Hum. Vaccin. Immunother. 1–8. https://doi.org/10.1080/21645515.2019.1710413. (2020).
Baker, F. L. et al. Systemic β-adrenergic receptor activation augments the ex vivo expansion and anti-tumor activity of Vγ9Vδ2 T-cells. Front. Immunol. 10, 3082 (2020).
Lee Chong, T., Ahearn, E. L. & Cimmino, L. Reprogramming the epigenome with vitamin C. Front. Cell. Dev. Biol. 7, 128 (2019).
Peters, C., Kouakanou, L. & Kabelitz, D. A comparative view on vitamin C effects on αβ- versus γδ T-cell activation and differentiation. J. Leukoc. Biol. 107, 1009–1022 (2020).
Kouakanou, L. et al. Vitamin C promotes the proliferation and effector functions of human γδ T cells. Cell. Mol. Immunol. 17, 462–473 (2020).
Dalessandri, T., Crawford, G., Hayes, M., Castro Seoane, R. & Strid, J. IL-13 from intraepithelial lymphocytes regulates tissue homeostasis and protects against carcinogenesis in the skin. Nat. Commun. 7, 12080 (2016).
Li, D. et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target Ther. 4, 35 (2019).
Mohanty, R. et al. CAR T cell therapy: a new era for cancer treatment (Review). Oncol. Rep. 42, 2183–2195 (2019).
Fisher, J. & Anderson, J. Engineering approaches in human gamma delta T cells for cancer immunotherapy. Front. Immunol. 9, 1409 (2018). (2018).
Capsomidis, A. et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol. Ther. 26, 354–365 (2018).
Himoudi, N. et al. Human γδ T lymphocytes are licensed for professional antigen presentation by interaction with opsonized target cells. J. Immunol. 188, 1708–1716 (2012).
Harrer, D. C. et al. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: a safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 17, 551 (2017).
Gründer, C. et al. γ9 and δ2CDR3 domains regulate functional avidity of T cells harboring γ9δ2TCRs. Blood 120, 5153–5162 (2012).
Straetemans, T. et al. GMP-grade manufacturing of T cells engineered to express a defined γδTCR. Front. Immunol. 9, 1062 (2018).
Liu, E. et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 382, 545–553 (2020).
Pereboeva, L., Harkins, L., Wong, S. & Lamb, L. S. The safety of allogeneic innate lymphocyte therapy for glioma patients with prior cranial irradiation. Cancer Immunol. Immunother. 64, 551–562 (2015).
Lamb, L. S. Jr et al. Engineered drug resistant γδ T cells kill glioblastoma cell lines during a chemotherapy challenge: a strategy for combining chemo- and immunotherapy. PLoS ONE 8, e51805 (2013).
Lang, J. M. et al. Pilot trial of interleukin-2 and zoledronic acid to augment γδ T cells as treatment for patients with refractory renal cell carcinoma. Cancer Immunol. Immunother. 60, 1447–1460 (2011).
Kunzmann, V. et al. Tumor-promoting versus tumor-antagonizing roles of γδ T cells in cancer immunotherapy: results from a prospective phase I/II trial. J. Immunother. 35, 205–213 (2012).
Pressey, J. G. et al. In vivo expansion and activation of γδ T cells as immunotherapy for refractory neuroblastoma: A phase 1 study. Medicine 95, e4909 (2016).
Dieli, F. et al. Targeting human γδ T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 67, 7450–7457 (2007).
Meraviglia, S. et al. In vivo manipulation of Vgamma9Vdelta2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin. Exp. Immunol. 161, 290–297 (2010).
Kalyan, S., Quabius, E. S., Wiltfang, J., Mönig, H. & Kabelitz, D. Can peripheral blood γδ T cells predict osteonecrosis of the jaw? An immunological perspective on the adverse drug effects of aminobisphosphonate therapy. J. Bone Miner. Res. 28, 728–735 (2013).
de Witte, M. A. et al. Early reconstitution of NK and γδ T cells and its implication for the design of post-transplant immunotherapy. Biol. Blood Marrow Transplant. 24, 1152–1162 (2018).
