
Abstract
Lung cancer is a common malignant tumor that occurs in the human body and poses a serious threat to human health and quality of life. The existing treatment methods mainly include surgical treatment, chemotherapy, and radiotherapy. However, due to the strong metastatic characteristics of lung cancer and the emergence of related drug resistance and radiation resistance, the overall survival rate of lung cancer patients is not ideal. There is an urgent need to develop new treatment strategies or new effective drugs to treat lung cancer. Ferroptosis, a novel type of programmed cell death, is different from the traditional cell death pathways such as apoptosis, necrosis, pyroptosis and so on. It is caused by the increase of iron-dependent reactive oxygen species due to intracellular iron overload, which leads to the accumulation of lipid peroxides, thus inducing cell membrane oxidative damage, affecting the normal life process of cells, and finally promoting the process of ferroptosis. The regulation of ferroptosis is closely related to the normal physiological process of cells, and it involves iron metabolism, lipid metabolism, and the balance between oxygen-free radical reaction and lipid peroxidation. A large number of studies have confirmed that ferroptosis is a result of the combined action of the cellular oxidation/antioxidant system and cell membrane damage/repair, which has great potential application in tumor therapy. Therefore, this review aims to explore potential therapeutic targets for ferroptosis in lung cancer by clarifying the regulatory pathway of ferroptosis. Based on the study of ferroptosis, the regulation mechanism of ferroptosis in lung cancer was understood and the existing chemical drugs and natural compounds targeting ferroptosis in lung cancer were summarized, with the aim of providing new ideas for the treatment of lung cancer. In addition, it also provides the basis for the discovery and clinical application of chemical drugs and natural compounds targeting ferroptosis to effectively treat lung cancer.
Similar content being viewed by others

Facts
-
Ferroptosis is a type of programmed cell death that differs from the traditional cell death pathways such as apoptosis, necrosis, pyroptosis etc.
-
The regulatory mechanism of ferroptosis is mainly related to iron metabolism, lipid metabolism and redox dynamic equilibrium.
-
Ferroptosis has considerable research value and application prospect for the treatment of lung cancer.
-
It is a promising direction for the future treatment of lung cancer to find chemical drugs and natural compounds that target ferroptosis.
Open questions
-
What are the characteristics of ferroptosis?
-
What is the link between ferroptosis and lung cancer?
-
What are the chemical and natural drugs that target ferroptosis? How do they modulate ferroptosis and thus exert their anti-lung cancer effects?
Introduction
Lung cancer is a common malignant tumor occurring in the human body, mainly originating in the trachea, bronchus, and lung. It is mainly divided into squamous cell carcinoma, adenocarcinoma, small cell carcinoma, and large cell carcinoma. According to the Global Cancer Statistics 2020, lung cancer is not only the most commonly diagnosed cancer after breast cancer, but also the leading cause of cancer death [1], In China, the death rate of lung cancer has remained the first from 2015 to 2020 [2, 3]. However, the cause of lung cancer is not completely clear up to now, it is mostly related to living habits, environment, heredity, and many other factors [4]. As there are no obvious pathological symptoms in the early stage of lung cancer, most of them are respiratory symptoms such as cough, dyspnea, chest pain, etc. [5], which leads to lung cancer patients easy to miss the optimal treatment time. At present, common treatments for lung cancer include surgery, radiotherapy, chemotherapy, and immunotherapy [6]. However, due to the special invasiveness of lung cancer [7, 8], as well as the dependence of appellate therapy on patients’ physical conditions and family conditions, the prognosis of lung cancer is poorly treated. At the same time, with the emergence of resistance of lung cancer cells to some therapeutic means and the emergence of different degrees of resistance to a variety of therapeutic drugs, the therapeutic effect of lung cancer is often unsatisfactory. Therefore, it is urgent to find and explore new therapeutic drugs and targets for lung cancer [9, 10].
Programmed cell death (PCD) is a cell death process mediated by molecular programs regulated by specific genes in cells, and plays an important role in the normal development process and homeostasis maintenance of multicellular organisms. To date, the most well-studied PCD includes apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy, among others [11]. Ferroptosis is a novel form of PCD first proposed in 2012, which is regulated by genes in the same way as many PCD modalities and results from the interaction of multiple cellular signaling pathways [12]. Its characteristics mainly include smaller mitochondria, increased membrane density, reduced cristae, no obvious morphological changes in the nucleus, increased lipid peroxidation, increased ROS, and changes in some characteristic genes (such as GPX4, TFR1, etc.) [13]. However, it differs from other forms of PCD in that ferroptosis is based on divalent iron and leads to cell death by promoting lethal accumulation of intracellular lipid peroxides [12]. Interestingly, cancer cells are more iron-addicted than normal cells, because iron is one of the essential elements of the body. These cancer cells need more iron during the production process, and the imbalance of iron metabolism can increase the risk of cancer and promote tumor growth [14]. As a consequence, by promoting ferroptosis in lung tumors, the development of lung cancer can be inhibited, and at the same time, the drug resistance and radiation resistance of lung cancer can be resisted to a certain extent [15]. And this may be just right to become a breakthrough in the current problem of lung cancer treatment facing. Therefore, this review will further explore the potential therapeutic targets of ferroptosis in lung cancer and summarize the existing drugs for the treatment of ferroptosis in lung cancer, in order to provide a theoretical basis for further research of ferroptosis in lung cancer.
Ferroptosis
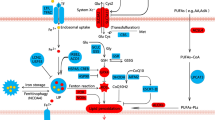
Cell death is an inevitable and irreversible process of life in the body [16, 17]. The existing cell death pathways include apoptosis, necroptosis, autophagy, ferroptosis, pyroptosis, and necrosis [18]. The concept of ferroptosis was first proposed in 2012 by Scott J Dixon et al., and it is iron-dependent and distinct from other forms of cell death [12]. When ferroptosis occurs, there is no cell shrinkage, chromatin condensation, formation of apoptotic bodies and disintegration of the cytoskeleton, which are the traditional phenomena of apoptosis [19, 20]. It is mainly manifested as mitochondrial morphological changes under the electron microscope, such as mitochondrial shrinkage, cristal reduction, membrane density increase, outer membrane rupture, etc. [12, 13]. The biochemical characteristics of ferroptosis are mainly manifested as the accumulation of intracellular iron, the production of reactive oxygen species (ROS), the increase of lipid peroxides, and the decreased expression of glutathione peroxidase 4 in antioxidant system (glutathione system) [21]. The regulatory mechanism of ferroptosis is mainly related to iron metabolism, lipid metabolism and redox dynamic equilibrium (Fig. 1).

TF transferrin, TFR1 transferrin receptor, SLC40A1 solute carrier family 40 member 1, DMT1 solute carrier family 11 member 2, STEAP3 STEAP3 metalloreductase, NCOA4 nuclear receptor coactivator 4, ROS reactive oxygen species, AA arachidonic acid, Ada adrenic acid, CoA coenzyme A, PE phosphoethanolamine, PE-OOH phosphatidylethanolamine hydroperoxide, PE-OH alcohol phospholipid hydroxide, ACLS4 acyl-CoA synthetase long chain family member 4, LPCAT3 lysophosphatidylcholine acyltransferase 3, ALOX15 arachidonate 15-lipoxygenase, GPX4 glutathione peroxidase 4, System Xc− cystine/glutamate antiporter system, Cys2 cystine, Glu glutamate, Cys cysteine, Gly glycine, SLC3A2 solute carrier family 3, member 2, SLC7A11 solute carrier family 7 (L-type amino acid transporter), member 11, GCL glutamate-cysteine ligase, γGC gamma-glutamylcysteine, GSS glutathione synthetase, GSH glutathione, GSSG oxidized glutathione, CoQ10 coenzyme Q10, FSP1 ferroptosis inhibitor protein 1, CoQH2 ubiquinol.
Iron overload
As a common trace element, iron is distributed in the liver, spleen and bone marrow of the human body, which is closely related to the hematopoietic function of body and is necessary to maintain the normal life activities of human [22, 23]. Most iron in the body is obtained from food and then absorbed by active transport in the duodenum and upper quarter of the small intestine. The absorbed Fe2+ is oxidized to Fe3+ in epithelial cells of small intestine, which is then bound to transferrin (TF) in plasma and transported to various sites of utilization or storage [24,25,26]. Iron is involved in oxygen transport, electron transfer reaction in mitochondria, DNA synthesis and other processes [27]. And all of these processes are based on iron homeostasis [28]. Conversely, when one of the components of iron uptake, storage, utilization and efflux is interfered [29], such as increased degradation of ferritin induced by nuclear receptor coactivator 4 (NCOA4) or inhibition of the iron export protein SLC40A1 [30, 31], intracellular iron homeostasis will be affected. Subsequently, with the increase of Fe2+, it will react with hydrogen peroxide (H2O2) in a Fenton reaction and produce large amounts of ROS, which is harmful to cells [32, 33], thereby breaking the redox balance in cells and causing oxidative stress [34]. Under normal circumstances, oxygen-free radical reaction and lipid peroxidation reaction are in a state of coordination and dynamic balance. When excessive ROS occurs, the dynamic balance of the two will be disordered and dysregulated, which will cause metabolic disorders and decreased immune function, form a chain reaction of oxygen free radicals, start lipid peroxidation reaction, and thus cause ferroptosis of cells [35, 36].
Lipid peroxidation
The cell membrane is mainly composed of lipids, proteins, and sugars, which are essential for maintaining intracellular and extracellular stability, regulating the movement of substances into and out of cells, and conducting intercellular signal transduction [37]. Lipid peroxide (LPO) is a product of the reaction of oxygen free radicals with polyunsaturated fatty acids (PUFA), and it is present in extremely low amounts under normal conditions [38]. However, when intracellular ROS increases, it will combine with the polyunsaturated fatty acid side chains of phospholipids, enzymes or membrane receptors, and lipid peroxidation will occur to generate large amounts of LPO, which will destroy the normal fluidity and permeability of cell membrane, leading to changes in cell structure and function, and then induce cell death [39, 40]. Arachidonic acid lipoxygenase 15 (ALOX15), acyl-coa synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are key enzymes in lipid metabolism and are required for ferroptosis. Acyls-arachidonoyl (AA) and adrenoyl (AdA) were firstly catalyzed by ACSL4 to generate acyl Co-A derivatives [41, 42]. LPCAT3 then esterified these derivatives to phosphatidylethanolamines (AA-PE and AdA-PE) [43]. Lipoxygenases (LOX) are a family of iron-containing enzymes [44], which can oxidize PUFA by phosphorylating enzyme kinase G2 (PHKG2) -dependent iron pools through the diallyl site [45]. Afterwards, AA-PE and AdA-PE catalyzed by LPCAT3 are oxidized by ALOX15 to generate LPO, leading to ferroptosis [46]. In addition, some studies have shown that cytochrome P450 reductase (POR) and cytochrome B5 reductase (CYB5R1) can also promote ferroptosis by catalyzing phospholipid peroxidation. POR may promote lipid peroxidation by accelerating the cycling between Fe2+ and Fe3+ in heme [47]. Meanwhile, POR and CYB5R1 can also transfer electrons from NAD(P)H to oxygen to generate hydrogen peroxide, which subsequently generates reactive hydroxyl radicals through the Fenton reaction and further promotes the production of LPO, destroying the integrity of the cell membrane [48]. However, the specific mechanism of action is still unclear and needs to be further explored.
Imbalance of the antioxidant system
Glutathione (GSH), a tripeptide containing sulfhydryl group consisting of glutamate (Glu), cysteine (Cys) and glycine (Gly), has antioxidant and free radical scavenging ability [49]. The cystine/glutamate antiporter system (system xc−) is an amino acid transporter expressed at the plasma membrane and consists of a light chain, xCT, and a heavy chain, 4F2 [50]. It can export Glu and import cystine (Cys2) in a 1:1 ratio, then reduce intracellular Cys2 to Cys and further synthesize GSH [51]. Glutathione peroxidase 4 (GPX4) is a key enzyme in the glutathione antioxidant system. It can use GSH as a substrate to convert LPO into lipid alcohols that are harmless to the cell membrane, thereby inhibiting the lipid peroxidation caused by the increase of ROS [52, 53]. However, the generation of substrate GSH is tightly regulated by system xc−. When Cys2 is deprived and Cys uptake is inhibited, GSH conversion is insufficient, which allows lipid peroxidation products to accumulate and lead to ferroptosis [54, 55]. However, inhibiting GPX4, inhibiting system xc−, or blocking GPX4 synthesis can all affect the intracellular redox balance, reduce the antioxidant capacity of cells, and reduce the elimination of lipid peroxides [56, 57]. At the same time, non-canonical GPX4 regulatory pathways can also regulate the occurrence of ferroptosis. Studies have shown that ferroptosis inhibitor protein 1 (FSP1) has a parallel effect with GPX4, which can reduce coenzyme Q10 (CoQ10) to ubiquinol (CoQH2) on the cell membrane. Then CoQH2 inhibits ferroptosis by trapping free radicals and acting as an antioxidant [58, 59]. Moreover, FSP1 can also reduce vitamin K to hydroquinone to play a role in trapping free radicals, thereby protecting cells from ferroptosis [60]. Further studies showed that mitochondrial dihydroorotate dehydrogenase (DHODH) also runs in parallel with mitochondrial GPX4 to oxidify dihydroorotate to orotate and reduce panquinol to pantanol, thus acting as an anti-ferroptosis protein in the inner mitochondrial membrane [61]. In addition, tetrahydrobiopterin (BH4), which is produced by guanosine triphosphate (GTP) conversion and synthesized using GTP cyclohydrolase 1 (GCH1) as the rate-limiting enzyme, has also been shown to possess endogenous antioxidant effects [62]. BH4 can act against ferroptosis by selectively preventing the depletion of phospholipids with two polyunsaturated fatty acyl groups [63, 64]. Therefore, with the deepening of research, the regulatory pathways of ferroptosis in the antioxidant system are not limited to GSH-dependent antioxidant pathways, but also can play a role in regulating ferroptosis through GSH-independent antioxidant pathways, which means that there may be more regulatory mechanisms related to ferroptosis waiting for us to further explore.
