
Abstract
The initiation of osteogenesis primarily occurs as mesenchymal stem cells undergo differentiation into osteoblasts. This differentiation process plays a crucial role in bone formation and homeostasis and is regulated by two intricate processes: cell signal transduction and transcriptional gene expression. Various essential cell signaling pathways, including Wnt, BMP, TGF-β, Hedgehog, PTH, FGF, Ephrin, Notch, Hippo, and Piezo1/2, play a critical role in facilitating osteoblast differentiation, bone formation, and bone homeostasis. Key transcriptional factors in this differentiation process include Runx2, Cbfβ, Runx1, Osterix, ATF4, SATB2, and TAZ/YAP. Furthermore, a diverse array of epigenetic factors also plays critical roles in osteoblast differentiation, bone formation, and homeostasis at the transcriptional level. This review provides an overview of the latest developments and current comprehension concerning the pathways of cell signaling, regulation of hormones, and transcriptional regulation of genes involved in the commitment and differentiation of osteoblast lineage, as well as in bone formation and maintenance of homeostasis. The paper also reviews epigenetic regulation of osteoblast differentiation via mechanisms, such as histone and DNA modifications. Additionally, we summarize the latest developments in osteoblast biology spurred by recent advancements in various modern technologies and bioinformatics. By synthesizing these insights into a comprehensive understanding of osteoblast differentiation, this review provides further clarification of the mechanisms underlying osteoblast lineage commitment, differentiation, and bone formation, and highlights potential new therapeutic applications for the treatment of bone diseases.
Similar content being viewed by others

Introduction
Adult bone comprises three main cell types: osteoblasts, osteocytes (both of which are derived from mesenchymal stem cells (MSCs)), and osteoclasts (which are derived from hematopoietic stem cells (HSCs)). Regarding bone homeostasis, osteoblasts function to form bone tissue, osteoclasts resorb and degrade bone tissue, while the primary role of osteocytes is to maintain the structural integrity and function of bone by controlling the activity of both osteoclasts and osteoblasts. These opposing cell types regulate the physiological bone turnover rate, which can be separated into two stages: modeling and remodeling. Bone modeling happens continuously during development, while remodeling is involved in tissue renewal over the course of a person’s life. Bone remodeling typically initiates with osteoclasts breaking down the matrix, consisting mainly of type I collagen and crystalline hydroxyapatite proteins. Once resorption occurs, osteoblasts are recruited to the area to produce and mineralize fresh matrix. In the bone matrix, osteoblasts create a critical assortment of macromolecules, including collagen and non-collagen proteins, which act as a platform for lattice mineralization by means of hydroxyapatite formation. Imbalances in the quantity and ratio of osteoblasts to osteoclasts, stemming from bone tissue remodeling, can give rise to conditions such as osteoporosis and other phenotypically akin disorders. As such, there has been continued research on osteoblast differentiation to elucidate mechanisms with therapeutic potential. This review examines the recent advancements in the understanding of cell signaling, transcription, epigenetics, extracellular interplay, and physiological mechanisms that underpin osteoblast differentiation and their application to the treatment of related diseases.
Osteoblast function
Osteoblasts, a major cellular component of bone and the terminal differentiation products of MSCs, have three main functions. First, osteoblasts play a pivotal role as the primary cellular regulator of bone formation. This involves synthesizing and releasing various proteins found in the extracellular matrix (ECM) of bone, along with orchestrating gene expression to facilitate ECM mineralization. With the assistance of β1 integrin, osteoblasts interface with the current matrix to shape cadherin-connected monolayers. Whenever osteoblasts are activated, they secrete a matrix consisting primarily of collagen type I and other vital components (Table 1), as well as significant developmental factors, such as bone morphogenetic protein (BMP) and transforming growth factor-β (TGF-β)1. Previous research also indicates that osteoblasts play important roles in glucose homeostasis via insulin signaling because it increases osteocalcin activity2.
Second, osteoclast differentiation can be directly regulated by osteoblast protein expression3. For example, macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor kappa β ligand (RANKL), two fundamental proteins for osteoclast differentiation, can be expressed by osteoblasts (Fig. 1)4. Osteoblast-produced M-CSF and RANKL have the ability to bind to C-FMS and RANK receptors (respectively) on osteoclast progenitors, triggering subsequent signals that promote osteoclast differentiation by activating the transcription factor nuclear factor of activated T cells 1 (NFATc1)5. Osteoblasts secrete osteoprotegerin (OPG) and can regulate osteoclasts by binding to RANKL, preventing RANKL from binding to RANK. Chemoattractants (CCL8, CCL6, and CCL12) that can influence osteoclast differentiation and recruit osteoclast precursors to bone are also regulated by osteoblast calcineurin/NFAT signaling6. Osteoclasts can also regulate osteoblast differentiation. Previous work in our had found that osteoclasts regulate osteoblast differentiation and tooth root formation via IGF/AKT/mTOR signaling7 (Fig. 2).

MSCs have three different differentiation fates — adipocytes, osteoblasts, and chondrocytes — which are regulated by different genes. In the differentiation process, some cells in the three differentiation pathways also have a reciprocal transformation relationship through the regulation of related genes, such as interactions between mature osteoblasts and mature osteoclasts or hypertrophic chondrocytes and early osteoblasts. Transcription factors have different functions in different stages of osteoblast differentiation. Runx2 is a vital factor in all osteoblast differentiation stages; Runx2 promotes osteoblast differentiation in the early stage while inhibiting mature osteoblast differentiation into osteocytes. Cbfβ is the major co-factor of Runx2 and Runx1. Runx3 can promote chondrocytes into hypertrophic chondrocytes. SIRT1 and FOXO1 can promote Runx2 expression. Osx and β-catenin also have important functions in the early stage of osteoblast differentiation. SATB2 and ATF4 are important in promoting the terminal differentiation stage of osteoblast. SATB2 inhibits Hoxa2 activity in the early stage of osteoblast differentiation. Runx1 plays an important role in inhibiting adipocyte differentiation and promoting chondrocyte differentiation. The interaction between osteoblasts and osteoclasts is also very important. Osteoblasts regulate osteoclast differentiation via RANKL signaling and inhibit osteoclast differentiation through OPG. Similarly, osteoclasts can regulate osteoblast differentiation through the Wnt10b, BMP6, or Ephrin signaling pathway.

Several canonical signaling pathways control the activity of key transcription factors to mediate osteoblast differentiation. Wnt, TGF-β, BMP, FGF, and Hedgehog pathways are the most classic pathways that have been studied during osteoblast differentiation. Wnt binds with FZD receptors, causing the β-catenin accumulation. β-catenin then moves to the nucleus, in which it causes target genes to be transcribed. Wnt signaling also regulates Runx1 and Runx2 functions. TGF-β and BMP signaling regulate osteoblast-specific gene expression through multiple Smad proteins. TGF-β signaling mainly activates Smad2/3, while BMP signaling activates Smad1/5/8. FGF and FGFR can also regulate osteoblast differentiation and osteoblast-specific gene expression via downstream pathways such as PI3K-AKT and ERK pathways. Runx2, Osterix, and several other transcription factors are also necessary for osteoblast differentiation, and these transcription factors are regulated by these classic pathways. Hedgehog signaling is activated through Hh ligand binding to the 12-transmembrane receptor Patched 1 (PTCH1), which relieves inhibition of the seven-pass transmembrane G protein-coupled receptor Smoothened (SMO). Actived SMO can initiate the intracellular cascade leading to the activation of three Gli transcription factors, and Gli can then translocate into the nucleus and regulate Osx activation, thereby modulating osteoblast-specific gene expression. PTH regulates osteoblast differentiation by binding with PTH1R, subsequently activating cAMP and PKA, leading to the phosphorylation of CREB to regulate osteoblast-specific gene expression. IGF-1 binds to its receptor IGF1R and activates PI3K-Akt pathway, resulting in the activation of mTOR and promotion of osteoblast differentiation.
Third, osteoblasts are inextricably linked to the behavior and function of HSCs8. Research has demonstrated the significant involvement of osteoblasts in the HSC microenvironment9,10,11. Based on studies of PTH/parathyroid-related protein receptor (PPR) and bone morphogenetic protein receptor 1a (Bmpr1a), osteoblasts that express N-cadherin form N-cadherin/β-catenin adhesion complexes with HSCs9. Osteoblasts also produce many essential molecules for HSC renewal and maintenance, such as osteopontin (OPN), thrombopoietin, annexin-2, and angiopoietin-112,13. Similarly, C-X-C Motif Chemokine Ligand 12 (CXCL12) in mesenchymal progenitor cells is required for the maintenance of HSCs and can maintain HSCs in an undifferentiated state14. These complexes may mediate the adhesion of hematopoietic stem cells within their niche15.
Osteoblast origin and cell lineage
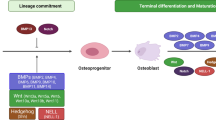
The process of osteoblast formation involves a series of distinct stages and cell types within the osteoblast lineage. Osteoblast lineage cells include mesenchymal progenitors, pre-osteoblasts, osteoblasts, osteocytes, and bone-lining cells (Fig. 1). The exact origin of osteoblasts can vary depending on the life stage and the specificity of the local environment, and therefore, the naming of progenitor cells has always been controversial16. Skeletal stem/progenitor cells (SSPCs) can broadly include all immature precursor cells located within the bone and can differentiate into bone tissue-forming cell types16. This includes cartilage stem cells, recently found in growth plates17,18, fetal perichondrium cells which give rise to bone lines19, and periosteum progenitor cells, in endochondral bone development which is important in fracture repair20,21, as well as distinct reticular and perivascular stromal cell subpopulations in bone marrow, commonly referred to as bone marrow stromal cell (BMSC) populations, which seem to have lipogenic potential in some cases22,23.
During development, osteoblast formation proceeds via two distinct pathways: intramembranous ossification or endochondral ossification (Fig. 1). In mammals, intramembranous ossification primarily affects only a portion of the clavicle and the skull, whereas endochondral ossification controls bone formation throughout the skeletal system24. During intramembranous ossification, MSCs can directly differentiate into osteoblasts, while osteoblasts are indirectly formed during endochondral ossification through the formation of the periosteum, which contains immature osteoprogenitor cells25. Osteoblastogenesis during intramembranous ossification occurs through three main stages: mineralization, proliferation, and matrix maturation. On the other hand, endochondral ossification occurs in two steps: building cartilage templates in a nonvascularized environment and steadily replacing the cartilage template with bone. Through mesenchymal condensations, endochondral ossification constructs a framework for subsequent bones and leads to the mass production of undifferentiated mesenchymal cells. Following the action of SOX9, condensing mesenchymal cells quickly transform into chondrocytes (Fig. 1), which will continue to proliferate and produce cartilage templates26. At this stage, the perichondrium forms a highly vascularized fibrous tissue adjacent to the cartilage template. In an area immediately adjacent to the pre-hypertrophic zone, the perichondrium produces the first osteoblasts in endochondral bones. Later, the perichondrium will develop into the bone collar and periosteum27.
During intramembranous ossification, lineage commitment of MSCs is dependent on the early activation of specific BMPs, peroxisome proliferator-activated receptor γ (PPAR-γ), and Wnt signaling, with alkaline phosphatase (ALP), collagen type I (COL1A1), OPN, osteocalcin (OCN), and PPR among the markers most highly expressed during the differentiation of osteoblasts (Table 1; Fig. 1)15,25,28. After initial lineage commitment, an MSC will divide into an osteoprogenitor and an additional stem cell; this division is important to maintain frequent proliferative activity and self-renewal29. The newly formed pre-osteoblast is an intermediate cell that can express the MSC marker STRO1, ALP, PPR, and collagen type I and possesses an extensive capacity for replication; however, it has no capacity for self-renewal30. Located near newly synthesized osteoid cells, ALP, OPN, bone sialoprotein (BSP), and OCN are expressed by mature osteoblasts. The pre-osteoblast cell is responsible for bone deposition and has limited replication potential31. Terminal differentiation and permanent stoppage in cell division are the next crucial steps. Post-mitotic osteocytes are the final stage of the bone lineage. They are typically found separated in the bone and may be embedded in developing osteoid cells.
Osteoblast signaling
Several essential signaling pathways regulate osteoblast differentiation (Figs. 2, 3). Wnt, TGF-β, Hedgehog, and FGF signaling pathways are canonical pathways known to affect osteoblast differentiation. Additionally, Notch, Hippo, and NF-κB signaling also affect osteoblast differentiation.

Notch, NF-κB, Hippo, Ephrin, and Piezo1/2 signaling pathways also play important roles in osteoblast differentiation. Osteoblast differentiation is inhibited by the Notch and NF-κB signaling pathways, with the Notch signaling pathway’s downstream factors NICD binding to CLS and inhibiting β-catenin. NICD/CLS/Foxo1 complex can promote Hey1 expression, then Hey1 can bind with Runx2 to inhibit Runx2 activity. When NF-κB signaling is stimulated, the P50 and P65 complex translocates into the nucleus and inhibits Smad protein activity. Hippo signaling is a crucial pathway in cell growth and development. The important downstream factors YAP/TAZ in Hippo signaling can regulate osteoblast differentiation-related gene expression. When Hippo signaling is in an “off” state, the YAP/TAZ complex will not be degraded and will translocate into the nucleus to regulate osteoblast-specific gene expression. The snail/slug complex can promote YAP/TAZ activity in nuclear and inhibit YAP/TAZ degradation in the cytoplasm. Ephrin signaling has different regulatory effects on osteoblast differentiation through different pathways and plays a specific role in the regulation of osteoblasts and osteoclasts. The EphrinA2 and EphrinB2 expressed on osteoclast membranes can interact with EphA2 and EphB4 expressed on osteoblast membranes to regulate osteoblast differentiation. Ephrin signaling regulated osteoblast-specific gene expression mainly through RhoA. EphrinA2 binds with EphA2 and promotes RhoA activity, while EphrinB1/2 binds with EphB4 and inhibits RhoA activity. When RhoA is activated, it will inhibit osteoblast differentiation. Piezo1/2 is a recently discovered pathway functioning in osteoblast differentiation. Piezo1/2 can regulate osteoblast-specific gene expression by regulating downstream pathways such as ERK and P38 and interacting with Hippo signaling. Piezo1/2 can regulate β-catenin activation to active Runx2 and regulate osteoblast gene expression. Piezo1/2 can also activate NFATc1 and YAP via calcium signal, and then NFATc1 and YAP can translocate into the nucleus and regulate osteoblast differentiation. Insulin signaling inhibits the production of FoxO1 and Twist2, which can inhibit the expression of Runx2 and Ocn. Ocn improves insulin sensitivity and energy expenditure through multiple mechanisms like activating to β-cells. The direct effect of osteocalcin as an insulin-sensitizing hormone is speculative and remains to be determined.
Wnt signaling pathway
Overview of Wnt signaling
The family of secretory glycoproteins known as Wnt contains numerous protein factors that are ligands of the 7-membrane-spanning frizzled (FZD) receptor family (Fig. 2). The secreted factors of the Wnt family are implicated in numerous cellular processes, such as regulating cell polarity, cell differentiation, cell proliferation and cell function. There are two different categories of Wnt proteins. The first type causes activation of canonical Wnt signaling by forming complexes among Wnt proteins, the low-density lipoprotein receptor-associated protein 5 or 6 (LRP5 or LRP6) and FZD32,33 (Fig. 2). Of note, LRP4 has been implicated in sclerosteosis and van Buchem disease due in part to its role in Wnt signaling, with deficiency of LRP4 in osteoblast-lineage cells causing higher cortical and trabecular bone mass in mutant mice, which is linked to increased bone formation and less bone resorption34. The second type of Wnt protein causes activation of the noncanonical pathway through a protein kinase C-dependent mechanism, where Wnt5a binds with FZD to activate heterotrimeric G proteins and subsequently raises intracellular calcium to promote osteoblast function35.
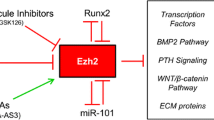
Both canonical and noncanonical Wnt signaling are crucial for bone remodeling36,37. Wnt proteins, whether displayed on the cell surface or secreted, have the capability to engage osteoblasts via binding with the FZD/LRP5/6 complex38. The PTH1 receptor, which binds PTH and forms a complex with LRP5/6, can also activate the Wnt signaling in the absence of the Wnt ligand. Disheveled, Axin39, and Frat-1 proteins stimulate the signal by destroying the protein complex and inhibiting glycogen synthase kinase 3 (GSK3) function, resulting in reduced phosphorylation of β-catenin40 (Fig. 2). By hindering GSK3 activity, the stability of β-catenin is improved and Wnt signaling is turned on (Fig. 2). MiR-346 promotes osteoblast differentiation by inhibiting glycogen synthetase kinase 3β (GSK-3β), preventing β-catenin degradation in human BMSC41 (Fig. 2). Recent studies have shown that 1-Azakenpaullone, a highly selective GSK-3β inhibitor, is a potent inducer for osteogenic differentiation and mineralization of human MSCs42. 1-Azakenpaulone significantly induces osteogenic differentiation and mineralization of human MSCs by activating Wnt signaling, leading to the up-regulation of Runt-related transcription factor 2 (Runx2), a key transcription factor that ultimately promotes osteogenic specific gene expression42. Once stabilized in the cytoplasm, β-catenin translocates to the nucleus to promote target gene expression. The nuclear partners of β-catenin are the lymphoid enhancer-binding factor/T cell factor (Lef/Tcf) family43,44. β-catenin substitutes Lef/Tcf corepressors, establishing heterodimers with Lef/Tcf proteins (Fig. 2). With the help of P300/CBP and other transcriptional co-activators, this heterodimer binds to DNA and promotes target gene transcription for osteoblast differentiation45 (Fig. 4).

Among the many transcription factors that participate in osteoblast differentiation, the Runx family, Osx, ATF4, Cbfβ, and SATB2 are significant. Cbfβ binds to Runx family proteins to form heterodimers, improving Runx2’s and Runx1’s stability and subsequently facilitating Runx2 or Runx1 binding to target DNA sequences. Runx1 positively regulates osteoblast lineage gene expression at various stages of differentiation. Runx1 plays a significant role in postnatal bone homeostasis by binding to ATF4, Ocn, and Runx2 promoters to activate the corresponding genes and promote osteoblast early differentiation. Runx1 promotes BMP7 and Alk3 expression to regulate BMP signaling. How Runx1 can regulate Wnt10b and TGF-β signaling remains unclear. Runx1 can also regulate osteoblast-adipocyte lineage via inducing Wnt/β-catenin signaling, TGF-β signaling, and restraining adipogenic gene transcription. Cbfβ is also crucial in stimulating osteogenesis by inhibiting the expression of the adipogenesis regulatory gene C/EBPα and activating Wnt10b/β-catenin signaling. Wnt binds with FZD receptors, causing the ß-catenin accumulation. Pizeo1/2 can also regulate ß-catenin. ß-catenin then moves to the nucleus, in which it causes target genes to be transcribed by interacting with P300/CBP and TCF/LEF. TGF-β and BMP regulate transcription factors through SMAD proteins to activate Runx1 and Runx2 activity, and the SMAD itself can also regulate osteoblast-specific gene expression. BMP signaling can also activate Dlx5 through ERK and promote Runx2, ATF4, and Osx expression. When the Hippo signaling is at the “off” state, YAP/TAZ can translocate into the nucleus and bind with Runx2 to inhibit its activity. NICD1 in Notch signaling can translocate into the nucleus and promote the expression Hey1, which in turn inhibits Runx2 function by binding with Runx2. The activation of β-catenin and Runx2 will also inhibit the expression of C/EBPα, PPAR-γ, and Fabp4; these genes are important in adipocyte differentiation.
β-catenin degradation can be facilitated through its interaction with protein complexes consisting of APC, Axin, and GSK3 if Wnts are either not expressed or are unable to bind to the receptor (Fig. 2). GSK3 phosphorylates β-catenin, after which the phosphorylated form undergoes ubiquitination by the β-TrCP ubiquitin E3 ligase and subsequent degradation via the ubiquitin-dependent proteasomal system46 (Fig. 2). Pannexins3 (Panx3) can promote the degradation of β-catenin by activating GSK3β, thus inhibiting the activity of the Wnt/β-catenin pathway and the differentiation process of osteoblasts47. In the nucleus, Tcf/Lef and its repressor bind to inhibit target genes downstream of the Wnt signaling. In the “off” state of Wnt signaling, characterized by low cytosolic and nuclear levels of β-catenin, osteoblast differentiation is reduced44 (Fig. 2). Similarly, promotion of APC degradation via genistein-induced autophagy initiated Wnt/β-catenin signaling pathway activation and β-catenin–driven osteoblast differentiation, rebalancing dysregulated osteogenesis within the femurs and tibias of OVX mice48. Taken together, these findings could indicate that targeting the degradation mechanisms for β-catenin may be a potential avenue for better treatment of diseases driven by improper osteoblast differentiation.
Wnt ligands can bind with a variety of inhibitors to limit pathway activation, like FZD-associated protein (sFRP) secretion and Wnt repressor SOST (Sclerosteosis gene product)49, Dickkopf (Dkk) family50, and Src (Fig. 2). These inhibitors like SOST and Dkk can bind to LRP5/6 and inhibit its coreceptor activity. Capable of interfering with canonical Wnt signaling, Dkk1 and Dkk2 bind to LRP5/6 and kremen (a transmembrane protein) (Fig. 2)51. The silencing of the FZD6 receptor influences osteoblast differentiation, restraining osteoblast differentiation through the canonical pathway of Wnt. Meanwhile, FZD4 silencing reduces the rate of osteoblast differentiation via reduced expression of Tcf/Lef52,53.
Conversely, the promotion of osteoblast differentiation through the stabilization of Wnt-signaling can be achieved through various mechanisms. The R-spondin family is comprised of four secreted glycoproteins (Rspo1–4) and functions to amplify Wnt signaling. Notably, Rspo3 variants are strongly linked to bone density54,55. A recent study indicated that in appendicular bones, insufficient Rspo3 haploid and loss of Rspo3 targeting in Runx2+ bone progenitor cells can lead to increased trabecular bone mass, osteoblasts, and bone formation numbers54. The inhibitory effect of Dkk1 on Wnt signaling activation and bone mass is compromised by Rspo3 deficiency54. Defects in Rspo3 result in the activation of extracellular signal-regulated kinase (ERK) signaling, which subsequently stabilizes β-catenin and activates Wnt signaling, thereby promoting osteoblastogenesis and bone formation54.
Wnt signaling performs various functions in different stages of osteoblast differentiation. Canonical Wnt signaling can enhance early osteoblast differentiation, while mature osteoblast mineralization induction is severely hampered by it56,57. During osteoblast differentiation, multiple Wnt signaling genes are changed, and endogenous Wnt signaling is suppressed58,59. Wnt16 can regulate Wnt/β-catenin signaling by antagonizing Wnt5a to suppress the non-canonical Wnt/Ca2+ pathway60. In a prior study, it was discovered that Wnt16 enhances the expression of osteoblast differentiation genes (BMPR1b, BMP7, and ENPP1), while simultaneously reducing the expression of specific osteoblast maturation and mineralization genes (ALP1 and RSPO2)61. However, another study found that Wnt16 positively regulates osteoblast differentiation and matrix mineralization60. Therefore, the mechanistic basis for how Wnt16 regulates osteoblast mineralization still needs further research.
Wnt canonical pathway
The canonical Wnt/β-catenin signaling pathway promotes bone formation by stimulating osteoblast development62,63,64. Bone formation and specific osteoblast gene expression are both enhanced by high levels of β-catenin62,65, while ectopic chondrogenesis and abnormal osteoblast differentiation resulted from conditional β-catenin knockdown at the initial stage of osteoblast development65,66,67 (Table 1). Additionally, Wnt is involved in all stages of bone development, as demonstrated by the process of bone formation exhibited by LRP5-deficient mice68,69,70. Altered bone mass was observed in mice with LRP5 deficiency71,72, indicating that Wnt signaling is crucial during bone development73,74. Furthermore, the functional acquisition of the LRP5 mutation increases Wnt signaling, leading to higher bone mineral density in mammalian models73,75,76,77 (Fig. 2).
Wnt-LRP5 signaling promotes bone formation in part by directly reprogramming glucose metabolism78. Mammalian target of rapamycin complex 2 (mTORC2) and protein kinase B (AKT) are activated by Wnt3a via LRP5 and ras-related C3 botulinum toxin substrate1 (RAC1), which causes the activity of glycolytic enzymes to increase78. In addition, the Warburg effect, induced by Wnt3a’s elevation of key glycolytic enzyme levels, can trigger aerobic glycolysis78. In vitro metabolic regulation aids Wnt-induced osteoblast differentiation and is linked to the bone-forming activity of LRP5 signaling in vivo78. Exosomes of adipose stem cells (ASCs-exos) have recently been found to promote osteogenic differentiation of BMSCs, with ASCs-exos improving bone healing ability in a rat model of nonunion fracture repair79. ASCs-exos play a role in activating the Wnt3a/β-catenin signaling pathway and promoting osteogenic differentiation of BMSCs79. By activating the Wnt3a/β-catenin signaling pathway, ASCs-exos enhance the osteogenesis potential of BMSCs and promote bone repair and regeneration in vivo, providing a new direction for the treatment of diabetic fracture nonunion79.
Bipotential mesenchymal precursors undergo osteoblastogenesis stimulation and adipogenesis inhibition via canonical Wnt signaling. By enhancing osteoclastogenic transcription factors like Runx2 and Osx while inhibiting adipogenic transcription factors, Wnt10b induces osteoblast differentiation62. The inhibition of PPAR-γ and C/EBPα expression is one way through which Wnt10b promotes osteoblastogenesis62. C/EBPα is also an essential regulator in osteoclast differentiation80,81,82. Indeed, Wnt10b–/– mice showed reduced trabecular bone density and serum osteocalcin, further illustrating that Wnt10b is an endogenous regulator of bone formation62.
