
Abstract
Chemokines secreted by dendritic cells (DCs) play a key role in the regulation of inflammation and autoimmunity through chemokine receptors. However, the role of chemokine receptor CXCR1 in inflammation-inducing experimental autoimmune encephalomyelitis (EAE) and acute respiratory distress syndrome (ARDS) remains largely enigmatic. Here we reported that compared with healthy controls, the level of CXCR1 was aberrantly increased in multiple sclerosis (MS) patients. Knockout of CXCR1 not only ameliorated disease severity in EAE mice but also suppressed the secretion of inflammatory factors (IL-6/IL-12p70) production. We observed the same results in EAE mice with DCs-specific deletion of CXCR1 and antibody neutralization of the ligand CXCL5. Mechanically, we demonstrated a positive feedback loop composed of CXCL5/CXCR1/HIF-1α direct regulating of IL-6/IL-12p70 production in DCs. Meanwhile, we found CXCR1 deficiency in DCs limited IL-6/IL-12p70 production and lung injury in LPS-induced ARDS, a disease model caused by inflammation. Overall, our study reveals CXCR1 governs DCs-mediated inflammation and autoimmune disorders and its potential as a therapeutic target for related diseases.
Similar content being viewed by others


Introduction
Inflammation is one of the most important components of the immune system because of its critical role in physiological response to injury and infection. However, unresolved or aberrantly activated inflammation can become pathogenic [1]. The pathogenic inflammation triggers complex, self-perpetuating immune responses and activates of receptors on immune cells. Derived signalling from activated receptor cascades that amplify local or systemic immune responses compromise the structural integrity and/or function of specific cell types, tissues, or organs [2,3,4]. Previous evidence has revealed that some inflammatory disorders (acute respiratory distress syndrome (ARDS), inflammatory bowel disease (IBD), and autoimmune diseases (type 1 diabetes, primary biliary cirrhosis, and multiple sclerosis (MS)) are associated with a cytokine storm (characterized by high levels of IL-6, IL12p70, and IL-1β, and tumor necrosis factor (TNF)) [5,6,7]. However, the massive production of inflammatory cytokines depends on the context, location, and timing, which makes it considerably difficult to develop effective therapeutics [8].
Dendritic cells (DCs) are the key link between innate immunity and adaptive immunity because of their promotion of immune defense and the maintenance of immune tolerance [9, 10]. On stimulation by antigens, DCs increase the expression of surface co-stimulatory molecules and pro-inflammatory or anti-inflammatory cytokines to direct differentiation of naïve CD4+ T cells [11,12,13]. Alterations in the functioning of DC may result in an imbalance of immune responses and even immune pathologies, like systemic lupus erythematosus (SLE), IBD, and MS. Thus, it is important to study molecular mechanisms that mediate DC activation and cytokines secretion function for identifying the underlying mechanisms [14,15,16]. Existing studies have reported that DCs produce polarizing and pro-inflammatory cytokines by complicated intracellular signaling pathways involving PI3K/AKT, MAPK/NF-κB, and JAK/STAT [17,18,19,20]. Furthermore, Beth et al. [21] suggested that transcription factor hypoxia-inducible factor-1α (HIF-1α) promotes DCs maturation and increases inflammatory gene expression as a metabolic switch in LPS-activated DCs. Although the intrinsic pathways of DCs secretion function have been well described, how instructive signals trigger and amplify the inflammatory responses in DCs remains poorly understood.
Chemokines are a family of inflammatory cytokines with the ability to regulate cell migration, adhesion, phagocytosis, cytokine secretion, proliferation, and apoptosis [22]. Increasing evidence suggests that chemokines and their receptors play a fundamental role in regulating DCs migration and homeostatic and inflammatory functions under both steady-state and inflammatory conditions [23,24,25,26]. However, very little is known about specific chemokines that mediate inflammatory cytokine by DCs in autoimmune diseases. Therefore, we focused on CXCR1, the first chemokine receptor cloned in 1991, and its mouse ligand CXCL5/LIX, which has been shown to mediate cell migration and granule release in neutrophils in response to bacterial infections and autoimmune diseases [27, 28]. Nevertheless, whether and how the CXCR1 plays critical roles in DCs-mediated inflammation and autoimmune disorders remains to be explored.