Targets of ferroptosis in lung cancer
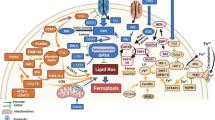
Targeting ferroptosis in lung cancer can inhibit the development process and metastasis of lung cancer, and also overcome the drug resistance and radiation resistance of lung cancer cells to a certain extent. According to the current research content, the targets of ferroptosis in lung cancer mainly focus on the regulation of antioxidant pathways, among which GPX4 and SLC7A11 are the core targets of the GSH-dependent antioxidant pathway. In addition, many studies have suggested that many non-coding RNAs play an important role in the regulation of ferroptosis in lung cancer. Furthermore, ferroptosis in lung cancer is also closely related to autophagy and ubiquitin-proteasome pathways. Therefore, we sorted out the relevant targets of ferroptosis in lung cancer therapy and their interactions, as shown in Fig. 2.

GPX4 glutathione peroxidase 4, SLC3A2 solute carrier family 3, member 2, SLC7A11 solute carrier family 7 (L-type amino acid transporter), member 11, RBMS1 RNA-binding protein, SOX2 stem cell transcriptional factor, AKR1B1 aldo-keto reductase family 1 member B1, STAT3 activator of transcription 3, METTL3 methyltransferase-like 3, CCT3 chaperonin containing TCP1 subunit 3, Nrf2 nuclear factor erythroid 2-related factor 2, GCL glutamate-cysteine ligase, γGC gamma-glutamylcysteine, MIB1 E3 ubiquitin ligase, p53 tumor suppressor protein, SAT1 spermidine/spermine N1-acetyltransferase 1, GLS2 glutaminase 2, FSP1 ferroptosis inhibitor protein 1, NFS1 NFS1 cysteine desulfurase, STYK1 Serine-threonine tyrosine kinase 1, LSH Lymph-specific helicase, GINS4 a member of GINS family proteins, Yap Yes associated transcriptional regulator, TFR1 transferrin receptor, FTH ferritin heavy chain, FTL ferritin light chain, BECN1 ecombinant human autophagy effector protein, USP7 ubiquitin-specific processing protease 7, USP11 ubiquitin-specific processing protease 11, CRL4DCAF8 cullin-RING E3 ligase complex, YTHDC2 N6-methyladenosine (m6A) RNA binding protein, A20 ubiquitin-regulated enzyme, SENP1 Small ubiquitin-like modifier specific protease 1, Notch3 the third subtype of Notch family, PRDX6 peroxiredoxin 6, CERK ceramide kinaseCeramide kinase, VDAC voltage-dependent anion-selective channel, MMP mitochondrial membrane potential, miRNA MicroRNA, circRNA Circular RNA, lncRNA Long non-coding RNA, METTL3 methyltransferase-like 3, FABP3 fatty acid binding protein 3, ELAVL1 RNA-binding protein, CBS cystathionine β-synthase, G3BP1 Ras GTPase activating protein binding protein 1.
GPX4
GPX4, also known as phospholipid hydroperoxide glutathione peroxidase (PHGPx), is the fourth member of the selenium-containing GPX family and is capable of removing lipid peroxides that accumulate in cells [65]. Due to its potent antioxidant activity, GPX4 exerts a negative effect on ferroptosis in a variety of cancer cells. For example, when GPX4 is overexpressed, HT-1080 cells are resistant to RSL3, whereas the knockdown of GPX4 makes HT-1080 cells sensitive to RSL3 [53]. In the study of lung cancer, the immunohistochemical staining results of clinical lung adenocarcinoma samples compared with normal alveolar epithelial cells and bronchial epithelial cells showed that GPX4 was strongly positive in lung adenocarcinoma tissues. And by targeting GPX4 can induce ferroptosis in lung adenocarcinoma cells and reverse their resistance to epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) [66]. Meanwhile, targeting GPX4 can also promote lapatinib (Lap) -induced ferroptosis in drug-resistant non-small cell lung cancer cells [67]. In addition to lapatinib, GPX4 was also positively correlated with chemoresistance to anticancer drugs L-685458, palbociclib, and topotecan [68, 69]. At the same time, inhibition of GPX4 expression can partially reduce the radiation resistance of lung cancer cells and promote the efficacy of radiotherapy [70]. In conclusion, GPX4 can affect the development of lung cancer by regulating ferroptosis. On the other hand, it is also related to the drug resistance and radiation resistance of lung cancer, and has great significance in the research of ferroptosis in lung cancer.
SLC7A11
Cystine transporter solute carrier family 7 member 11 (SLC7A11) encodes the light chain subunit xCT of system xc− and promotes GSH synthesis by mediating Cys uptake and Glu release [51, 71]. SLC7A11 is overexpressed in multiple human cancers and promotes tumor survival [72,73,74]. Study has shown that controlling H3K9me3 demethylation in the promoter region of SLC7A11 can increase SLC7A11 expression, inhibit ferroptosis, and promote osteosarcoma cell lung metastasis [75]. Through RNAseq and miRNAseq data analysis of lung adenocarcinoma (LUAD) in the Cancer Genome Atlas (TCGA) data, it was found that SLC7A11 is a potential biomarker for the prognosis of patients with LUAD [76]. Further studies showed that SLC7A11 regulated ferroptosis in lung cancer mainly by regulating metabolic demand [77]. An RNA-binding protein, RBMS1, interacts with eIF3d to bridge the 3’- and 5’-UTR of SLC7A11, facilitating its translation. When RBMS1 was ablated, SLC7A11 translation was inhibited and ferroptosis was induced [78]. Moreover, SLC7A11 expression is positively correlated with the stem cell transcription factor SOX2, which showed a high binding affinity for sequences with CTTTGTT located on the SLC7A11 promoter. When SOX2 is overexpressed, endogenous SLC7A11 expression is enhanced at both mRNA and protein levels. This will maintain a certain level of Cys in the cells to promote ferroptosis resistance of lung cancer cells [79]. It has also been shown that aldo-keto reductase family 1 member B1 (AKR1B1) interacts with signal transducer and activator of transcription 3 (STAT3) to up-regulate SLC7A11, which leads to increased Cys2 uptake in lung cancer cell lines and xenograft mouse models, and further promotes lung cancer cell resistance [80]. Methyltransferase-like 3 (METTL3) can directly target the m6 modification of SLC7A11, which enables m6A reading protein YTHDF1 to recognize the mRNA of SLC7A11, and then promote the expression of SLC7A11 by enhancing its stability and translation, thereby inhibiting ferroptosis [81]. Meanwhile, the chaperone protein containing TCP1 subunit 3 (CCT3) can also inhibit ferroptosis by promoting SLC7A11 expression in lung cancer cells [82]. In conclusion, the inhibition of ferroptosis caused by SLC7A11 up-regulation is very unfavorable for the treatment of lung cancer, but it also reflects its important regulatory role in the ferroptosis of lung cancer. Therefore, it is very beneficial to find more upstream targets of SLC7A11 regulation to promote ferroptosis of lung cancer.
Nrf2
Nuclear factor erythroid 2-related factor 2 (Nfr2), a member of the Cap’n’Collar (CNC) subfamily of transcription factors, is a key factor in cellular oxidative stress [83]. Under normal conditions, Keel-like ECH-associated protein 1 (KEAP1) binds to Nrf2 and undergoes ubiquitin-mediated degradation to maintain the Nrf2 level at a low level [84]. However, when cells are exposed to oxidative stress, the nuclear accumulation and constitutive activation of Nrf2 will increase, which will promote cancer progression and is also an important cause of tumor drug resistance [85, 86]. Inhibition of Nrf2 expression or activity can increase the sensitivity of tumors to anticancer drugs [87]. It also has a strong correlation with ferroptosis in lung cancer. Study has shown that Nrf2 is highly expressed in cancer tissues of NSCLC patients and can regulate the sensitivity of NSCLC cells to ferroptosis induced by Cys2 deprivation [88]. Nrf2 promotes γ-glutamyl-peptide (γGC) synthesis and inhibits ferroptosis caused by Cys2 deficiency by regulating the catalytic subunit of glutamate-cysteine ligase (GCLC) [89]. It can also promote chemoresistance of non-small cell lung cancer brain metastases by targeting GPX4 and binding to the GPX4 promoter region [90]. When Nrf2 is degraded by E3 ubiquitin ligase (MIB1), the antioxidant capacity of lung cancer cells is weakened, and the sensitivity of lung cancer cells to ferroptosis inducers is enhanced [91]. Therefore, inhibiting Nrf2 or promoting its degradation may be beneficial for ferroptosis induction in lung cancer.
ACSL4
Acyl-CoA synthetase long-chain family member 4 (ACSL4), a member of the long-chain acyl-CoA synthetase (ACS) family, is involved in the regulation of arachidonic acid (AA) and eicosapentaenoic acid [41]. ACSLA can specifically esterify arachidonic acid (AA) and epinephrine (AdA) to phosphatidyleolamine (PE) [92]. Based on lipidomics analysis, activated ACSL4 can catalyze the synthesis of lipid organisms containing polyunsaturated fatty acids (PUFA), leading to the accumulation of lipid peroxides, which in turn promotes ferroptosis [93]. When ACSL4 is highly expressed, it can improve the sensitivity of breast cancer to neoadjuvant chemotherapy [94]. Is it also a potential therapeutic target for ferroptosis in lung cancer? Through TCGA database analysis and immunohistochemical staining, ACSL4 is down-regulated in lung adenocarcinoma. When ACSL4 is knocked down in vitro, the survival rate and invasiveness of lung cancer cells are enhanced, on the contrary, overexpression would promote ferroptosis of lung cancer cells [95]. In addition, ACSL4 was positively correlated with erastin-induced ferroptosis in NSCLC cells [96]. When ACSL4 is upregulated, lipid peroxidation products and lethal reactive oxygen species accumulate, thus sensitiating lung cancer cells to ferroptosis [97, 98]. Therefore, targeting ACSL4 is an effective way to promote ferroptosis in lung cancer.
P53
P53 is a tumor suppressor, and the cell signal transduction pathway mediated by p53 plays a crucial role in regulating the normal life activities of cells [99]. Activation of p53 could not directly induce ferroptosis, but it can inhibit the expression of SLC7A11 by promoting nuclear translocation of the deubiquitinase USP7 and then decreasing the occupancy of monoubiquitination of histone H2B on lysine 120 (H2Bub1) in the regulatory region of SLC7A11 gene [100]. Alternatively, P53 enhances the sensitivity of cells to ferroptosis by enhancing the expression of spermidine/spermine N1-acetyltransferase 1 (SAT1) and glutaminase 2 (GLS2) and inhibiting cystine uptake [101,102,103]. Levobupivacaine, a local anesthetic, induces ferroptosis in NSCLC cells by upregulating p53 [104]. Meanwhile, erastin also inhibited the corresponding target gene SLC7A11 by activating p53 to promote ROS accumulation [105]. Furthermore, some researchers have used nano-cascade engineering to construct metal-organic supermolecules with p53-activating peptides and CeO2 nanoparticles, which can activate the p53 signaling pathway and down-regulate the downstream gene GPX4 in vitro and in vivo, thereby inhibiting the development and progression of lung cancer [106]. Therefore, the ferroptosis-promoting effect of targeting p53 in lung cancer should be well utilized.
FSP1
Inhibitor of ferroptosis 1 (FSP1), also known as flavoprotein apoptosis-inducing factor mitochondria-associated 2 (AIFM2), is a glutathione-independent inhibitor of ferroptosis [107]. FSP1 can use reduced coenzyme II (NAD(P)H) to catalyze the regeneration of ubiquinone (CoQ10) and inhibit ferroptosis by reducing ubiquinone to panthol and capturing free radicals that mediate lipid peroxidation [59]. There is a positive correlation between the expression of FSP1 and ferroptosis resistance of lung cancer cells and xenografts [58]. A recent study identified FSP1 as a transcriptional target of Nrf2. When the CoQ-FSP1 axis is inhibited, the sensitivity of KEAP1-deficient lung cancer cells and patient-derived xenograft tumors to radiation is enhanced [108]. And a plasma-activated medium (PAM) also promotes cell death by depleting FSP1 in human lung cancer cells and increasing intracellular lipid ROS [109]. Therefore, FSP1 has a good application prospect in the ferroptosis of lung cancer. Inhibition of FSP1 expression or depletion of FSP1 can promote ferroptosis of lung cancer.