Liu, J. et al. Immunosuppressant indulges EBV reactivation and related lymphoproliferative disease by inhibiting Vδ2+ T cells activities after hematopoietic transplantation for blood malignancies. J. Immunother. Cancer 8, e000208 (2020).
Bertaina, A. et al. Zoledronic acid boosts γδ T-cell activity in children receiving αβ+ T and CD19+ cell-depleted grafts from an HLA-haplo-identical donor. Oncoimmunology 6, e1216291 (2016).
Merli, P. et al. Immune modulation properties of zoledronic acid on TcRγδ T-lymphocytes after TcRαβ/CD19-depleted haploidentical stem cell transplantation: an analysis on 46 pediatric patients affected by acute leukemia. Front. Immunol. 11, 699 (2020).
Bell, C. J. et al. Sustained in vivo signaling by long-lived IL-2 induces prolonged increases of regulatory T cells. J. Autoimmun. 56, 66–80 (2015).
Bennouna, J. et al. Phase I study of bromohydrin pyrophosphate (BrHPP, IPH 1101), a Vgamma9Vdelta2 T lymphocyte agonist in patients with solid tumors. Cancer Immunol. Immunother. 59, 1521–1530 (2010).
Blazquez, J. L., Benyamine, A., Pasero, C. & Olive, D. New insights into the regulation of γδ T cells by BTN3A and other BTN/BTNL in tumor immunity. Front. Immunol. 9, 1601 (2018).
Garber, K. γδ T cells bring unconventional cancer-targeting to the clinic–again. Nat. Biotechnol. 38, 389–391 (2020).
Oberg, H. H. et al. γδ T cell activation by bispecific antibodies. Cell. Immunol. 296, 41–49 (2015).
Oberg, H. H. et al. Novel bispecific antibodies increase γδ T-cell cytotoxicity against pancreatic cancer cells. Cancer Res. 74, 1349–1360 (2014).
de Bruin, R. et al. Highly specific and potently activating Vγ9Vδ2-T cell specific nanobodies for diagnostic and therapeutic applications. Clin. Immunol. 169, 128–138 (2016).
Labrijn, A. F., Janmaat, M. L., Reichert, J. M. & Parren, P. W. H. I. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 18, 585–608 (2019).
de Bruin, R. et al. A bispecific nanobody approach to leverage the potent and widely applicable tumor cytolytic capacity of Vγ9Vδ2-T cells. Oncoimmunology 7, e1375641 (2017).
Bonaventura, P. et al. Cold tumors: a therapeutic challenge for immunotherapy. Front. Immunol. 10, 168 (2019).
Capietto, A. H., Martinet, L. & Fournié, J. J. Stimulated γδ T cells increase the in vivo efficacy of trastuzumab in HER-2+ breast cancer. J. Immunol. 187, 1031–1108 (2011).
Hoeres, T. et al. Improving immunotherapy against B-cell malignancies using γδ T-cell-specific stimulation and therapeutic monoclonal antibodies. J. Immunother. 42, 331–344 (2019).
Iwasaki, M. et al. Expression and function of PD-1 in human γδ T cells that recognize phosphoantigens. Eur. J. Immunol. 41, 345–355 (2011).
Hoeres, T., Holzmann, E., Smetak, M., Birkmann, J. & Wilhelm, M. PD-1 signaling modulates interferon-γ production by Gamma Delta (γδ) T-Cells in response to leukemia. Oncoimmunology 8, 1550618 (2018).
Lin, M. et al. Pembrolizumab plus allogeneic NK cells in advanced non-small cell lung cancer patients. J. Clin. Invest. 130, 2560–2569 (2020).
Angelini, D. F. et al. NKG2A inhibits NKG2C effector functions of γδ T cells: implications in health and disease. J. Leukoc. Biol. 89, 75–84 (2011).
André, P. et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175, 1731–1743 (2018).
van Hall, T. et al. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 7, 263 (2019).
Todaro, M. et al. Chemotherapy sensitizes colon cancer initiating cells to Vγ9Vδ2 T cell-mediated cytotoxicity. PLoS ONE 8, e65145 (2013).