Herein, we found the mRNA level of CXCR1 was markedly upregulated in the peripheral blood leukocytes of MS patients. The deletion of CXCR1 and inhibition of CXCL5 significantly reduced the production of inflammatory cytokines, including IL-6 and IL-12p70, by DCs and restricted EAE progression. Moreover, activation of CXCR1 signaling amplified inflammation production through the CXCL5/CXCR1/HIF-1α positive feedback in DCs. Significantly, CXCR1 was also found to be functionally involved in LPS-induced ARDS. Therefore, this study reveals the role and mechanism of CXCR1 in regulating DCs mediating inflammation and autoimmunity. Furthermore, our data also shed light on uncovering new strategies for the treatment of autoimmune and inflammatory diseases.
Materials and methods
Human subjects
The blood samples of MS patients and healthy donors were obtained from Shanghai East Hospital, affiliated with Tongji University in Shanghai. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (the institutional review board at Tongji University) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all subjects to be included in the study. All patient data were anonymized before study inclusion. See Table 1 for full patient information, including age, sex, and health status. All MS participants were examined for expanded disability status scale measures (EDSS) [29]. Informed consent was obtained from all subjects to be included in the study. All patient’s data were anonymized before study inclusion.
Peripheral blood mononuclear cells (PBMC) were isolated from the fresh peripheral blood of healthy volunteers (Stem Express, Inc.) by density gradient centrifugation using Ficoll Paque PLUS (GE Healthcare). CD14+ monocytes were purified by magnetic selection (Stem Cell Technology) according to the manufacturer’s protocol and 106 cells/ml of CD14+ monocytes were cultured in complete RPMI 1640 supplemented with 10% FBS, l-glutamine (2 mM), 2-ME (50 mM), human recombinant GM-CSF (20 ng/ml), and human recombinant IL-4 (1 ng/ml) for 7 days. Thereafter, the cells were collected and seeded into 24-well plates (1 × 106 cells per well per milliliter) in the presence of anti-CXCR1 (3, 10, and 30 μg/ml) and KC7F2 (HIF-1α inhibitor) [30] for 30 min.
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee of Tongji University. OT-II C57BL/6J mice were obtained from Zhongjun Dong, Tsinghua University. GFP-OT-II C57BL/6J mice were generated by breeding GFP mice with OT-II mice. Cxcr1−/− C57BL/6J mice were constitutive knockout mice and generated by TALNEN-mediated gene targeting, DC(WT) C57BL/6J mice were purchased from GemPharmatech Co., Ltd (T051927). Conditional deficiency of CXCR1 in DCs was generated by crossing Cxcr1fl/fl mice with Itgax-cre C57BL/6J mice, which were purchased from The Jackson Laboratory (stock No. J008068). All mice were bred and maintained in the laboratory animal center of Tongji University in a specific pathogen-free (SPF) facility at room temperature and were 6–10 weeks at the beginning of the experiments.
Cell line
HEK293T cell line was provided by P. Kang (School of Life Sciences and Technology, Tongji University, Shanghai, China). HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 5% (v/v) FBS and 1% (v/v) penicillin/streptomycin at 37 °C in 5% CO2 at 100% humidity.
Reagents
MOG35–55 was purchased from GL Biochem with a purity > 95%. Complete Freund’s adjuvant (CFA) was purchased from Sigma-Aldrich. Anti-mouse CXCL5 antibody (MAB433–500) and isotype control (MAB0061) were purchased from R&D. Anti-mouse CD4-PE-Cy7 (1/100, 25-0042-82) was purchased from eBioscience. anti-mouse FOXP3-PE (1/100, 126404), anti-mouse IL-17A-PE (1/100, 506904), anti-mouse IFN-γ-APC (1/100, 505810) were purchased from BioLegend. OVA323–339 and lipopolysaccharides (LPS) from Escherichia coli O55:B5 (LPS 055: B5, L2880) were purchased from Sigma. Anti-mouse HIF-1α-PE (1/100, IC1935P) was purchased from R&D. U0126 (A1337) was purchased from Apexbio Technology. CoCl2 and KC7F2 were purchased from Sigma. Anti-mouse CXCR1 antibody (MAB330-100) was purchased from R&D.