NFS1
Iron-sulfur cluster synthase (NFS1) is an essential enzyme in eukaryotes to obtain sulfur from Cys for the synthesis of iron-sulfur clusters (ISC) [110]. Study has shown that when ISC synthesis is reduced, iron load is increased, making cancer cells sensitive to ferroptosis [111]. Inhibition of NFS1, which is required for primary lung tumor growth, limits the availability of ISC and strongly activates the iron deficiency response [112]. More recently, it has been suggested that NFS1 inhibition may act synergistically with glutathione (GSH) synthesis inhibition to promote cell death through ferroptosis [113]. An anti-cancer drug, Eprenetapopt, inhibits ISC synthesis by inhibiting cysteine desulfurase activity, thereby inducing ferroptosis and limiting cell proliferation [114]. In conclusion, affecting the synthesis of ISC by inhibiting NFS1 and thus altering the sensitivity of cells to ferroptosis is a potential therapeutic tool to promote ferroptosis in lung cancer, but the precise mechanism remains to be further investigated.
STYK1/NOK
Serine-threonine tyrosine kinase 1 (STYK1), also known as a novel oncogene with kinase domain (NOK), belongs to the receptor protein tyrosine kinases (RPTKs) subfamily [115], and has a strong ability to promote tumor formation and metastatic capacity [116]. Studies have found that NOK is positively expressed in tumor cells of NSCLC patients, and it is significantly correlated with the degree of lung tumor differentiation and lymphatic metastasis [117]. When STYK1 is overexpressed, it promotes proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) of NSCLC cells [118]. Excitingly, the overexpression of STYK1 in lung cancer cell line SW900 upregulates GPX4 expression, promotes cell proliferation and attenuates ferroptosis-specific mitochondrial abnormalities, while downregulation of GPX4 exacerbates this attenuation without affecting cell proliferation [119]. In summary, the current study highlights that STYK1 expression is upregulated in NSCLC, that overexpression of STYK1 is associated with worse prognosis, that STYK1 is positively correlated with GPX4, and that overexpression of STYK1 inhibits ferroptosis in NSCLC cells, which provides a new opportunity to study the oncogenic mechanisms of STYK1.
LSH
Lymphoid-specific helicase (LSH) is a member of the ATP-dependent chromatin remodeling protein SNF2 helicase family [120], which is involved in a variety of epigenetic regulation including nucleosome remodeling, DNA methylation, and histone modification [121]. LSH, as an oncogene, has been found to be highly expressed in lung cancer tissues, and is positively correlated with the expression of GINS4. Further studies have found that LSH can increase the expression of GINS4 by stabilizing its post-transcriptional mRNA level, thus promoting the progression of lung cancer [122]. In addition, LSH can activate genes related to glucose transporter (GLUTs) and fatty acid dessaturase (FADSs), and affect the metabolism of lung cancer cells. It has been demonstrated that LSH can interact with WDR76 to restrain ferroptosis through activating lipid metabolism-related genes (including GLUT1) and ferroptosis-related genes FADS2 and sterol-coenzyme A desaturase 1 (SCD1), and thus participate in the Warburg effect [123]. In short, the above studies provide ample evidence that LSH, as an oncogene, is participated in ferroptosis and is a possible therapeutic target for lung cancer due to its key effect in ferroptosis. However, current studies are still limited and more experiments are needed in the future to further demonstrate whether LSH has a regulatory effect on ferroptosis in lung cancer.
Yap
Yes-associated protein (Yap) is the key transcription factor of the Hippo tumor suppressor pathway, which is closely related to cell growth, tissue homeostasis and organ size [124, 125]. Yap has a significant promoting effect on cancer progression, drug resistance and metastasis of NSCLC [126, 127]. On the contrary, reducing the expression or inhibiting the activity of Yap can inhibit the growth and metastasis of NSCLC tumors [128]. A recent study has shown that Yap is associated with the activation of ferroptosis, Yap can promote an iron environment in cancer cells through stimulating transcription of TEAF-based members of the Transferrin receptor (TFRC) and Acyl-CoA synthetase long chain family 4 (ACSL4) [129, 130]. Activation of the Hippo pathway can damage an iron environment. Surprisingly, the accumulate of endogenous glutamate after system xc− inhibition can determine the sensitivity of ferroptosis by inhibiting Yap in LUAD cells. When GFPT1 is impaired, Yap O-GlcNAcylation and expression are not sustained in LUAD cells. As a result, Hippo pathway like ubiquitination and phosphorylation of YAP are strengthened [130]. Furthermore, Yap can also control intracellular iron levels by affecting the transcription of ferritin heavy chain (FTH) and ferritin light chain (FTL), thereby regulating cell ferroptosis sensitivity [131]. In a word, Yap can not only regulate tumor metastasis and progression on the one hand, but also affect ferroptosis on the other hand. The above studies demonstrate that YAP is crucial in regulating both the initiation and development of ferroptosis, and Yap can be a good candidate target for lung cancer therapy.
The relationship with autophagy
Autophagy is a process that engulfs its own cytoplasmic proteins or organelles and encapsulates them into vesicles, then unites with lysosomes to remove the encapsulated contents [132]. Ferritin is very important for maintaining iron homeostasis in cells. Recent studies have shown that the occurrence of iron death is dependent on autophagy and that many iron death regulators are considered to be potential regulators of autophagy [133]. It has been reported that ROS generated by ferroptosis inducer erastin can induce autophagy, promote ferritin degradation, and induce TFR1 expression, thereby leading to ferroptosis [134]. Moreover, there is increasing evidence that ferroptosis is an autophagy-dependent form of cell death and that either intracellular transition activation or abnormally elevated lysosomal activity can cause the accumulation of intracellular iron ions and lipid peroxides, which in turn promotes ferroptosis. Particularly, ferritin phagocytosis promoted by NCOA4, BECN1-mediated system xc− inhibition, lysosomal membrane penetration induced by STAT3, and chaperon-mediated autophagy of HSP90 related molecules can all play a role in promoting ferroptosis [135]. NCOA4 was identified as a cargo receptor that mediates ferritin phagocytosis and maintains intracellular iron homeostasis [136]. By directly binding to SLC7A11, recombinant human autophagy effector protein (BECN1) inhibits the activity of system xc−, promotes GSH consumption and lipid peroxidation, and induces ferroptosis in cancer cells [137]. In short, the above studies fully demonstrate that there is a connection between the regulation of ferroptosis and autophagy, and the multiple fine regulatory mechanisms between them will be gradually revealed in the future.
The relationship with the ubiquitin-proteasome system
The ubiquitin-proteasome system (UPS) is an important regulatory system for protein degradation in cells. It can affect or regulate a variety of life activities in cells through polyubiquitination of substrate proteins and degradation by the proteasome [138]. There is growing evidence that that the dysregulation of UPS is related to the development and treatment of cancer, and these enzymes can interact with ferroptosis related proteins, thereby mediating the sensitivity of cancer cells to ferroptosis [139, 140]. Pyridinolone palladium complex (PdPT), a broad-spectrum deubiquitinase inhibitor, can promote the death of non-small cell lung cancer cells by inducing the degradation of GPX4 protein [141]. Ubiquitin specific protease 35 (USP35) can directly interact with ferroportin to maintain the stability of ferroportin and prevent iron overload. Knockdown of USP35 can inhibit the progression of lung cancer, promote ferroptosis, and increase the sensitivity of lung cancer cells to cisplatin and paclitaxel chemotherapy [142]. Ubiquitin-specific processing protease 11 (USP11) can interact with Nrf2 to deubiquitinate Nrf2. Conversely, depletion of USP11 inhibited H1299 cell proliferation and induced ferroptosis [143]. What’s more, cullin-RING E3 ligase complex CRL4DCAF8 regulates ferroptosis by controlling the stability of LSH [144]. In conclusion, targeting USP can indirectly regulate ferroptosis by controlling the stability of ferroptosis related proteins, which brings new opportunities and challenges for the treatment of lung cancer. However, due to the complexity of USP regulation, the precise mechanism of its interaction with ferroptosis is still unknown, which requires more future studies to confirm.
Non-coding RNA
Non-coding RNAs refer to the RNAs transcribed from the genome, which can exercise their own biological functions without coding proteins. More and more studies have shown that non-coding RNAs play an important regulatory role in tumor development [145, 146]. In recent years, as researchers have deepened their research on ferroptosis, more and more non-coding RNAs have been proven to regulate ferroptosis in tumor cells [96, 147, 148].
MicroRNAs (miRNAs) are a group of endogenous non-coding RNAs that regulate gene expression, which can regulate the cell cycle, metastasis, angiogenesis, metabolism and apoptosis, and are considered as promising biomarkers in tumor diagnosis and disease prognosis [149, 150]. Many studies have shown that miRNAs are closely related to lung cancer drug resistance. Exosomal miR-4443 can reduce the expression level of methyltransferase-like 3 (METTL3) by targeting METTL3, and increase the level of FSP1 by N6-methyladenosine (m6A) methylation to inhibit ferroptosis and make NSCLC cells resistant to CDDP [151]. At the same time, miR-6077 could protect LUAD cells from cell death induced by CDDP combined with pemetrexed (PEM) by regulating the KEAP1-Nrf2-SLC7A11/NQO1 axis to mediate ferroptosis [147]. When miR-27a-3p is overexpressed, it directly binds to the 3’-UTR of SLC7A11, leading to its inhibition and subsequently reducing the ferroptosis induced by erastin. On the contrary, when miR-27a-3p is inhibited, NSCLC cells become more sensitive to erastin [152]. Moreover, by targeting ferroportin, miR-302a-3p can induce lipid peroxidation and iron overload, inhibit the growth and colony formation of lung cancer cells, and increase their sensitivity to CDDP and paclitaxel chemotherapy [153]. In addition, bioinformatics analysis has been used by some scholars to find that miR-17-5p may lead to ferroptosis-related brain metastasis in lung adenocarcinoma patients by down-regulating HOXA7 [154], but further experiments are lacking to verify this. Taken together, these results suggest that microRNAs may be involved in lung cancer drug resistance by regulating ferroptosis.
Circular RNAs (circRNAs) are a class of single-stranded non-coding RNAs that form circular conformations through non-canonical splicing or back-splicing events, and play a key role in the occurrence and development of a variety of tumors [155, 156]. Many circRNAs can act as microRNA sponges to relieve the inhibitory effect of micrornas on their target genes and increase the expression level of corresponding genes [157, 158]. Some studies have found that circ FOXP1 was significantly overexpressed in the serum of NSCLC patients [159], and the down-regulation of circ FOXP1 can inhibit the proliferation of lung cancer cells [160, 161]. Further studies showed that circ FOXP1 can increase the expression of SLC7A11 by directly sponging miR-520a-5p in lung cancer cells to promote lung cancer tumor growth [148]. Circ DTL is also up-regulated in NSCLC cells, which can play a carcinogenic role by targeting the miR-1287-5p/GPX4 axis. On the contrary, silencing circ DTL can promote the sensitivity of lung cancer cells to chemotherapy drugs and inhibit tumor growth in vivo [162]. And circ P4HB could regulate SLC7A11 by regulating miR-1184 to trigger GSH synthesis, which can protect lung cancer cells from erastin-induced ferroptosis, and promote tumor growth in vivo [163]. In addition, exosomal circRNA 101093 regulates overall arachidonic acid (AA) levels and induces N-arachidonyl taurine (NAT) production through interaction with fatty acid binding protein 3 (FABP3), resulting in desensitization of LUAD cells to ferroptosis [164]. In other words, circRNA can interact with microRNA to regulate the corresponding target of ferroptosis and play an anti-ferroptosis role.