Wnt noncanonical pathway
Previous research has extensively studied how the canonical Wnt signaling pathway can influence osteoblast differentiation. However, the noncanonical Wnt signaling pathway also plays a significant role in mediating osteoblast differentiation. In comparison to canonical Wnt signaling, the non-canonical Wnt signaling pathway regulates osteoblast differentiation not through β-catenin but rather through various other transcriptional and signaling mechanisms.
Wnt5a
Wnt5a is a significant marker of noncanonical Wnt signaling, with Wnt5a inhibiting the expression of PPAR-γ through SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) to promote osteoblast differentiation83. Although TAZ (Table 1) is an important transcription factor that promotes MSC differentiation by trans-inhibiting PPAR-γ function, noncanonical Wnt signaling via Wnt5a activation does not appear to modulate TAZ expression during osteoblast differentiation83. Following Wnt5a-induced activation of noncanonical Wnt signaling, SETDB1 interacts with chromodomain-helicase-DNA-binding protein 7 (CHD7) and phosphorylated nemo-like kinase (NLK) to form a multi-protein complex83. This complex methylates the histone H3 lysine 9 (H3K9) of the PPAR-γ promoter, resulting in chromatin inactivation and the failure of PPAR-γ to initiate transcription83. By suppressing the transformation of bone marrow MSCs into adipocytes via a reduction in PPAR-γ’s transcriptional effect, Wnt5a can stimulate osteoblast production83.
Wnt/calcium pathway
In the process of osteoblast differentiation, the Wnt/calcium pathway operates as a noncanonical Wnt signaling pathway. The Wnt/calcium pathway can raise intracellular calcium levels to activate calcineurin, protein kinase C (PKC), and calmodulin-dependent protein kinase II (CaMKII), which in turn induces the expression of activating protein 1 (AP-1) transcription factors. The combination of integrin receptors and collagen I may cause CaMKII to be activated in osteoblasts, resulting in ERK phosphorylation, thus promoting Runx2 activation and subsequent osteoblast differentiation84. Therefore, the noncanonical Wnt signaling pathway can also promote osteoblast differentiation by regulating the expression of Runx2, which is mechanistically similar to canonical Wnt/β-catenin signaling.
Wnt7b
In osteoblast precursors, the noncanonical Wnt pathway plays a role in promoting bone formation. This involves Wnt-mediated activation of G protein-linked phosphatidylinositol signaling, leading to the activation of G protein-linked protein kinase C δ (PKCδ). This process, facilitated by Dvl, remains unaffected by Dkk185. In mouse embryos, the elimination of either Wnt7b or PKCδ led to impaired bone formation85. Wnt 7b induces osteoblast differentiation via PKCδ (Fig. 2)85. Wnt-induced PKC activation requires the Gq subunit, which is required for osteoblast differentiation induced by Wnt7b. Wnt7b activates Runx2 through PKCδ signaling to regulate osteoblast-specific gene expression. Although PKCδ signaling activates osteoblast differentiation, in vitro experiments demonstrated that Wnt7b or PKCδ mutant mice can still form bone85. This suggests that this pathway can act as a mechanism to augment other osteogenic signaling pathways, but it may not be enough for osteoblast differentiation85.
Prior research has suggested a connection between increased glycolysis and osteoblast differentiation triggered by Wnt signaling86. However, direct genetic confirmation of the role of glucose metabolism in Wnt-induced bone formation has been lacking. A recent study revealed that overexpression of Wnt7b significantly enhanced bone formation, yet this effect was largely negated in the absence of Glucose Transporter 1 (Glut1), despite the transient deletion of Glut1 itself not affecting normal bone accrual87. Wnt7b was also observed to elevate Glut1 expression and glucose consumption in primary cultures of osteoblast lineage cells, with Glut1 deletion impairing osteoblast differentiation in vitro87. Thus, Wnt7b appears to contribute to bone formation partly by stimulating glucose metabolism in osteoblast lineage cells87.
Role of β-catenin in osteoblast differentiation
β-catenin is required for mature osteoblast differentiation (Table 1)65,66,88. Without β-catenin, osteoblast progenitors will instead develop into chondrocytes, with chondrogenic interstitial precursor fate being determined by β-catenin activity65,66. Runx2 promoter activity and expression are both enhanced by β-catenin/TCF189. Experimental findings have shown that mice lacking β-catenin develop osteopenia (Table 1), while bone mass was increased when β-catenin function was rescued in osteoblasts67,90. Previous work in our lab also found that Cbfβ/Runx2 complex promotes Wnt10b/β-catenin to improve osteoblast differentiation and inhibit c/ebpα expression to inhibit lineage switch into adipocytes91.
Regulation of Wnt signaling pathway
MTSS1 plays a significant role in osteoblast differentiation and bone homeostasis by controlling Src-Wnt/β-catenin signaling. In osteoblast differentiation, Src acts as a negative regulator by phosphorylating LRP6 at several conserved tyrosine residues. This phosphorylation disrupts the removal of LRP6 from the osteoblast cell surface and the formation of LRP6 signalosomes, thereby inhibiting canonical Wnt signal transduction92. When MTSS1 increases, Src diminishes the content of phosphorylated-GSK3β, non-phospho-β-catenin, and transcription factor 7 like 2 (TCF7L2)93. Thus, Src inhibits Wnt/β-catenin signaling activation in osteoblasts, whereas MTSS1 stimulates Wnt/β-catenin signaling by inhibiting the non-receptor tyrosine kinase Src.
Similar to MTSS1, Tenascin-C also positively regulates Wnt signaling. Deletion of Tenascin-C significantly inhibits osteoblast differentiation, indicating its role as a positive regulator of Wnt signaling94. Likewise, β-catenin overexpression can significantly reverse the inhibition of osteoblast differentiation that is caused by Tenascin-C deficiency94. Tenascin-C can bind to DKK-1, the primary inhibitor of Wnt signaling, suppressing DKK-1 function94. Previous work has shown that DKK-1 expression is increased when Tenascin-C is knocked down by siRNA, thereby reducing the transcriptional activity of Wnt signaling94.
TNF receptor-associated factor 3 (TRAF3), a TNF receptor family adaptor protein, positively regulates osteoblast differentiation95. Mice deficient in TRAF3 exhibited reduced bone formation and increased bone resorption, displaying a phenotype indicative of early-onset osteoporosis95. It was also discovered that TRAF3 promoted osteoblast formation and prevented the degradation of β-catenin in MSCs96. TGF-β1, which is released when bone resorption occurs, induces TRAF3 degradation and thus inhibits osteoblast differentiation through GSK-3β-mediated β-catenin degradation96.
Hormones can regulate the canonical Wnt signaling pathway by controlling important proteins in Wnt signaling. In osteoblast progenitors that express Osterix1 (Osx1), Wnt/β-catenin signaling is boosted by estrogen receptor α (Erα), causing an increase in periosteal cell proliferation and differentiation97. Estrogen-related receptor α (ERRα), in conjunction with the coactivator peroxisome proliferator-activated receptor-gamma coactivator (PGC-1α), acts as a positive regulator of Wnt signaling in osteoblast differentiation. This occurs via a cell-intrinsic mechanism that remains unaffected by β-catenin nuclear translocation98. Activated ERRα can bind with the TCF/LEF complex and stimulate osteoblast-specific gene expression similar to that induced by β-catenin98. Thus, it can be understood that TCF/LEF is activated to promote the expression of osteoblast-specific genes in the absence of β-catenin nuclear translocation.
TGF-β and BMP signaling
TGF-β and bone morphogenetic proteins (BMPs) are members of the TGF-β superfamily, playing crucial roles in osteoblast differentiation99,100. There are two main types of TGF-β and BMP signaling: one is Smad-dependent and the other is Smad-independent (Fig. 2)101. There are three Smad types. The first is the receptor-regulated Smads (R-Smads), which can either be triggered by BMPs (Smad1/5/8) or activated by TGF-β (Smad2/3). The second is co-Smads, such as Smad4, which can be co-mediated by BMP and TGF-β. The third is inhibitory Smads, like Smad6 and Smad7, which can negatively regulate BMP and TGF-β signaling (Fig. 2)102,103. R-Smads and co-Smads combine to form a complex that moves to the nucleus and controls osteoblast-specific gene expression101. Runx2 expression is not directly induced by the BMP signaling pathway104, but it can be induced by promoting distal-less homeobox 5 (DLX5) expression (Fig. 2)105,106.
Overview of TGF-β signaling
TGF-β signaling pathways govern osteoblast and chondrocyte differentiation, influencing bone formation at different stages of development and within disease pathology. Smad proteins are critical in TGF-β signaling pathways (Fig. 2)102,107. In conjunction with Smads, TGF-β signaling regulates osteoblast and chondrocyte differentiation108,109. TGF-β signaling primarily activates Smad2/3 to regulate osteoblast differentiation. However, TGF-β can also bind to ALK-1, transducing SMAD-1, 5, and 8 signaling, typically activated by BMPs (Fig. 2)110,111. During the early stage of differentiation, TGF-β signaling promotes osteoprogenitor proliferation and osteogenesis. However, at later stages, it inhibits bone formation102. Previous work in our lab also found that TGF-β1 signaling pathway also is critical in tooth root development and odontoblast differentiation112. Moreover, in vitro studies have demonstrated that TGF-β, along with SMAD3 and SMAD2, inhibits osteogenesis113,114,115,116. Excessive TGF-β signaling was also found to be involved in the pathogenesis of osteogenesis imperfecta in mouse models, with anti-TGF-β treatment correcting altered bone phenotype117. These findings were validated in a small clinical study, where the administration of the monoclonal antibody fresolimumab to participants showed an absence of severe side effects. Furthermore, it was noted to enhance the areal bone mineral density in the lumbar spine of individuals diagnosed with osteogenesis imperfecta type IV118. Deletion of Transforming growth factor beta receptor 2 (TGFBR2), the sole type II receptor for TGF-βs, effectively abolishes TGF-β signaling. This deletion leads to severe defects in calvarial, appendicular, and axis bones119.
TGF-β signaling regulation
Higenamine (HG) has been recognized as a promising candidate for treating osteoporosis through its influence on SMAD2/3 signaling. A novel target for HG has been identified in IQ motif-containing GTPase activating protein 1 (IQGAP1), with HG binding to the Glu-1019 site of IQGAP1, facilitating its osteogenic effects120. Through this mechanism, HG induces Smad2/3 phosphorylation and modulates the Smad2/3 pathway by inhibiting Smad4 ubiquitination120. Consequently, HG emerges as a potential novel small-molecule drug to stimulate bone formation in osteoporosis via the Smad2/3 pathway120.
Recent research has suggested that intraflagellar transport 20 (IFT20) modulates osteoblast differentiation and bone formation by regulating the TGF-β signaling pathway121,122. IFT20 controls MSC lineage allocation by regulating glucose metabolism during bone development123. In MSCs, the absence of IFT20 results in a significant reduction in glucose tolerance and inhibits glucose uptake, lactic acid production, and ATP production123. Deleting IFT20 markedly reduced the signaling activity of TGF-β-Smad2/3, decreased the binding activity of Smad2/3 to the Glut1 promoter, and down-regulated Glut1 expression123. These findings suggest that IFT20 plays a vital role in preventing the allocation of MSC lineages to fat cells through the TGF-β-Smad2/3-Glut1 axis123. Moreover, GATA binding protein 2 (GATA2), necessary for HSC differentiation, can impede osteoblast differentiation by inhibiting Smad1/5/8 activation124.
Overview of BMP signaling
BMP signaling (an evolutionarily conserved group of signaling proteins belonging to the TGF-β superfamily) also has an effective role in osteoblast differentiation. Indeed, BMPs display effective osteogenic effects, similar to the physiological effect mediated by TGF-β125. BMP-2, -4, and -6 are signaling molecules that are highly expressed in both osteoblast cultures and bone tissue126. BMP molecules like BMP-2, -6, -7, and -9 aid in bone formation127. Of note, BMP3B negatively regulates bone formation128, in which BMP3B and BMP-2 may antagonize each other through competition with the availability of Smad4129.
Although BMP signaling can easily regulate the differentiation of osteoblasts through Smad proteins, it is also capable of this regulatory function by activating other signaling pathways controlled by Smad proteins. Prior research indicates that BMP-2 facilitates the interaction between Dvl-1 and Smad1, resulting in the inhibition of β-catenin nuclear accumulation130. Because Dvl-1 is an important inhibitor of GSK-3β, β-catenin is degraded when Dvl-1 binds to Smad1, thus inhibiting the activity of the Wnt signaling pathway130. BMP-2 can inhibit the proliferation of the Wnt signaling in this way, thus enabling MSCs to differentiate into osteoblasts130. Persicae semen (PS) originates from the dried and mature seeds of peach (Prunus persica, L.). PS facilitates mineralization and up-regulates Runx2 via BMP-2 and Wnt signaling pathways. Consequently, this leads to the expression of various osteoblast genes, including Alp, bone gamma-carboxyglutamate protein (Bglap), and integrin binding sialoprotein (Ibsp)131. Research has demonstrated that PS promotes fracture recovery by enhancing osteoblast differentiation and bone formation. Consequently, it could serve as a potential treatment option for patients with fractures131.
Aside from Smad-dependent pathways, BMP can regulate osteoblast differentiation through various other signaling mechanisms. BMP2 stimulates the expression of ALP and OCN by activating the mitogen-activated protein kinase (MAPK) signaling pathway132,133. By triggering Runx2 expression and activating c-Jun N-terminal kinase (JNK), BMP2 promotes osteoblast differentiation through the protein kinase D (PKD) pathway134. DLX5 has been shown to be a BMP2 signaling upstream target and can increase Runx2 expression downstream (Fig. 2)135. By activating DLX5 through the p38 signaling pathway, BMP2 can also induce Osx expression in addition to Runx2 expression (Fig. 2)136. In the bone microenvironment, BMP and other growth factors, like IGF-I, can co-activate Osx expression137. Recent research has also revealed that BMP2 can inhibit the expression and activity of salt-inducible kinase 1 (SIK1) through protein kinase A (PKA)-dependent pathways, thereby promoting osteogenesis138. SIK1 functions as a critical regulator, suppressing preosteoblast proliferation and osteoblast differentiation. The repression of SIK1 is crucial for facilitating BMP2 signaling in osteogenesis138.
BMP signaling regulation
The regulation of BMP signaling concerning osteoblast differentiation is influenced by numerous factors, all of which play distinctive roles in osteogenic processes. For instance, LIM homeobox transcription factor 1 beta (Lmx1b) hinders osteoblast differentiation by negatively controlling BMP2139. Lmx1b can decrease Runx2’s recruitment in the promoter sequence of the target gene and when combined with Runx2, inhibiting Runx2 activity139. Through the BMP2 promoter’s Tcf/Lef response elements, cWnt signals can trigger BMP2 activity140. BMP3 is a negative feedback regulator of osteoblast generation that is induced by cWnt, with decreased BMP3 expression possibly increasing the activity of the cWnt signal, encouraging osteoblast differentiation141. Hey1, which is involved in Notch signaling, can also be expressed by BMP2 signaling (Table 1)142. Particularly noteworthy is that kisspeptin-10 (KP-10), acting as a ligand for GPR54, has previously been identified to initiate the binding of NFATc4 to the BMP2 promoter via its interaction with GPR54. As a result of this interaction, BMP2 upregulates the expression of osteogenic genes by phosphorylating Smad1/5/9, thereby stimulating osteoblast differentiation in vitro143. Subsequent research unveiled that KP-10 encourages osteogenic differentiation of osteoblast progenitors and inhibits bone resorption in human cultured cells, while its administration to healthy male subjects resulted in a notable increase in the bone formation marker osteocalcin without corresponding effects on resorption markers144. Overall, the regulation of BMP2 activity and its signaling pathways represent a potential therapeutic target for conditions such as osteoporosis and other pathologically similar disorders.
Ubiquitin-mediated proteasome degradation also affects BMP signaling, with Smurf proteins regulating the interaction between osteoblasts and osteoclasts145,146. When Smurf1 is deficient, MEKK2 builds up, which triggers JNK activation, a necessary and sufficient event for osteocyte BMP sensitization146,147. Studies have demonstrated that Smurf2-deficient mice exhibit severe osteoporosis148. Osteoblasts deficient in Smurf2 exhibit elevated expression of RANKL. This phenomenon is attributed to Smurf2’s regulation of Smad3’s ubiquitination status and its disruption of the interaction between Smad3 and vitamin D receptors, ultimately resulting in alterations in RANKL expression148.
Hedgehog signaling
Indian Hedgehog (Ihh)
Hh signaling plays an important role in cell proliferation and differentiation, and there are three Hedgehog homologs known to exist in mammals: Sonic Hedgehog (Shh), (Ihh), and Desert Hedgehog (Dhh). In cells gathered from mutant mice lacking smoothened (Smo), an Hh signal, osteoblastic differentiation is impossible149. As such, it is clear that in vitro mesenchymal and bone-forming cells rely heavily on the activity of Smo and the Hh signaling pathway (Fig. 2)149. Previous studies have demonstrated that Ihh is important in chondrocyte differentiation150,151. Ihh is produced by hypertrophic pre-chondrocytes, a group of cells in the inner perichondrium where osteoblast progenitors first appear149,152. The close proximity of these cell types shows the significant and interdependent effect of Ihh signaling on both osteoblast and chondrocyte differentiation153. In vitro experiments have demonstrated that osteoblast differentiation is compromised in mice lacking Ihh, and there is a deficiency in Runx2 expression. These findings suggest that Ihh signaling is capable of initiating osteoblastogenesis154. Additionally, a variety of mesenchymal and bone-forming cells have been stimulated in vitro to develop into osteoblasts by Ihh155,156.
In vitro studies have revealed that Speckle-type POZ protein (Spop), a component of the Cullin-3 (Cul3) ubiquitin ligase complex, positively regulates Hedgehog signaling157,158. Spop-deficient mutant mice exhibited defects in chondrocyte and osteoblast differentiation158. Parathyroid hormone-like peptide (PTHLH) expression was diminished in Spop mutants, while GLI family zinc finger 3 (Gli3) repressor form expression was up-regulated and GLI family zinc finger 2 (Gli2) expression remained unchanged, demonstrating that the Hh signaling was impaired159. Consistent with this finding, the Spop mutant’s skeletal defects were greatly ameliorated by decreasing Gli3 dosage159. A reduction in Gli3 dosage averted the formation of brachydactyly and osteopenia caused by Spop loss159. Therefore, Spop is an important positive regulator of Ihh signaling and skeletal development159.
Shh
The association between focal adhesion kinase (FAK) Tyr (397) expression and Sonic Hedgehog (Shh) expression suggests a potential link between FAK-regulated Osterix and the early differentiation of osteoblasts160. However, to better understand how FAK is controlled at the end of osteoblast differentiation, more research is needed. Purmorphamine, an Hh agonist, is a 2,6,9-trisubstituted purine that targets Smo transmembrane proteins161. The expression of Hh mediators such as Smo, patched 1 (PTCH1), Gli1, and Gli2 can be increased by purmorphamine (Fig. 2). Runx2 and BMP expression is also increased after Hh is activated by purmorphamine162.
From a clinical perspective, Shh has been implicated in the survival of tumor metastases. Research has suggested that signal peptide-CUB domain-EGF-related 2 (SCUBE2) operates in an autocrine fashion on tumor cells, releasing Shh that remains bound to the cell membrane163,164,165,166. This mechanism initiates the activation of Hedgehog signaling, consequently prompting the differentiation of osteogenic cells166. Notably, osteoblasts play a role in this mechanism by secreting collagen, which, in turn, activates the inhibitory leukocyte-associated immunoglobulin-like receptor 1 (LAIR1) signal in natural killer (NK) cells166. This cascade ultimately results in immune suppression and promotes the survival of tumor cells within the bone166.
Regulation of Hedgehog signaling
Pregnane X receptor (PXR) inhibits Hh signaling inducer genes like Gli1 and hedgehog-interacting protein (Hhip) while also inducing the expression of Hh signaling suppressor genes like cell adhesion associated (CDON), BOC cell adhesion associated (BOC), and growth arrest-specific 1 (GAS1)167. After treatment with Smo agonists, osteoblast differentiation and Gli-mediated transcriptional activity was significantly restored when Smo-mediated signaling was activated in these cells167. During osteoblast differentiation, Hh signaling significantly increases insulin-like growth factor 2 (IGF2) expression, which triggers the mTORC2-AKT signaling cascade. In turn, IGF2-AKT signaling stabilizes full-length Gli2, thereby enhancing the output of Hh transcriptional activation168,169.
In osteoblasts, the transmembrane protein SLIT and NTRK-like protein-5 (Slitrk5) are negative regulators of Hh signaling with a few known functions170. Osteoblasts exhibit a distinctive expression pattern of Slitrk5, where overexpression of Slitrk5 in osteoblasts impedes the activation of targeted genes involved in Hh signaling. Conversely, Slitrk5 deficiency in vitro enhances Hh signaling171. Slitrk5 has an extracellular domain that binds to hedgehog ligands and an intracellular domain that interacts with PTCH1171. Through binding with hedgehog ligands and PTCH1, Slitrk5 can inhibit Hh signaling activation, thus reducing osteoblastic differentiation.
Interaction between Hh signaling and Wnt signaling
The interplay between Hh signaling and Wnt signaling is intricate. Previous research has demonstrated that in Ihh-knockout embryos, β-catenin is not translocated into the nucleus and that the Wnt canonical signaling target genes are not expressed88. Thus, the Wnt signaling pathway can only be activated, in part, due to the presence of Hh signaling. Hh signaling also affects how Wnt9a and Wnt7b are expressed, where a noted decrease in their expression accompanies Ihh knockout88. Alternatively, Hh and Wnt signaling pathways could have intracellular crosstalk through common regulators, such as the fusion repressor172 and GSK3173,174. Some studies have demonstrated that BMP is important for Hh-actuated osteoblast differentiation, in addition to Hh’s interaction with the Wnt signaling pathway175,176. For example, during the development of long bones, Ihh and BMP signals are jointly regulated to promote the differentiation of osteoblasts149. Therefore, studying the interaction between Hh signaling and other signaling pathways could provide a new understanding of the regulatory process behind osteoblast differentiation.
FGF signaling
Overview of FGF signaling
Fibroblast growth factors (FGFs) are a series of secreted peptides that control many developmental processes and are essential for controlling endochondral and intramembranous ossification (Fig. 2)177. For instance, craniosynostosis — a condition that presents with the early onset of osseous occlusion of the cranial suture — is usually acquired via mutations in FGF receptors 1-3178,179 (Fig. 2). During growth plate development, both FGF receptor 1 (Fgfr1) and FGF receptor 2 (Fgfr2) are expressed in the condensing stroma, contributing to cartilage formation180. Reserve chondrocytes express Fgfr2, and proliferating chondrocytes down-regulate it, while hypertrophic chondrocytes express Fgfr1180. At the final developmental stages, the tissues that produce osteoblasts and cortical bone, the perichondrium and periosteum, express both Fgfr1 and Fgfr2180. Osteoprogenitor cells typically use Fgfr1 signaling to encourage differentiation, but matured osteoblasts use it to prevent further differentiation180. As a result, Fgfr1 signaling influences osteoblast maturation in a stage-specific manner180. Unlike Fgfr1/2, Fgfr3 significantly regulates proliferating chondrocytes’ growth and differentiation181 while also mediating cortical thickness and bone mineral density in differentiated osteoblasts182,183. As such, human craniosynostosis and achondroplasia syndromes are primarily caused by Fgfrs mutations177,184,185. Additionally, exogenous FGF7 stimulates embryonic stem cell (ESC) differentiation by activating ERK-Runx2 signaling but does not affect the proliferation of osteoblasts186. FGF8 can stimulate Connexin 43 (Cx43) expression, which mainly occurs in osteoblasts and regulates osteoblast proliferation and differentiation187. Therefore, by controlling Cx43 expression, FGF8 can influence osteoblast differentiation. Furthermore, research indicates that FGF18 can also positively regulate osteoblast differentiation by mediating Fgfr1 and Fgfr2 activation via ERK1/2 and PI3K (Fig. 2)188,189.
Role of FGF signaling in osteoblast differentiation
Through phosphorylation of the MAPK signaling pathway, FGF2 can stimulate Runx2 expression, indicating that FGF2 participates in osteoblast differentiation190. Additionally, FGF2 can regulate the Wnt signaling pathway’s activity. Expressions of Wnt10b, LRP6, and β-catenin were reduced in FGF2-deficient BMSCs in vitro, and the inactivated GSK3β was also significantly reduced, suggesting that β-catenin degradation is possible191. By directly or indirectly controlling GSK3’s activity to regulate β-catenin stability, FGF2 may influence the Wnt signaling191.