Chitadze, G. et al. NKG2D- and T-cell receptor-dependent lysis of malignant glioma cell lines by human γδ T cells: Modulation by temozolomide and A disintegrin and metalloproteases 10 and 17 inhibitors. Oncoimmunology 5, e1093276 (2015).
Joalland, N. et al. Combined chemotherapy and allogeneic human Vγ9Vδ2 T lymphocyte-immunotherapies efficiently control the development of human epithelial ovarian cancer cells in vivo. Oncoimmunology 8, e1649971 (2019).
de Weerdt, I. et al. Improving CLL Vγ9Vδ2-T-cell fitness for cellular therapy by ex vivo activation and ibrutinib. Blood 132, 2260–2272 (2018).
Kejík, Z. et al. Epigenetic agents in combined anticancer therapy. Future Med. Chem. 10, 1113–1130 (2018).
Wang, S. et al. Valproic acid combined with zoledronate enhance γδ T cell-mediated cytotoxicity against osteosarcoma cells via the accumulation of mevalonate pathway intermediates. Front. Immunol. 9, 377 (2018).
Bhat, J. et al. Histone deacetylase inhibitor modulates NKG2D receptor expression and memory phenotype of human gamma/delta T cells upon interaction with tumor cells. Front. Immunol. 10, 569 (2019).
Wang, Z. et al. Decitabine enhances Vγ9Vδ2 T cell-mediated cytotoxic effects on osteosarcoma cells via the NKG2DL-NKG2D axis. Front. Immunol. 9, 1239 (2018).
Bhat, J., Kouakanou, L., Peters, C., Yin, Z. & Kabelitz, D. Immunotherapy with human gamma delta T cells-synergistic potential of epigenetic drugs? Front. Immunol. 9, 512 (2018).
Klug, F. et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 24, 589–602 (2013).
Suto, A. et al. Increase of tumor infiltrating γδ T-cells in pancreatic ductal adenocarcinoma through remodeling of the extracellular matrix by a hyaluronan synthesis suppressor, 4-methylumbelliferone. Pancreas 48, 292–298 (2019).
Javaid, N. & Choi, S. Toll-like receptors from the perspective of cancer treatment. Cancers 12, 297 (2020).
Serrano, R., Wesch, D. & Kabelitz, D. Activation of human γδ T cells: modulation by toll-like receptor 8 ligands and role of monocytes. Cells 9, E713 (2020).
Li, A. et al. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J. Hematol. Oncol. 12, 35 (2019).
Wang, D., Jiang, W., Zhu, F., Mao, X. & Agrawal, S. Modulation of the tumor microenvironment by intratumoral administration of IMO-2125, a novel TLR9 agonist, for cancer immunotherapy. Int. J. Oncol. 53, 1193–1203 (2018).
McCall, K. D., Muccioli, M. & Benencia, F. Toll-like receptors signaling in the tumor microenvironment. Adv. Exp. Med. Biol. 1223, 81–97 (2020).
Harabuchi, S. et al. Intratumoral STING activations overcome negative impact of cisplatin on antitumor immunity by inflaming tumor microenvironment in squamous cell carcinoma. Biochem. Biophys. Res. Commun. 522, 408–414 (2020).
Jonescheit, H. et al. Influence of indoleamine-2,3-dioxygenase and its metabolite kynurenine on γδ T cell cytotoxicity against ductal pancreatic adenocarcinoma cells. Cells 9, E1140 (2020).
Gonnermann, D. et al. Galectin-3 released by pancreatic ductal adenocarcinoma suppresses γδ T-cell proliferation but not their cytotoxicity. Front. Immunol. 11, 1328 (2020).
Siegers, G. M., Dutta, I., Lai, R. & Postovit, L. M. Functional plasticity of gamma delta T cells and breast tumor targets in hypoxia. Front. Immunol. 9, 1367 (2018).
Chou, F. C., Chen, H. Y., Kuo, C. C. & Sytwu, H. K. Role of galectins in tumors and in clinical immunotherapy. Int. J. Mol. Sci. 9, E430 (2018).