Long non-coding RNAs (lncRNAs) are non-coding RNAs with a length of more than 200 nucleotides, which play an important role in many life activities such as regulating energy metabolism, controlling gene expression and protein synthesis [165, 166]. However, more and more studies have identified lncRNAs as key mediators regulating ferroptosis and iron metabolism in lung cancer [167]. LncRNA NEAT1 can target ACSL4 and inhibit the protein expression of ACSL4, thus regulating the sensitivity of lung cancer cells to ferroptosis [96]. Knockdown of lncRNA OGFRP1 can promote lipid peroxidation and iron accumulation in lung cancer, thereby promoting cell ferroptosis. It regulates lung cancer cell proliferation and ferroptosis mainly by inhibiting miR-299-3p and enhancing solute carrier family 38 member 1 (SLC38A1) expression [168]. LncRNA ASMTL-AS1, which is down-regulated in lung cancer cells, can stimulate ferroptosis in lung cancer cells by binding to U2AF2 and stabilizing spermidine/spermine N1-acetyltransferase 1 (SAT1) [169]. LncRNA P53RRA can bind to Ras GTPase activating protein binding protein 1 (G3BP1) to activate the p53 pathway and retain more p53 in the nucleus, thereby exerting a pro-ferroptosis effect [170]. LncRNA C00336 is up-regulated in lung cancer, and it can bind to the RNA-binding protein ELAVL1 to inhibit ferroptosis. Meanwhile, it can also act as an endogenous sponge for microRNA 6852 to regulate the expression of cystathionine β-synthase (CBS), a ferroptosis surrogate marker [171]. While ectopic expression of lncRNA MT1DP can stabilize miR-365a-3p and downregulate Nrf2, sensitizing A549 and H1299 cells to erastin-induced ferroptosis [172]. LncRNA T-UCR Uc.33 is up-regulated in lung adenocarcinoma patients, which can competitively bind to pri-miR-339, inhibit the production of mature miR-339, and further promote the expression of SCL7A11 [173]. In summary, these results suggest that non-coding RNA plays an important regulatory role in ferritin formation in lung cancer. However, the role of non-coding RNA in regulating ferroptosis in lung cancer and its precise mechanism are still much incompletely understood. Therefore, a full understanding of the regulatory process of non-coding RNA in the process of ferroptosis in the occurrence and metastasis of lung cancer, which is of great significance for further elucidating the mechanism of ferroptosis in the process of lung cancer metastasis.
Other targets
In addition to the regulatory targets described above, there are some ferroptosis targets in lung cancer as follows. N6-methyladenosine (m6A) RNA binding protein YTHDC2 can directly inhibit SLC7A11 on the one hand, and destabilize the mRNA of transcription factor HOXA13 in an m6A dependent manner, thereby inhibiting HOXA13-mediated transcription of SLC3A2, thereby damaging tumor growth. At the same time, it can trigger lipid peroxidation and induce ferroptosis in lung cancer [174]. A20 is a ubiquitin-regulated enzyme, which has considerable application prospect in inflammation and tumors [175]. Small ubiquitin-like modifier (SUMO) specific protease 1(SENP1) can regulate A20 by desumoylation, and then inhibit ferroptosis of lung cancer cells by affecting the interaction between A20 and ACSL4, SLC7A11 [176]. Notch3, the third subtype of the Notch family, can regulate tumor maintenance and affect tumor chemotherapy resistance [177]. Knockdown of Notch3 can increase the ROS level in lung cancer cells, reduce the expression levels of GPX4 and peroxiredoxin 6 (PRDX6), and then induce lipid peroxidation to trigger cell ferroptosis [178]. Ceramide kinase (CERK) inhibits dysregulated voltage-dependent anion-selective channel (VDAC) -mediated mitochondrial function-driven ferroptosis in KRAS mutant lung cancer. Therefore, when CERK is downregulated or inhibited, MMP increases and VDAC modulation occur, sensitizing NSCLC cells with oncogenic KRAS to CDDP [179].
Drugs targeting ferroptosis in lung cancer
Chemical drugs
Currently, there are many drugs for tumor ferroptosis, and most of them inhibit system xc−and GPX4 to promote ferroptosis [53, 54, 180, 181]. The structural formula of the current chemical drugs targeting ferroptosis for lung cancer is shown in Fig. 3. The targets and mechanisms of these chemical drugs regulating ferroptosis for lung cancer are summarized in Fig. 4 and Table 1.

The names, structural formulas, and molecular formulas of these chemical agents that target ferroptosis for lung cancer treatment are shown in the figure.

CDDP Cisplatin, SOR Sorafenib, AF Auranofin, LEV Levobupivacaine, AFC Ammonium Ferric Citrate, PdPT Palladium pyrithione complex, FLD Falnidamol, APAP Acetaminophen, SAHA Vorinostat, IKE Imidazole ketone erastin, ZVI-NP Zero-valent-iron nanoparticle, FTG/L&SMD Multifunctional “ball-rod” Janus nanoparticles, FAF2 fatty acid synthase-associated factor 2, HMOX1 Heme oxygenase 1, GOD glucose oxidase, OH• reactive hydroxyl radical, TrxR thioredoxin reductase 1, DUSP26 Dual-specificity phosphatase 26, GSK3 phospho-glycogen synthase kinase 3, β-TrCP beta-Transducin repeats-containing protein, Nrf2 nuclear factor erythroid 2-related factor 2, p53 tumor suppressor protein, GPX4 glutathione peroxidase 4, System Xc− cystine/glutamate antiporter system, Cys2 cystine, Glu glutamate, Cys cysteine, Gly glycine, GCL glutamate-cysteine ligase, SLC3A2 solute carrier family 3, member 2, SLC7A11 solute carrier family 7 (L-type amino acid transporter), member 11, GSS glutathione synthetase, GSH glutathione, GSSG oxidized glutathione, GSR glutathione reductase, ROS reactive oxygen species.
Cisplatin (CDDP) is a traditional platinum-based chemotherapy drug, existing studies have shown that CDDP can induce ferroptosis when the concentration is higher than 5 μg/mL and the action time is longer than 48 h [182]. Sorafenib (SOR) is a novel multi-targeted oral cancer agent that is approved for the treatment of gastrointestinal stromal tumors and metastatic renal cell carcinoma that do not respond to or are not tolerated by standard therapies [183]. But a recent study has shown that SOR also has therapeutic effects on lung cancer and can promote ferroptosis in lung cancer [184, 185]. Olanprofen (AF), an antirheumatic drug, can promote apoptosis by targeting the thioredoxin reductase 1 (TrxR) system, and according to transcriptome analysis, it is also involved in gene regulation of ferroptosis in lung cancer [186]. Orlistat is currently the only OTC weight-loss drug in the world, which can inhibit the progression of lung cancer by targeting FAF2, significantly inhibit GPX4 expression, and induce lipid peroxidation in cells [187]. Levobupivacaine (LEV), a local anesthetic, can inhibit the expression of GPX4 and SLC7A11 by activating p53, thereby exerting a pro-ferroptosis effect and inhibiting the growth of tumors in vivo [104]. Treatment of A549 and HCC827 cells with the food additive ferric ammonium citrate (AFC) for 24 h resulted in a significant decrease in the expression of cell proliferation-related proteins (Ki67, CDK2 and CCND), autophagy-related proteins (ATG3 and LC3A/B) and ferroptosis negative regulators (GPX4 and FTH1), followed by a decrease in intracellular autophagy and induction of cellular ferroptosis [188]. XAV-939, a Tankyrase inhibitor, has been shown to promote ferroptosis in lung cancer cells by inducing the down-regulation of SLC7A11, and inhibiting the progression of lung cancer by down-regulating lncRNA MIR503HG, sponging miR-1273c and regulating SOX4 expression [189].
Compared with the use of a single drug, the existing research trend is to pay more attention to the combination of drugs, on the one hand, to improve the efficacy, on the other hand, to avoid the occurrence of cell resistance. Although cisplatin is a traditional anticancer drug, it is easy to cause cell resistance, which will lead to unsatisfactory anticancer efficacy [190, 191]. PRLX93936, an elastine analogue, can induce lipid peroxidation and ferroptosis in lung cancer cells when combined with CDPP. At the same time, the combination of them can also target Nrf2/Keap1 to regulate the drug sensitivity of lung cancer cells [192]. The combination of faridamol (FLD) and CDPP significantly induced ROS production, free iron accumulation and lipid peroxidation, which greatly triggered ferroptosis [193]. In addition, the combination with ferroptosis inducers is also a current research hotspot. Acetaminophen (APAP) is a traditional antipyretic and analgesic drug [194], which can combine with erastin to promote cell ferroptosis by regulating Nrf2 nuclear translocation and inhibits the growth of lung cancer xenograft tumors [195]. Vorinostat (SAHA), a histone deacetylase inhibitor [196], can inhibit the expression of system xc− and significantly enhance the ferroptosis effect of Erastin on EGFR-TKI resistant LUAD cells. Meanwhile, SAHA could block the compensatory upregulation of SCL7A11 induced by Erastin treatment alone and improve the efficacy of the drug [197]. Combination of Erastin or SOR with low-dose CDDP can effectively inhibit the growth of CDDP-resistant NSCLC cells (N5CP) in vivo, and promote ferroptosis by inducing the accumulation of intracellular lipid peroxides [198]. In addition, it has been shown that when the ferroptosis inducer imidazole ketone erastin (IKE), SOR and cytoplasmic proton radiation act synergistically, tumor killing is significantly enhanced in mouse xenograft models and human patient-derived lung adenocarcinoma models [199].
Besides, nanomedicine has also been applied to ferroptosis in lung cancer. A zero-valent-iron nanoparticle (ZVI-NP) can promote ferroptosis in lung cancer cells by enhancing phosphorylation-dependent ubiquitination and Nrf2 degradation, and preferentially accumulate in tumor and lung tissues. At the same time, it can significantly inhibit tumor growth and metastasis, stimulate the in vitro immunity of macrophages and lymphocytes, and weaken the ability of tumor self-renewal [200]. Multifunctional “ball-rod” Janus nanoparticles (FTG/L&SMD) can promote ferroptosis process by initiating the Fenton reaction cycle while effectively inhibiting tumor growth with negligible toxicity in vivo [201].
Therefore, there are still few chemotherapeutic drugs targeting ferroptosis in lung cancer, and most of them are combination drugs. Most of the studies on drugs used alone are drugs originally used for other purposes, and the specific mechanisms of ferroptosis induced by drugs are still less studied. Among the combined drugs, cisplatin and ferroptosis inducer erastin are commonly used, and the synergistic effect with cytoplasmic proton radiation has also been studied. To our surprise, the combination had an excellent anti-drug effect on lung cancer drug resistance. In addition, there are few combined applications with nanotechnology, and its role in promoting ferroptosis of lung cancer is also very impressive.
Natural medicines
Natural compounds have also been shown to be useful in the treatment of cancer ferroptosis due to their rich biological activities [202]. Currently, the natural compounds for ferroptosis in lung cancer mainly include terpenoids, alkaloids, phenols, quinones and saponins, and also contain few steroids and flavonoids. Figure 5 lists the sources and chemical formulas of natural products currently acting on ferroptosis for lung cancer treatment, and their regulatory targets and mechanisms are shown in Fig. 6 and Table 2.

Natural products of plant origin that target ferroptosis for lung cancer treatment are shown in the figure and it mainly includes terpenoids, alkaloids, phenols, quinones, saponins, flavonoids, and others.

ART Artesunate, DHA Dihydroartemisinin, CUL Curcumenol, CEL Celastrol, BE Betulin, SAG Sanguinarine, SI Sinapine, SS Solasonine, CAP Capsaicin, ERI Erianin, DT Dihydroisotanshinone I, CTS Cryptotanshinone, OP-B Ophiopogonin-B, GK Ginkgetin, BT Bufotalin, SFN Sulforaphane, RGP Red ginseng polysaccharide, SLC40A1 solute carrier family 40 member 1, HO-1 Heme oxygenase 1, LC3 microtubule-associated protein 1 light chain, ATG5 autophagy-related protein 5, ATG7 autophagy-related protein 7, PRIM2 DNA primase polypeptide 2, FIS1 mitochondrial adaptor fission 1, DRP1 dynamin related protein 1, FTH1 ferritin heavy chain 1, AURKA Aurora kinase A, USP11 ubiquitin-specific processing protease 11, MMP mitochondrial membrane potential, miRNA MicroRNA, lncRNA Long non-coding RNA, DMT1 solute carrier family 11 member 2, Nrf2 nuclear factor erythroid 2-related factor 2, p53 tumor suppressor protein, GPX4 glutathione peroxidase 4, System Xc− cystine/glutamate antiporter system, Cys2 cystine, Glu glutamate, Cys cysteine, Gly glycine, GCL glutamate-cysteine ligase, SLC3A2 solute carrier family 3, member 2, SLC7A11 solute carrier family 7 (L-type amino acid transporter), member 11, GSS glutathione synthetase, GSH glutathione, GSSG oxidized glutathione, GSR glutathione reductase, ROS reactive oxygen species.