The differentiation of osteoblasts also depends on FGFR. For example, Fgfr2-deficient mice exhibit a phenotype of decreased bone mineral density192. Different ligands of Fgfr2 include FGF7 and 10 (which are capable of activating FGFR2b) and FGF2, 4, 6, 8, 9 (which are capable of activating Fgfr2c)193,194. Adult Fgfr3-deficient mice displayed osteopenia, indicating that Fgfr3 also participates in osteoblast differentiation182. FGF18 can act as a physiological ligand for Fgfr3 to co-regulate osteoblast differentiation195. Furthermore, FGF18–/– mice exhibited diminished endochondral and intramembranous bone formation, indicating that FGF18 can promote osteoblast differentiation independently of Fgfr3195. Moreover, FGF signaling has been implicated in cranial development, as mutations in FGFR2 have been linked to the autosomal dominant condition Crouzon syndrome. Mouse strains harboring the FGFR2 mutation (p.Cys342Arg) have demonstrated an enhancement in the osteogenic differentiation of MC3T3-E1 cells. This enhancement is achieved through the upregulation of the AMP-activated protein kinase (AMPK)-Erk1/2 signaling pathway196. Furthermore, the FGFR2 p.Cys342Arg mutation increased oxidative phosphorylation and altered mitochondrial dynamics from fusion to fission in MC3T3-E1 cells. This shift facilitated osteogenic differentiation and contributed to craniosynostosis in Crouzon syndrome196. A recent study showed that the stability of FGFR2 is maintained by OTU Deubiquitinase, Ubiquitin Aldehyde Binding 1 (OTUB1)197. OTUB1 diminishes the E3 ligase activity of Smurf1, which mediates FGFR2 ubiquitination, by hindering the binding of Smurf1 to E2197. When OTUB1 is absent, Smurf1 excessively ubiquitinates FGFR2, leading to its degradation in the lysosomes197. These discoveries suggest that OTUB1 actively participates in regulating osteogenic differentiation and mineralization in bone homeostasis by modulating the stability of FGFR2. This highlights OTUB1 as a potential therapeutic target for mitigating osteoporosis197.
Hereditary hypophosphatemic disorders are also associated with FGF signaling pathways, stemming from excess FGF23 — a phosphate (Pi)-regulating hormone produced by bone — which results in impaired skeletal growth and osteomalacia. A recent study explored the impact of Pi repletion and bone-specific deletion of FGF23 on bone and mineral metabolism in the dentin matrix acidic phosphoprotein 1 (Dmp1) knockout mouse model of autosomal recessive hypophosphatemic rickets (ARHR)198. The bone defects observed in Dmp1 knockout mice are only partially attributable to FGF23-induced hypophosphatemia198. The study proposed that simultaneous restoration of DMP1 levels and inhibition of FGF23 could effectively rectify mineral and bone disorders associated with ARHR. Moreover, furin demonstrated the ability to cleave FGF23 in vitro, suggesting a potential therapeutic avenue198. Indeed, inactivation of furin in osteoblasts and osteocytes increased circulating intact FGF23 by 25%199 without significantly impacting serum phosphate levels. Therefore, therapeutically targeting excess FGF23 in patients suffering from hereditary hypophosphatemic disorders could represent a novel treatment modality.
Regulation of FGF signaling
Engrailed homeobox 1 (En1) can regulate FGFR-mediated signaling. ERK activation is inhibited when En1 is mutated, and the ERK activation is confined to mature intracranial osteoblasts of the wild-type skull200. The outer periosteal osteoblasts affected by En1 ablation lost the FGF target gene sprouty RTK signaling antagonist 2 (SPRY2)200. P38, MAPK, and PKC, the effectors of FGF signaling known to influence osteoblast differentiation, may also be influenced by En1200,201,202. En1 and FGF’s coordination of osteoblast differentiation will be better understood if these regulatory pathways are precisely described in a spatiotemporal manner.
The Sprouty family functions as an inhibitor of FGF signaling (Fig. 2). Basic FGF stimulation causes Sprouty2 expression to rise to high levels203. The increased presence of Sprouty2 directs ERK1/2 phosphorylation after basal FGF activation and Smad1/5/8 after BMP activation (Fig. 2)203. Expressions of Ocn, ALP, and Osterix mRNA were all inhibited by Sprouty2. Additionally, osteoblast matrix mineralization was stifled by Sprouty2. By inhibiting the expression of markers in differentiated osteoblasts, like Runx2 and ALP, and reducing FGF-ERK1/2 and BMP-Smad signaling, osteoblast differentiation and proliferation are negatively regulated by Sprouty2203.
Noncanonical signaling pathways in osteoblast differentiation
Ephrin signaling
Overview of Ephrin signaling
Ephrins have bidirectional signal transduction capabilities. This class of signaling proteins falls into two categories: class A (ephrins A1 through A5), which bind to GPI-anchored EphA receptors (A1 through A10), and class B (ephrins B1 through B3), which bind to EphB tyrosine kinase receptors B1 through B6204. The interaction between Ephrin B and EphB, a transmembrane protein with a cytoplasmic domain, facilitate bidirectional signaling in cells. EphrinB2 from osteoclasts and EphB4 from osteoblasts, for instance, combine to create the signal between the two cells205. EphrinB2 on the surface of osteoclasts mediates EphB4 activation and promotes osteoblast differentiation206 (Fig. 1). In contrast, activation of EphrinB2-fold EphB4 on the surface of osteoclasts inhibits C-Fos/NFATc1 signaling of osteoclasts, thereby preventing osteoclast differentiation206. EphB4 signaling can prompt osteoblasts to express transcription factors like DLX5, Osx, and Runx2, underscoring the significance of Ephrin signaling in osteoblast differentiation25. Additionally, the inactivation of ras homolog family member A (RhoA) in osteoblasts may be required for Ephrin signaling to promote osteoblast differentiation207 (Fig. 3), with a study finding that suppression of osteoblast differentiation by EphrinA2(EfnA2)-EphA2 is mediated by increased RhoA activity208.
Ephrin signaling regulation
The effects of stage-specific EphrinB2 deletion on bone strength vary. The early loss of EphrinB2 in osteoblasts results in osteoblast apoptosis, delayed onset of mineralization, and increased bone flexibility. Subsequent deletion of EphrinB2 in osteocytes targeted by the cell can lead to a fragile bone phenotype and heightened osteocyte autophagy209. When drugs were administered to cultured osteoblasts to prevent EphrinB2 from interacting with EphB4, the final stages of osteoblast differentiation and mineralization were repressed210. When inhibiting the interaction of EphrinB2 with EphB4 in vivo, early-stage osteoblast numbers increased while late-stage osteoblast numbers decreased, suggesting the existence of an EphrinB2:EphB4-dependent checkpoint in the process of osteoblast differentiation210. When targeting this EphrinB2:EphB4 checkpoint, the final osteoblast differentiation is blocked, interrupting the initiation of mineralization210. Therefore, these studies suggest that this crucial checkpoint controls the onset of bone mineralization during the late stage of osteoblast differentiation.
Additionally, in osteoblast lineages, the interaction between EphrinB1 and EphB2 impacts the expression from Runx2 to Osx, whereas the interaction between EphrinB2 and EphB4 allows for ALPl expression210. The interaction between EphrinB1 and EphB2 improves their function in osteoblast differentiation and bone formation211. EphrinB1 is necessary for early osteoblast marker expression like Osx and Runx2, with TAZ — a transcriptional coactivator with a PDZ-binding motif — causing Osx expression when translocated into the nucleus through stimulating EphrinB1 reverse signaling with aggregated EphB2211. In the osteoblast lineage, the interaction between EphB2 and EphB4 not only facilitates the differentiation of late osteoblasts but also prevents the formation of osteoclasts211.
The TGF-β1-Scx-EfnA2 axis can negatively regulate periodontal ligament (PDL) tension-induced osteoblast differentiation. TGF-β1-Smad3 signaling and EfnA2 are upstream and downstream regulators of scleraxis (Scx) in PDL cell response to tension, respectively212. Previous work has shown that Scx knockdown completely inhibits tension-induced EfnA2 expression, suggesting that tension-induced Scx inhibits PDL osteoblast differentiation by inducing EfnA2 expression212. This example of the co-regulation of osteoblast differentiation by coordinating the TGF-β and Hh signaling pathways exemplifies the importance of studying the intersection of various signaling pathways of osteoblast differentiation.
TLR/NF-κB signaling
Overview of TLR/NF-κB signaling
RelA (p65), RelB, cRel, NF-κB1 (P50), and NF-κB (P52) form the components of NF-κB signaling, usually kept sequestered by κB inhibitors within the cytoplasm213 (Fig. 3). During bone repair, inflammation is common and has been shown to prevent bone regeneration214. Osteoblast differentiation was inhibited by NF-κB signaling stimulation in previous research215. Cytokines such as tumor necrosis factor (TNF), interleukin-1 (IL-1), interleukin-6 (IL-6), and interleukin-7 (IL-7) facilitate NF-κB activation and hinder osteoblast function in osteoporosis213. Furthermore, TNF-α and IL-1 can regulate osteoblast differentiation by down-regulating the promoter function of osteocalcin, a key gene in osteoblasts214,216. The expression of those cytokines is elevated when NF-κB signaling is stimulated, which will inhibit osteoblast differentiation217.
TLR/NF-κB signaling regulation
IKK-NF-κB stimulation promotes β-catenin ubiquitination and degradation through Smurf1 and Smurf2, inhibiting the Wnt signaling pathway and stimulating Runx2 degradation (Fig. 3). Furthermore, TNF can induce p65 to bind to Smurf1 and Smurf2 promoters in MSCs215. Therefore, TNF may hinder osteoblast differentiation by activating NF-κB signaling. In the healing of occlusion injuries in rodents, the levels of β-catenin and Runx2 decreased alongside increased expression of p65 and IκBα215. However, after hindering IKK-NF-κB signaling, there was a significant increase in the expression of β-catenin, OCN, and Runx2215. Thus, IKK-NF-κB activation is involved in the degradation of β-catenin, ultimately preventing osteoblast differentiation and bone formation215. Similarly, melatonin was found to counteract the activation of the NF-κB pathway by inhibiting TNF-α, thereby reducing osteogenic differentiation and inflammation in bone marrow-derived mesenchymal stem cells (BMSCs)218.
Rocaglamide-A, a suppressor of NF-κB signaling, inhibits phosphorylation of NF-κB to promote osteoblast differentiation219. Conversely, p65 overexpression prevents the promotion of osteoblast differentiation by rocaglamide-A219. Rocaglamide-A inhibits p65 protein phosphorylation and the accumulation of p65 in the nucleus, reducing the transcriptional activity for NF-κB219. According to this work, the NF-κB p65 protein is engaged with the enactment of the NF-κB signaling, and recaglamide-A can forestall the NF-κB inhibitory impact on osteoblast differentiation by preventing the phosphorylation of p65219.
Osteoblasts’ autophagy-related function changes significantly when the NF-κB signaling pathway is disabled, demonstrating a possible connection between the two mechanisms220. However, according to some studies, NF-κB appears to play two roles in autophagy. NF-κB activation has the potential to encourage autophagy in some kinds of cells, such as myocardial cells221, and can also hinder autophagy in other cells like porcine granulosa cells220,222. Therefore, the mechanism of NF-κB in the regulation of autophagy during osteoblast differentiation still needs further research.
Toll-like receptor 4 (TLR4) is also a key factor in NF-κB signaling, with osteoblast expression of TLR4 necessary to activate the NGF-TrkA signaling required for stress-induced bone formation223,224. For example, in TLR4-conditioned knockout mice with normal adult bone mass and strength, loss of TLR4 signaling significantly reduced lamellar bone formation after stress loading225. Inhibition of TLR4 signaling decreased the expression of Ngf in primary osteoblasts225. Bone RNA sequencing in TLR4-conditioned knockout mice and wild-type pups revealed dysregulated inflammatory signaling three days after mechanical osteogenic loading, revealing the important role of osteoblast TLR4 in bone adaptation to mechanical forces225.
Notch signaling
Notch is cleaved by ADAM, a disintegrin and γ-secretase complex containing presenilin 1 or 2 that is formed by Notch ligand interactions226. Upon cleavage, the cytoplasmic Notch intracellular domain (NICD) is liberated and translocated to the nucleus (Figs. 3, 4)226. There, it forms a complex that controls gene transcription via binding with CSL family members226. This complex can activate HEY1 expression, with HEY1 then binding to Runx2 to prevent osteoblast differentiation (Fig. 4)227. NICD can block NFAT signaling by interacting with Foxo1, which also results in the inhibition of osteoblast generation. NICD also impedes Wnt/β-catenin signaling228. Hes1 is one of the downstream target genes of the Notch signaling pathway228,229 (Table 1). Hes1 silencing in vitro suggests that NICD inhibits Wnt/β-catenin signaling primarily through Hes1, where the binding of Hes1 to LEF-1 or transducin-like enhancer protein (TLE) may inhibit Wnt signaling228,229.
Notch signaling exhibits a bidirectional effect on osteoblast differentiation, as it can either inhibit or promote osteoblast differentiation depending on the stage of osteoblast differentiation and the timing of Notch activation230. Studies have shown that activation of Notch signaling can promote the mineralization process of osteoblasts231,232, while it has also been shown that overexpression of Notch1 can inhibit osteoblast differentiation by inhibiting the Wnt/β-catenin signaling pathway233. It was shown that Notch1 not only inhibits terminal osteoblast maturation but also promotes immature osteoblast formation234. Moreover, it was observed that Notch signaling enhances the effectiveness of BMP9-induced BMP/Smad signaling, leading to an upregulation in the gene expression of critical osteogenic factors induced by BMP9 in MSCs, such as Runx2, Colla1, and inhibitor of differentiation234.
Forkhead box protein O1 (Foxo1) inhibits osteoblast production through Notch signaling. Foxo1 forms complexes with NICD and Mastermind to inhibit gene expression235,236 (Figs. 3, 4). As an illustration, Foxo1 inhibits nuclear factor of activated T cells (NFAT) signaling in activated T cells. NFATc1 and NFATc2, in turn, promote the production of osteoblasts, bone formation, and osteoclasts235. Through its interaction with Foxo1, Notch directly and indirectly inhibits the generation of osteoblasts and osteoclasts236. GSK3 can phosphorylate NFATc, resulting in its nuclear export237. The co-localization of Notch2’s NICD with GSK3 in the nucleus can inhibit Wnt/β-catenin signaling235 and suppress osteoblast differentiation. The spalt-like transcription factor 4 (SALL4) can also regulate osteoblast differentiation by inhibiting Notch2’s ability to translocate into the nucleus238.
Epigenetic molecular processes dynamically modify both DNA and histone tails, influencing the spatial organization of chromatin and fine-tuning the outcome of Notch1 transcriptional response239. Although researchers have examined the interaction between histone deacetylase 1 (HDAC1) and Notch in vitro and in Drosophila wing development, the precise role of this interaction in mammalian skeletal development and disorders remains uncertain240,241. In a murine model of osteosclerosis, HDAC1/2 has been identified as a contributor to the disease pathogenesis, which has been attributed to the conditionally cre-activated expression of the Notch1 intracellular domain in immature osteoblasts242. Significantly, targeted homozygous deletions of HDAC1/2 in osteoblasts result in partial alleviation of osteosclerotic phenotypes242. When HDAC1/2 was specifically deleted in osteoblasts of male and female mice, there was an absence of overt bone phenotypes, even in the absence of the Notch1 gain-of-function allele242. These findings provide evidence supporting the idea that HDAC1/2 contributes to the pathogenic signaling of Notch1 in the mammalian skeleton242.
Hippo signaling
Cell proliferation, apoptosis, and stem cell development rely heavily on the Hippo signaling cascade. When Hippo signaling is at an “on” state, YAP and TAZ undergo phosphorylation and are inhibited downstream243. YAP/TAZ are broken down in the cytoplasm, either through interaction with 14-3-3 proteins or through degradation mediated by the proteasome (Fig. 3)243. Yet, when Hippo signaling is inactive, YAP/TAZ translocates into the nucleus where they bind to the TEA domain transcription factor (TEAD), jointly regulating gene expression (Fig. 3)244. By initiating the downstream RhoGTPase-actomyosin signaling cascade, matrix metallopeptidase 14 (MMP14) can trigger β1-integrin activation and thereby promote YAP/TAZ nuclear transfer to regulate gene expression245.
YAP is an important component within Hippo signaling (Table 1). It regulates cell proliferation and apoptosis via cooperating with the Tead/Tef family and activating target genes in the Hippo signaling pathway246 (Fig. 3). In vitro experiments have exhibited that YAP can inhibit bone marrow MSC osteogenesis, subsequently reducing the rate of osteoblast differentiation247. According to previous research, YAP is a mediator of the Src/Yes tyrosine kinase pathway, which can interact with Runx2 to suppress Runx2 transcriptional activity to inhibit osteocalcin expression247.
YAP’s nuclear localization is influenced by numerous factors, which, in turn, limit its transcriptional activity. For instance, the MAPK signaling pathway’s expression of ERK and JNK controls the degree of YAP phosphorylation248. However, YAP phosphorylation can be accelerated by Src/Yes kinase249. Conversely, the Runx2-YAP complex separates when Src/Yes kinase is inhibited249. By blocking the interaction between YAP and 14-3-3 protein through YAP phosphorylation, which is mediated by AKT kinase, YAP can remain in the cytoplasm and not be degraded, ultimately preventing the expression of osteoblast-related genes250.
Transformation-associated protein 53 (Trp53) is a key regulator that regulates the activation of TAZ to regulate the osteoblast differentiation251. Elimination of Trp53 in osteoblast lineages markedly boosts osteogenesis, bone formation, and bone remodeling252. The lack of Trp53 significantly increases the transcriptional activity of TAZ by hindering TAZ phosphorylation and its translocation into the nucleus, while this activity is notably reduced upon enforced expression of Trp53251. Trp53 is linked to TAZ and reduces the stability of the TAZ protein, promoting its degradation through ubiquitination mediated by beta-transducin repeats-containing proteins (β-TRCP)251. To sum up, Trp53 governs chondrogenesis and intrachondral ossification by inhibiting TAZ activity and stability. This implies that manipulating Trp53 signaling might offer a promising avenue for addressing fracture healing, ectopic ossification, arthritis, and various other bone-related disorders251.
Hippo signaling also crosstalks with other signaling pathways, including the Wnt and TGF-β pathways, to modulate osteoblast differentiation. Hippo signaling stimulates TAZ to cooperate with disheveled (DVL) in the cytoplasm, which limits Wnt/β-catenin signaling253. By preventing DVL phosphorylation, TAZ can block Wnt/β-catenin signaling253. Similarly, the Hippo signaling pathway inhibits TGF-β signaling by preventing Smad complexes like Smad2/3 from translocating into the nucleus through the cytoplasmic localization of TAZ/YAP254,255. Clinically, the TAZ/YAP signaling pathway has recently been linked to abnormal differentiation of mesenchymal lineage (MLin) cells, which leads to the formation of bone within soft tissues of the musculoskeletal system following traumatic injury256. Recent studies have demonstrated that discoidin domain receptor 2 (DDR2) is significantly and specifically upregulated in collagen-expressing heterotopic ossification MLin cells256. DDR2 acts as a cell surface receptor for fibrillar collagen and serves as a crucial regulator of MLin cell function in heterotopic ossification formation256. Mechanistically, perturbation of DDR2 alters focal adhesion orientation and subsequent matrix organization, thereby modulating the YAP/TAZ-mediated signaling of MLin cells through focal adhesion kinase FAK and YAP/TAZ256. Therefore, the interactions between ECM and DDR2 play a crucial role in driving heterotopic ossification, presenting a potential novel therapeutic target for addressing this pathological condition256.
Piezo1 and Piezo2 signaling
Bone tissue responds to mechanical stress stimulation, with both unloading and loading of mechanical stress exerting significant effects on osteoclast differentiation and function, as well as osteoblast differentiation and function257. The bone microenvironment and metabolic activity is influenced by mechanical stress257. At present, the understanding of mechanoreceptors is thought to involve ion channels, extracellular matrix adhesion molecules, and the cytoskeleton, yet the current understanding of these receptors is incomplete258. Piezo1 and Piezo2 are important mechanosensitive channels. Piezo1 and Piezo2 have been identified as crucial players in regulating osteoblast differentiation and function259,260,261. Deletion of Piezo1 or Piezo2 in osteoblast or osteoclast lineage cells causes a severe osteoporosis phenotype with numerous spontaneous fractures, specifically in osteoblast lineage cells, indicating that Piezo1 and Piezo2 are important in bone formation and bone function262.
Piezo1 and Piezo2 mechanosensitive channels have been recognized as critical force sensors essential for bone development and osteoblast differentiation263. Depletion of Piezo1, and particularly Piezo1/2, in mesenchymal or osteoblast progenitor cells results in spontaneous bone fractures in newborn mice263. This outcome was attributed to the suppression of osteoblast differentiation and increased bone resorption263. Furthermore, the absence of Piezo1/2 conferred resistance to additional bone loss induced by unloading in both bone development and homeostasis263. Mechanistically, Piezo1/2 channels transmit signals from fluid shear stress and extracellular matrix stiffness, triggering calcium influx that activates Calcineurin263. This activation leads to the coordinated activation of NFATc1, YAP1, and β-catenin by inducing their dephosphorylation, along with the formation of NFAT/YAP1/β-catenin complexes (Fig. 3)263. Piezo1 and Piezo1/2 mutant mouse bones have reduced Yap1 and β-catenin activities, and enhanced β-catenin activity partially rescues the phenotype263.
Zou et al. discovered that PIEZO1 functions in osteoblast lineage cells to regulate bone remodeling by sensing mechanical loading264. Their research demonstrated that PIEZO1 regulates YAP signaling, which in turn controls the expression of various bone matrix proteins, including several types of collagens (Fig. 3)264. These collagens’ expression is contingent upon a PIEZO1-dependent response to mechanical stimulation264. This suggests that PIEZO1 may also play a role in regulating bone mass and bone strength in humans in vivo265.
For bone growth to occur, a specific type of highly angiogenic blood vessels known as type H vessels is required. These vessels provide a pathway for osteoblasts surrounding them266. Towards the end of adolescence, type H vessels undergo a transition to a quiescent type L endothelium, losing their ability to support bone growth267,268. Dzamukova et al. discovered that mechanical forces, linked to increased body weight at the end of adolescence, activate the mechanoreceptor PIEZO1, leading to heightened production of FAM20C (Golgi-associated secretory pathway kinase) in osteoblasts266. FAM20C, the primary kinase of the secreted phosphoproteome, phosphorylates DMP1, previously recognized as a crucial factor in bone mineralization269,270. Following this, osteoblasts release dentin matrix protein 1 in a pulsatile fashion266. Extracellular DMP1 inhibits vascular endothelial growth factor (VEGF) signaling by preventing the phosphorylation of vascular endothelial growth factor receptor 2 (VEGFR2)266. This process results in the conversion of type H vessels into type L vessels, decreasing bone growth activity and fostering heightened bone mineralization266. Consequently, targeting PIEZO1 may offer a novel approach for addressing bone-related diseases266.
Hormone regulation
Hormones also play significant role in regulating osteoblast differentiation. Thyroid-stimulating hormone (TSH) is an important hormone that can act on the thyroid gland to produce two thyroid hormones, thyroxine (T4) and tri-iodothyronine (T3)271. A study found that a novel form of the TSHβ subunit, TSHβv, can promote osteoblast differentiation272.
The sympathetic nervous system boosts bone breakdown by increasing Rankl in early bone cell precursors, a process needing ATF4 phosphorylation. In mice lacking Adrb2 and gonads, bone breakdown decreases, whereas in hypogonadal mice with low sympathetic activity, bone breakdown increases. This discrepancy might be due to CART, a neuropeptide influenced by leptin, which regulates RankL to curb bone breakdown. Leptin-driven neural pathways manage two aspects of bone remodeling273,274.
Insulin signaling can regulate osteoblast differentiation2,275,276 (Fig. 3). Study found that insulin signaling can inhibit Twist2 to regulate osteoblast differentiation277. Twist2 is one of the inhibitor of Runx2276 (Fig. 3). The transcription factor forkhead box O1 (FoxO1) acts as an inhibitor of insulin signaling. Once Ocn is synthesized, it undergoes γ-carboxylation, followed by export and storage bound to the mineralized bone extracellular matrix (ECM). The inactivation of Ocn can be hindered by the cellular import of glutamate, which inhibits the activity of the γ-carboxylase enzyme278 (Fig. 3). Alternatively, γ-carboxylated Ocn bound to the ECM can undergo partial decarboxylation and be released into the general circulation through bone resorption2,278. Besides, Ocn could activate β-cells, and the activated β-cells can promote insulin signaling to promote osteoblast differentiation.
Osteoblast transcriptional factors
Several homeodomain proteins are included in the group of transcription factors that control osteoblasts: Smads, CCAAT/enhancer binding proteins β (C/EBβ), C/EBPδ (Table 1), lymphatic enhancer factor (Wnt effector), Twist, Runx1/Cbfβ, activating transcription factor 4 (ATF4), Runx2, and Osterix, of which the latter four are the major transcription factors that function within osteoblast differentiation (Fig. 4; Table 1).
Transcriptional modulators that function as “master switches” coordinate the transformation of MSCs into tissue-specific cell types. Runx2 is pivotal in bone formation279. During differentiation, it plays a major role in regulating cell growth and the activation or repression of genes279. Even though osteoblast differentiation mainly relies on Runx2 and Osterix, other important genes like Runx1 and Cbfβ are also required for differentiation (Fig. 4)3.
Runx2
Runx2, also known as Cbfa1 or PEBP2, is a member of the RUNT-related transcription factor family, which also includes Runx1 and Runx3280. Runx2 is particularly important for osteoblast differentiation. All Runx family proteins can bind to Cbfβ to form stable heterodimers with a DNA-recognition sequence of TGTGGT281. Runx2 has an activation domain that turns on the genes for osteocalcin and COL1A1 in addition to its conserved DNA-binding domain280,282,283. As a result, Runx2 is an early marker of osteoblast differentiation, which is indicated by the relative timing of its expression284 (Fig. 4). At the stage where MSCs first differentiate into osteoblast precursor cells before transforming into immature osteoblasts, Runx2 expression is upregulated284. However, by the final stages of osteoblast differentiation and formation, Runx2 expression gradually decreases (Fig. 4)284,285. Glucose, the main nutrient for osteoblasts, is transported into these cells through Glut1, which is expressed before Runx2. Glucose uptake facilitates osteoblast differentiation by suppressing AMPK-dependent proteasomal degradation of Runx2 and also enhances bone formation by inhibiting another aspect of AMPK function286.