EAE model
Male mice at 10–12 weeks of age were immunized with MOG35–55 (200 μg) in CFA containing 5 mg/ml heat-killed mycobacterium tuberculosis H37RA. Mice received 200 ng of pertussis toxin (PTX, Calbiochem) by i.p. injection on days 0 and 2 [31]. Clinical appearance was assessed daily and scored as follows: 0, no clinical signs; 1, paralyzed tail; 2, paresis; 3, paraplegia; 4, paraplegia with forelimb weakness or paralysis; and 5, moribund or death. Mice received isotype control or anti-CXCL5 antibody by i.p. injection on days 0, 4, 8, 12, and 16.
After 3–4 weeks, all the mice were euthanized, mononuclear cells in the central nervous system (CNS) were removed for flow cytometry analysis, lumbar spinal cord was collected for hematoxylin and eosin (H&E) staining or luxol fast blue (LFB) staining and histology analysis.
LPS-induced ARDS
Ten to twelve weeks-old male C57BL/6 mice, Cxcr1−/−, DC (WT), and DC (Cxcr1−/−), were subjected to LPS-induced ARDS as described [32, 33]. Briefly, 10 mg/kg LPS or vehicle was i.p. injected. Survival was monitored for up to 6 days, and survival rate was determined by Kaplan–Meier analysis. For acute lung injury, mice were injected with i.p. of 10 mg/kg LPS. Subsequently, 24 h after the LPS instillation, mice were euthanized, and bronchoalveolar lavage fluid (BALF) was collected with 1 ml of PBS and then centrifuged at 1000g for 10 min. Leukocytes in BALF were processed for CD4+IFN-γ+IL-17A− (Th1 cells) and CD4+IFN-γ−IL-17A+ (Th17 cells) by flow cytometry. Multiple soluble cytokines: IL-6, CXCL5, and IL-12p70 in BALF supernatant, were measured by ELISA kits.
To obtain lung lymphocytes, lung tissue was minced into 1–2 mm pieces and enzymatically digested for 30 min at 37 °C in 15 ml of digestion buffer (RPMI 1640, 5% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate, 1 mg/ml collagenase, 30 μg/ml DNase). After erythrocyte lysis using RBC lysis for 3 min, cells were washed, filtered over 70 μm cell strainer, and centrifuged for 30 min at 2000×g in the presence of 20% Percoll (Sigma-Aldrich) to separate leukocytes from cell debris and epithelial cells [34]. One leukocyte sample was isolated and processed for CD4+IFN-γ+IL-17A− (Th1 cells) and CD4+IFN-γ−IL-17A+ (Th17 cells) by flow cytometry, and the other was isolated for the mRNA level of Cxcl5, Hif1α, Il12a, and Il6 by qRT-PCR.
In all cases, at the experimental endpoint, the lungs were inflated with cold PBS, excised, and fixed in 4% paraformaldehyde (pH 7.2) for histological evaluation.
Histopathology
Mice were euthanized and perfused with 25 ml of PBS with 2 mM EDTA by heart puncture to remove blood from internal organs. Cervical spinal cords or lungs were collected, fixed by 10% neutral buffered formalin solution, and decalcified. Tissues were then embedded in paraffin, sectioned, and stained with standard histological methods for hematoxylin and eosin (H&E) staining for inflammation. Cervical spinal cords stained with LFB staining for demyelination.
CD4+ T cell isolation and differentiation in vitro
The methods were prepared as previously reported [35]. Briefly, splenocytes from 6–8week-old WT mice and Cxcr1−/− mice were obtained and purified Naïve CD4+ T cells by a CD4+ T cells sorting kit (Dynabeads® UntouchedTM Mouse CD4 Cells Isolation Kit, Invitrogen). Cells were activated with anti-CD28 (2 μg/ml) and anti-CD3 (2 μg/ml) and then differentiated under Treg, Th1, or Th17 differentiation conditions. For Treg differentiation, hTGF-β1 (5 ng/ml), rmIL-2 (10 ng/ml), and anti-interferon gamma (anti-IFN-γ) (10 μg/ml) were added. For Th1 differentiation, anti-IL-4 (10 μg/ml) and IL-12 (10 ng/ml) were added. For Th17 differentiation, TGF-β1 (3 ng/ml), anti-IFN-γ (10 μg/ml), anti-IL-4 (10 μg/ml), IL-6 (30 ng/ml), IL-1β (10 ng/ml), and TNF-α (10 ng/ml) were added. Cells were collected on the fourth day for flow cytometry analysis.