Terpenoids
Artemisinin and its derivatives are well known for their ability to treat malaria [203], but more and more studies have shown that artemisinin is also effective in promoting tumor ferroptosis [204, 205]. Artesunate (ART) and dihydroartemisinin (DHA), derivatives of artemisinin [204, 206], can induce ROS-dependent ferroptosis, down-regulate the protein level of voltage-dependent anion channel (VDAC), and promote cell apoptosis [207]. DHA can down-regulate PRIM2 and regulate the expression of SLC7A11 and β-catenin, thereby inhibiting the proliferation and cloning ability of lung cancer cells and promoting ferroptosis [208]. In addition, DHA can also combat the resistance of lung cancer cells to chlorinated protein e6 (Ce6) -mediated photodynamic therapy (PDT) by promoting ferroptosis [209]. Curcuma (CUL) is a terpenoid compound derived from Curcuma zedoaria (Christm.) Roscoe [210], which can promote the ferroptosis process of lung cancer in vitro and in vivo, and has good anticancer effect. lncRNA H19 can act as a competitive endogenous RNA to bind to miR-19b-3p, thereby enhancing the transcriptional activity of its endogenous target ferritin heavy chain 1(FTH1), and CUL plays a role in promoting ferroptosis by down-regulating lncRNA H19 in lung cancer cells [211]. Celastrol (Cel), a natural triterpenoid compound [212], can increase the production of reactive oxygen species (ROS), disrupt mitochondrial membrane potential, promote mitochondrial fission, and further activate ATG5/ ATG7-dependent autophagy, and inhibit tumor growth in vivo when combined with erastin [213]. Betulin (BE) is a triterpenoid compound in the bark of Betula platyphylla Sukaczev [214], which can overcome gefitinib resistance in EGFR wild-type /KRAS mutant NSCLC cells by inducing ferrodeath when used in combination with gefitinib [215].
Alkaloids
Sanguinarine (SAG), an isoquinoline alkaloid, can promote the production of reactive oxygen species and inhibit the JAK/STAT pathway to promote apoptosis of lung cancer cells [216]. In addition to this, treatment of A549 and H3122 cells with SAG caused an increase in intracellular Fe2+ concentration, ROS levels and MDA content as well as a decrease in GSH content, which was thus shown to also trigger ferroptosis in lung cancer [217]. Sinapine (SI), an alkaloid isolated from rapeseed and cruciferous plants, targets mitochondria to regulate mitochondrial oxidative stress and induces ferroptosis in lung cancer cells [218, 219]. Solanine (SS), a compound derived from Solanum nigrum L. Steroid glycoside alkaloids [220], which can cause iron overload and redox imbalance in Calu-1 and A549 cells, and then trigger ferroptosis [221].
Phenols
6-gingerol is a monophenolic compound derived from rhizome of Zingiber officinale Roscoe [222], which can promote ferroptosis of lung cancer in vitro and in vivo by inhibiting the expression of autophagy-related protein ubiquitin-specific peptidase 14 (USP14) and regulating the downstream of autophagy-dependent ferroptosis [223]. Capsaicin (CAP), a natural active ingredient in Capsicum annuum L. and the source of their pungent taste, has anticancer activity [224, 225]. In the ferroptosis related studies of lung cancer, CAP can inhibit the proliferation of A549 and NCI-H23 cells and induce ferroptosis by inactivating SLC7A11/GPX4 signaling pathway [226]. Erianin, an extract from Dendrobium nobile Lindl. can promote G2/M cell cycle arrest, inhibit cell migration and induce ferroptosis in lung cancer cells, while inhibiting tumor growth in vivo [227, 228].
Quinones
Dihydroisotanshinone I (DT) is a bioactive compound derived from the rhizome of Salvia miltiorrhiza Bunge [229], which can inhibit the growth of lung cancer cells, promote the apoptosis and ferroptosis of lung cancer cells, and inhibit the metastasis of A549 cells in vivo [230]. Meanwhile, another kind of cryptotanshinone (CTS) extracted from the rhizome of Salvia miltiorrhiza Bunge can not only prevent pulmonary fibrosis [231], but also promote ferroptosis of lung cancer by inhibiting GPX4 activity, and activate caspase-3 to promote apoptosis of lung cancer [232].
Saponins
Ophiopogon B (OP-B) is a saponin compound derived from the root of Ophiopogon japonicus (Thunb.) Ker, and its inhibitory effect on lung cancer has been studied to some extent [233, 234]. According to proteomic sequencing analysis, AURKA, a classical cell cycle regulatory protein kinase, is highly expressed in NSCLC, and OP-B promotes ferroptosis in lung cancer in vitro and vivo by targeting AURKA [235]. α-heguelin is a monodesmotic triterpenoid saponin isolated from Hedera nepalensis K.Koch [236], which induces apoptosis at high doses, disrupts the redox system by significantly affecting glutathione metabolism at safe and low toxic doses to induce ferroptosis in NSCLC, and also increases the sensitivity of NSCLC cells to cisplatin [237].
Others
Ginkgetin (GK), a diflavone isolated from Ginkgo biloba L. [238], when combined with cisplatin, can induce Nrf2/HO-1 inactivation, increase unstable iron pool and lipid peroxidation, on the one hand, promote ferroptosis in lung cancer, on the other hand, significantly enhance the therapeutic effect [239]. Bufotalin (BT) is a steroid lactone derived from bufalin, which has anti-tumor activity [240]. It can not only promote ferroptosis of lung cancer cells in vitro, but also inhibit the growth process of xenograft tumors in vivo [241]. Sulforaphane (SFN) is a natural isothiocyanate present in a variety of vegetables [242], which can target Nrf2 and has potential anti-cancer activity [243]. At the same time, SFN can also inhibit the expression of SLC7A11, leading to the decrease of GSH and the increase of ROS level in lung cancer cells [244]. Red ginseng polysaccharide (RGP) is the active ingredient in Panax ginseng C.A.Mey. [245], which can induce the release of lactate dehydrogenase (LDH) in A549 cells, inhibit the expression of GPX4, and play a role in promoting ferroptosis [246].
Chinese medicine preparations
In addition, some studies have shown that some Chinese medicine preparations can also have potential therapeutic effects on lung cancer by promoting ferroptosis. For example, hedyotis diffusa injection can induce ferroptosis in lung cancer by inhibiting Bcl2, promoting Bax and activating VDAC2/3 channel, thereby increasing the release of ROS [247]. Fuzheng Kangai Decoction (FZKA) can induce ferroptosis of lung cancer cells by inhibiting GPX4, increasing lipid peroxidation and cellular Fe2+ level, and inhibit tumor growth in vivo [248]. Furthermore, there are no more studies on the ferroptosis of lung cancer about Chinese medicine preparations, which is also a research blank at present. How to combine traditional Chinese medicine compound with new programmed death pathway to overcome the shortcomings of lung cancer treatment is a problem that many researchers of Chinese medicine preparations need to think about.
Conclusions and perspectives
Targeting the cell death process is a common approach in cancer therapy, and ferroptosis, a new form of programmed cell death, is also thought to play an important role in anti-cancer treatment. Ferroptosis, a hot topic of research in recent years, has been gradually enriched by its corresponding regulatory mechanisms. Nevertheless, there are still many research gaps regarding the regulatory mechanism of ferroptosis, which needs to be further explored and solved in future ferroptosis-related studies. Also, the increased reactive oxygen species-induced lipid peroxidation response exhibited as a result of the regulation of ferroptosis is present in other cell death pathways [249], How to clarify that the cells undergo ferroptosis and find more exclusive physiological and biochemical indicators of ferroptosis is the focus in current research.
In contrast, in the existing lung cancer studies, the targets related to ferroptosis are more often targeted to the system xc− or GPX4 system, and there is a lack of studies on non-classical GPX4 regulatory pathways, with only few studies on FSP1. There exist some studies on Nrf2, a key factor of oxidative stress, but on the one hand, Nrf2 has preventive oxidative and anti-inflammatory effects on normal cells, on the other hand, it can promote tumor metastasis and induce drug resistance [250, 251]. In addition, the tumor suppressor P53 also plays a regulatory role in ferroptosis of lung cancer by inhibiting the expression of SLC7A11, the key component of system xc− [101]. However, it has also been shown that P53 inhibits erastin-induced ferroptosis by blocking dipeptidyl peptidase-4 (DPP4) activity in a transcriptionally non-dependent manner [252]. How to correctly view the role of Nrf2 and P53 in ferroptosis of lung cancer is to be investigated. What’s more, there are a few studies related to NFS1, STYK1, LSH and other regulatory targets, but the relevant research content is not in-depth. It is important to mention that studies promoting ferroptosis by targeting iron metabolism and lipid peroxide formation are clearly lacking. Meanwhile, non-coding RNAs affect the expression level of target genes through the competitive endogenous RNA regulation mechanism, and also have an appreciable regulatory role in lung cancer ferroptosis [253]. Furthermore, it is worth noting that although ferroptosis is a new programmed cell death pathway, many studies define ferroptosis as an autophagy-dependent death pathway, which has some potential connection with autophagy [133, 135, 213, 254, 255], and there is also a good research prospect to explore the regulatory relationship between ferroptosis and autophagy.
In the research of drugs that promote tumor ferroptosis, most of them also target system xc− or GPX4 drugs [256]. However, the existing researches on ferroptosis in lung cancer mainly focus on chemical drugs, natural drugs and the combination of drugs and drugs, and there is also a small amount of synergistic effect of drugs with certain therapies. Fortunately, some drugs can not only promote ferroptosis, apoptosis, induce cell cycle arrest or inhibit cell growth and migration in vitro, but also inhibit tumor progression in vivo. At the same time, when the two drugs are combined or a drug combined with a certain therapy, it can also play a better therapeutic effect and resist lung cancer drug resistanc. Compared with chemical drugs, there are more studies on natural drugs in lung cancer ferroptosis, and the regulatory pathways involved are broader, which also indicates that natural compounds have considerable research value in lung cancer ferroptosis. However, it cannot be ignored that although ferroptosis has a positive regulatory effect on tumors, it is extremely unfavorable for normal cells. In the cardiovascular system and nervous system, inhibition of ferroptosis is required to play a protective role [257, 258]. How to selectively apply ferroptosis to the treatment of lung cancer, combat the drug resistance and radiation resistance of lung cancer, inhibit the development and metastasis of lung cancer, and protect normal cells from ferroptosis is a difficulty in the future research of ferroptosis in tumors.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Xia C, Dong X, Li H, Cao M, Sun D, He S, et al. Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin Med J (Engl). 2022;135:584–90.
Cao W, Chen HD, Yu YW, Li N, Chen WQ. Changing profiles of cancer burden worldwide and in China: a secondary analysis of the global cancer statistics 2020. Chin Med J (Engl). 2021;134:783–91.
Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med 2020;41:1–24.
Nielsen AH, Fredberg U. Earlier diagnosis of lung cancer. Cancer Treat Res Commun. 2022;31:100561.
Wu F, Wang L, Zhou C. Lung cancer in China: current and prospect. Curr Opin Oncol. 2021;33:40–46.
Toki MI, Harrington K, Syrigos KN. The role of spread through air spaces (STAS) in lung adenocarcinoma prognosis and therapeutic decision making. Lung Cancer. 2020;146:127–33.
Rosell R, Karachaliou N, Arrieta O. Novel molecular targets for the treatment of lung cancer. Curr Opin Oncol. 2020;32:37–43.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–54.
Lim ZF, Ma PC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol. 2019;12:134.
Liu J, Hong M, Li Y, Chen D, Wu Y, Hu Y. Programmed cell death tunes tumor immunity. Front Immunol. 2022;13:847345.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88.
Manz DH, Blanchette NL, Paul BT, Torti FM, Torti SV. Iron and cancer: recent insights. Ann N. Y Acad Sci. 2016;1368:149–61.
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34.
Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N. Engl J Med. 2009;361:1570–83.
Strasser A, Vaux DL. Cell death in the origin and treatment of cancer. Mol Cell. 2020;78:1045–54.
Moujalled D, Strasser A, Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021;28:2029–44.
Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–45.
Suzanne M, Steller H. Shaping organisms with apoptosis. Cell Death Differ. 2013;20:669–75.
Chen X, Comish PB, Tang D, Kang R. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. 2021;9:637162.
Hunt JR. Bioavailability of iron, zinc, and other trace minerals from vegetarian diets. Am J Clin Nutr. 2003;78:633s–639s.
Georgieff MK. Iron deficiency in pregnancy. Am J Obstet Gynecol. 2020;223:516–24.
Milto IV, Suhodolo IV, Prokopieva VD, Klimenteva TK. Molecular and cellular bases of iron metabolism in humans. Biochem (Mosc). 2016;81:549–64.
Lane DJ, Merlot AM, Huang ML, Bae DH, Jansson PJ, Sahni S, et al. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim Biophys Acta. 2015;1853:1130–44.
Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell 2010;142:24–38.
Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: an intimate relationship. Biochim Biophys Acta Mol Cell Res. 2019;1866:118535.
Nakamura T, Naguro I, Ichijo H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim Biophys Acta Gen Subj. 2019;1863:1398–409.
Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:590226.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–8.
Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200.
Kruszewski M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat Res. 2003;531:81–92.
Dunford HB. Oxidations of iron(II)/(III) by hydrogen peroxide: from aquo to enzyme. Coord Chem Rev. 2002;233-4:311–8.
Hamaï A, Mehrpour M. [Autophagy and iron homeostasis]. Med Sci (Paris). 2017;33:260–7.
Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843.
Doll S, Conrad M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life. 2017;69:423–34.
Zalba S, Ten Hagen TL. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat Rev. 2017;52:48–57.
Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162–76.
Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419–25.
Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5:108.
Kuwata H, Hara S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 2019;144:106363.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–9.