Runx2 is abundant in bone and calcified cartilage but is also expressed in the thymus and testicles287,288. Runx2-deficient mice failed to form bone, indicating that Runx2 is necessary for the formation of both membranous and endochondral bone280,289,290. Osteoblast-specific genes were expressed when Runx2 was produced in cultured skin fibroblasts, suggesting that Runx2 could promote osteoblast differentiation280. Runx2 may govern the transition from the growth phase to the postproliferative phase by inhibiting progenitor cell proliferation, thus serving as a critical element in the development of cells within the osteogenic lineage291. Thus, Runx2 may induce the gene expression needed for lineage identification and mesenchymal differentiation in early bone progenitor cells.
Due to this critical role in bone homeostasis, Runx2 has also been implicated in the symptomatic progression of bone-associated cancers. A recent study found that increased expression of Runx2 is responsible for bone destruction found in multiple myeloma (MM)292. The paper demonstrated that Runx2 promotes the suppression of osteoblast activity and enhancement of osteoclast activity by multiple myeloma cells using in vitro and in vivo approaches292. Notably, the data suggests that therapeutic blockade of Runx2 could safeguard against bone degradation by preserving the equilibrium between osteoblast and osteoclast functions in MM292.
Co-activators of Runx2
Numerous transcriptional coactivators can engage with Runx2. Cbfβ is the most important of these factors because it can bind to Runx family proteins to form heterodimers, improving Runx2’s stability and subsequently facilitating Runx2 binding to target DNA sequences (Fig. 4). Cbfβ settles Runx2 by restraining ubiquitination, thus preventing Runx2 degradation and further promoting the expression of osteoblast-specific genes293. Cbfβ is also crucial in stimulating osteogenesis by inhibiting the expression of the adipogenesis regulatory gene C/EBPα and activating Wnt10b/β-catenin signaling91 (Table 1). Wnt10b expression was down-regulated in Cbfβ-deficient cells, and another analysis showed that Cbfβ increased the transcriptional expression of Wnt10b91. Furthermore, adipocyte development was suppressed in the presence of Wnt3L conditioned medium containing Wnt3a, but osteoblast differentiation was not reversed91. According to these findings, Runx/Cbfβ controls osteoblast differentiation independently of Wnt10b/β-catenin signaling and suppresses the transformation of MSCs and osteoblasts into adipocytes91.
In addition to Cbfβ, co-activators such as monocytic leukemia zinc finger protein (MOZ), MOZ-related factor (MORF), and others play a crucial role in facilitating Runx2’s control over osteoblastogenesis. MOZ and MORF belong to the MYST family that comprises histone acetyltransferases, which can interact with Runx2’s activation domain to increase osteocalcin expression294. Grg5, pRb, and TAZ are also effective Runx2 activators295,296 that can increase Runx2’s transcriptional activity through their interaction with the protein297,298. These co-activators interact with Runx2 to enhance the transcriptional activity of Runx2 and promote the regulation of Runx2 on osteoblast-specific gene expression, thus promoting osteoblast differentiation297,298.
Vestigial-like family member 4 (VGLL4) disrupts TEAD-mediated Runx2 transcriptional repression, thereby facilitating osteoblast differentiation and bone development (Table 2)299. TEAD transcription factors are inhibitors of Runx2 transcriptional activity, which can act independently of YAP binding, severely inhibiting osteoblast differentiation299. In addition, VGLL4 attenuates TEAD transcriptional repression by directly competing with Runx2 for binding of TEAD through its two TDU domains299. VGLL4 can remove the inhibitory function of TEADs by disrupting its interaction with Runx2299. Additionally, the absence of VGLL4 in MSCs has shown a skeletal defect similar to global VGLL4-deficient mice, along with the knockdown of TEAD or overexpression of Runx2 in VGLL4-mutant osteoblasts reversing osteoblast differentiation inhibition299. These results indicate that VGLL4 can control the Runx2-TEADS transcription complex to control osteoblast differentiation and bone formation299.
Runx2 can also promote gene expression by interacting with the myeloid Elf-1-like factor (MEF) and binding to an osteoblast markers’ promoter, like OPN, close to MEF (Table 2)300. The presence of these two transcription factor response elements (RRE and ERE) near the promoter’s proximal region is necessary for MEF and Runx2 to function on the promoter of bone marker genes300. When getting close to these two elements, MEF and Runx2 show significant synergistic effects on promoter activity. MEF and Runx2 synergistically affect OPN, OCN, and ALP promoters300.
Sirtuin 1 (SIRT1) has the potential to positively regulate Runx2 (Table 2). Runx2’s activity is enhanced by SIRT1, a Class III histone deacetylase301. In vitro experiments showed that Runx2 expression did not change when SIRT1 was absent, but osteoblast differentiation was inhibited, and Runx2 target protein expression was decreased301. By interacting with Runx2, SIRT1 can enhance its trans-activation activity301. As Osx is one of Runx2’s downstream targets, SIRT1 can trigger Runx2’s activation and promote Osx expression, which in turn targets osteoblast-specific gene expression and promotes osteoblast differentiation301.
Co-repressors of Runx2
One particularly important regulatory mechanism that is utilized to help govern the transcription of osteoblast-linked genes is protein degradation. As Runx2 is a critical transcription factor in promoting osteoblast differentiation, the degradation of this protein through the proteasome pathway has been shown to inhibit the rate of osteoblastogenesis302. Smad6 can initiate Smurf1-induced Runx2 degradation, triggered by the ubiquitin isopeptide ligase (E3) Smurf1303. E3 ligase can interact with Hsc/HSP70, which negatively regulates osteoblast differentiation by promoting Runx2 ubiquitination degradation304. Additionally, a novel AMPK pathway has been identified to modulate Runx2 degradation via ubiquitin degradation305,306,307. However, the extent to which Runx2, and any subsequent osteoblast differentiation, is altered by AMPK via ubiquitin degradation or rather is more significantly mediated by alternative mechanisms requires further investigation due to recent conflicting findings307,308.
The epidermal growth factor receptor (EGFR) signaling pathway plays an important role in tissue development and tumor development and has been previously shown to regulate the expression of Runx2 and Osterix (Table 2). EGFR diminished Runx2 and Osx expression and increased the expression of inhibitors HDAC4 and HDAC6 in differentiated osteoblasts309. In vitro overexpression of HDAC4 diminishes Runx2 levels because HDAC deacetylates lysine residues in histones, preventing transcriptional machinery from accessing DNA and repressing the transcription of target genes309. Subsequently, the mechanism of EGFR inhibiting Runx2 activity may be accomplished through the upregulation of HDAC4 and HDAC6 expression309.
Chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) is an orphan nuclear receptor belonging to the steroid thyroid hormone receptor superfamily. It physically interacts with Runx2310. Specifically, COUP-TFII suppresses Runx2-dependent osteocalcin promoter activation310. COUP-TFII inhibits Runx2 DNA binding to the osteocalcin gene. Conversely, Runx2 suppresses COUP-TFII expression by directly binding to the COUP-TFII promoter310. Hence, this indicates that COUP-TFII may negatively regulate osteoblast differentiation through its interaction with Runx2310.
Runx1
Runx1, also called AML1 or Cbfa2, belongs to the RUNT-related transcription factor family. Runx1 and Cbfβ form a heterodimer and are highly expressed in osteoblasts and pre-osteoblasts311. Runx1 controls how HSCs become mature blood cells312 and is key in the development of pain-transmitting neurons313 (Fig. 1). Additionally, in vitro studies have proved that Runx1 is fundamental for early chondrocyte differentiation314 and osteoblast differentiation315. Runx1 positively regulates osteoblast lineage gene expression at various stages of differentiation, promoting bone formation and inhibiting adipogenesis (Fig. 1)315. Runx1 primarily participates in the early stage of osteoblast differentiation (Table 1). In our previous study, we observed that during this period, when bone marrow-derived mesenchymal stem cells (BMSCs) differentiate into chondrocytes and osteoblasts, Runx1 and Runx2 are co-expressed314. Runx1 stimulates chondrocyte commitment to the osteoblast lineage and augments bone formation by increasing both chondrogenesis and osteogenesis. Furthermore, overexpression of Runx1 in BMSCs initiates chondrocyte differentiation, while a deficiency in Runx1 results in reduced expression of Runx2 and impaired differentiation of both chondrocytes and osteoblasts316. Besides, previous study also found that loss of Runx1 will decrease the expression of SOX9, Ihh, PTC in the growth plate, while the expression of OCN, Osx, Runx2 and ATF4 are also decreased315. These results demonstrated that Runx1 is important in osteoblast differentiation and bone formation.
Runx1 is very important for coordinating several pathways implicated in bone formation and homeostasis, including BMP and Wnt/β-catenin signaling (Fig. 4). Our lab found that Runx1 plays a significant role in postnatal bone homeostasis by binding to BMP7, activin-like kinase-3 (ALK3), and ATF4 promoters to activate the corresponding genes311. Expression of these genes produces critical transcription factors in osteoblast differentiation, which, after activation, can further regulate osteoblast-specific gene expression to promote osteoblastogenesis.
In addition, our lab found that Runx1 regulates osteoblast-adipocyte lineage via inducing Wnt/β-catenin signaling and restraining adipogenic gene transcription311. Previous research in our lab has demonstrated that Wnt10b/β-catenin signaling is crucial to the composition of the osteoblast-adipocyte lineage91 (Fig. 4). In vitro research has previously established that β-catenin signaling is diminished with Runx1 deficiency311. When the Wnt/β-catenin signaling pathway is inhibited, osteoblast cell lines will differentiate into adipocytes311. Therefore, Runx1 can regulate the differentiation pathway of osteoblasts by controlling the expression of β-catenin in the Wnt signaling pathway.
Along with adipogenic gene regulation, Runx1 can directly interact with the Ihh promoter to mediate its expression, up-regulate the transfer of chondrocytes to osteoblasts, and promote the expression of chondrogenesis/osteogenesis-related genes315. Additionally, our lab found that Runx1 also mediates osteoblast differentiation and bone formation by binding to Runx2 and OCN gene promoters and multiple bone-specific genes, leading to increased Runx2 expression317. The Runx2 promoter’s high binding efficiency at binding site 4 is due to the acetylation of chromatin-associated histones due to Runx1’s interaction with histone acetyltransferase, which alters chromatin conformation and activates transcription317. Runx1 can partially replace the function of Runx2 and positively regulate osteoblast differentiation311. Overexpression of Runx1 in Runx2-deficient osteoblasts can salvage the expression of key regulatory proteins (ATF4 and OCN), rescue bone formation in cultured osteoblasts, and restore the activity of osteoblasts by repairing the damage caused by Runx2-deficiency311.
Runx3
Runx3 is another member of the RUNT family318. Similar to Runx2, Runx3 also binds DNA in a heterodimer form with Cbfβ319 (Table 1). Runx3 is expressed in hematopoietic cells, skin appendages, and bone tissue320,321. Ablation of Runx3 controls early and late chondrocyte differentiation in Runx3-deficient embryos322 (Fig. 1). In addition, overexpression of Runx3 in chondrocytes leads to ectopic mineralization of the rib cage within mice, indicating that Runx3 plays a noteworthy role in cartilage development323. In contrast, Runx3-mutant mice developed severe congenital osteopenia, suggesting that Runx3 positively regulates osteoblast differentiation324 (Table 1).
In MSCs, BMP9 up-regulates endogenous Runx3 expression325. Runx3 overexpression increases BMP9-induced transcription factor expression, like Runx2 or DLX5, in osteoblasts, while Runx3 knockdown decreases the expression of these transcription factors325. In addition, Runx3 overexpression also leads to increased Smad1/5/8 phosphorylation induced by BMP, whereas Runx3 knockdown leads to a decrease325. As a result, Runx3 controls the induction of osteoblast differentiation by its interaction with BMP9325.
Osterix
Osx has two effects on osteoblast differentiation, where it can both promote and inhibit osteoblast differentiation, depending on the situation (Fig. 4). Osx’s C-terminus comprises a DNA domain highly similar to the sequences found in Sp1, Sp3, and Sp4326. Osx also possesses a proline- and serine-rich domain that is capable of activating the expression of OCN and COL1A1326. Numerous MyF5-binding sequences and myoblast determination protein 2 (MyoD2)-binding sequences can be found in the proximal Osx promoter327. These basic helix-loop-helix structural transcription factors are essential for osteoblast differentiation, and Osx transcription may be controlled by these two sequences327. Zinc finger and BTB domain-containing 16, downstream target genes of Osx, act as late markers of osteoblast differentiation and thereby play a role in regulating osteogenesis in MSCs328. Fibrillin-2 and periosteal were also identified as candidate targets of Osx in osteoblast differentiation329. The transcription factor early B cell factor 1 (Ebf1) — known to regulate B cell, neuronal cell, and adipocyte differentiation — has also been shown to modulate osteoblast activity in a stage-dependent manner through its interaction with Osx, with Ebf1 promoting early osteoblast differentiation but hampering proper mature osteoblast function330.
While Runx2 is indispensable for osteoblast differentiation, certain mechanisms operate independently of Runx2331,332. For example, even though Runx2 is necessary for BMP-mediated Osx expression, Runx2 on its own is not enough to elicit Osx expression136. This suggests that DLX5, a transcription factor downstream of BMP, can also induce Osx expression136. The MAPK signaling pathway can also regulate Osx expression induced by BMP2 and IGF-1333. The transcription factor NFAT, which is particularly important to osteoclasts, can work with Osx in osteoblasts to speed up the process of differentiation334. Overexpression of NFATc1 has been shown to encourage the Osx-dependent activation of the COL1A1 promoter335. Through the activation of SOST and DKK1 expression, two significant inhibitors of the Wnt signaling pathway, Osx can suppress osteoblast activity336. By specifically binding to SOST gene promoter sequences, Osx can trigger SOST expression337. Thus, the Wnt signaling is inhibited, and osteoblast differentiation is disrupted. This intricate regulatory network is further underscored by the involvement of En1, which, due to Osx expression being delayed in En1 deficient mice, is expected that En1 is located upstream of Osx338. The manifestation of delayed skull ossification in mouse models with the En1 mutation indicates that En1 also plays a significant role in osteoblast differentiation338. In addition, ALP activity in osteoblast cells lacking En1 is reduced, with ALP being an essential enzyme in osteoblast differentiation339. OCN and BSP, two genes involved in the final stage of osteoblast differentiation, also have reduced expression when En1 is deleted339.
Recent investigations have delved into the impact of osterix stability on osteoblast differentiation, particularly in the context of E3 ligase complexes and ubiquitination processes. Within the evolutionarily conserved SCF ubiquitin ligase complex, F-box and WD repeat domain-containing 7 (FBW7) plays a crucial role as the substrate recognition element340,341. The phosphorylation of Osx at S73/77 by P38 facilitates its interaction with Fbw7, leading to subsequent Osx ubiquitination341. Furthermore, the depletion of Fbw7 in human mesenchymal stem cells and primary mouse calvarial cells resulted in an augmented osteogenic capacity341. Similarly, members of the Casitas B-lineage lymphoma family, Cbl-b and c-Cbl, have been identified as regulators that diminish the protein stability of Osx during BMP2-induced osteoblast differentiation through ubiquitin–proteasome-mediated degradation342. More recently, ubiquitin-specific protease 17 (USP17) has been found to increase Osx protein levels via modulating deubiquitylation, positively impacting osteoblast differentiation as evidenced by enhanced alkaline phosphatase (ALP) expression and activity343. These findings open a novel avenue for direct intervention and manipulation of Osx stability, providing a potential approach to enhance or inhibit osteoblast differentiation.
Co-activators of Osx
By binding to and activating CCACCC sites upstream of its promoter, Osx can control its own expression, forming a positive feedback mechanism344. BMP2 and Msh homeobox 2 (Msx2) trigger the expression of Osx in Runx2-mutant MSCs, and a mutation in Msx2 prevents the induction of Osx in Runx2-deficient MSCs (Table 1). This suggests that BMP2 independently controls Osx expression via Msx2345. Similarly, BMP2 and TGFβ-mediated Osx expression also require a novel factor, TGFβ-induced early gene 1 (Tieg1), to optimize Osx expression in osteoblasts346,347,348. Additionally, BMP2 directly regulates Osx expression by connecting with its proximal promoter, thus promoting osteoblast differentiation346,347,348.
Jumonji domain-containing 3 (Jmjd3) is also important in osteoblast differentiation and controls BSP and OCN expression through Runx2 and Osx349. When Jmjd3 was knocked out in mutant mouse models, Runx2 and Osx promoter activity decreased while H3K27me3 levels increased (Fig. 5)349. As a result, both DLX5 and Msx2 were unable to get close to these binding sites, preventing Runx2 and Osx from being activated349. Osteoblast differentiation was inhibited by increasing H3K27me3 occupancy of Runx2 and Osx promoter regions when Jmjd3 expression was silenced349. According to these findings, Jmjd3 positively regulates Osx function to exert transcriptional control over osteoblast lineage commitment349.

a The non-coding RNAs have a regulatory effect on osteoblast differentiation; miRNA-346 can inhibit GSK3β to promote Wnt signaling activation, and miRNA-181a can inhibit TGFβ1 and TGFβR1 to promote TGFβ signaling activation to promote osteoblast differentiation. b The methylation and acetylation of histone H3 control the promoter of the transcriptional genes. The enrichment of H3K4me1, H3K9me3, and H3K27me3 leads to transcriptional silencing of gene promoters, while the enrichment of H3K4me3 promotes transcriptional activation. Demethylation of H3K9me3 and H3K27me3 also induces transcriptional activation of gene promoters. This group of proteins regulates the enrichment of different methylation groups and their methylation degree. Jarid1/Kdm5 can convert H3K4me3 and H3K4me2 to H3K4me1, while MLL3/4-COMPASS-like can catalyze the deposition of H3K4me1 on the enhancer elements. Set1-COMPASS and HoxA10 promote H3K4 methylation. Jmjd2a, Kdm4a and Kdm4B promote H3K9 demethylation. Kmt1e/Setdb1/ESET can mediate H3K9 trimethylation. Osteoblast differentiation is facilitated by the acetylation of H3K9, H3K14, and H3K27. HDAC1/2/5 promotes deacetylation of H3K9, H3K14, and H3K27, while GCN5 promotes H3K9 and H3K14 histone acetylation, KATA5 promotes H3K14 acetylation and CBP/P300 promotes H3K27 acetylation. c SWI/SNF regulates osteoblast differentiation through chromatin remodeling. BRG1 is one of the subunits of the SWI/SNF complex. Baf45d also belongs to SWI/SNF complex. When activated, it can regulate osteoblast early differentiation. SWI/SNF chromatin remodeling complex is required for Runx2-dependent initiation of skeletal gene expression. d The degree of DNA methylation affects the gene promoters. The Dnmts family, such as Dnmt1, can regulate DNA methylation while Tet1/2 can demethylate 5mCpG to promote Runx2 and Osx gene transcription, thereby promoting osteoblast differentiation. BMP2 can induce Tet1/2 activity to convert 5mCpG to 5hmCpG on Osx promoter.
Panx3 acts as a highly expressed gap junction protein in bone cells that can regulate the initiation of osteoblast precursor cell differentiation. Panx3 can indirectly regulate the expression of Cx43, a key gene in osteoblast differentiation, by regulating the expression of Osx350. Panx3 acts as a hemichannel to release ATP, which can activate the PI3K/AKT signaling pathway350. Further opening of Panx3 ER Ca2+ channels leads to increased intracellular Ca2+ concentration, activation of CaM/NFAT signal transduction, and increased Osx expression334. Cx43 is the target gene of Osx, so Panx3 can regulate the differentiation process of osteoblasts by regulating the expression of Osx351.
Co-repressors of Osx
Osteoblast differentiation and Osx expression can be induced by inhibiting protein phosphatase 2Acα (PP2Acα), which can negatively regulate Osx expression352. Indeed, previous work has indicated that the silencing of PP2Acα expression led to an increased level of Osx expression, promoting the up-regulation of other bone-related genes, such as BSP and OCN352. Through MAPK and PKCδ signaling pathways, BMP2 and IGF-I can mediate Osx expression in MSCs. In addition, inhibition of PP2Acα significantly up-regulates Osx expression and its related genes, leading to the sustained differentiation of osteoblasts352.
NO66 is a protein containing a Jumonji C (JmjC) domain that directly interacts with Osx to inhibit Osx-mediated promoter activation353. Osteoblast differentiation and mineralization were accelerated when NO66 was knocked out of preosteoblasts, and Osx- targeted gene expression was significantly increased353. NO66 shows JmjC-subordinate histone demethylase movement, which is explicit for H3K4me and H3K36me (Fig. 5)353. Osx interacts with NO66 to regulate osteoblast-specific genes’ histone methylation status. This interaction between NO66 and Osx acts as histone demethylase to negatively regulate osteoblast gene expression353.
ATF4
ATF4 is necessary for the timely onset of terminal osteoblast differentiation and controls osteoblast-specific gene expression (Table 1)354,355. The ATF/CREB protein family includes ATF4, the basic leucine zipper transcription factor356. Through the use of leucine domains, these proteins bind to DNA and transform into numerous dimers that coordinate the cellular regulation of signals from multiple pathways357 (Fig. 4). RSK2 and ATF4 direct type I collagen synthesis, an essential component of the bone matrix, following transcription354. Osteoblast-specific osteocalcin gene expression is triggered by the synergistic interaction between ATF4 and Runx2/Cbfa358. OCN is activated by the synergistic mutual effect between ATF4 and Runx2 at the 657 bp mOG2 promoter’s OSE1- and OSE2-binding sites, as determined by mutation analysis358.
Vimentin prevents osteoblast differentiation and OCN transcription, both of which are dependent on ATF4359. Vimentin binds to ATF4 in osteoblasts through the leucine zipper space, preventing osteoblast differentiation and ATF4-dependent OCN transcription359. This prevents ATF4 from binding to the OCN promoter’s cognate DNA, OSE1359. Additionally, the TGF-β signaling pathway regulates ATF4. Vimentin can be produced through the PI3K-Akt-mTOR signaling pathway, thereby inhibiting ATF4-dependent OCN transcription and osteoblast differentiation, even though it does not directly affect ATF4 expression359.
During bone formation, the dimerization of ATF4 with the C/EBP family is crucial. The heterodimerization of C/EBP with ATF4 is linked to one of the defining mechanisms of the delayed bone formation seen in C/EBP-deficient mice360. OCN transcription is triggered by these two proteins binding to the promoter of the OCN gene close to the ATF4-binding site360. C/EBP causes ATF4 to bind with Runx2, resulting in OCN expression360.
SATB2
Matrix attachment region (MAR)-dependent transcription can be initiated by special AT-rich sequence-binding protein-2 (SATB2), which belongs to a distinct AT-rich family of binding proteins that bind to the nuclear MAR361,362. To regulate the chromatin structure, SATB2 collaborates with transfer-associated protein 2 (MAT2) and histone deacetylase 1 (HDAC1) (Table 1)363. In addition to directly activating or inhibiting DNA expression, SATB2 promotes DNA-binding protein activity by acting as a protein bracket364 (Fig. 4). SATB2 may play a significant role as a key node in the transcriptional regulatory network since it controls several important aspects of skeletal development365.
The BSP and OCN genes, which are essential for osteoblast differentiation, can be bound and controlled by SATB2. In addition, SATB2 prevented Hoxa2, a bone formation inhibitor, from being expressed (Fig. 1)366,367. Through physical interaction, SATB2 strengthened ATF4’s and Runx2’s ability to bind to similar DNA recognition elements368. By binding to the SATB2 promoter, Osx can directly control the expression of SATB2369. BMP4 may regulate upstream of SATB2, as evidenced by the fact that SATB2 was up-regulated in the mandible of mice overexpressing BMP4 but down-regulated in the mandible of mice lacking BMP4370. SATB2 expression was also increased when Osx was overexpressed371. Mechanistically, Osx can bind directly to the 130-bp GC-rich SATB2 promoter binding site at the 368 proximal end, activating SATB2 expression371. As a result, Osx is an upstream regulator of SATB2, with SATB2 transcription occurring after BMP signaling371. SATB2 expression was inversely correlated with TNF-α content369. The mesenchymal cell line C2C12 osteoblasts can be constrained by TNF-α, reducing the expression of SATB2 induced by BMP369. Therefore, TNF-α is a negative regulator of osteoblast differentiation and can inhibit the differentiation of osteoblasts by inhibiting the expression of SATB2369.
TAZ/YAP
A transcriptional regulator known as TAZ has a domain that binds to PDZ (Table 1). YAP is a significant downstream effector in the Hippo signaling pathway (Table 1)372. Bone formation is one area in which YAP/TAZ plays a significant role. TAZ can interact with Runx2 and PPAR, promoting Runx2 transcription and inhibiting PPAR, thereby preventing adipocyte generation and promoting osteoblast differentiation373. The transcriptional regulator LEF is crucial for osteoblast-specific gene expression within the Wnt/β-catenin signaling pathway374. TAZ combines with LEF to create a complex that promotes Wnt3a signaling and regulates Runx2 function375. By binding to Runx2, YAP can stop Runx2 activation. The Runx2-YAP complex can be separated by hindering Src/Yes kinase or obstructing tyrosine phosphorylation of YAP247. Because the interaction between YAP and Runx2 is facilitated by tyrosine phosphorylation of endogenous YAP, Runx2 can recruit phosphorylated YAP to the OCN promoter and the subnuclear site of Runx2, inhibiting OCN promoter activity247. Snail (SNAI1) and Slug (SNAI2) form a binary complex with YAP/TAZ that regulates the protein level of YAP/TAZ (Fig. 3)376. This protects YAP/TAZ from proteolytic hydrolysis and controls the expression of YAP/TAZ-targeted genes, positively regulating osteoblast differentiation376.