DCs and T cells co-culture
DCs were separated from the spleen of Cxcr1−/− or WT mice with MOG35–55 induction on day 12 by using MagniSort® Mouse CD11c Positive Selection Kit (8802-6861-74, eBioscience). Naïve CD4+ T cells were purified from the spleen of Cxcr1−/− or WT mice by using magnetic cell separation (Invitrogen). DCs and T cells were co-cultured at a ratio of 1:10. T cells were subjected to intracellular cytokine staining (ICS) and flow cytometry analysis on day 3 as follows: Th1 cells (CD4+ IFN-γ+) and Th17 cells (CD4+ IL-17A+). The supernatant was collected to measure the amount of IFN-γ, IL-17A, IL-6, IL-12p70, and TGF-β1.
To detect the proportion of T cell differentiation in co-culture after replenishing inflammatory factors (IL-6 and IL-12p70). For Th1 cell differentiation, WT and Cxcr1−/−splenic DCs were stimulated by LPS (100 ng/ml) for 12 hr, for washed and then cocultured with naïve CD4+ T cell in the presence of anti-IL4 (10 μg/ml) alone or supplemented with IL-12p70. After 3 days of co-culture, Th1 (CD4+ IFN-γ+) cells were analyzed by flow cytometry, and supernatants were collected to measure the amount of IFN-γ. For Th17 cell differentiation, WT and Cxcr1−/− splenic DCs were stimulated by LPS (100 ng/ml) for 12 h, washed, and then cocultured with naïve CD4+ T cell in the presence of anti-IL4 (10 μg/ml) and anti-IFN-γ (10 μg/ml) or supplemented with IL-6. After 3 days of co-culture, Th17 (CD4+ IL-17A+) cells were analyzed by flow cytometry, and the supernatant was collected to measure the amount of IL-17A.
In vitro permeability assay
For permeability assay, coculture transwell assay was performed using the 12-mm transwell with 0.4-mm pore size (Corning) In vitro. DCs were separated from the bone marrow of WT or Cxcr1−/−mice and cultured in complete RPMI 1640 supplemented with murine GM-CSF (20 ng/ml) and murine IL-4 (1 ng/ml) for 7 days. After 7 days, DCs (1 × 106) were stimulated by LPS (100 ng/ml) for 24 h and precultured in the upper chamber of the Transwell. During the same time, naïve CD4+ T cells were purified from the spleen of WT mice by using magnetic cell separation (Invitrogen). In total, 3 × 105 naïve CD4+ T cells were added to the bottom chamber and activated with anti-CD28 (2 μg/ml) and anti-CD3 (2 μg/ml), then differentiated under Th1 or Th17 differentiation conditions. For Th1 differentiation, anti-IL-4 (10 μg/ml) was added. For Th17 differentiation, anti-IFN-γ (10 μg/ml), and anti-IL-4 (10 μg/ml) were added. Cells were collected on the fourth day for flow cytometry analysis.
Cell adoptive transfer
Naïve GFP OT-II T cells (2 × 106) were injected into C57BL/6 mice by the lateral tail vein injecting for one day. In total, 2 × 105 splenic DCs isolated from WT and Cxcr1−/− mice were pulsed with 50 μg/ml OVA and 500 ng/ml LPS. After 8 h, the cells were then injected into recipient mice. Seven days later, mice were killed to analyze the Th1 cell (CD4+ IFN-γ+ IL-17A−) and Th17 cell (CD4+ IFN-γ− IL-17A+) proportion in the spleen.
Immunoblot analysis
Cells were lysed with SDS-PAGE loading buffer at 95 °C for 5 min and separated in 10% Bis–Tris gel, then transferred to PVDF membranes (Millipore) by electroblotting. The following primary antibodies were used: total-AKT (1/1000, CY5561), AKT phosphorylated at Ser473 (1/1000, CY6016) and Thr308 (1/1000, CY6017), ERK phosphorylated at T202/Y204 and T185/Y187 (1/1000, CY5277), total-JNK (1/1000, CY5490), GAPDH (1/1000, AB0037) were purchased by Abways Technology; total-ERK (1/1000, #9102), total-p38 (1/1000, #8690), p38MAPK phosphorylated at Thr180 and Tyr182 (1/1000, #4511), JNK phosphorylated at Thr183 and Tyr185 (1/1000, #4668), total-S6 (1/1000, #2217), S6 phosphorylated at Ser235/236 (1/1000, #4858) were purchased by Cell Signaling Technology, CXCR1 (1/500, 55450-1-AP) was purchased by Proteintech; anti-HIF-1α (1/1000, SC13515) was purchased by Santa Cruz Biotechnology.