Vishnupriya P, Aparna A, Viswanadha VP. Lipoxygenase (LOX) pathway: a promising target to combat cancer. Curr Pharm Des. 2021;27:3349–69.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–4975.
Singh NK, Rao GN. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res. 2019;73:28–45.
Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–9.
Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. 2021;81:355–369.
Lv H, Zhen C, Liu J, Yang P, Hu L, Shang P. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid Med Cell Longev. 2019;2019:3150145.
Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18:522–55.
Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids. 2012;42:231–46.
Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–70.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317–31.
Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599–620.
Li Y, Wang X, Huang Z, Zhou Y, Xia J, Hu W, et al. CISD3 inhibition drives cystine-deprivation induced ferroptosis. Cell Death Dis. 2021;12:839.
Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17.
Kong R, Wang N, Han W, Bao W, Lu J. IFNγ-mediated repression of system xc(-) drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J Leukoc Biol. 2021;110:301–14.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608:778–83.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16:1351–60.
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6:41–53.
Vasquez-Vivar J, Shi Z, Tan S. Tetrahydrobiopterin in cell function and death mechanisms. Antioxid Redox Signal. 2022;37:171–83.
Brigelius-Flohé R. Glutathione peroxidases and redox-regulated transcription factors. Biol Chem. 2006;387:1329–35.
Zhang C, Wang C, Yang Z, Bai Y, Shukuya T, Poh ME, et al. Identification of GPX4 as a therapeutic target for lung adenocarcinoma after EGFR-TKI resistance. Transl Lung Cancer Res. 2022;11:786–801.
Ni J, Chen K, Zhang J, Zhang X. Inhibition of GPX4 or mTOR overcomes resistance to Lapatinib via promoting ferroptosis in NSCLC cells. Biochem Biophys Res Commun. 2021;567:154–60.
Zhang X, Sui S, Wang L, Li H, Zhang L, Xu S, et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J Cell Physiol. 2020;235:3425–37.
Wang Z, Zhang X, Tian X, Yang Y, Ma L, Wang J, et al. CREB stimulates GPX4 transcription to inhibit ferroptosis in lung adenocarcinoma. Oncol Rep. 2021;45:88.
Pan X, Lin Z, Jiang D, Yu Y, Yang D, Zhou H, et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol Lett. 2019;17:3001–8.
Li FJ, Long HZ, Zhou ZW, Luo HY, Xu SG, Gao LC. System X(c) (-)/GSH/GPX4 axis: an important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharm. 2022;13:910292.
Fan Z, Yang G, Zhang W, Liu Q, Liu G, Liu P, et al. Hypoxia blocks ferroptosis of hepatocellular carcinoma via suppression of METTL14 triggered YTHDF2-dependent silencing of SLC7A11. J Cell Mol Med. 2021;25:10197–212.
Feng L, Zhao K, Sun L, Yin X, Zhang J, Liu C, et al. SLC7A11 regulated by NRF2 modulates esophageal squamous cell carcinoma radiosensitivity by inhibiting ferroptosis. J Transl Med. 2021;19:367.
Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond). 2018;38:12.
Chen M, Jiang Y, Sun Y. KDM4A-mediated histone demethylation of SLC7A11 inhibits cell ferroptosis in osteosarcoma. Biochem Biophys Res Commun. 2021;550:77–83.
Wu X, Sui Z, Zhang H, Wang Y, Yu Z. Integrated analysis of lncRNA-Mediated ceRNA network in lung adenocarcinoma. Front Oncol. 2020;10:554759.
Ji X, Qian J, Rahman SMJ, Siska PJ, Zou Y, Harris BK, et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018;37:5007–19.
Zhang W, Sun Y, Bai L, Zhi L, Yang Y, Zhao Q, et al. RBMS1 regulates lung cancer ferroptosis through translational control of SLC7A11. J Clin Invest. 2021;131:e152067.
Wang X, Chen Y, Wang X, Tian H, Wang Y, Jin J, et al. Stem cell factor SOX2 confers ferroptosis resistance in lung cancer via upregulation of SLC7A11. Cancer Res. 2021;81:5217–29.
Zhang KR, Zhang YF, Lei HM, Tang YB, Ma CS, Lv QM, et al. Targeting AKR1B1 inhibits glutathione de novo synthesis to overcome acquired resistance to EGFR-targeted therapy in lung cancer. Sci Transl Med. 2021;13:eabg6428.
Xu Y, Lv D, Yan C, Su H, Zhang X, Shi Y, et al. METTL3 promotes lung adenocarcinoma tumor growth and inhibits ferroptosis by stabilizing SLC7A11 m(6)A modification. Cancer Cell Int. 2022;22:11.
Wang K, He J, Tu C, Xu H, Zhang X, Lv Y, et al. Upregulation of CCT3 predicts poor prognosis and promotes cell proliferation via inhibition of ferroptosis and activation of AKT signaling in lung adenocarcinoma. BMC Mol Cell Biol. 2022;23:25.
He F, Antonucci L, Karin M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020;41:405–16.
Panieri E, Saso L. Inhibition of the NRF2/KEAP1 Axis: a promising therapeutic strategy to alter redox balance of cancer cells. Antioxid Redox Signal. 2021;34:1428–83.
Wang J, Yang J, Cao M, Zhao Z, Cao B, Yu S. The potential roles of Nrf2/Keap1 signaling in anticancer drug interactions. Curr Res Pharm Drug Disco. 2021;2:100028.
Sajadimajd S, Khazaei M. Oxidative stress and cancer: The Role of Nrf2. Curr Cancer Drug Targets. 2018;18:538–57.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016;63:173–84.
Liu P, Wu D, Duan J, Xiao H, Zhou Y, Zhao L, et al. NRF2 regulates the sensitivity of human NSCLC cells to cystine deprivation-induced ferroptosis via FOCAD-FAK signaling pathway. Redox Biol. 2020;37:101702.
Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. 2021;33:174–189.
Liu W, Zhou Y, Duan W, Song J, Wei S, Xia S, et al. Glutathione peroxidase 4-dependent glutathione high-consumption drives acquired platinum chemoresistance in lung cancer-derived brain metastasis. Clin Transl Med. 2021;11:e517.
Wang H, Huang Q, Xia J, Cheng S, Pei D, Zhang X, et al. The E3 Ligase MIB1 promotes proteasomal degradation of nrf2 and sensitizes lung cancer cells to ferroptosis. Mol Cancer Res. 2022;20:253–64.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98.
Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24:88–98.
Sha R, Xu Y, Yuan C, Sheng X, Wu Z, Peng J, et al. Predictive and prognostic impact of ferroptosis-related genes ACSL4 and GPX4 on breast cancer treated with neoadjuvant chemotherapy. EBioMedicine 2021;71:103560.
Zhang Y, Li S, Li F, Lv C, Yang QK. High-fat diet impairs ferroptosis and promotes cancer invasiveness via downregulating tumor suppressor ACSL4 in lung adenocarcinoma. Biol Direct. 2021;16:10.
Wu H, Liu A. Long non-coding RNA NEAT1 regulates ferroptosis sensitivity in non-small-cell lung cancer. J Int Med Res. 2021;49:300060521996183.
Hou J, Jiang C, Wen X, Li C, Xiong S, Yue T, et al. ACSL4 as a potential target and biomarker for anticancer: from molecular mechanisms to clinical therapeutics. Front Pharm. 2022;13:949863.
Yang X, Hu X, Guo N. Clinical and biological significances of a ferroptosis-related gene signature in lung cancer based on deep learning. Comput Math Methods Med. 2022;2022:6495301.
Guan YS, He Q, Zou Q. Status quo of p53 in the treatment of tumors. Anticancer Drugs. 2016;27:811–8.
Wang Y, Yang L, Zhang X, Cui W, Liu Y, Sun QR, et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019;20:e47563.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Liu Y, Gu W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 2022;29:895–910.
Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162–8.
Meng M, Huang M, Liu C, Wang J, Ren W, Cui S, et al. Local anesthetic levobupivacaine induces ferroptosis and inhibits progression by up-regulating p53 in non-small cell lung cancer. Aging (Albany NY). 2021;13.
Huang C, Yang M, Deng J, Li P, Su W, Jiang R. Upregulation and activation of p53 by erastin‑induced reactive oxygen species contribute to cytotoxic and cytostatic effects in A549 lung cancer cells. Oncol Rep. 2018;40:2363–70.
Wang J, Yang W, He X, Zhang Z, Zheng X. Assembling p53 activating peptide With CeO(2) nanoparticle to construct a metallo-organic supermolecule toward the synergistic ferroptosis of tumor. Front Bioeng Biotechnol. 2022;10:929536.
Marshall KR, Gong M, Wodke L, Lamb JH, Jones DJ, Farmer PB, et al. The human apoptosis-inducing protein AMID is an oxidoreductase with a modified flavin cofactor and DNA binding activity. J Biol Chem. 2005;280:30735–40.
Koppula P, Lei G, Zhang Y, Yan Y, Mao C, Kondiparthi L, et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun. 2022;13:2206.
Jo A, Bae JH, Yoon YJ, Chung TH, Lee EW, Kim YH, et al. Plasma-activated medium induces ferroptosis by depleting FSP1 in human lung cancer cells. Cell Death Dis. 2022;13:212.
Song JY, Marszalek J, Craig EA. Cysteine desulfurase Nfs1 and Pim1 protease control levels of Isu, the Fe-S cluster biogenesis scaffold. Proc Natl Acad Sci USA. 2012;109:10370–5.
Terzi EM, Sviderskiy VO, Alvarez SW, Whiten GC, Possemato R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv. 2021;7:eabg4302.
Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017;551:639–43.
Alvarez SW, Possemato R. Leveraging the iron-starvation response to promote ferroptosis. Oncotarget 2018;9:10830–1.
Fujihara KM, Zhang BZ, Jackson TD, Ogunkola MO, Nijagal B, Milne JV, et al. Eprenetapopt triggers ferroptosis, inhibits NFS1 cysteine desulfurase, and synergizes with serine and glycine dietary restriction. Sci Adv. 2022;8:eabm9427.
Ye X, Ji C, Huang Q, Cheng C, Tang R, Xu J, et al. Isolation and characterization of a human putative receptor protein kinase cDNA STYK1. Mol Biol Rep. 2003;30:91–96.
Liu L, Yu XZ, Li TS, Song LX, Chen PL, Suo TL, et al. A novel protein tyrosine kinase NOK that shares homology with platelet- derived growth factor/fibroblast growth factor receptors induces tumorigenesis and metastasis in nude mice. Cancer Res. 2004;64:3491–9.
Chen P, Li WM, Lu Q, Wang J, Yan XL, Zhang ZP, et al. Clinicopathologic features and prognostic implications of NOK/STYK1 protein expression in non-small cell lung cancer. BMC Cancer. 2014;14:402.
Ma Z, Liu D, Li W, Di S, Zhang Z, Zhang J, et al. STYK1 promotes tumor growth and metastasis by reducing SPINT2/HAI-2 expression in non-small cell lung cancer. Cell Death Dis. 2019;10:435.
Lai Y, Zhang Z, Li J, Li W, Huang Z, Zhang C, et al. STYK1/NOK correlates with ferroptosis in non-small cell lung carcinoma. Biochem Biophys Res Commun. 2019;519:659–66.
Ho L, Crabtree GR. Chromatin remodelling during development. Nature 2010;463:474–84.
Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol. 2017;18:407–22.
Yang R, Liu N, Chen L, Jiang Y, Shi Y, Mao C, et al. LSH interacts with and stabilizes GINS4 transcript that promotes tumourigenesis in non-small cell lung cancer. J Exp Clin Cancer Res. 2019;38:280.
Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu X, et al. EGLN1/c-Myc induced lymphoid-specific helicase inhibits ferroptosis through lipid metabolic gene expression changes. Theranostics 2017;7:3293–305.
Misra JR, Irvine KD. The hippo signaling network and its biological functions. Annu Rev Genet. 2018;52:65–87.
Hansen CG, Ng YL, Lam WL, Plouffe SW, Guan KL. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015;25:1299–313.
Hsu PC, Jablons DM, Yang CT, You L. Epidermal growth factor receptor (EGFR) pathway, yes-associated protein (YAP) and the regulation of programmed death-ligand 1 (PD-L1) in non-small cell lung cancer (NSCLC). Int J Mol Sci. 2019;20:3821.
Jin D, Guo J, Wu Y, Du J, Yang L, Wang X, et al. m(6)A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2019;12:135.
Jin D, Guo J, Wu Y, Yang L, Wang X, Du J, et al. m(6)A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2-mediated YAP activity in NSCLC. Mol Cancer. 2020;19:40.
Magesh S, Cai D. Roles of YAP/TAZ in ferroptosis. Trends Cell Biol. 2022;32:729–32.
Zhang X, Yu K, Ma L, Qian Z, Tian X, Miao Y, et al. Endogenous glutamate determines ferroptosis sensitivity via ADCY10-dependent YAP suppression in lung adenocarcinoma. Theranostics. 2021;11:5650–74.