Cbfβ
Core binding factors (Cbfs) are heterodimeric transcription factors with two subunits: Cbfα (Runx proteins) and Cbfβ. The Cbfβ subunit functions as a non-DNA binding factor that can bind to Runx (Cbfα) and encourage gene expression (Table 1). Numerous developmental processes, including the formation of the skeletal system, are influenced by the Runx/Cbfβ complex (Fig. 4). Cbfβ can not only bind to the Runx protein, but also able to interact with other osteoblast differentiation-related transcription factors like ATF4 and Osx377. This interaction has the potential to significantly up-regulate the transcriptional expression of ATF4 and Osx377. Cbfβ deficiency can lead to the down-regulation of chondrocyte differentiation-related gene expression, impairing chondrogenesis378. Cbfβ-mediated chondrocyte maturation is also important for trabecular bone morphogenesis378. For instance, Cbfβ-deficient mice suffer from bone defects and die during embryonic development as a result of a lack of hematopoiesis and subsequent hemorrhaging379,380. Furthermore, not only does down-regulation of the Cbfβ gene delay the development of the sternum, but it also impacts the growth of numerous bones, such as the skull, tibia, femur, spine, and ribs381.
Through the formation of a binding complex, Cbfβ stabilizes Runx2 in osteoblasts and promotes Runx2’s regulation of osteoblast-specific gene expression (Fig. 4). By inhibiting the ability of WW domain-containing E3 ubiquitin protein ligase 1 (WWP1) to bind to Runx2, the Runx2/Cbfβ complex can prevent Runx2 from being ubiquitinated and subsequently degraded379. Additionally, the disruption of the Runx2/Cbfβ complex via small osteoblast vesicles containing micro-RNA that suppresses the expression of Cbfβ was shown to inhibit the transformation of pre-osteoblast cells to mature osteoblast cells382. The Runx/Cbfβ complex can directly increase transcriptional Ihh expression and promote osteoblast differentiation by combining with the Runx binding site in the Ihh promoter, controlling Runx2 acetylation, phosphorylation, and isomerization, as well as other posttranslational modifications380. The maturation of osteoblasts and their transformation into osteocytes are slowed down when Runx2 and Cbfβ are overexpressed in osteoblasts379.
Epigenetic modification of transcriptional gene regulation
The study of epigenetic modification includes DNA methylation, acetylation, and ubiquitination, as well as the chromatin regulatory proteins that control changes in gene expression. The epigenetic regulation during osteoblast differentiation has progressed significantly in recent years. Epigenetic inheritance plays a crucial role in gene expression by regulating chromatin structure to dynamically control gene expression via permitting or declining access of transcriptional machinery to chromatin. Non-coding RNAs, DNA modifications, chromatin remodeling, and the dynamic epigenetic regulatory process are all essential for osteoblast development. The methylation and acetylation of DNA and histones have received the most attention (Fig. 5; Table 3).
Non-coding RNAs
MicroRNAs (miRNAs) are noncoding RNA molecules that exert a negative regulatory effect on target gene expression through mRNA degradation or translational repression383. Past studies have found that miRNAs play an important role in osteoblast differentiation384,385. A recent study found that miR-346 can directly bind to the 3’-untranslated region (UTR) of GSK-3β mRNA, and miR-346 can promote osteogenic differentiation by repressing GSK-3β and activating the Wnt/β-catenin pathway41 (Fig. 5a). Another study found that miR-181a is highly upregulated during BMP-induced osteoblastic differentiation. They demonstrated that miR-181a targets the negative regulator of osteoblastic differentiation Tgfbi (Tgf-beta induced) and TβR-I/Alk5 (TGF-β type I receptor), thereby promoting osteoblastic differentiation via repression of TGF-β signaling molecules386 (Fig. 5a). Therefore, the miRNAs are important in osteoblast differentiation via regulating osteoblast differentiation-specific genes386.
Histone methylation
Chromatin reorganization and gene transcription are both regulated by methylation of the histone. Lysine (K) methylation of H3K9387 and H3K27388 is associated with transcriptionally silenced regions. By removing H3K9me3 and H3K27me3, the histone demethylases KDM4B and KDM6B are important in MSC differentiation (Fig. 5b; Table 3)389. Osteogenic differentiation was significantly reduced when lysine demethylase 4B (KDM4B) or lysine demethylase 6B (KDM6B) were depleted390,391. The epigenetic switch for human MSC differentiation can be regulated by the mutual and concerted effect of enhancer of zeste homolog 2 (EZH2) and lysine demethylase 6 A (KDM6A)391. EZH2, the catalytic subunit of Polycomb repressive complex 2 (PRC2), plays a crucial role in gene repression by catalyzing the trimethylation of H3K27392. H3K27me3 is specifically trimethylated by EZH2, whereas the methyl group is removed by KDM6A392. The histone H3K27 methyltransferase EZH2 negatively regulates osteogenesis, and the expression of Runx2 and OCN is inhibited in the presence of EZH2 during osteogenic differentiation because H3K27me3 is dramatically increased on Runx2 and OCN transcription start sites (TSS) (Fig. 5b; Table 3)393. Cyclin-dependent kinase 1 (CDK1) can phosphorylate EZH2 at Thr 487. This phosphorylation event not only hinders the binding of EZH2 to its targets on the promoter but also inhibits the histone methyltransferase (HMTase) activity of EZH2 by disrupting the PRC2 complex392. The removal of repression of the putative EZH2-target genes, facilitated by the phosphorylation of EZH2 at Thr 487 by CDK1, results in the promotion of MSC osteogenic differentiation392. Kmt1e/Setdb1/ESET can mediate H3K9 trimethylation394.
Methylation of H3K4 is associated with transcriptionally active regions395. Trxg-mediated activity encompasses monomethylation, dimethylation, and trimethylation of lysine-4 residues of histone H3 (H3K4me1, H3K4me2, and H3K4me3)396. The Jarid1/Kdm5 family (Jarid1a, b, c, and d) is capable of converting H3K4me3 and H3K4me2 to H3K4me1 (Fig. 5b; Table 3)390. H3K4me3 is commonly found in transcriptionally active chromatin (euchromatin), primarily concentrated around the transcription initiation site (TSS) of promoters394. The global genomic deposition of H3K4me3 in most mammalian cells is regulated by Set1-COMPASS, thereby closely linking its function to the transcriptional activation of a large number of genes397. MLL3/4-COMPASS-like complexes can catalyze the deposition of H3K4me1 on the enhancer elements of mammalian cells398,399. The reduction of MLL3/4 results in the transcriptional activation of specific MLL3/4 target genes. This happens because both Set1a/b-COMPASS and MLL1-COMPASS can reach these genes, aiding in the shift of associated transcription from H3K4me1 to H3K4me3400. Enrichment of H3K4me1 typically results in the inhibition of osteoblast differentiation. Furthermore, the accumulation of H3K4me1 suppresses the expression of key transcription factors in osteoblasts, such as Runx2 and Osx (Fig. 5b; Table 3)401,402. Methylation of H3K4, particularly the enrichment of H3K4me3, facilitates osteoblast differentiation. In this context, crucial transcription factors of osteoblasts like Runx2 and Osx become transcriptionally active (Fig. 5b; Table 3)402.
Histone acetylation
Histone acetylation is regulated by histone deacetylase (HDAC) and histone acetyltransferases, which affect chromatin conformation and regulate gene transcription. Acetylation of a histone typically facilitates gene transcription by allowing transcription factors and RNA polymerase II access to the DNA via relaxing the chromatin structure. In this manner, histone acetylation encourages gene transcription, while histone deacetylation causes chromatin to become compacted and prevents gene expression403. During osteoblast differentiation, H3K9Ac is linked to genes that are activated, and H3K9me2 is linked to genes that are silenced (Fig. 5b; Table 3)394. Sirtuin 1 (SIRT1) is a Class III histone deacetylase301. SIRT1 activity can promote osteoblast differentiation301. By interacting with Runx2, SIRT1 can enhance its trans-activation activity301. As Osx is one of Runx2’s downstream targets, SIRT1 can trigger Runx2’s activation and promote Osx expression, which in turn targets osteoblast-specific gene expression and promotes osteoblast differentiation301.
HDACs, or histone deacetylases, are a class of enzymes responsible for removing acetyl groups from both histone and non-histone proteins. This action facilitates the tightening of DNA wrapping around histones404,405. HDAC1 exerts a negative regulatory effect on osteoblast generation by reducing the acetylation levels of histones H3 and H4 on the promoters of key osteoblast-related genes such as Osx and OCN (Fig. 5b; Table 3)403. HDAC1 can bind to the promoter of Runx2, thereby abolishing its transcriptional activity and consequently exerting its inhibitory effect on osteogenesis in the absence of osteogenic signals403. During osteogenesis, rapidly proliferating osteogenic progenitors exit the cell cycle to differentiate into functional osteoblasts. The subsequent arrest of the cell cycle is crucial for osteoblast differentiation406. HDAC1 interacts with promoters of cell cycle inhibitors such as P27, impeding the progression of the osteogenic lineage407. HDAC2 is broadly expressed in bone tissue and enhances Akt expression. This upregulation of Akt inhibits FoxO1 transcription, consequently promoting RANKL-induced osteoblast generation403. HDAC5 binds to MEF2C, leading to the inhibition of SOST expression. This action reduces H3K27 acetylation levels at the SOST gene site, thereby promoting Wnt/β-catenin signaling408.
General control non-repressed protein5 (GCN5) is a histone acetyltransferase (HAT) that plays a crucial role in normal embryogenesis. Its function involves gene transcriptional activation and the regulation of a specific chromatin program characterized by histone acetylation409. Gcn5 is associated with the acetylation of histones at residues K9 and K14, which are responsible for gene activation410. Previous work has studied the epigenetic regulation mechanism of Gcn5 on osteoblast differentiation in an inflammatory environment410. During inflammation, Gcn5 expression is reduced, which leads to Wnt/β-catenin development and impingement of PDLSC osteogenesis. Gcn5 regulates osteoblast differentiation by depositing acetyl markers on histone H3 at lysine 9 and lysine 14 (H3K9 and H3K14) in the DKK1 promoter region (Fig. 5b; Table 3)410. DKK1 inhibits Wnt/β-catenin signaling by upregulating DKK1 in periodontal ligament stem cells (PDLSCs), thereby promoting osteogenesis with support from Gcn5410. KAT6A, an HAT belonging to the MYST family, is responsible for acetylating histone H3 at lysine 9 and lysine 14 (H3K9 and H3K14) (Fig. 5b; Table 3)411. In the context of cellular senescence, the levels of KAT6A protein and Nrf2/ARE signaling decrease as a result of stem cell dysfunction. Overexpression of KAT6A leads to the promotion of the Nrf2/ARE pathway through altered histone H3 acetylation patterns, thereby restoring cellular stemness411. Besides, CREB-binding protein (CBP) (CREBBP) and p300 (EP300) are multifunctional HATs with broad homology (Fig. 5b; Table 3)412. One study reported that conditional knockout of CBP or p300 in osteoblasts inhibited bone formation, resulting in reduced bone mass and strength412. Mechanistically, CBP/p300 HAT regulates the expression of osteogenic genes partly by activating β-catenin transcriptionally and suppressing Stat1. The acetylation of histone H3K27 and the transcription factor Foxo1 have been demonstrated to play roles in the CBP/p300 HAT-mediated transcriptional regulation of β-catenin and STAT1, respectively412(Fig. 5b; Table 3). These findings indicate that the acetyltransferase CBP/p300 could be a key regulator in promoting osteoblast differentiation412.
Chromatin remodeling
The switch/sucrose non-fermenting (SWI/SNF) chromatin remodeling complex comprises multiple subunits engaged in diverse cellular processes, such as gene regulation, cell cycle modulation, development, and differentiation413. The mammalian SWI/SNF complex contains a catalytic subunit, BRG1 or BRM, which includes ATPase activity414,415. Mutations in the ATPase domain of BRG1 or BRM disrupt ATP binding, leading to the formation of inactive SWI/SNF complexes414,416,417. When mutant BRG1 or BRM proteins were expressed in NIH3T3 cells, it hindered the cells’ capability to activate endogenous stress response genes and to undergo differentiation into muscle or fat cells in the presence of arsenite414,416. Mutant BRG1 protein in NIH3T3 cell lines has been shown to inhibit BMP2-induced osteoblast lineage differentiation418. SWI/SNF is required for BMP2-induced alkaline phosphatase (APase) expression, an early marker reflecting Runx2’s control of osteoblast differentiation418. BRG1 is expressed in developing bone cells in mouse embryos and in vitro osteoblasts418. These results suggested that MP2-mediated osteogenesis requires Runx2 and suggest that the SWI/SNF chromatin remodeling complex is required for BMP2-induced, Runx2-dependent initiation of skeletal gene expression (Fig. 5c; Table 3)418.
Baf45a and Baf45d, two homologs involved in chromatin remodeling, have been demonstrated to be expressed in osteoblasts during maturation (Fig. 5c; Table 3). BAF45A is associated with the polybromo-associated BAF (PBAF) complex, while the BAF45D subunit belongs to the polymorphic canonical BRG1-associated factor (cBAF) complex419,420. Both BAF45A and BAF45D belong to the subunits of SWI/SNF complex415. During BMSCs differentiation, chromatin immunoprecipitation sequencing (ChIP-seq) revealed elevated histone H3K9 and H3K27 acetylation modifications in the promoter regions of Baf45a and Baf45d, accompanied by enhanced binding of RUNX2420. The overexpression of Baf45a in osteoblasts triggered the activation of genes crucial for osteoblast maturation and mineralization420. Conversely, Baf45a knockdown in odontoblasts led to significant changes in the expression of genes associated with proliferation, apoptosis, DNA repair, and a modest reduction in dentinogenic marker gene expression420. Additionally, Baf45a knockout osteoblasts exhibited a noticeable decrease in chromatin accessibility for osteoblast and odontoblast-specific genes, including the transcription factor Atf4420. These findings suggest that BAF45A-dependent chromatin remodeling, facilitated through PBAF-RUNX2 crosstalk, plays a pivotal role in the transcriptional activation essential for the early differentiation and matrix maturation of mineralized tissues420.
DNA methylation and demethylation
DNA methylation entails the epigenetic process of adding a methyl group to the C5 position of cytosine, resulting in the formation of 5-methylcytosine421. DNA methylation is mainly regulated by a family of DNA methyltransferases (Dnmts), which are found to be important in osteoblast differentiation421. A recent study found that Dnmt3a may inhibit osteoblast differentiation via regulating Wnt/β-catenin signaling and Runx2, Ocn activity (Fig. 5d; Table 3)422. Demethylation of the promoter’s CG region is linked to the ten-eleven translocases (TET) protein, which may alter chromatin status and encourage osteoblast differentiation422. The TET protein forms a complex with Runx2 to mediate the demethylation of the Runx2 promoter and subsequently promote Runx2 expression423 (Fig. 5d; Table 3). By recruiting the TET protein into the Runx2 promoter region, the resulting hypomethylation of DNA can provide a more accessible chromatin environment for RNA Pol II, promoting gene expression423. These findings demonstrate that TET enzymes function to regulate Runx2 activity and maintain skeletal homeostasis423. During BMP2-induced osteoblast differentiation, DNA demethylation of the Osx promoter is mediated by a Tet1/Tet2-containing complex that converts 5mCpG to 5hmCpG424. A recent study also found that Tet1 recruits Tet2 to mediate 5mCpG demethylation and gene transcription during BM-MSC differentiation425 (Fig. 5d; Table 3).
The expression of osteoblast genes may also be controlled via the methylation mechanism of Vitamin C (ascorbic acid, AA), a well-known regulator of bone and cartilage metabolism. A recent study confirmed that AA exerts differential effects on osteoblast differentiation, with AA increasing levels of 5-hydroxymethylcytosine (5-hmC) and inducing DNA demethylation via the ten-eleven translocases (TETs)426. Prolyl hydroxylase domain-containing protein 2 (PHD2), recognized as a mediator of AA’s effects in these tissues, belongs to the same enzyme family as the TETs426. Blocking PHD2 resulted in decreased 5-hmC levels in promoters of genes associated with chondrocyte differentiation426. Knockdown of Phd2 in chondrocytes also led to a reduction in global 5-hmC levels, indicating that PHD2 might directly contribute to increases in 5-hmC in genes related to chondrocyte and osteoblast functions426.
Summary and future perspectives
Embryonic bone formation and the dynamic remodeling of adult bone are processes by which mesenchymal cells of the osteoblast lineage transform into mature osteocytes in mineralized connective tissue. The intricate interaction of signaling proteins, transcription factors, and co-regulatory proteins supports these processes279. Numerous transcription factors and signaling pathways that occupy important positions in osteoblast differentiation have been elucidated over the previous decades thanks to advances in biomolecular techniques. Numerous studies and data support the hypothesis that Runx family proteins and the Wnt signaling pathway regulate osteoblast differentiation. Different signaling pathways contain a series of molecular interactions, which provide potential targets for clinical drug therapy. Many signaling pathways have been implicated in osteoblast differentiation, such as Wnt, Notch, and Hippo, making them attractive targets for the research of drugs to treat bone diseases. With the progress of technology, the factors that participate in osteoblast differentiation have been gradually discovered. However, the regulation of osteoblast differentiation is a large and complex network that involves not only intracellular proteins and genetic interactions but also communication between cells. As such, osteoblast differentiation research faces many unanswered questions and challenges that still need to be overcome, such as:
-
(1)
The mechanisms behind the temporal and spatial regulation of many essential signaling and transcription factors (such as BMP or Runx2) in osteoblast differentiation during different stages remain unclear.
-
(2)
Some signaling pathways have bi-directional effects on osteoblast differentiation, like the Notch signaling pathway and TGFβ signaling pathway. How this bidirectional effect regulates osteoblast differentiation and under what circumstances it promotes or inhibits osteoblast differentiation still needs to be determined.
-
(3)
In addition to the effect of signaling pathways on osteoblast differentiation, cell−cell interactions, such as osteoblast and osteoclast interaction, and the genetic regulation that controls the mutual transformation between osteoblasts, chondrocytes, and adipocytes, still requires further investigation.
-
(4)
With developments in molecular biology and genetics, epigenetic regulation has become a major source of intrigue within the study of osteoblast differentiation. A major focus has been afforded to how histones are modified, the regulation of chromatin structure, and how small RNAs function in regulating gene expression. The interaction and coordination of these regulatory processes are also worth studying, such as how chromatin is “turned on” to expose genes, enabling transcription factors to control the expression of pivotal genes. Epigenetic changes at various destinations will produce different outcomes, and the epigenetic control at the various phases of osteoblast differentiation should be examined.
-
(5)
Bioinformatic technology can also be used to study the transcriptional regulation of osteoblast differentiation. For example, bioinformatic skills such as ATAC-seq can be used to study how enhancers regulate gene expression427.
-
(6)
In the regulation of osteoblast signaling, many molecules are involved in skeleton-related diseases. Therefore, many proteins in the Wnt signaling pathway are promising targets for clinical therapy. Determining which targets can be used and how to design efficacious pharmaceutical drugs that can treat these skeleton-related diseases with minimal side effects is also a current challenge in this area of clinical research.
-
(7)
Osteoporosis has been studied in different animal models; yet these models do not satisfactorily resemble the features of the human disease phenotype428. Although experimental modeling of human bone diseases represents a breakthrough by allowing greater insight into the cellular and molecular mechanisms involved in bone pathology, there are shortcomings in using MSC models. Limited availability of MSCs in patients, extreme heterogeneity, limited proliferative capacity, and loss of function are the most common limitations when using MSCS in vitro429. These limitations on clinical treatment are also issues that need to be addressed.
-
(8)
As our comprehension of cellular dynamics during bone remodeling advances, a plethora of therapeutic targets has emerged from preclinical studies, encompassing membrane expression, cellular crosstalk, and gene regulation. These targets show promising therapeutic effects, offering diverse options for drug development in treating bone-related diseases. Additionally, drugs utilizing carrier transport mechanisms have been devised for this purpose430. Nonetheless, our current understanding of bone biology and its interplay with other systems remains incomplete. The potential impacts on the entire bone remodeling process and other physiological systems, rather than solely focusing on the bone microenvironment or individual cell−cell interactions, represent a fertile area for investigation. Further research is needed to identify more precise targets in order to optimize therapeutic outcomes.
-
(9)
Bone tissue has numerous vital functions and is closely tied to many important tissues and organs’ biochemical, homeostatic, and pathophysiological functions. Because of this critical link, many diseases are also associated with musculoskeletal diseases, such as obesity, cancer, bone metastases, and kidney diseases. Therefore, interdisciplinary research on musculoskeletal diseases, including obesity, cancer, kidney, and other diseases, may provide new insights into bone-related diseases and vice versa.
Currently, the drugs used in the clinical treatment of osteoporosis-related diseases focus on inhibiting osteoclastic bone resorption. In contrast, the drugs targeted at osteoblasts are mainly sclerostin inhibitors that maintain osteoblasts’ function by inhibiting sclerostin’s inhibition on the Wnt signaling pathway431. However, a more recently developed inhibitor of Fap has also been found to be an effective drug for osteoporosis432. The classical signaling pathway is an appealing target for clinical drug development. Wnt, BMP, and other classical signaling pathways have many known key signaling factors, such as β-catenin and Smad proteins. These factors can also be used as clinical targets to make corresponding activators or inhibitors. Researching compelling small-molecule activators or inhibitors to manage the pathway might emphatically affect the treatment of bone-related diseases like osteoporosis. Therefore, more therapeutic targets and new pharmaceutical research strategies can be developed to treat a diverse range of disease pathologies by studying the regulation of signaling pathways and transcription factors during the differentiation of osteoblasts.
References
Murshed, M., Harmey, D., Millán, J. L., McKee, M. D. & Karsenty, G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 19, 1093–1104 (2005).
Ferron, M. et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142, 296–308 (2010).
Karsenty, G. & Wagner, E. F. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2, 389–406 (2002).
Bar-Shavit, Z. The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J. Cell Biochem. 102, 1130–1139 (2007).
Soltanoff, C. S., Yang, S., Chen, W. & Li, Y. P. Signaling networks that control the lineage commitment and differentiation of bone cells. Crit. Rev. Eukaryot. Gene Expr. 19, 1–46 (2009).
Winslow, M. M. et al. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev. Cell 10, 771–782 (2006).
Huang, H. et al. Bone resorption deficiency affects tooth root development in RANKL mutant mice due to attenuated IGF-1 signaling in radicular odontoblasts. Bone 114, 161–171 (2018).
Taichman, R. S. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood 105, 2631–2639 (2005).
Zhang, J. et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425, 836–841 (2003).
Calvi, L. M. et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846 (2003).
Lim, J. et al. Dual function of Bmpr1a signaling in restricting preosteoblast proliferation and stimulating osteoblast activity in mouse. Development 143, 339–347 (2016).
Galán-Díez, M. & Kousteni, S. The osteoblastic niche in hematopoiesis and hematological myeloid malignancies. Curr. Mol. Biol. Rep. 3, 53–62 (2017).
Pinho, S. & Frenette, P. S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 20, 303–320 (2019).
Omatsu, Y. et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33, 387–399 (2010).
Greenbaum, A. et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230 (2013).
Sipp, D., Robey, P. G. & Turner, L. Clear up this stem-cell mess. Nature 561, 455–457 (2018).
Mizuhashi, K. et al. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 563, 254–258 (2018).
Newton, P. T. et al. A radical switch in clonality reveals a stem cell niche in the epiphyseal growth plate. Nature 567, 234–238 (2019).
Maes, C. et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 19, 329–344 (2010).
Duchamp de Lageneste, O. et al. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat. Commun. 9, 773 (2018).
Debnath, S. et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139 (2018).
Chen, K. G., Johnson, K. R., McKay, R. D. G. & Robey, P. G. Concise Review: Conceptualizing paralogous stem-cell niches and unfolding bone marrow progenitor cell identities. Stem Cells 36, 11–21 (2018).
Kfoury, Y. & Scadden, D. T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell 16, 239–253 (2015).
Olsen, B. R., Reginato, A. M. & Wang, W. Bone development. Annu Rev. Cell Dev. Biol. 16, 191–220 (2000).
Huang, W., Yang, S., Shao, J. & Li, Y. P. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front Biosci. 12, 3068–3092 (2007).
Bi, W., Deng, J. M., Zhang, Z., Behringer, R. R. & de Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 22, 85–89 (1999).
Kronenberg, H. M. The role of the perichondrium in fetal bone development. Ann. N. Y. Acad. Sci. 1116, 59–64 (2007).
Pino, A. M., Rosen, C. J. & Rodríguez, J. P. In osteoporosis, differentiation of mesenchymal stem cells (MSCs) improves bone marrow adipogenesis. Biol. Res. 45, 279–287 (2012).
Watt, F. M. Stem cell fate and patterning in mammalian epidermis. Curr. Opin. Genet Dev. 11, 410–417 (2001).
Gronthos, S. et al. Differential cell surface expression of the STRO-1 and alkaline phosphatase antigens on discrete developmental stages in primary cultures of human bone cells. J. Bone Min. Res. 14, 47–56 (1999).
Stein, G. S., Lian, J. B., Stein, J. L., Van Wijnen, A. J. & Montecino, M. Transcriptional control of osteoblast growth and differentiation. Physiol. Rev. 76, 593–629 (1996).