Li, X. et al. Tim-3 suppresses the killing effect of Vγ9Vδ2 T cells on colon cancer cells by reducing perforin and granzyme B expression. Exp. Cell Res. 386, 111719 (2020).
Chen, L. et al. First in class immunotherapy targeting Galectin-9 promotes T-cell activation and anti-tumor response against pancreatic cancer and other solid tumors. Cancer Res. 79(13 Suppl), 1551 (2019).
Lim, A. R., Rathmell, W. K. & Rathmell, J. C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 9, e55185 (2020).
Varesano, S., Zocchi, M. R. & Poggi, A. Zoledronate triggers Vδ2 T cells to destroy and kill spheroids of colon carcinoma: quantitative image analysis of three-dimensional cultures. Front. Immunol. 9, 998 (2018).
Gundermann, S. et al. A comprehensive analysis of primary acute myeloid leukemia identifies biomarkers predicting susceptibility to human allogeneic Vγ9Vδ2 T cells. J. Immunother. 37, 321–330 (2014).
Demaria, O. et al. Harnessing innate immunity in cancer therapy [published correction appears in Nature . Nature 574, 45–56 (2019).
Kalyan, S. et al. Distinct inactivated bacterial-based immune modulators vary in their therapeutic efficacies for treating disease based on the organ site of pathology. Sci. Rep. 10, 5901 (2020).
Kalyan, S., Bazett, M., Mullins, D. W. & Gunn, H. Novel Microbial-based Immunotherapy Approach Enables Application of Adoptive Cell Therapy for Solid Tumors by Enhancing Both Immune Access to Tumor Microenvironment and Cancer Cell Immunogenecity [abstract]. Fifth International Cancer Immunotherapy Conference “Translating Science into Survival”. September 25–28, Paris, France: CRI-CIMT-EATI-AACR 2019 Abstract A055 (2019).
Magrì, A. et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 12, 532 (2020).
van Gorkom, G. N. Y., Lookermans, E. L., Van Elssen, C. H. M. J. & Bos, G. M. J. The effect of vitamin C (Ascorbic Acid) in the treatment of patients with cancer: a systematic review. Nutrients 11, E977 (2019).
Acknowledgements
The results from D.K.’s laboratory discussed in this article were supported by grants from the Deutsche Forschungsgemeinschaft (DFG Ka 502/16–1, Ka 502 19–1) and the Wilhelm Sander-Stiftung Foundation (grant 2018.045.1). R.S. and L.K. are the recipients of long-term fellowships from the German Academic Exchange Service (DAAD). Figures were created with Biorender.com.
Author information
Authors and Affiliations
Contributions
D.K. wrote a first draft of the manuscript; all authors discussed and finalized the manuscript.
Corresponding author
Ethics declarations
Competing interests
D.K. is a member of the Scientific Advisory Board of Incysus Therapeutics, Inc.; Imcheck Therapeutics; Lava Therapeutics B.V.; and Qu Biologics, Inc. S.K. is the Scientific Director of Qu Biologics, Inc. The remaining authors declare no competing interests.
Additional information
The original version of this article was revised due to a retrospective Open Access order
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kabelitz, D., Serrano, R., Kouakanou, L. et al. Cancer immunotherapy with γδ T cells: many paths ahead of us. Cell Mol Immunol 17, 925–939 (2020). https://doi.org/10.1038/s41423-020-0504-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-020-0504-x
- Springer Nature Limited
Keywords
This article is cited by
-
Novel insights into regulation of butyrophilin molecules: critical components of cancer immunosurveillance by γδ T cells
Cellular & Molecular Immunology (2024)
-
γδ T cells as critical anti-tumor immune effectors
Nature Cancer (2024)
-
Unsynchronized butyrophilin molecules dictate cancer cell evasion of Vγ9Vδ2 T-cell killing
Cellular & Molecular Immunology (2024)
-
γδ T cells in oral diseases
Inflammation Research (2024)
-
Lymphocyte subsets and soluble forms of MIC-A and MIC-B are prognostic factors in non-Hodgkin lymphoma patients
Annals of Hematology (2024)