Wang Y, Qiu S, Wang H, Cui J, Tian X, Miao Y, et al. Transcriptional repression of ferritin light chain increases ferroptosis sensitivity in lung adenocarcinoma. Front Cell Dev Biol. 2021;9:719187.
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12.
Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-dependent ferroptosis: machinery and regulation. Cell. Chem Biol. 2020;27:420–35.
Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822.
Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100.
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014;509:105–9.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-Mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X(c)(-) activity. Curr Biol. 2018;28:2388–2399.
Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J Biosci. 2006;31:137–55.
Park J, Cho J, Song EJ. Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch Pharm Res. 2020;43:1144–61.
Meng Y, Sun H, Li Y, Zhao S, Su J, Zeng F, et al. Targeting ferroptosis by ubiquitin system enzymes: a potential therapeutic strategy in cancer. Int J Biol Sci. 2022;18:5475–88.
Yang L, Chen X, Yang Q, Chen J, Huang Q, Yao L, et al. Broad spectrum deubiquitinase inhibition induces both apoptosis and ferroptosis in cancer cells. Front Oncol. 2020;10:949.
Tang Z, Jiang W, Mao M, Zhao J, Chen J, Cheng N. Deubiquitinase USP35 modulates ferroptosis in lung cancer via targeting ferroportin. Clin Transl Med. 2021;11:e390.
Meng C, Zhan J, Chen D, Shao G, Zhang H, Gu W, et al. The deubiquitinase USP11 regulates cell proliferation and ferroptotic cell death via stabilization of NRF2 USP11 deubiquitinates and stabilizes NRF2. Oncogene 2021;40:1706–20.
Huang D, Li Q, Sun X, Sun X, Tang Y, Qu Y, et al. CRL4(DCAF8) dependent opposing stability control over the chromatin remodeler LSH orchestrates epigenetic dynamics in ferroptosis. Cell Death Differ. 2021;28:1593–609.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74.
He Y, Jiang X, Duan L, Xiong Q, Yuan Y, Liu P, et al. LncRNA PKMYT1AR promotes cancer stem cell maintenance in non-small cell lung cancer via activating Wnt signaling pathway. Mol Cancer. 2021;20:156.
Bi G, Liang J, Zhao M, Zhang H, Jin X, Lu T, et al. miR-6077 promotes cisplatin/pemetrexed resistance in lung adenocarcinoma via CDKN1A/cell cycle arrest and KEAP1/ferroptosis pathways. Mol Ther Nucleic Acids. 2022;28:366–86.
Wang W, Xie Y, Malhotra A. Potential of curcumin and quercetin in modulation of premature mitochondrial senescence and related changes during lung carcinogenesis. J Environ Pathol Toxicol Oncol. 2021;40:53–60.
Ho PTB, Clark IM, Le LTT. MicroRNA-based diagnosis and therapy. Int J Mol Sci. 2022;23:7167.
Iqbal MA, Arora S, Prakasam G, Calin GA, Syed MA. MicroRNA in lung cancer: role, mechanisms, pathways and therapeutic relevance. Mol Asp Med. 2019;70:3–20.
Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 2021;276:119399.
Lu X, Kang N, Ling X, Pan M, Du W, Gao S. MiR-27a-3p promotes non-small cell lung cancer through SLC7A11-mediated-ferroptosis. Front Oncol. 2021;11:759346.
Wei D, Ke YQ, Duan P, Zhou L, Wang CY, Cao P. MicroRNA-302a-3p induces ferroptosis of non-small cell lung cancer cells via targeting ferroportin. Free Radic Res. 2021;55:821–30.
Chen Q, Pan Q, Gao H, Wang Y, Zhong X. miR-17-5p/HOXA7 is a potential driver for brain metastasis of lung adenocarcinoma related to ferroptosis revealed by bioinformatic analysis. Front Neurol. 2022;13:878947.
Kristensen LS, Hansen TB, Venø MT, Kjems J. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene. 2018;37:555–65.
Chen L, Shan G. CircRNA in cancer: fundamental mechanism and clinical potential. Cancer Lett. 2021;505:49–57.
Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675–91.
Karreth FA, Pandolfi PP. ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Disco. 2013;3:1113–21.
Luo Y, Zhang Q, Lv B, Shang Y, Li J, Yang L, et al. CircFOXP1: a novel serum diagnostic biomarker for non-small cell lung cancer. Int J Biol Markers. 2022;37:58–65.
Li O, Kang J, Zhang JJ, Wang J, Hu LW, Li L, et al. Circle RNA FOXP1 promotes cell proliferation in lung cancer by regulating miR-185-5p/Wnt1 signaling pathway. Eur Rev Med Pharm Sci. 2020;24:6767–78.
Li H, Liu L. Zinc moderates circular RNA CircFOXP1 expression in order to regulate ferroptosis during lung adenocarcinoma. Chem Biol Interact. 2022;352:109760.
Shanshan W, Hongying M, Jingjing F, Yiming Y, Yu R, Rui Y. CircDTL functions as an oncogene and regulates both apoptosis and ferroptosis in non-small cell lung cancer cells. Front Genet. 2021;12:743505.
Pan C, Wei K, Ma Z, He Y, Huang J, Guo Z, et al. CircP4HB regulates ferroptosis via SLC7A11-mediated glutathione synthesis in lung adenocarcinoma. Transl Lung Cancer Res. 2022;11:366–80.
Zhang X, Xu Y, Ma L, Yu K, Niu Y, Xu X, et al. Essential roles of exosome and circRNA_101093 on ferroptosis desensitization in lung adenocarcinoma. Cancer Commun (Lond). 2022;42:287–313.
Peng WX, Koirala P, Mo YY. LncRNA-mediated regulation of cell signaling in cancer. Oncogene. 2017;36:5661–7.
Yang M, Lu H, Liu J, Wu S, Kim P, Zhou X. lncRNAfunc: a knowledgebase of lncRNA function in human cancer. Nucleic Acids Res. 2022;50:D1295–d1306.
Yao J, Chen X, Liu X, Li R, Zhou X, Qu Y. Characterization of a ferroptosis and iron-metabolism related lncRNA signature in lung adenocarcinoma. Cancer Cell Int. 2021;21:340.
Liu L, Su S, Ye D, Yu Z, Lu W, Li X. Long non-coding RNA OGFRP1 regulates cell proliferation and ferroptosis by miR-299-3p/SLC38A1 axis in lung cancer. Anticancer Drugs. 2022;33:826–39.
Sui X, Hu N, Zhang Z, Wang Y, Wang P, Xiu G. ASMTL-AS1 impedes the malignant progression of lung adenocarcinoma by regulating SAT1 to promote ferroptosis. Pathol Int. 2021;71:741–51.
Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018;78:3484–96.
Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019;26:2329–43.
Gai C, Liu C, Wu X, Yu M, Zheng J, Zhang W, et al. MT1DP loaded by folate-modified liposomes sensitizes erastin-induced ferroptosis via regulating miR-365a-3p/NRF2 axis in non-small cell lung cancer cells. Cell Death Dis. 2020;11:751.
Zhang N, Huang J, Xu M, Wang Y. LncRNA T-UCR Uc.339/miR-339/SLC7A11 axis regulates the metastasis of ferroptosis-induced lung adenocarcinoma. J Cancer. 2022;13:1945–57.
Ma L, Zhang X, Yu K, Xu X, Chen T, Shi Y, et al. Targeting SLC3A2 subunit of system X is essential for mA reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic Biol Med. 2021;168:25–43.
Priem D, van Loo G, Bertrand MJM. A20 and cell death-driven inflammation. Trends Immunol. 2020;41:421–35.
Gao C, Xiao F, Zhang L, Sun Y, Wang L, Liu X, et al. SENP1 inhibition suppresses the growth of lung cancer cells through activation of -mediated ferroptosis. Ann Transl Med. 2022;10:224.
Aburjania Z, Jang S, Whitt J, Jaskula-Stzul R, Chen H, Rose JB. The role of Notch3 in cancer. Oncologist. 2018;23:900–11.
Li Z, Xiao J, Liu M, Cui J, Lian B, Sun Y, et al. Notch3 regulates ferroptosis via ROS-induced lipid peroxidation in NSCLC cells. FEBS open bio. 2022;12:1197–205.
Vu N, Kim M, Stephenson D, MacKnight H, Chalfant C. Ceramide kinase inhibition drives ferroptosis and sensitivity to cisplatin in mutant KRAS lung cancer by dysregulating VDAC-mediated mitochondria function. Mol Cancer Res. 2022;20:1429–42.
Li Y, Zeng X, Lu D, Yin M, Shan M, Gao Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum Reprod (Oxf, Engl). 2021;36:951–64.
Liu S, Yan S, Zhu J, Lu R, Kang C, Tang K, et al. Combination RSL3 treatment sensitizes ferroptosis- and EGFR-inhibition-resistant HNSCCs to cetuximab. Int J Mol Sci. 2022;23:9014.
Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50:445–60.
Abdelgalil AA, Alkahtani HM, Al-Jenoobi FI. Sorafenib. Profiles Drug Subst Excip Relat Methodol. 2019;44:239–66.
Blumenschein G Jr. Sorafenib in lung cancer: clinical developments and future directions. J Thorac Oncol. 2008;3:S124–127.
Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34:6417–22.
Freire Boullosa L, Van Loenhout J, Flieswasser T, De Waele J, Hermans C, Lambrechts H, et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol. 2021;42:101949.
Zhou W, Zhang J, Yan M, Wu J, Lian S, Sun K, et al. Orlistat induces ferroptosis-like cell death of lung cancer cells. Front Med. 2021;15:922–32.
Wu W, Geng Z, Bai H, Liu T, Zhang B. Ammonium ferric citrate induced ferroptosis in non-small-cell lung carcinoma through the inhibition of GPX4-GSS/GSR-GGT axis activity. Int J Med Sci. 2021;18:1899–909.
Yu H, Han Z, Xu Z, An C, Xu L, Xin H. RNA sequencing uncovers the key long non-coding RNAs and potential molecular mechanism contributing to XAV939-mediated inhibition of non-small cell lung cancer. Oncol Lett. 2019;17:4994–5004.
Kryczka J, Kryczka J, Czarnecka-Chrebelska KH, Brzeziańska-Lasota E. Molecular mechanisms of chemoresistance induced by cisplatin in NSCLC cancer therapy. Int J Mol Sci. 2021;22:8885.
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene 2012;31:1869–83.
Liang Z, Zhao W, Li X, Wang L, Meng L, Yu R. Cisplatin synergizes with PRLX93936 to induce ferroptosis in non-small cell lung cancer cells. Biochem Biophys Res Commun. 2021;569:79–85.
Cui Z, Li D, Zhao J, Chen K. Falnidamol and cisplatin combinational treatment inhibits non-small cell lung cancer (NSCLC) by targeting DUSP26-mediated signal pathways. Free Radic Biol Med. 2022;183:106–24.
Abdel Shaheed C, Ferreira GE, Dmitritchenko A, McLachlan AJ, Day RO, Saragiotto B, et al. The efficacy and safety of paracetamol for pain relief: an overview of systematic reviews. Med J Aust. 2021;214:324–31.
Gai C, Yu M, Li Z, Wang Y, Ding D, Zheng J, et al. Acetaminophen sensitizing erastin-induced ferroptosis via modulation of Nrf2/heme oxygenase-1 signaling pathway in non-small-cell lung cancer. J Cell Physiol. 2020;235:3329–39.
Laengle J, Kabiljo J, Hunter L, Homola J, Prodinger S, Egger G, et al. Histone deacetylase inhibitors valproic acid and vorinostat enhance trastuzumab-mediated antibody-dependent cell-mediated phagocytosis. J Immunother Cancer. 2020;8:e000195.
Zhang T, Sun B, Zhong C, Xu K, Wang Z, Hofman P, et al. Targeting histone deacetylase enhances the therapeutic effect of Erastin-induced ferroptosis in EGFR-activating mutant lung adenocarcinoma. Transl Lung Cancer Res. 2021;10:1857–72.
Li Y, Yan H, Xu X, Liu H, Wu C, Zhao L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol Lett. 2020;19:323–33.
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. 2020;15:469–84.
Hsieh CH, Hsieh HC, Shih FS, Wang PW, Yang LX, Shieh DB, et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics. 2021;11:7072–91.
Zhu G, Chi H, Liu M, Yin Y, Diao H, Liu Z, et al. Multifunctional “ball-rod” Janus nanoparticles boosting Fenton reaction for ferroptosis therapy of non-small cell lung cancer. J Colloid Interface Sci. 2022;621:12–23.
Ge C, Zhang S, Mu H, Zheng S, Tan Z, Huang X, et al. Emerging mechanisms and disease implications of ferroptosis: potential applications of natural products. Front Cell Dev Biol. 2021;9:774957.
Neill US. From branch to bedside: Youyou Tu is awarded the 2011 Lasker~DeBakey Clinical Medical Research Award for discovering artemisinin as a treatment for malaria. J Clin Invest. 2011;121:3768–73.