Tamai, K. et al. LDL-receptor-related proteins in Wnt signal transduction. Nature 407, 530–535 (2000).
He, X., Semenov, M., Tamai, K. & Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development 131, 1663–1677 (2004).
Leupin, O. et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 286, 19489–19500 (2011).
Veeman, M. T., Axelrod, J. D. & Moon, R. T. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev. Cell 5, 367–377 (2003).
Westendorf, J. J., Kahler, R. A. & Schroeder, T. M. Wnt signaling in osteoblasts and bone diseases. Gene 341, 19–39 (2004).
Rawadi, G. & Roman-Roman, S. Wnt signalling pathway: a new target for the treatment of osteoporosis. Expert Opin. Ther. Targets 9, 1063–1077 (2005).
Nabhan, A. N. et al. Targeted alveolar regeneration with Frizzled-specific agonists. Cell 186, 2995–3012.e2915 (2023).
Shu, B. et al. Inhibition of Axin1 in osteoblast precursor cells leads to defects in postnatal bone growth through suppressing osteoclast formation. Bone Res. 8, 31 (2020).
Hay, E. et al. Interaction between LRP5 and Frat1 mediates the activation of the Wnt canonical pathway. J. Biol. Chem. 280, 13616–13623 (2005).
Wang, Q., Cai, J., Cai, X. H. & Chen, L. miR-346 regulates osteogenic differentiation of human bone marrow-derived mesenchymal stem cells by targeting the Wnt/β-catenin pathway. PLoS One 8, e72266 (2013).
AlMuraikhi, N. et al. Inhibition of GSK-3β enhances osteoblast differentiation of human mesenchymal stem cells through Wnt signalling overexpressing Runx2. Int. J. Mol. Sci. 24, 7164 (2023).
Clevers, H. Wnt signaling: Ig-norrin the dogma. Curr. Biol. 14, R436–R437 (2004).
Nelson, W. J. & Nusse, R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303, 1483–1487 (2004).
Lévy, L. et al. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol. Cell Biol. 24, 3404–3414 (2004).
Latres, E., Chiaur, D. S. & Pagano, M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene 18, 849–854 (1999).
Zhang, C. et al. Inhibition of Wnt signaling by the osteoblast-specific transcription factor Osterix. Proc. Natl. Acad. Sci. USA 105, 6936–6941 (2008).
Ge, J. et al. Activating Wnt/β-catenin signaling by autophagic degradation of APC contributes to the osteoblast differentiation effect of soy isoflavone on osteoporotic mesenchymal stem cells. Acta Pharmacol. Sin. 44, 1841–1855 (2023).
Semënov, M., Tamai, K. & He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 280, 26770–26775 (2005).
Mao, B. et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature 417, 664–667 (2002).
Mao, B. et al. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 411, 321–325 (2001).
Abuna, R. P. F. et al. Frizzled 6 disruption suppresses osteoblast differentiation induced by nanotopography through the canonical Wnt signaling pathway. J. Cell Physiol. 235, 8293–8303 (2020).
Abuna, R. P. F. et al. The Wnt/β-catenin signaling pathway is regulated by titanium with nanotopography to induce osteoblast differentiation. Colloids Surf. B Biointerfaces 184, 110513 (2019).
Nagano, K. et al. R-spondin 3 deletion induces Erk phosphorylation to enhance Wnt signaling and promote bone formation in the appendicular skeleton. Elife 11, e84171 (2022).
Alhazmi, N. et al. Synergistic roles of Wnt modulators R-spondin2 and R-spondin3 in craniofacial morphogenesis and dental development. Sci. Rep. 11, 5871 (2021).
De Boer, J., Wang, H. J. & Van Blitterswijk, C. Effects of Wnt signaling on proliferation and differentiation of human mesenchymal stem cells. Tissue Eng. 10, 393–401 (2004).
van der Horst, G. et al. Downregulation of Wnt signaling by increased expression of Dickkopf-1 and -2 is a prerequisite for late-stage osteoblast differentiation of KS483 cells. J. Bone Min. Res. 20, 1867–1877 (2005).
Cho, H. H. et al. Endogenous Wnt signaling promotes proliferation and suppresses osteogenic differentiation in human adipose derived stromal cells. Tissue Eng. 12, 111–121 (2006).
Eijken, M. et al. Wnt signaling acts and is regulated in a human osteoblast differentiation dependent manner. J. Cell Biochem. 104, 568–579 (2008).
Jin, Y., Sun, X., Pei, F., Zhao, Z. & Mao, J. Wnt16 signaling promotes osteoblast differentiation of periosteal derived cells in vitro and in vivo. PeerJ 8, e10374 (2020).
Sebastian, A., Hum, N. R., Morfin, C., Murugesh, D. K. & Loots, G. G. Global gene expression analysis identifies Mef2c as a potential player in Wnt16-mediated transcriptional regulation. Gene 675, 312–321 (2018).
Bennett, C. N. et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 102, 3324–3329 (2005).
Stambolic, V., Ruel, L. & Woodgett, J. R. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664–1668 (1996).
Jackson, A. et al. Gene array analysis of Wnt-regulated genes in C3H10T1/2 cells. Bone 36, 585–598 (2005).
Day, T. F., Guo, X., Garrett-Beal, L. & Yang, Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750 (2005).
Hill, T. P., Später, D., Taketo, M. M., Birchmeier, W. & Hartmann, C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727–738 (2005).
Holmen, S. L. et al. Essential role of beta-catenin in postnatal bone acquisition. J. Biol. Chem. 280, 21162–21168 (2005).
Abe, E. et al. TSH is a negative regulator of skeletal remodeling. Cell 115, 151–162 (2003).
Yang, Y., Topol, L., Lee, H. & Wu, J. Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development 130, 1003–1015 (2003).
Yamaguchi, T. P., Bradley, A., McMahon, A. P. & Jones, S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 126, 1211–1223 (1999).
Gong, Y. et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107, 513–523 (2001).
Ai, M., Holmen, S. L., Van Hul, W., Williams, B. O. & Warman, M. L. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass-associated missense mutations in LRP5 affect canonical Wnt signaling. Mol. Cell Biol. 25, 4946–4955 (2005).
Boyden, L. M. et al. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med. 346, 1513–1521 (2002).
Van Wesenbeeck, L. et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am. J. Hum. Genet. 72, 763–771 (2003).
Babij, P. et al. High bone mass in mice expressing a mutant LRP5 gene. J. Bone Min. Res. 18, 960–974 (2003).
Bain, G., Müller, T., Wang, X. & Papkoff, J. Activated beta-catenin induces osteoblast differentiation of C3H10T1/2 cells and participates in BMP2 mediated signal transduction. Biochem. Biophys. Res. Commun. 301, 84–91 (2003).
Rawadi, G., Vayssière, B., Dunn, F., Baron, R. & Roman-Roman, S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J. Bone Min. Res. 18, 1842–1853 (2003).
Esen, E. et al. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 17, 745–755 (2013).
Zhang, D. et al. Exosomes derived from adipose stem cells enhance bone fracture healing via the activation of the Wnt3a/β-catenin signaling pathway in rats with Type 2 diabetes mellitus. Int. J. Mol. Sci. 24, 4852 (2023).
Chen, W., Zhu, G., Tang, J., Zhou, H. D. & Li, Y. P. C/ebpalpha controls osteoclast terminal differentiation, activation, function, and postnatal bone homeostasis through direct regulation of Nfatc1. J. Pathol. 244, 271–282 (2018).
Chen, W. et al. C/EBPalpha regulates osteoclast lineage commitment. Proc. Natl. Acad. Sci. USA 110, 7294–7299 (2013).
Chen, W., Zhu, G., Jules, J., Nguyen, D. & Li, Y. P. Monocyte-specific knockout of C/ebpalpha results in osteopetrosis phenotype, blocks bone loss in ovariectomized mice, and reveals an important function of C/ebpalpha in osteoclast differentiation and function. J. Bone Min. Res. 33, 691–703 (2018).
Takada, I., Suzawa, M., Matsumoto, K. & Kato, S. Suppression of PPAR transactivation switches cell fate of bone marrow stem cells from adipocytes into osteoblasts. Ann. N. Y. Acad. Sci. 1116, 182–195 (2007).
Zayzafoon, M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J. Cell Biochem. 97, 56–70 (2006).
Tu, X. et al. Noncanonical Wnt signaling through G protein-linked PKCdelta activation promotes bone formation. Dev. Cell 12, 113–127 (2007).
Esen, E. & Long, F. Aerobic glycolysis in osteoblasts. Curr. Osteoporos. Rep. 12, 433–438 (2014).
Chen, H. et al. Increased glycolysis mediates Wnt7b-induced bone formation. FASEB J. 33, 7810–7821 (2019).
Hu, H. et al. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 132, 49–60 (2005).
Gaur, T. et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 280, 33132–33140 (2005).
Glass, D. A. 2nd et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8, 751–764 (2005).
Wu, M. et al. Cbfβ governs osteoblast-adipocyte lineage commitment through enhancing β-catenin signaling and suppressing adipogenesis gene expression. Proc. Natl. Acad. Sci. USA 114, 10119–10124 (2017).
Chen, Q. et al. Tyrosine phosphorylation of LRP6 by Src and Fer inhibits Wnt/β-catenin signalling. EMBO Rep. 15, 1254–1267 (2014).
Chen, M. et al. Metastasis suppressor 1 controls osteoblast differentiation and bone homeostasis through regulating Src-Wnt/β-catenin signaling. Cell Mol. Life Sci. 79, 107 (2022).
Chen, Y., Chen, Z. F. & He, F. Tenascin-C knockdown suppresses osteoblast differentiation and promotes osteoporosis in mice by inhibiting Wnt signaling. Nan Fang Yi Ke Da Xue Xue Bao 36, 1117–1122 (2016).
Yao, Z., Xing, L. & Boyce, B. F. NF-kappaB p100 limits TNF-induced bone resorption in mice by a TRAF3-dependent mechanism. J. Clin. Invest. 119, 3024–3034 (2009).
Boyce, B. F., Xing, L. & Chen, D. Osteoprotegerin, the bone protector, is a surprising target for beta-catenin signaling. Cell Metab. 2, 344–345 (2005).
Almeida, M. et al. Estrogen receptor-α signaling in osteoblast progenitors stimulates cortical bone accrual. J. Clin. Invest. 123, 394–404 (2013).
Auld, K. L. et al. Estrogen-related receptor α regulates osteoblast differentiation via Wnt/β-catenin signaling. J. Mol. Endocrinol. 48, 177–191 (2012).
Lee, C. W., Ito, K. & Ito, Y. Role of RUNX3 in bone morphogenetic protein signaling in colorectal cancer. Cancer Res. 70, 4243–4252 (2010).
Wu, M., Wu, S., Chen, W. & Li, Y. P. The roles and regulatory mechanisms of TGF-β and BMP signaling in bone and cartilage development, homeostasis and disease. Cell Res. 34, 101–123 (2024).
Derynck, R., Akhurst, R. J. & Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 29, 117–129 (2001).
Chen, G., Deng, C. & Li, Y. P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J. Biol. Sci. 8, 272–288 (2012).
Yi, J. J., Barnes, A. P., Hand, R., Polleux, F. & Ehlers, M. D. TGF-beta signaling specifies axons during brain development. Cell 142, 144–157 (2010).
Lee, K. S. et al. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol. Cell Biol. 20, 8783–8792 (2000).
Miyama, K. et al. A BMP-inducible gene, dlx5, regulates osteoblast differentiation and mesoderm induction. Dev. Biol. 208, 123–133 (1999).
Lee, M. H. et al. BMP-2-induced Runx2 expression is mediated by Dlx5, and TGF-beta 1 opposes the BMP-2-induced osteoblast differentiation by suppression of Dlx5 expression. J. Biol. Chem. 278, 34387–34394 (2003).
Wu, M., Chen, G. & Li, Y. P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 4, 16009 (2016).
Canalis, E., Economides, A. N. & Gazzerro, E. Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr. Rev. 24, 218–235 (2003).
Chen, D., Zhao, M. & Mundy, G. R. Bone morphogenetic proteins. Growth Factors 22, 233–241 (2004).
Finnson, K. W., Parker, W. L., ten Dijke, P., Thorikay, M. & Philip, A. ALK1 opposes ALK5/Smad3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Miner. Res. : Off. J. Am. Soc. Bone Miner. Res. 23, 896–906 (2008).
Goumans, M. J. et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 12, 817–828 (2003).
Wang, J. et al. Atp6i deficient mouse model uncovers transforming growth factor-β1 /Smad2/3 as a key signaling pathway regulating odontoblast differentiation and tooth root formation. Int J. Oral. Sci. 15, 35 (2023).
Li, J. et al. Smad2 overexpression enhances Smad4 gene expression and suppresses CBFA1 gene expression in osteoblastic osteosarcoma ROS17/2.8 cells and primary rat calvaria cells. J. Biol. Chem. 273, 31009–31015 (1998).
Lin, H. T. et al. Dynamic expression of SMAD3 is critical in osteoblast differentiation of PDMCs. Int. J. Mol. Med. 43, 1085–1093 (2019).
Kang, J. S., Alliston, T., Delston, R. & Derynck, R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 24, 2543–2555 (2005).
Hjelmeland, A. B., Schilling, S. H., Guo, X., Quarles, D. & Wang, X. F. Loss of Smad3-mediated negative regulation of Runx2 activity leads to an alteration in cell fate determination. Mol. Cell. Biol. 25, 9460–9468 (2005).
Grafe, I. et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 20, 670–675 (2014).
Song, I. W. et al. Targeting TGF-β for treatment of osteogenesis imperfecta. J. Clin. Invest. 132, e152571 (2022).
Seo, H. S. & Serra, R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev. Biol. 310, 304–316 (2007).
Dong, H. et al. Higenamine promotes osteogenesis via IQGAP1/SMAD4 signaling pathway and prevents age- and estrogen-dependent bone loss in mice. J. Bone Min. Res. 38, 775–791 (2023).
Yang, H. et al. IFT20 mediates the transport of cell migration regulators from the trans-golgi network to the plasma membrane in breast cancer cells. Front Cell Dev. Biol. 9, 632198 (2021).
Lim, J. et al. Primary cilia control cell alignment and patterning in bone development via ceramide-PKCζ-β-catenin signaling. Commun. Biol. 3, 45 (2020).
Li, Y., Yang, S., Liu, Y., Qin, L. & Yang, S. IFT20 governs mesenchymal stem cell fate through positively regulating TGF-β-Smad2/3-Glut1 signaling mediated glucose metabolism. Redox Biol. 54, 102373 (2022).
Tolkachov, A. et al. Loss of the hematopoietic stem cell factor GATA2 in the osteogenic lineage impairs trabecularization and mechanical strength of bone. Mol. Cell Biol. 38, e00599–17 (2018).
Cao, X. & Chen, D. The BMP signaling and in vivo bone formation. Gene 357, 1–8 (2005).
Anderson, H. C., Hodges, P. T., Aguilera, X. M., Missana, L. & Moylan, P. E. Bone morphogenetic protein (BMP) localization in developing human and rat growth plate, metaphysis, epiphysis, and articular cartilage. J. Histochem. Cytochem. 48, 1493–1502 (2000).
Peng, Y. et al. Transcriptional characterization of bone morphogenetic proteins (BMPs)-mediated osteogenic signaling. J. Cell Biochem. 90, 1149–1165 (2003).
Daluiski, A. et al. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat. Genet. 27, 84–88 (2001).
Matsumoto, Y. et al. Bone morphogenetic protein-3b (BMP-3b) inhibits osteoblast differentiation via Smad2/3 pathway by counteracting Smad1/5/8 signaling. Mol. Cell Endocrinol. 350, 78–86 (2012).
Liu, Z., Tang, Y., Qiu, T., Cao, X. & Clemens, T. L. A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells. J. Biol. Chem. 281, 17156–17163 (2006).
Jun, J. Y. et al. Persicae semen promotes bone union in rat fractures by stimulating osteoblastogenesis through BMP-2 and Wnt signaling. Int. J. Mol. Sci. 24, 7388 (2023).
Guicheux, J. et al. Activation of p38 mitogen-activated protein kinase and c-Jun-NH2-terminal kinase by BMP-2 and their implication in the stimulation of osteoblastic cell differentiation. J. Bone Min. Res. 18, 2060–2068 (2003).
Lai, C. F. & Cheng, S. L. Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-beta in normal human osteoblastic cells. J. Biol. Chem. 277, 15514–15522 (2002).
Lemonnier, J., Ghayor, C., Guicheux, J. & Caverzasio, J. Protein kinase C-independent activation of protein kinase D is involved in BMP-2-induced activation of stress mitogen-activated protein kinases JNK and p38 and osteoblastic cell differentiation. J. Biol. Chem. 279, 259–264 (2004).
Lee, K. S., Hong, S. H. & Bae, S. C. Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-beta and bone morphogenetic protein. Oncogene 21, 7156–7163 (2002).
Lee, M. H., Kwon, T. G., Park, H. S., Wozney, J. M. & Ryoo, H. M. BMP-2-induced Osterix expression is mediated by Dlx5 but is independent of Runx2. Biochem. Biophys. Res. Commun. 309, 689–694 (2003).
Celil, A. B., Hollinger, J. O. & Campbell, P. G. Osx transcriptional regulation is mediated by additional pathways to BMP2/Smad signaling. J. Cell Biochem. 95, 518–528 (2005).
Kim, M. K. et al. Salt-inducible kinase 1 regulates bone anabolism via the CRTC1-CREB-Id1 axis. Cell Death Dis. 10, 826 (2019).
Kim, K. et al. Transcription factor Lmx1b negatively regulates osteoblast differentiation and bone formation. Int. J. Mol. Sci. 23, 5225 (2022).
Zhang, R. et al. Wnt/β-catenin signaling activates bone morphogenetic protein 2 expression in osteoblasts. Bone 52, 145–156 (2013).
Kokabu, S. & Rosen, V. BMP3 expression by osteoblast lineage cells is regulated by canonical Wnt signaling. FEBS Open Bio 8, 168–176 (2018).
de Jong, D. S. et al. Identification of novel regulators associated with early-phase osteoblast differentiation. J. Bone Min. Res 19, 947–958 (2004).
Son, H. E., Kim, K. M., Kim, E. J. & Jang, W. G. Kisspeptin-10 (KP-10) stimulates osteoblast differentiation through GPR54-mediated regulation of BMP2 expression and activation. Sci. Rep. 8, 2134 (2018).
Comninos, A. N. et al. Acute effects of kisspeptin administration on bone metabolism in healthy men. J. Clin. Endocrinol. Metab. 107, 1529–1540 (2022).
Izzi, L. & Attisano, L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene 23, 2071–2078 (2004).
Datto, M. & Wang, X. F. Ubiquitin-mediated degradation a mechanism for fine-tuning TGF-beta signaling. Cell 121, 2–4 (2005).
Dupont, S. et al. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 121, 87–99 (2005).
Xu, Z. et al. SMURF2 regulates bone homeostasis by disrupting SMAD3 interaction with vitamin D receptor in osteoblasts. Nat. Commun. 8, 14570 (2017).
Long, F. et al. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 131, 1309–1318 (2004).
Deng, A. et al. The inhibitory roles of Ihh downregulation on chondrocyte growth and differentiation. Exp. Ther. Med. 15, 789–794 (2018).
Minina, E. et al. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 128, 4523–4534 (2001).
St-Jacques, B., Hammerschmidt, M. & McMahon, A. P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086 (1999).
Chung, U. I., Schipani, E., McMahon, A. P. & Kronenberg, H. M. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J. Clin. Invest. 107, 295–304 (2001).
Razzaque, M. S., Soegiarto, D. W., Chang, D., Long, F. & Lanske, B. Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J. Pathol. 207, 453–461 (2005).
Nakamura, T. et al. Induction of osteogenic differentiation by hedgehog proteins. Biochem. Biophys. Res. Commun. 237, 465–469 (1997).
van der Horst, G., Farih-Sips, H., Löwik, C. W. & Karperien, M. Hedgehog stimulates only osteoblastic differentiation of undifferentiated KS483 cells. Bone 33, 899–910 (2003).
Chen, M. H. et al. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 23, 1910–1928 (2009).
Wen, X. et al. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell Biol. 30, 1910–1922 (2010).
Cai, H. & Liu, A. Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc. Natl. Acad. Sci. USA 113, 14751–14756 (2016).
Horikiri, Y. et al. Sonic hedgehog regulates osteoblast function by focal adhesion kinase signaling in the process of fracture healing. PLoS One 8, e76785 (2013).
Sinha, S. & Chen, J. K. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat. Chem. Biol. 2, 29–30 (2006).
Oliveira, F. S. et al. Hedgehog signaling and osteoblast gene expression are regulated by purmorphamine in human mesenchymal stem cells. J. Cell Biochem. 113, 204–208 (2012).
Jakobs, P. et al. Scube2 enhances proteolytic Shh processing from the surface of Shh-producing cells. J. Cell Sci. 127, 1726–1737 (2014).
Parchure, A., Vyas, N. & Mayor, S. Wnt and Hedgehog: Secretion of lipid-modified morphogens. Trends Cell Biol. 28, 157–170 (2018).
Wierbowski, B. M. et al. Hedgehog pathway activation requires coreceptor-catalyzed, lipid-dependent relay of the sonic hedgehog ligand. Dev. Cell 55, 450–467.e458 (2020).
Wu, Q. et al. SCUBE2 mediates bone metastasis of luminal breast cancer by modulating immune-suppressive osteoblastic niches. Cell Res. 33, 464–478 (2023).
Saeki, N. et al. Pregnane X receptor (PXR) represses osteoblast differentiation through repression of the Hedgehog signaling pathway. Exp. Cell Res. 416, 113156 (2022).
Shi, Y., Chen, J., Karner, C. M. & Long, F. Hedgehog signaling activates a positive feedback mechanism involving insulin-like growth factors to induce osteoblast differentiation. Proc. Natl. Acad. Sci. USA 112, 4678–4683 (2015).
Shi, Y. et al. Gli1(+) progenitors mediate bone anabolic function of teriparatide via Hh and Igf signaling. Cell Rep. 36, 109542 (2021).
Aruga, J. & Mikoshiba, K. Identification and characterization of Slitrk, a novel neuronal transmembrane protein family controlling neurite outgrowth. Mol. Cell Neurosci. 24, 117–129 (2003).
Sun, J. et al. SLITRK5 is a negative regulator of hedgehog signaling in osteoblasts. Nat. Commun. 12, 4611 (2021).
Meng, X. et al. Suppressor of fused negatively regulates beta-catenin signaling. J. Biol. Chem. 276, 40113–40119 (2001).
Jia, J. et al. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 416, 548–552 (2002).
Price, M. A. & Kalderon, D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 108, 823–835 (2002).
Spinella-Jaegle, S. et al. Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocytic differentiation. J. Cell Sci. 114, 2085–2094 (2001).
Yuasa, T. et al. Sonic hedgehog is involved in osteoblast differentiation by cooperating with BMP-2. J. Cell Physiol. 193, 225–232 (2002).
Ornitz, D. M. & Marie, P. J. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 16, 1446–1465 (2002).
Webster, M. K. & Donoghue, D. J. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 15, 520–527 (1996).
Wilkie, A. O. Craniosynostosis: genes and mechanisms. Hum. Mol. Genet. 6, 1647–1656 (1997).
Jacob, A. L., Smith, C., Partanen, J. & Ornitz, D. M. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev. Biol. 296, 315–328 (2006).
Naski, M. C., Colvin, J. S., Coffin, J. D. & Ornitz, D. M. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 125, 4977–4988 (1998).
Valverde-Franco, G. et al. Defective bone mineralization and osteopenia in young adult FGFR3-/- mice. Hum. Mol. Genet. 13, 271–284 (2004).
Xiao, L. et al. Stat1 controls postnatal bone formation by regulating fibroblast growth factor signaling in osteoblasts. J. Biol. Chem. 279, 27743–27752 (2004).
Chen, L. & Deng, C. X. Roles of FGF signaling in skeletal development and human genetic diseases. Front Biosci. 10, 1961–1976 (2005).
Ornitz, D. M. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 16, 205–213 (2005).
Jeon, Y. M. et al. Fibroblast growth factor-7 facilitates osteogenic differentiation of embryonic stem cells through the activation of ERK/Runx2 signaling. Mol. Cell Biochem. 382, 37–45 (2013).
Lin, J. M. et al. Actions of fibroblast growth factor-8 in bone cells in vitro. Am. J. Physiol. Endocrinol. Metab. 297, E142–E150 (2009).
Jeon, E. et al. Investigating the role of FGF18 in the cultivation and osteogenic differentiation of mesenchymal stem cells. PLoS One 7, e43982 (2012).
Hamidouche, Z. et al. Autocrine fibroblast growth factor 18 mediates dexamethasone-induced osteogenic differentiation of murine mesenchymal stem cells. J. Cell Physiol. 224, 509–515 (2010).
Xiao, G., Jiang, D., Gopalakrishnan, R. & Franceschi, R. T. Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J. Biol. Chem. 277, 36181–36187 (2002).
Fei, Y., Xiao, L., Doetschman, T., Coffin, D. J. & Hurley, M. M. Fibroblast growth factor 2 stimulation of osteoblast differentiation and bone formation is mediated by modulation of the Wnt signaling pathway. J. Biol. Chem. 286, 40575–40583 (2011).
Miraoui, H., Ringe, J., Häupl, T. & Marie, P. J. Increased EFG- and PDGFalpha-receptor signaling by mutant FGF-receptor 2 contributes to osteoblast dysfunction in Apert craniosynostosis. Hum. Mol. Genet. 19, 1678–1689 (2010).
Ornitz, D. M. & Itoh, N. Fibroblast growth factors. Genome Biol. 2, Reviews3005 (2001).
Ornitz, D. M. et al. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 271, 15292–15297 (1996).
Liu, Z., Xu, J., Colvin, J. S. & Ornitz, D. M. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 16, 859–869 (2002).
Wang, Y. et al. FGFR2 Mutation p.Cys342Arg enhances mitochondrial metabolism-mediated osteogenesis via FGF/FGFR-AMPK-Erk1/2 axis in Crouzon syndrome. Cells 11, 3129 (2022).
Zhu, Q. et al. OTUB1 promotes osteoblastic bone formation through stabilizing FGFR2. Signal Transduct. Target Ther. 8, 142 (2023).
Courbon, G. et al. FGF23 directly inhibits osteoprogenitor differentiation in Dmp1 knockout mice. JCI Insight 8, e156850 (2023).
Al Rifai, O. et al. In vivo analysis of the contribution of proprotein convertases to the processing of FGF23. Front Endocrinol. (Lausanne) 12, 690681 (2021).
Kozawa, O., Tokuda, H., Matsuno, H. & Uematsu, T. Involvement of p38 mitogen-activated protein kinase in basic fibroblast growth factor-induced interleukin-6 synthesis in osteoblasts. J. Cell Biochem. 74, 479–485 (1999).
Lemonnier, J. et al. The Ser252Trp fibroblast growth factor receptor-2 (FGFR-2) mutation induces PKC-independent downregulation of FGFR-2 associated with premature calvaria osteoblast differentiation. Exp. Cell Res. 256, 158–167 (2000).
Lomri, A., Lemonnier, J., Delannoy, P. & Marie, P. J. Increased expression of protein kinase Calpha, interleukin-1alpha, and RhoA guanosine 5’-triphosphatase in osteoblasts expressing the Ser252Trp fibroblast growth factor 2 receptor Apert mutation: identification by analysis of complementary DNA microarray. J. Bone Min. Res. 16, 705–712 (2001).
Taketomi, T. et al. Sprouty2 is involved in the control of osteoblast proliferation and differentiation through the FGF and BMP signaling pathways. Cell Biol. Int. 42, 1106–1114 (2018).
Mundy, G. R. & Elefteriou, F. Boning up on ephrin signaling. Cell 126, 441–443 (2006).
Arthur, A. & Gronthos, S. Eph-Ephrin signaling mediates cross-talk within the bone microenvironment. Front Cell Dev. Biol. 9, 598612 (2021).
Tonna, S. et al. EphrinB2 signaling in osteoblasts promotes bone mineralization by preventing apoptosis. FASEB J. 28, 4482–4496 (2014).
Zhao, C. et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 4, 111–121 (2006).
Irie, N. et al. Bidirectional signaling through ephrinA2-EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J. Biol. Chem. 284, 14637–14644 (2009).
Vrahnas, C. et al. Increased autophagy in EphrinB2-deficient osteocytes is associated with elevated secondary mineralization and brittle bone. Nat. Commun. 10, 3436 (2019).
Blank, M. & Sims, N. A. Cellular processes by which osteoblasts and osteocytes control bone mineral deposition and maturation revealed by stage-specific EphrinB2 knockdown. Curr. Osteoporos. Rep. 17, 270–280 (2019).
Tonna, S. & Sims, N. A. Talking among ourselves: paracrine control of bone formation within the osteoblast lineage. Calcif. Tissue Int. 94, 35–45 (2014).
Kawatsu, M. et al. Scleraxis upregulated by transforming growth factor-β1 signaling inhibits tension-induced osteoblast differentiation of priodontal ligament cells via ephrin A2. Bone 149, 115969 (2021).
Oeckinghaus, A. & Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1, a000034 (2009).
Li, Y. P. & Stashenko, P. Proinflammatory cytokines tumor necrosis factor-alpha and IL-6, but not IL-1, down-regulate the osteocalcin gene promoter. J. Immunol. 148, 788–794 (1992).
Chang, J. et al. NF-κB inhibits osteogenic differentiation of mesenchymal stem cells by promoting β-catenin degradation. Proc. Natl. Acad. Sci. USA 110, 9469–9474 (2013).
Li, Y. P. & Stashenko, P. Characterization of a tumor necrosis factor-responsive element which down-regulates the human osteocalcin gene. Mol. Cell Biol. 13, 3714–3721 (1993).
Chang, J. et al. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat. Med. 15, 682–689 (2009).
Hu, Y. et al. Melatonin promotes BMSCs osteoblastic differentiation and relieves inflammation by suppressing the NF-κB pathways. Stem Cells Int. 2023, 7638842 (2023).
Li, A. et al. Rocaglamide-A potentiates osteoblast differentiation by inhibiting NF-κB signaling. Mol. Cells 38, 941–949 (2015).
Qin, H., Xu, H. Z. & Gong, Y. Q. Mechanism of NF-κB signaling pathway and autophagy in the regulation of osteoblast differentiation. Mol. Membr. Biol. 33, 138–144 (2016).
Guo, X. et al. Radioprotective 105 kDa protein attenuates ischemia/reperfusion-induced myocardial apoptosis and autophagy by inhibiting the activation of the TLR4/NF-κB signaling pathway in rats. Int. J. Mol. Med. 38, 885–893 (2016).
Gao, H., Lin, L., Haq, I. U. & Zeng, S. M. Inhibition of NF-κB promotes autophagy via JNK signaling pathway in porcine granulosa cells. Biochem. Biophys. Res. Commun. 473, 311–316 (2016).
Bleedorn, J. A. et al. Temporal mechanically-induced signaling events in bone and dorsal root ganglion neurons after in vivo bone loading. PLoS One 13, e0192760 (2018).
Tomlinson, R. E. et al. NGF-TrkA signaling in sensory nerves is required for skeletal adaptation to mechanical loads in mice. Proc. Natl. Acad. Sci. USA 114, E3632–e3641 (2017).
Rajpar, I., Kumar, G., Fortina, P. & Tomlinson, R. E. Toll-like receptor 4 signaling in osteoblasts is required for load-induced bone formation in mice. iScience 26, 106304 (2023).
Zhou, B. et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 7, 95 (2022).
Xu, J., Li, Z., Hou, Y. & Fang, W. Potential mechanisms underlying the Runx2 induced osteogenesis of bone marrow mesenchymal stem cells. Am. J. Transl. Res. 7, 2527–2535 (2015).
Canalis, E. Notch signaling in osteoblasts. Sci. Signal. 1, pe17 (2008).
Liu, Z. H., Dai, X. M. & Du, B. Hes1: a key role in stemness, metastasis and multidrug resistance. Cancer Biol. Ther. 16, 353–359 (2015).
Luo, Z. et al. Notch signaling in osteogenesis, osteoclastogenesis, and angiogenesis. Am. J. Pathol. 189, 1495–1500 (2019).
Tezuka, K. et al. Stimulation of osteoblastic cell differentiation by Notch. J. Bone Min. Res. 17, 231–239 (2002).
Xu, Y. et al. Notch activation promotes osteoblast mineralization by inhibition of apoptosis. J. Cell Physiol. 233, 6921–6928 (2018).
Deregowski, V., Gazzerro, E., Priest, L., Rydziel, S. & Canalis, E. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. J. Biol. Chem. 281, 6203–6210 (2006).
Cao, J. et al. Notch signaling pathway promotes osteogenic differentiation of mesenchymal stem cells by enhancing BMP9/Smad signaling. Int. J. Mol. Med. 40, 378–388 (2017).
Lin, G. L. & Hankenson, K. D. Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J. Cell. Biochem. 112, 3491–3501 (2011).
Kode, A. et al. FoxO1-dependent induction of acute myeloid leukemia by osteoblasts in mice. Leukemia 30, 1–13 (2016).
Neal, J. W. & Clipstone, N. A. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J. Biol. Chem. 276, 3666–3673 (2001).
Yu, W. et al. SALL4 promotes osteoblast differentiation by deactivating NOTCH2 signaling. Biomed. Pharmacother. 98, 9–17 (2018).
Torres, H. M. et al. Hdac1 and Hdac2 positively regulate Notch1 gain-of-function pathogenic signaling in committed osteoblasts of male mice. Birth Defects Res. 116, e2266 (2024).
Wang, Z. et al. The histone deacetylase HDAC1 positively regulates Notch signaling during Drosophila wing development. Biol. Open 7, bio029637 (2018).
Tang, Y., Boucher, J. M. & Liaw, L. Histone deacetylase activity selectively regulates notch-mediated smooth muscle differentiation in human vascular cells. J. Am. Heart Assoc. 1, e000901 (2012).
Torres, H. M. et al. Hdac1 and Hdac2 positively regulate Notch1 gain-of-function pathogenic signaling in committed osteoblasts of male mice. Birth Defects Res 116, e2266 (2023).
Hansen, C. G., Moroishi, T. & Guan, K. L. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 25, 499–513 (2015).
Hong, W. & Guan, K. L. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev. Biol. 23, 785–793 (2012).
Tang, Y. et al. MT1-MMP-dependent control of skeletal stem cell commitment via a β1-integrin/YAP/TAZ signaling axis. Dev. Cell 25, 402–416 (2013).
Zheng, Y. & Pan, D. The hippo signaling pathway in development and disease. Dev. Cell 50, 264–282 (2019).
Zaidi, S. K. et al. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 23, 790–799 (2004).
Zhu, W. Q. et al. Regulation of osteoblast behaviors via cross-talk between Hippo/YAP and MAPK signaling pathway under fluoride exposure. J. Mol. Med (Berl.) 97, 1003–1017 (2019).
Chuang, L. S. H. & Ito, Y. The multiple interactions of RUNX with the Hippo-YAP pathway. Cells 10, 2925 (2021).
Franceschi, R. T. & Ge, C. Control of the osteoblast lineage by mitogen-activated protein kinase signaling. Curr. Mol. Biol. Rep. 3, 122–132 (2017).
Li, Y., Yang, S. T. & Yang, S. Trp53 controls chondrogenesis and endochondral ossification by negative regulation of TAZ activity and stability via β-TrCP-mediated ubiquitination. Cell Death Discov. 8, 317 (2022).
Velletri, T. et al. Loss of p53 in mesenchymal stem cells promotes alteration of bone remodeling through negative regulation of osteoprotegerin. Cell Death Differ. 28, 156–169 (2021).
Varelas, X. et al. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev. Cell 18, 579–591 (2010).
Varelas, X. et al. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 19, 831–844 (2010).
Varelas, X. et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 10, 837–848 (2008).
Pagani, C. A. et al. Discoidin domain receptor 2 regulates aberrant mesenchymal lineage cell fate and matrix organization. Sci. Adv. 8, eabq6152 (2022).
Liu, P. et al. Effects of mechanical stress stimulation on function and expression mechanism of osteoblasts. Front Bioeng. Biotechnol. 10, 830722 (2022).
Yavropoulou, M. P. & Yovos, J. G. The molecular basis of bone mechanotransduction. J. Musculoskelet. Neuronal Interact. 16, 221–236 (2016).
Sun, W. et al. The mechanosensitive Piezo1 channel is required for bone formation. Elife 8, e47454 (2019).
Li, X. et al. Stimulation of Piezo1 by mechanical signals promotes bone anabolism. Elife 8, e49631 (2019).
Assaraf, E. et al. Piezo2 expressed in proprioceptive neurons is essential for skeletal integrity. Nat. Commun. 11, 3168 (2020).
Hendrickx, G. et al. Piezo1 inactivation in chondrocytes impairs trabecular bone formation. J. Bone Min. Res. 36, 369–384 (2021).
Zhou, T. et al. Piezo1/2 mediate mechanotransduction essential for bone formation through concerted activation of NFAT-YAP1-ß-catenin. Elife 9, e49631 (2020).
Wang, L. et al. Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat. Commun. 11, 282 (2020).
Morris, J. A. et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet. 51, 258–266 (2019).
Dzamukova, M. et al. Mechanical forces couple bone matrix mineralization with inhibition of angiogenesis to limit adolescent bone growth. Nat. Commun. 13, 3059 (2022).
Kusumbe, A. P., Ramasamy, S. K. & Adams, R. H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507, 323–328 (2014).
Romeo, S. G. et al. Endothelial proteolytic activity and interaction with non-resorbing osteoclasts mediate bone elongation. Nat. Cell Biol. 21, 430–441 (2019).
Tagliabracci, V. S. et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 336, 1150–1153 (2012).
Tagliabracci, V. S. et al. A single kinase generates the majority of the secreted phosphoproteome. Cell 161, 1619–1632 (2015).
Gurr, J. A., Catterall, J. F. & Kourides, I. A. Cloning of cDNA encoding the pre-beta subunit of mouse thyrotropin. Proc. Natl. Acad. Sci. USA 80, 2122–2126 (1983).
Baliram, R. et al. Thyroid and bone: macrophage-derived TSH-β splice variant increases murine osteoblastogenesis. Endocrinology 154, 4919–4926 (2013).
Elefteriou, F. et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 434, 514–520 (2005).
Takeda, S. et al. Leptin regulates bone formation via the sympathetic nervous system. Cell 111, 305–317 (2002).
Fulzele, K. et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 185, 746 (2022).
Bialek, P. et al. A twist code determines the onset of osteoblast differentiation. Dev. Cell 6, 423–435 (2004).
Fulzele, K. et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142, 309–319 (2010).
Kode, A. et al. FoxO1 protein cooperates with ATF4 protein in osteoblasts to control glucose homeostasis. J. Biol. Chem. 287, 8757–8768 (2012).
Lian, J. B. et al. Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit. Rev. Eukaryot. Gene Expr. 14, 1–41 (2004).
Ducy, P., Zhang, R., Geoffroy, V., Ridall, A. L. & Karsenty, G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89, 747–754 (1997).
Wang, Q. et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell 87, 697–708 (1996).
Kania, M. A., Bonner, A. S., Duffy, J. B. & Gergen, J. P. The Drosophila segmentation gene runt encodes a novel nuclear regulatory protein that is also expressed in the developing nervous system. Genes Dev. 4, 1701–1713 (1990).
Kern, B., Shen, J., Starbuck, M. & Karsenty, G. Cbfa1 contributes to the osteoblast-specific expression of type I collagen genes. J. Biol. Chem. 276, 7101–7107 (2001).
Salhotra, A., Shah, H. N., Levi, B. & Longaker, M. T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell Biol. 21, 696–711 (2020).
Stein, G. S. et al. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene 23, 4315–4329 (2004).
Wei, J. et al. Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell 161, 1576–1591 (2015).
Ogawa, E. et al. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila runt gene and the human AML1 gene. Proc. Natl. Acad. Sci. USA 90, 6859–6863 (1993).
Banerjee, C. et al. Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone-specific complex. J. Cell. Biochem. 66, 1–8 (1997).
Komori, T. et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764 (1997).
Otto, F. et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765–771 (1997).
Pratap, J. et al. Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Res. 63, 5357–5362 (2003).
Huang, B. et al. RUNX2 promotes the suppression of osteoblast function and enhancement of osteoclast activity by multiple myeloma cells. Med. Oncol. 40, 115 (2023).
Komori, T. Runx2 an inducer of osteoblast and chondrocyte differentiation. Histochem Cell Biol. 149, 313–323 (2018).
Pelletier, N., Champagne, N., Stifani, S. & Yang, X. J. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene 21, 2729–2740 (2002).
Thomas, D. M. et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol. Cell 8, 303–316 (2001).
Cui, C. B., Cooper, L. F., Yang, X., Karsenty, G. & Aukhil, I. Transcriptional coactivation of bone-specific transcription factor Cbfa1 by TAZ. Mol. Cell Biol. 23, 1004–1013 (2003).
Wang, W. et al. Groucho homologue Grg5 interacts with the transcription factor Runx2-Cbfa1 and modulates its activity during postnatal growth in mice. Dev. Biol. 270, 364–381 (2004).
Byun, M. R. et al. (-)-Epicatechin gallate (ECG) stimulates osteoblast differentiation via Runt-related transcription factor 2 (RUNX2) and transcriptional coactivator with PDZ-binding motif (TAZ)-mediated transcriptional activation. J. Biol. Chem. 289, 9926–9935 (2014).
Suo, J. et al. VGLL4 promotes osteoblast differentiation by antagonizing TEADs-inhibited Runx2 transcription. Sci. Adv. 6 (2020).
Lee, J. M., Libermann, T. A. & Cho, J. Y. The synergistic regulatory effect of Runx2 and MEF transcription factors on osteoblast differentiation markers. J. Periodontal Implant Sci. 40, 39–44 (2010).
Zainabadi, K., Liu, C. J. & Guarente, L. SIRT1 is a positive regulator of the master osteoblast transcription factor, RUNX2. PLoS One 12, e0178520 (2017).
Tintut, Y., Parhami, F., Le, V., Karsenty, G. & Demer, L. L. Inhibition of osteoblast-specific transcription factor Cbfa1 by the cAMP pathway in osteoblastic cells. Ubiquitin/proteasome-dependent regulation. J. Biol. Chem. 274, 28875–28879 (1999).
Shen, R. et al. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J. Biol. Chem. 281, 3569–3576 (2006).
Tosa, I. et al. Postnatal Runx2 deletion leads to low bone mass and adipocyte accumulation in mice bone tissues. Biochem. Biophys. Res. Commun. 516, 1229–1233 (2019).
Chava, S., Chennakesavulu, S., Gayatri, B. M. & Reddy, A. B. M. A novel phosphorylation by AMP-activated kinase regulates RUNX2 from ubiquitination in osteogenesis over adipogenesis. Cell Death Dis. 9, e49631 (2018).
Dai, X. et al. Epigenetic upregulation of H19 and AMPK inhibition concurrently contribute to S-Adenosylhomocysteine hydrolase deficiency-promoted atherosclerotic calcification. Circ. Res. 130, 1565–1582 (2022).
Liu, H. et al. Rho A/ROCK1 signaling-mediated metabolic reprogramming of valvular interstitial cells toward Warburg effect accelerates aortic valve calcification via AMPK/RUNX2 axis. Cell Death Dis. 14, 108 (2023).
Kim, K. M., Lim, Y. J. & Jang, W. G. Policosanol stimulates osteoblast differentiation via adenosine monophosphate-activated protein kinase-mediated expression of insulin-induced genes 1 and 2. Cells 12, 108 (2023).
Zhu, J., Shimizu, E., Zhang, X., Partridge, N. C. & Qin, L. EGFR signaling suppresses osteoblast differentiation and inhibits expression of master osteoblastic transcription factors Runx2 and Osterix. J. Cell Biochem. 112, 1749–1760 (2011).
Lee, K. N. et al. Orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) protein negatively regulates bone morphogenetic protein 2-induced osteoblast differentiation through suppressing runt-related gene 2 (Runx2) activity. J. Biol. Chem. 287, 18888–18899 (2012).
Tang, C. Y. et al. Runx1 is a central regulator of osteogenesis for bone homeostasis by orchestrating BMP and WNT signaling pathways. PLoS Genet 17, e1009233 (2021).
Okuda, T., Nishimura, M., Nakao, M. & Fujita, Y. RUNX1/AML1: a central player in hematopoiesis. Int J. Hematol. 74, 252–257 (2001).
Chen, C. L. et al. Runx1 determines nociceptive sensory neuron phenotype and is required for thermal and neuropathic pain. Neuron 49, 365–377 (2006).
Lian, J. B. et al. Runx1/AML1 hematopoietic transcription factor contributes to skeletal development in vivo. J. Cell Physiol. 196, 301–311 (2003).
Tang, C. Y. et al. Runx1 up-regulates chondrocyte to osteoblast lineage commitment and promotes bone formation by enhancing both chondrogenesis and osteogenesis. Biochem J. 477, 2421–2438 (2020).
Wang, Y. et al. Runx1/AML1/Cbfa2 mediates onset of mesenchymal cell differentiation toward chondrogenesis. J. Bone Min. Res. 20, 1624–1636 (2005).
Tang, J. et al. Runt-related transcription factor 1 is required for murine osteoblast differentiation and bone formation. J. Biol. Chem. 295, 11669–11681 (2020).
Levanon, D. & Groner, Y. Structure and regulated expression of mammalian RUNX genes. Oncogene 23, 4211–4219 (2004).
de Bruijn, M. F. & Speck, N. A. Core-binding factors in hematopoiesis and immune function. Oncogene 23, 4238–4248 (2004).
Levanon, D. et al. Spatial and temporal expression pattern of Runx3 (Aml2) and Runx1 (Aml1) indicates non-redundant functions during mouse embryogenesis. Mech. Dev. 109, 413–417 (2001).
Yamashiro, T., Aberg, T., Levanon, D., Groner, Y. & Thesleff, I. Expression of Runx1, -2 and -3 during tooth, palate and craniofacial bone development. Mech. Dev. 119, S107–S110 (2002).
Soung do, Y. et al. Runx3/AML2/Cbfa3 regulates early and late chondrocyte differentiation. J. Bone Min. Res. 22, 1260–1270 (2007).
Sato, S. et al. The distinct role of the Runx proteins in chondrocyte differentiation and intervertebral disc degeneration: findings in murine models and in human disease. Arthritis Rheum. 58, 2764–2775 (2008).
Bauer, O. et al. Loss of osteoblast Runx3 produces severe congenital osteopenia. Mol. Cell Biol. 35, 1097–1109 (2015).
Wang, Y. et al. RUNX3 plays an important role in mediating the BMP9-induced osteogenic differentiation of mesenchymal stem cells. Int. J. Mol. Med. 40, 1991–1999 (2017).
Nakashima, K. et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29 (2002).
Hewitt, J., Lu, X., Gilbert, L. & Nanes, M. S. The muscle transcription factor MyoD promotes osteoblast differentiation by stimulation of the Osterix promoter. Endocrinology 149, 3698–3707 (2008).
Onizuka, S. et al. ZBTB16 as a downstream target gene of Osterix regulates osteoblastogenesis of human multipotent mesenchymal stromal cells. J. Cell. Biochem. 117, 2423–2434 (2016).
Lee, S. J., Lee, E. H., Park, S. Y. & Kim, J. E. Induction of fibrillin-2 and periostin expression in Osterix-knockdown MC3T3-E1 cells. Gene 596, 123–129 (2017).
Nieminen-Pihala, V. et al. Early B-cell Factor1 (Ebf1) promotes early osteoblast differentiation but suppresses osteoblast function. Bone 146, 115884 (2021).
Kato, M. et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 157, 303–314 (2002).
Cheng, S. L., Shao, J. S., Charlton-Kachigian, N., Loewy, A. P. & Towler, D. A. MSX2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. J. Biol. Chem. 278, 45969–45977 (2003).
Celil, A. B. & Campbell, P. G. BMP-2 and insulin-like growth factor-I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. J. Biol. Chem. 280, 31353–31359 (2005).
Ishikawa, M. et al. Pannexin 3 functions as an ER Ca(2+) channel, hemichannel, and gap junction to promote osteoblast differentiation. J. Cell Biol. 193, 1257–1274 (2011).
Koga, T. et al. NFAT and Osterix cooperatively regulate bone formation. Nat. Med. 11, 880–885 (2005).
Colditz, J., Picke, A. K., Hofbauer, L. C. & Rauner, M. Contributions of dickkopf-1 to obesity-induced bone loss and marrow adiposity. JBMR 4, e10364 (2020).
Chen, D. et al. Synergistic inhibition of Wnt pathway by HIF-1α and osteoblast-specific transcription factor osterix (Osx) in osteoblasts. PLoS One 7, e52948 (2012).
Deckelbaum, R. A., Majithia, A., Booker, T., Henderson, J. E. & Loomis, C. A. The homeoprotein engrailed 1 has pleiotropic functions in calvarial intramembranous bone formation and remodeling. Development 133, 63–74 (2006).
Fedde, K. N. et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J. Bone Min. Res. 14, 2015–2026 (1999).
Welcker, M. & Clurman, B. E. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 83–93 (2008).
Hoshikawa, S. et al. Phosphorylation-dependent osterix degradation negatively regulates osteoblast differentiation. FASEB J. 34, 14930–14945 (2020).
Choi, Y. H. et al. Cbl-b and c-Cbl negatively regulate osteoblast differentiation by enhancing ubiquitination and degradation of Osterix. Bone 75, 201–209 (2015).
Kim, M. J., Piao, M., Li, Y., Lee, S. H. & Lee, K. Y. Deubiquitinase USP17 regulates osteoblast differentiation by increasing Osterix protein stability. Int. J. Mol. Sci. 24, 15257 (2023).
Barbuto, R. & Mitchell, J. Regulation of the osterix (Osx, Sp7) promoter by osterix and its inhibition by parathyroid hormone. J. Mol. Endocrinol. 51, 99–108 (2013).
Matsubara, T. et al. BMP2 regulates Osterix through Msx2 and Runx2 during osteoblast differentiation. J. Biol. Chem. 283, 29119–29125 (2008).
Subramaniam, M., Pitel, K. S., Withers, S. G., Drissi, H. & Hawse, J. R. TIEG1 enhances Osterix expression and mediates its induction by TGFβ and BMP2 in osteoblasts. Biochem. Biophys. Res. Commun. 470, 528–533 (2016).
Rajamannan, N. M. TIEG1 is upregulated in Lrp5/6-mediated valve osteogenesis. J. Cell Biochem. 120, 3362–3366 (2019).
Liu, Q. et al. Recent advances of Osterix transcription factor in osteoblast differentiation and bone formation. Front. Cell Dev. Biol. 8, 601224 (2020).
Yang, D., Okamura, H., Nakashima, Y. & Haneji, T. Histone demethylase Jmjd3 regulates osteoblast differentiation via transcription factors Runx2 and osterix. J. Biol. Chem. 288, 33530–33541 (2013).