Efferth T. From ancient herb to modern drug: artemisia annua and artemisinin for cancer therapy. Semin cancer Biol. 2017;46:65–83.
Chen G, Benthani F, Wu J, Liang D, Bian Z, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27:242–54.
Dai X, Zhang X, Chen W, Chen Y, Zhang Q, Mo S, et al. Dihydroartemisinin: a potential natural anticancer drug. Int J Biol Sci. 2021;17:603–22.
Zhang Q, Yi H, Yao H, Lu L, He G, Wu M, et al. Artemisinin derivatives inhibit non-small cell lung cancer cells through induction of ROS-dependent apoptosis/ferroptosis. J Cancer. 2021;12:4075–85.
Yuan B, Liao F, Shi Z, Ren Y, Deng X, Yang T, et al. Dihydroartemisinin inhibits the proliferation, colony formation and induces ferroptosis of lung cancer cells by inhibiting PRIM2/SLC7A11 Axis. Onco Targets Ther. 2020;13:10829–40.
Han N, Li L, Peng X, Ma Q, Yang Z, Wang X, et al. Ferroptosis triggered by dihydroartemisinin facilitates chlorin e6 induced photodynamic therapy against lung cancerthrough inhibiting GPX4 and enhancing ROS. Eur J Pharm. 2022;919:174797.
Ahmed Hamdi OA, Syed Abdul Rahman SN, Awang K, Abdul Wahab N, Looi CY, Thomas NF, et al. Cytotoxic constituents from the rhizomes of Curcuma zedoaria. ScientificWorldJournal 2014;2014:321943.
Zhang R, Pan T, Xiang Y, Zhang M, Xie H, Liang Z, et al. Curcumenol triggered ferroptosis in lung cancer cells via lncRNA H19/miR-19b-3p/FTH1 axis. Bioact Mater. 2022;13:23–36.
Ng SW, Chan Y, Chellappan DK, Madheswaran T, Zeeshan F, Chan YL, et al. Molecular modulators of celastrol as the keystones for its diverse pharmacological activities. Biomed Pharmacother. 2019;109:1785–92.
Liu M, Fan Y, Li D, Han B, Meng Y, Chen F, et al. Ferroptosis inducer erastin sensitizes NSCLC cells to celastrol through activation of the ROS-mitochondrial fission-mitophagy axis. Mol Oncol. 2021;15:2084–105.
Alakurtti S, Mäkelä T, Koskimies S, Yli-Kauhaluoma J. Pharmacological properties of the ubiquitous natural product betulin. Eur J Pharm Sci. 2006;29:1–13.
Yan WY, Cai J, Wang JN, Gong YS, Ding XB. Co-treatment of betulin and gefitinib is effective against EGFR wild-type/KRAS-mutant non-small cell lung cancer by inducing ferroptosis. Neoplasma. 2022;69:648–56.
Prabhu KS, Bhat AA, Siveen KS, Kuttikrishnan S, Raza SS, Raheed T, et al. Sanguinarine mediated apoptosis in non-small cell lung cancer via generation of reactive oxygen species and suppression of JAK/STAT pathway. Biomed Pharmacother. 2021;144:112358.
Xu R, Wu J, Luo Y, Wang Y, Tian J, Teng W, et al. Sanguinarine represses the growth and metastasis of non-small cell lung cancer by facilitating ferroptosis. Curr Pharm Des. 2022;28:760–8.
Boulghobra D, Grillet PE, Laguerre M, Tenon M, Fauconnier J, Fança-Berthon P, et al. Sinapine, but not sinapic acid, counteracts mitochondrial oxidative stress in cardiomyocytes. Redox Biol. 2020;34:101554.
Shao M, Jiang Q, Shen C, Liu Z, Qiu L. Sinapine induced ferroptosis in non-small cell lung cancer cells by upregulating transferrin/transferrin receptor and downregulating SLC7A11. Gene 2022;827:146460.
Zhu L, Lu Y, Sun Z, Han J, Tan Z. The application of an aqueous two-phase system combined with ultrasonic cell disruption extraction and HPLC in the simultaneous separation and analysis of solanine and Solanum nigrum polysaccharide from Solanum nigrum unripe fruit. Food Chem. 2020;304:125383.
Zeng YY, Luo YB, Ju XD, Zhang B, Cui YJ, Pan YB, et al. Solasonine causes redox imbalance and mitochondrial oxidative stress of ferroptosis in lung adenocarcinoma. Front Oncol. 2022;12:874900.
Koch W, Kukula-Koch W, Marzec Z, Kasperek E, Wyszogrodzka-Koma L, Szwerc W, et al. Application of chromatographic and spectroscopic methods towards the quality assessment of ginger (Zingiber officinale) rhizomes from ecological plantations. Int J Mol Sci. 2017;18:452.
Tsai Y, Xia C, Sun Z. The Inhibitory effect of 6-gingerol on ubiquitin-specific peptidase 14 enhances autophagy-dependent ferroptosis and anti-tumor in vivo and in vitro. Front Pharmacol. 2020;11:598555.
Merritt JC, Richbart SD, Moles EG, Cox AJ, Brown KC, Miles SL, et al. Anti-cancer activity of sustained release capsaicin formulations. Pharm Ther. 2022;238:108177.
Srinivasan K. Biological activities of red pepper (capsicum annuum) and its pungent principle capsaicin: a review. Crit Rev Food Sci Nutr. 2016;56:1488–1500.
Liu XY, Wei DG, Li RS. Capsaicin induces ferroptosis of NSCLC by regulating SLC7A11/GPX4 signaling in vitro. Sci Rep. 2022;12:11996.
Chen P, Wu Q, Feng J, Yan L, Sun Y, Liu S, et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct Target Ther. 2020;5:51.
Zhang HQ, Xie XF, Li GM, Chen JR, Li MT, Xu X, et al. Erianin inhibits human lung cancer cell growth via PI3K/Akt/mTOR pathway in vitro and in vivo. Phytother Res. 2021;35:4511–25.
Wang X, Morris-Natschke SL, Lee KH. New developments in the chemistry and biology of the bioactive constituents of Tanshen. Med Res Rev. 2007;27:133–48.
Wu CY, Yang YH, Lin YS, Chang GH, Tsai MS, Hsu CM, et al. Dihydroisotanshinone I induced ferroptosis and apoptosis of lung cancer cells. Biomed Pharmacother. 2021;139:111585.
Zhang Y, Lu W, Zhang X, Lu J, Xu S, Chen S, et al. Cryptotanshinone protects against pulmonary fibrosis through inhibiting Smad and STAT3 signaling pathways. Pharm Res. 2019;147:104307.
Li X, Li W, Yang P, Zhou H, Zhang W, Ma L. Anticancer effects of Cryptotanshinone against lung cancer cells through ferroptosis. Arab J Chem. 2021;14:103177.
Chen M, Guo Y, Zhao R, Wang X, Jiang M, Fu H, et al. Ophiopogonin B induces apoptosis, mitotic catastrophe and autophagy in A549 cells. Int J Oncol. 2016;49:316–24.
Nazim UM, Jeong JK, Park SY. Ophiopogonin B sensitizes TRAIL-induced apoptosis through activation of autophagy flux and downregulates cellular FLICE-like inhibitory protein. Oncotarget. 2018;9:4161–72.
Li L, Gao Q, Wang J, Gu L, Li Z, Zhang S, et al. Induction of ferroptosis by ophiopogonin-B through regulating the gene signature AURKA in NSCLC. Front Oncol. 2022;12:833814.
Gepdiremen A, Mshvildadze V, Süleyman H, Elias R. Acute anti-inflammatory activity of four saponins isolated from ivy: alpha-hederin, hederasaponin-C, hederacolchiside-E and hederacolchiside-F in carrageenan-induced rat paw edema. Phytomedicine. 2005;12:440–4.
Wu Y, Wang D, Lou Y, Liu X, Huang P, Jin M, et al. Regulatory mechanism of α-hederin upon cisplatin sensibility in NSCLC at safe dose by destroying GSS/GSH/GPX2 axis-mediated glutathione oxidation-reduction system. Biomed Pharmacother. 2022;150:112927.
Adnan M, Rasul A, Hussain G, Shah MA, Zahoor MK, Anwar H, et al. Ginkgetin: a natural biflavone with versatile pharmacological activities. Food Chem Toxicol. 2020;145:111642.
Lou JS, Zhao LP, Huang ZH, Chen XY, Xu JT, Tai WC, et al. Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer. Phytomedicine 2021;80:153370.
Waiwut P, Inujima A, Inoue H, Saiki I, Sakurai H. Bufotalin sensitizes death receptor-induced apoptosis via Bid- and STAT1-dependent pathways. Int J Oncol. 2012;40:203–8.
Zhang W, Jiang B, Liu Y, Xu L, Wan M. Bufotalin induces ferroptosis in non-small cell lung cancer cells by facilitating the ubiquitination and degradation of GPX4. Free Radic Biol Med. 2022;180:75–84.
Vanduchova A, Anzenbacher P, Anzenbacherova E. Isothiocyanate from broccoli, sulforaphane, and its properties. J Med Food. 2019;22:121–6.
Russo M, Spagnuolo C, Russo GL, Skalicka-Woźniak K, Daglia M, Sobarzo-Sánchez E, et al. Nrf2 targeting by sulforaphane: a potential therapy for cancer treatment. Crit Rev Food Sci Nutr. 2018;58:1391–405.
Iida Y, Okamoto-Katsuyama M, Maruoka S, Mizumura K, Shimizu T, Shikano S, et al. Effective ferroptotic small-cell lung cancer cell death from SLC7A11 inhibition by sulforaphane. Oncol Lett. 2021;21:71.
Lee SM, Bae BS, Park HW, Ahn NG, Cho BG, Cho YL, et al. Characterization of Korean Red Ginseng (Panax ginseng Meyer): history, preparation method, and chemical composition. J Ginseng Res. 2015;39:384–91.
Zhai FG, Liang QC, Wu YY, Liu JQ, Liu JW. Red ginseng polysaccharide exhibits anticancer activity through GPX4 downregulation-induced ferroptosis. Pharm Biol. 2022;60:909–14.
Huang F, Pang J, Xu L, Niu W, Zhang Y, Li S, et al. Hedyotis diffusa injection induces ferroptosis via the Bax/Bcl2/VDAC2/3 axis in lung adenocarcinoma. Phytomedicine 2022;104:154319.
Zhao YY, Yang YQ, Sheng HH, Tang Q, Han L, Wang SM, et al. GPX4 plays a crucial role in Fuzheng Kang’ai decoction-induced non-small cell lung cancer cell ferroptosis. Front Pharm. 2022;13:851680.
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin D-Mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24:97–108.
Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of bach1. Cell. 2019;178:316–329.
Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 System: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. 2018;98:1169–203.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–704.
Zhang X, Wang L, Li H, Zhang L, Zheng X, Cheng W. Crosstalk between noncoding RNAs and ferroptosis: new dawn for overcoming cancer progression. Cell Death Dis. 2020;11:580.
Zhang Z, Guo M, Li Y, Shen M, Kong D, Shao J, et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy. 2020;16:1482–505.
Yang M, Chen P, Liu J, Zhu S, Kroemer G, Klionsky DJ, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5:eaaw2238.
Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31:e1904197.
Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. 2021;11:3052–9.
Mahoney-Sánchez L, Bouchaoui H, Ayton S, Devos D, Duce JA, Devedjian JC. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog Neurobiol. 2021;196:101890.
Funding
This study was supported by the National Natural Science Foundation of China (82104534, 81903902) and the Sichuan Province Science Research Special Funding Project for Postdoctoral Fellows (2022BSH044).
Author information
Authors and Affiliations
Contributions
NX conducted a literature search and wrote the draft manuscript. QD, SG and GX conducted a literature search. YZ, XM and LX revised and edited the words in the manuscript. SW provided ideas for the manuscript and introduced the theme. SW and LX gave the fnancial supports. All authors agreed with the current version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xing, N., Du, Q., Guo, S. et al. Ferroptosis in lung cancer: a novel pathway regulating cell death and a promising target for drug therapy. Cell Death Discov. 9, 110 (2023). https://doi.org/10.1038/s41420-023-01407-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01407-z
- Springer Nature Limited
This article is cited by
-
Analysis of the incidence and influencing factors of abdominal distension in postoperative lung cancer patients in ICU based on real-world data: a retrospective cohort study
BMC Surgery (2024)
-
Recent advances in the potential effects of natural products from traditional Chinese medicine against respiratory diseases targeting ferroptosis
Chinese Medicine (2024)
-
Exploring the Role of Ferroptosis-Related Circular RNAs in Subarachnoid Hemorrhage
Molecular Biotechnology (2024)
-
Leveraging a disulfidptosis/ferroptosis-based signature to predict the prognosis of lung adenocarcinoma
Cancer Cell International (2023)
-
The dual role of ferroptosis in anthracycline-based chemotherapy includes reducing resistance and increasing toxicity
Cell Death Discovery (2023)