Ishikawa, M. & Yamada, Y. The role of Pannexin 3 in bone biology. J. Dent. Res. 96, 372–379 (2017).
Ishikawa, M. et al. Pannexin 3 and connexin 43 modulate skeletal development through their distinct functions and expression patterns. J. Cell Sci. 129, 1018–1030 (2016).
Okamura, H., Yoshida, K., Ochiai, K. & Haneji, T. Reduction of protein phosphatase 2A Cα enhances bone formation and osteoblast differentiation through the expression of bone-specific transcription factor Osterix. Bone 49, 368–375 (2011).
Sinha, K. M., Yasuda, H., Coombes, M. M., Dent, S. Y. & de Crombrugghe, B. Regulation of the osteoblast-specific transcription factor Osterix by NO66, a Jumonji family histone demethylase. EMBO J. 29, 68–79 (2010).
Yang, X. et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 117, 387–398 (2004).
Elefteriou, F. et al. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 4, 441–451 (2006).
Karpinski, B. A., Morle, G. D., Huggenvik, J., Uhler, M. D. & Leiden, J. M. Molecular cloning of human CREB-2: an ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proc. Natl. Acad. Sci. USA 89, 4820–4824 (1992).
Brindle, P. K. & Montminy, M. R. The CREB family of transcription activators. Curr. Opin. Genet Dev. 2, 199–204 (1992).
Xiao, G. et al. Cooperative interactions between activating transcription factor 4 and Runx2/Cbfa1 stimulate osteoblast-specific osteocalcin gene expression. J. Biol. Chem. 280, 30689–30696 (2005).
Lian, N. et al. Transforming growth factor β suppresses osteoblast differentiation via the vimentin activating transcription factor 4 (ATF4) axis. J. Biol. Chem. 287, 35975–35984 (2012).
St-Arnaud, R. & Hekmatnejad, B. Combinatorial control of ATF4-dependent gene transcription in osteoblasts. Ann. N. Y. Acad. Sci. 1237, 11–18 (2011).
Dobreva, G., Dambacher, J. & Grosschedl, R. SUMO modification of a novel MAR-binding protein, SATB2, modulates immunoglobulin mu gene expression. Genes Dev. 17, 3048–3061 (2003).
Britanova, O., Akopov, S., Lukyanov, S., Gruss, P. & Tarabykin, V. Novel transcription factor Satb2 interacts with matrix attachment region DNA elements in a tissue-specific manner and demonstrates cell-type-dependent expression in the developing mouse CNS. Eur. J. Neurosci. 21, 658–668 (2005).
Gyorgy, A. B., Szemes, M., de Juan Romero, C., Tarabykin, V. & Agoston, D. V. SATB2 interacts with chromatin-remodeling molecules in differentiating cortical neurons. Eur. J. Neurosci. 27, 865–873 (2008).
Roy, S. K., Shrivastava, A., Srivastav, S., Shankar, S. & Srivastava, R. K. SATB2 is a novel biomarker and therapeutic target for cancer. J. Cell. Mol. Med. 24, 11064–11069 (2020).
Dobreva, G. et al. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 125, 971–986 (2006).
Kanzler, B., Kuschert, S. J., Liu, Y. H. & Mallo, M. Hoxa-2 restricts the chondrogenic domain and inhibits bone formation during development of the branchial area. Development 125, 2587–2597 (1998).
Trainor, P. A. & Krumlauf, R. Hox genes, neural crest cells and branchial arch patterning. Curr. Opin. Cell Biol. 13, 698–705 (2001).
Huang, X. et al. SATB2: A versatile transcriptional regulator of craniofacial and skeleton development, neurogenesis and tumorigenesis, and its applications in regenerative medicine. Genes Dis. 9, 95–107 (2022).
Zuo, C. et al. TNF-α inhibits SATB2 expression and osteoblast differentiation through NF-κB and MAPK pathways. Oncotarget 9, 4833–4850 (2018).
Bonilla-Claudio, M. et al. Bmp signaling regulates a dose-dependent transcriptional program to control facial skeletal development. Development 139, 709–719 (2012).
Tang, W., Li, Y., Osimiri, L. & Zhang, C. Osteoblast-specific transcription factor Osterix (Osx) is an upstream regulator of Satb2 during bone formation. J. Biol. Chem. 286, 32995–33002 (2011).
Varelas, X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141, 1614–1626 (2014).
Hong, J. H. et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309, 1074–1078 (2005).
Li, Z. et al. Role of TCF/LEF transcription factors in bone development and osteogenesis. Int. J. Med Sci. 15, 1415–1422 (2018).
Kida, J. et al. Interaction of LEF1 with TAZ is necessary for the osteoblastogenic activity of Wnt3a. Sci. Rep. 8, 10375 (2018).
Tang, Y., Feinberg, T., Keller, E. T., Li, X. Y. & Weiss, S. J. Snail/Slug binding interactions with YAP/TAZ control skeletal stem cell self-renewal and differentiation. Nat. Cell Biol. 18, 917–929 (2016).
Chen, W. et al. Cbfβ deletion in mice recapitulates cleidocranial dysplasia and reveals multiple functions of Cbfβ required for skeletal development. Proc. Natl. Acad. Sci. USA 111, 8482–8487 (2014).
Wu, M. et al. Chondrocyte-specific knockout of Cbfβ reveals the indispensable function of Cbfβ in chondrocyte maturation, growth plate development and trabecular bone formation in mice. Int J. Biol. Sci. 10, 861–872 (2014).
Lim, K. E. et al. Core binding factor β of osteoblasts maintains cortical bone mass via stabilization of Runx2 in mice. J. Bone Min. Res. 30, 715–722 (2015).
Tian, F. et al. Core binding factor beta (Cbfβ) controls the balance of chondrocyte proliferation and differentiation by upregulating Indian hedgehog (Ihh) expression and inhibiting parathyroid hormone-related protein receptor (PPR) expression in postnatal cartilage and bone formation. J. Bone Min. Res. 29, 1564–1574 (2014).
Wu, M. et al. Deletion of core-binding factor β (Cbfβ) in mesenchymal progenitor cells provides new insights into Cbfβ/Runxs complex function in cartilage and bone development. Bone 65, 49–59 (2014).
Uenaka, M. et al. Osteoblast-derived vesicles induce a switch from bone-formation to bone-resorption in vivo. Nat. Commun. 13, 1066 (2022).
Valencia-Sanchez, M. A., Liu, J., Hannon, G. J. & Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 20, 515–524 (2006).
Laine, S. K., Alm, J. J., Virtanen, S. P., Aro, H. T. & Laitala-Leinonen, T. K. MicroRNAs miR-96, miR-124, and miR-199a regulate gene expression in human bone marrow-derived mesenchymal stem cells. J. Cell. Biochem. 113, 2687–2695 (2012).
Huang, S. et al. Upregulation of miR-22 promotes osteogenic differentiation and inhibits adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells by repressing HDAC6 protein expression. Stem Cells Dev. 21, 2531–2540 (2012).
Bhushan, R. et al. miR-181a promotes osteoblastic differentiation through repression of TGF-β signaling molecules. Int J. Biochem Cell Biol. 45, 696–705 (2013).
Nicetto, D. & Zaret, K. S. Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr. Opin. Genet Dev. 55, 1–10 (2019).
Xiang, Y. et al. JMJD3 is a histone H3K27 demethylase. Cell Res. 17, 850–857 (2007).
Huang, B., Li, G. & Jiang, X. H. Fate determination in mesenchymal stem cells: a perspective from histone-modifying enzymes. Stem Cell Res. Ther. 6, 35 (2015).
Kooistra, S. M. & Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 13, 297–311 (2012).
Hillringhaus, L. et al. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J. Biol. Chem. 286, 41616–41625 (2011).
Wei, Y. et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 13, 87–94 (2011).
Hemming, S. et al. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells 32, 802–815 (2014).
Montecino, M., Carrasco, M. E. & Nardocci, G. Epigenetic control of osteogenic lineage commitment. Front. Cell Dev. Biol. 8, 611197 (2020).
Yamane, K. et al. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell 25, 801–812 (2007).
Vermeulen, M. et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 (2007).
Denissov, S. et al. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 141, 526–537 (2014).
Hu, D. et al. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell Biol. 33, 4745–4754 (2013).
Yan, J. et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 28, 204–220 (2018).
Cheng, J. et al. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol. Cell 53, 979–992 (2014).
Sepulveda, H. et al. Epigenetic signatures at the RUNX2-P1 and Sp7 gene promoters control osteogenic lineage commitment of umbilical cord-derived mesenchymal stem cells. J. Cell Physiol. 232, 2519–2527 (2017).
Aguilar, R., Bustos, F. J., Nardocci, G., van Zundert, B. & Montecino, M. Epigenetic silencing of the osteoblast-lineage gene program during hippocampal maturation. J. Cell. Biochem. 122, 367–384 (2021).
McGee-Lawrence, M. E. & Westendorf, J. J. Histone deacetylases in skeletal development and bone mass maintenance. Gene 474, 1–11 (2011).
Seto, E. & Yoshida, M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6, a018713 (2014).
Milazzo, G. et al. Histone deacetylases (HDACs): Evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes (Basel) 11, 556 (2020).
Adithya, S. P., Balagangadharan, K. & Selvamurugan, N. Epigenetic modifications of histones during osteoblast differentiation. Biochim Biophys. Acta Gene Regul. Mech. 1865, 194780 (2022).
Lee, H. W. et al. Histone deacetylase 1-mediated histone modification regulates osteoblast differentiation. Mol. Endocrinol. 20, 2432–2443 (2006).
Wein, M. N. et al. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J. Bone Min. Res. 30, 400–411 (2015).
Grant, P. A. et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 11, 1640–1650 (1997).
Li, B. et al. GCN5 modulates osteogenic differentiation of periodontal ligament stem cells through DKK1 acetylation in inflammatory microenvironment. Sci. Rep. 6, 26542 (2016).
Fei, D. et al. KAT6A regulates stemness of aging bone marrow-derived mesenchymal stem cells through Nrf2/ARE signaling pathway. Stem Cell Res. Ther. 12, 104 (2021).
Zhang, L. et al. Acetyltransferases CBP/p300 control transcriptional switch of β-catenin and Stat1 promoting osteoblast differentiation. J. Bone Min. Res. 38, 1885–1899 (2023).
Peterson, C. L. & Workman, J. L. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr. Opin. Genet. Dev. 10, 187–192 (2000).
de la Serna, I. L., Carlson, K. A. & Imbalzano, A. N. Mammalian SWI/SNF complexes promote MyoD-mediated muscle differentiation. Nat. Genet. 27, 187–190 (2001).
Navickas, S. M., Giles, K. A., Brettingham-Moore, K. H. & Taberlay, P. C. The role of chromatin remodeler SMARCA4/BRG1 in brain cancers: a potential therapeutic target. Oncogene 42, 2363–2373 (2023).
de la Serna, I. L., Roy, K., Carlson, K. A. & Imbalzano, A. N. MyoD can induce cell cycle arrest but not muscle differentiation in the presence of dominant negative SWI/SNF chromatin remodeling enzymes. J. Biol. Chem. 276, 41486–41491 (2001).
Salma, N., Xiao, H., Mueller, E. & Imbalzano, A. N. Temporal recruitment of transcription factors and SWI/SNF chromatin-remodeling enzymes during adipogenic induction of the peroxisome proliferator-activated receptor gamma nuclear hormone receptor. Mol. Cell Biol. 24, 4651–4663 (2004).
Young, D. W. et al. SWI/SNF chromatin remodeling complex is obligatory for BMP2-induced, Runx2-dependent skeletal gene expression that controls osteoblast differentiation. J. Cell. Biochem. 94, 720–730 (2005).
Krasteva, V., Crabtree, G. R. & Lessard, J. A. The BAF45a/PHF10 subunit of SWI/SNF-like chromatin remodeling complexes is essential for hematopoietic stem cell maintenance. Exp. Hematol. 48, 58–71.e15 (2017).
Busby, T. et al. Baf45a mediated chromatin remodeling promotes transcriptional activation for osteogenesis and odontogenesis. Front Endocrinol. (Lausanne) 12, 763392 (2021).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013).
Zhang, M. et al. Downregulation of DNA methyltransferase-3a ameliorates the osteogenic differentiation ability of adipose-derived stem cells in diabetic osteoporosis via Wnt/beta-catenin signaling pathway. Stem Cell Res. Ther. 13, 397 (2022).
Wang, L. et al. TET enzymes regulate skeletal development through increasing chromatin accessibility of RUNX2 target genes. Nat. Commun. 13, 4709 (2022).
Sepulveda, H., Villagra, A. & Montecino, M. Tet-mediated DNA demethylation is required for SWI/SNF-dependent chromatin remodeling and histone-modifying activities that trigger expression of the Sp7 osteoblast master gene during mesenchymal lineage commitment. Mol. Cell Biol. 37, e00177–17 (2017).
Cakouros, D. et al. Specific functions of TET1 and TET2 in regulating mesenchymal cell lineage determination. Epigenet. Chromatin 12, 3 (2019).
Lindsey, R. C., Cheng, S. & Mohan, S. Vitamin C effects on 5-hydroxymethylcytosine and gene expression in osteoblasts and chondrocytes: Potential involvement of PHD2. PLoS One 14, e0220653 (2019).
Blayney, J. W. et al. Super-enhancers include classical enhancers and facilitators to fully activate gene expression. Cell (2023).
Paschalis, E. P., Gamsjaeger, S., Condon, K., Klaushofer, K. & Burr, D. Estrogen depletion alters mineralization regulation mechanisms in an ovariectomized monkey animal model. Bone 120, 279–284 (2019).
Mitxitorena, I., Infante, A., Gener, B. & Rodríguez, C. I. Suitability and limitations of mesenchymal stem cells to elucidate human bone illness. World J. Stem Cells 11, 578–593 (2019).
Cheng, H. et al. Development of nanomaterials for bone-targeted drug delivery. Drug Discov. Today 22, 1336–1350 (2017).
Baron, R. & Gori, F. Targeting WNT signaling in the treatment of osteoporosis. Curr. Opin. Pharm. 40, 134–141 (2018).
Wei, H. et al. Identification of fibroblast activation protein as an osteogenic suppressor and anti-osteoporosis drug target. Cell Rep. 33, 108252 (2020).
Behrens, A. et al. Impaired intervertebral disc formation in the absence of Jun. Development 130, 103–109 (2003).
Eferl, R. et al. The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J. 23, 2789–2799 (2004).
Brault, V. et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264 (2001).
Du, Y., Liu, Y., Zhou, Y. & Zhang, P. Knockdown of CDC20 promotes adipogenesis of bone marrow-derived stem cells by modulating β-catenin. Stem Cell Res. Ther. 13, 443 (2022).
Pannone, G. et al. Expression of beta-catenin, cadherins and P-Runx2 in fibro-osseous lesions of the Jaw: Tissue microarray study. Biomolecules 12, 587 (2022).
Pang, X. et al. Sox9CreER-mediated deletion of β-catenin in palatal mesenchyme results in delayed palatal elevation accompanied with repressed canonical Wnt signaling and reduced actin polymerization. Genesis 59, e23441 (2021).
Gutierrez, S. et al. CCAAT/enhancer-binding proteins (C/EBP) beta and delta activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J. Biol. Chem. 277, 1316–1323 (2002).
Hata, K. et al. A CCAAT/enhancer binding protein beta isoform, liver-enriched inhibitory protein, regulates commitment of osteoblasts and adipocytes. Mol. Cell Biol. 25, 1971–1979 (2005).
Nam, B. et al. TGFβ1 suppressed matrix mineralization of osteoblasts differentiation by regulating SMURF1-C/EBPβ-DKK1 axis. Int. J. Mol. Sci. 21, 9771 (2020).
Smink, J. J. et al. Transcription factor C/EBPbeta isoform ratio regulates osteoclastogenesis through MafB. EMBO J. 28, 1769–1781 (2009).
Tominaga, H. et al. CCAAT/enhancer-binding protein beta promotes osteoblast differentiation by enhancing Runx2 activity with ATF4. Mol. Biol. Cell 19, 5373–5386 (2008).
Iyer, V. V., Kadakia, T. B., McCabe, L. R. & Schwartz, R. C. CCAAT/enhancer-binding protein-beta has a role in osteoblast proliferation and differentiation. Exp. Cell Res. 295, 128–137 (2004).
Henriquez, B. et al. C/EBPβ binds the P1 promoter of the Runx2 gene and up-regulates Runx2 transcription in osteoblastic cells. J. Cell Physiol. 226, 3043–3052 (2011).
Hassan, M. Q. et al. Dlx3 transcriptional regulation of osteoblast differentiation: temporal recruitment of Msx2, Dlx3, and Dlx5 homeodomain proteins to chromatin of the osteocalcin gene. Mol. Cell Biol. 24, 9248–9261 (2004).
Li, D., Yuan, Q., Xiong, L., Li, A. & Xia, Y. The miR-4739/DLX3 axis modulates bone marrow-derived mesenchymal stem cell (BMSC) osteogenesis affecting osteoporosis progression. Front Endocrinol. (Lausanne) 12, 703167 (2021).
Zhao, N. et al. Senescence: novel insight into DLX3 mutations leading to enhanced bone formation in Tricho-Dento-Osseous syndrome. Sci. Rep. 6, 38680 (2016).
Duverger, O. et al. In vivo impact of Dlx3 conditional inactivation in neural crest-derived craniofacial bones. J. Cell Physiol. 228, 654–664 (2013).
Shirakabe, K., Terasawa, K., Miyama, K., Shibuya, H. & Nishida, E. Regulation of the activity of the transcription factor Runx2 by two homeobox proteins, Msx2 and Dlx5. Genes Cells 6, 851–856 (2001).
Robledo, R. F., Rajan, L., Li, X. & Lufkin, T. The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev. 16, 1089–1101 (2002).
Kim, C. Y. et al. Dlx5 represses the transcriptional activity of PPARγ. Biol. Pharm. Bull. 44, 1303–1308 (2021).
You, L. et al. Dysbacteriosis-derived lipopolysaccharide causes embryonic osteopenia through retinoic-acid-regulated DLX5 expression. Int. J. Mol. Sci. 21, 2518 (2020).
Rattanasopha, S. et al. Absent expression of the osteoblast-specific maternally imprinted genes, DLX5 and DLX6, causes split hand/split foot malformation type I. J. Med. Genet. 51, 817–823 (2014).
Sato, M. et al. Transcriptional regulation of osteopontin gene in vivo by PEBP2alphaA/CBFA1 and ETS1 in the skeletal tissues. Oncogene 17, 1517–1525 (1998).
Itoh, T., Takeda, S. & Akao, Y. MicroRNA-208 modulates BMP-2-stimulated mouse preosteoblast differentiation by directly targeting V-ets erythroblastosis virus E26 oncogene homolog 1. J. Biol. Chem. 285, 27745–27752 (2010).
Itoh, T., Ando, M., Tsukamasa, Y. & Akao, Y. Expression of BMP-2 and Ets1 in BMP-2-stimulated mouse pre-osteoblast differentiation is regulated by microRNA-370. FEBS Lett. 586, 1693–1701 (2012).
McLarren, K. W. et al. The mammalian basic helix loop helix protein HES-1 binds to and modulates the transactivating function of the runt-related factor Cbfa1. J. Biol. Chem. 275, 530–538 (2000).
Ishibashi, M. et al. Targeted disruption of mammalian hairy and Enhancer of split homolog-1 (HES-1) leads to up-regulation of neural helix-loop-helix factors, premature neurogenesis, and severe neural tube defects. Genes Dev. 9, 3136–3148 (1995).
Zamurovic, N., Cappellen, D., Rohner, D. & Susa, M. Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J. Biol. Chem. 279, 37704–37715 (2004).
Zeng, Z., Yin, X., Zhang, X., Jing, D. & Feng, X. Cyclic stretch enhances bone morphogenetic protein-2-induced osteoblastic differentiation through the inhibition of Hey1. Int. J. Mol. Med. 36, 1273–1281 (2015).
Kusuyama, J. et al. BMP9 prevents induction of osteopontin in JNK-inactivated osteoblasts via Hey1-Id4 interaction. Int J. Biochem. Cell Biol. 116, 105614 (2019).
Galceran, J., Sustmann, C., Hsu, S. C., Folberth, S. & Grosschedl, R. LEF1-mediated regulation of Delta-like1 links Wnt and Notch signaling in somitogenesis. Genes Dev. 18, 2718–2723 (2004).
Sowa, H. et al. Menin is required for bone morphogenetic protein 2- and transforming growth factor beta-regulated osteoblastic differentiation through interaction with Smads and Runx2. J. Biol. Chem. 279, 40267–40275 (2004).
Troka, I. et al. Effect of menin deletion in early osteoblast lineage on the mineralization of an in vitro 3D osteoid-like dense collagen gel matrix. Biomimetics (Basel) 7 (2022).
Abi-Rafeh, J. et al. Genetic deletion of menin in mouse mesenchymal stem cells: An experimental and computational analysis. JBMR 6, e10622 (2022).
Lee, S. et al. Deletion of Menin in craniofacial osteogenic cells in mice elicits development of mandibular ossifying fibroma. Oncogene 37, 616–626 (2018).
Liu, P. et al. Loss of menin in osteoblast lineage affects osteocyte-osteoclast crosstalk causing osteoporosis. Cell Death Differ. 24, 672–682 (2017).
Satokata, I. et al. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet. 24, 391–395 (2000).
Ichida, F. et al. Reciprocal roles of MSX2 in regulation of osteoblast and adipocyte differentiation. J. Biol. Chem. 279, 34015–34022 (2004).
Liu, F., Liang, Y. & Lin, X. MiR-151b inhibits osteoblast differentiation via downregulating Msx2. Connect. Tissue Res. 63, 112–123 (2022).
Cao, Y. et al. Osterix, a transcription factor for osteoblast differentiation, mediates antitumor activity in murine osteosarcoma. Cancer Res. 65, 1124–1128 (2005).
Jeon, M. J. et al. Activation of peroxisome proliferator-activated receptor-gamma inhibits the Runx2-mediated transcription of osteocalcin in osteoblasts. J. Biol. Chem. 278, 23270–23277 (2003).
Lecka-Czernik, B. et al. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPARgamma2. J. Cell. Biochem. 74, 357–371 (1999).
Cao, J. et al. Deletion of PPARγ in mesenchymal lineage cells protects against aging-induced cortical bone loss in mice. J. Gerontol. A Biol. Sci. Med. Sci. 75, 826–834 (2020).
Yoshida, C. A. et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 18, 952–963 (2004).
Li, B., Chen, S., Sun, K., Xu, R. & Wu, Y. Genetic analyses identified a SALL4 gene mutation associated with Holt-Oram syndrome. DNA Cell Biol. 37, 398–404 (2018).
Zhang, Y. W. et al. A RUNX2/PEBP2alpha A/CBFA1 mutation displaying impaired transactivation and Smad interaction in cleidocranial dysplasia. Proc. Natl. Acad. Sci. USA 97, 10549–10554 (2000).
Alliston, T., Choy, L., Ducy, P., Karsenty, G. & Derynck, R. TGF-beta-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J. 20, 2254–2272 (2001).
Kim, S. et al. Stat1 functions as a cytoplasmic attenuator of Runx2 in the transcriptional program of osteoblast differentiation. Genes Dev. 17, 1979–1991 (2003).
Alvandi, Z. & Opas, M. c-Src kinase inhibits osteogenic differentiation via enhancing STAT1 stability. PLoS One 15, e0241646 (2020).
Mizuno, N. et al. Optineurin regulates osteoblastogenesis through STAT1. Biochem. Biophys. Res. Commun. 525, 889–894 (2020).
Mouillé, M. et al. SATB2-associated syndrome: characterization of skeletal features and of bone fragility in a prospective cohort of 19 patients. Orphanet J. Rare Dis. 17, 100 (2022).
Kegelman, C. D. et al. YAP and TAZ mediate osteocyte perilacunar/canalicular remodeling. J. Bone Min. Res. 35, 196–210 (2020).
Kegelman, C. D. et al. Skeletal cell YAP and TAZ combinatorially promote bone development. FASEB J. 32, 2706–2721 (2018).
Kegelman, C. D. et al. YAP and TAZ promote periosteal osteoblast precursor expansion and differentiation for fracture repair. J. Bone Min. Res. 36, 143–157 (2021).
Xiong, J., Almeida, M. & O’Brien, C. A. The YAP/TAZ transcriptional co-activators have opposing effects at different stages of osteoblast differentiation. Bone 112, 1–9 (2018).
Acknowledgements
We thank Ms. Abigail McVicar for her excellent assistance with the editing of this paper. We apologize to the many researchers whose important primary papers could not be cited in this review due to space limitations. This work was supported by the National Institutes of Health (AR-070135 and AG-056438 to W.C., and AR075735, AR074954, DE023813, and DE028264 to Y.P.L).
Author information
Authors and Affiliations
Contributions
S.Z., W.C. and Y.P.L. discussed and conceived the framework of the manuscript. S.Z., W.C. and Y.P.L. searched and collected papers related to the topic. S.Z., W.C. and Y.P.L. drafted the manuscript. S.Z. and Y.P.L draw the figures and tables. S.Z., A.M., Y.P.L. and W.C. revised the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, S., Chen, W., Masson, A. et al. Cell signaling and transcriptional regulation of osteoblast lineage commitment, differentiation, bone formation, and homeostasis. Cell Discov 10, 71 (2024). https://doi.org/10.1038/s41421-024-00689-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41421-024-00689-6
- Springer Nature Singapore Pte Ltd.