RNA isolation and qRT-PCR (quantitative real-time PCR)
For qRT-PCR were subjected as previously described [36]. Briefly, total RNA was isolated using TRIzol reagent (Molecular Research Center, Inc.) and subjected to cDNA synthesis using M-MLV Reverse Transcriptase (Promega) with random hexamer primers. Gene expression was analyzed by real-time PCR performed with BiotoolTM 2× SYBR Green qPCR Master Mix. The expression of individual genes was calculated with a standard curve method and normalized to the expression of GAPDH. The gene-specific PCR primers (all mouse and human genes) are shown in Supplementary Table 1.
Enzyme-linked immunosorbent assay
Cytokines in culture supernatants after in vitro stimulation, in BALF, and in peripheral blood were quantified by ELISA according to the manufacturer’s instructions as follows: for mouse IL-17A, IL-6, IL-12p70, TGF-β1, and IFN-γ, Invitrogen; for mouse CXCL5/LIX, R&D Systems; for human IL-6, IL-12p70, Invitrogen.
Chromatin immunoprecipitation
The chromatin immunoprecipitation (ChIP) assay was performed with lysates from bone marrow-derived DCs after LPS stimulation by using SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling Technology). Anti-HIF-1α (Cat. #14179, Cell Signaling Technology) was used for the ChIP assay.
The following primer pairs were used:
Cxcl5 HRE-1 Forward: 5′-TGCCAAAACTAATTTGTATGCTGT-3′
Reverse: 5′-CTGGGGAGTGGTAATCTGTAACAA-3′
Cxcl5 HRE-2 Forward: 5′-TCATCTGCCCCAAGGAAGCCAAAA-3′
Reverse: 5′-ATGCAGGACCACATTGCCACATTG-3′
Vegf Forward: 5′-CTGGGCAGCTGGCCTACCTACCTT-3′
Reverse: 5′-GTTAGTCAGTGGCGGGGAGTGAGA-3′.
Plasmids
The mCXCL5 promoter was amplified from genomic DNA isolated from splenocyte of C57BL/6 mice using primers 5′-GGGGTACCATGACAAATGGATACAGGGAACAAC and 5′-GGAATTCCATATGTCCCGGACCAGGGAGCACCA and then subcloned into the pGL3 plasmid with KpnI and NdeI to create pGL3 mCXCL5 promoter-luciferase. The coding sequence of HIF-1α was amplified from the cDNA of mice splenocytes using primers 5′-CGGGATCCATGGAGGGCGCCGGCGGCGAGA and 5′-CGGAATTCCGCTCAGTTAACTTGATCCAAAGCT, then subcloned into the pcDNA3.0 plasmid with BamHI and EcoRI. The coding sequence of HIF-1β was amplified from the cDNA of mice splenocytes using primers 5′-CGGGATCCATGGCGGCGACTACAGCTAACCC and 5′-GCTCTAGATTCTATTCGGAAAAGGGGGGAA, then subcloned into the pcDNA3.0 plasmid with BamHI and XbaI.
Transfection and luciferase assays
HEK293T cells were transfected with 2 μg of each plasmid by using the calcium phosphate precipitation method. A firefly luciferase reporter gene construct (200 ng per well) and pRL-SV40 Renilla luciferase construct (1 ng per well; for normalization) were co-transfected. Cell extracts were prepared 24 h after transfection, and luciferase activity was measured with the Dual-Luciferase Reporter Assay system (Promega). Wild-type or mutated 3′ UTR sequences of Ets1 or Smo (encoding smoothened) were cloned into the modified pGL3-control vector (Promega) as described [37].
Statistical analysis
Statistical analysis was performed with GraphPad Prism (GraphPad Software). An unpaired, one-tailed parametric t-test was used for comparison of datasets. Data are presented as mean ± SEM. A Mann–Whitney U test was used to assess the significance of EAE clinical scores throughout the disease course. Spearman’s correlation coefficient, R2, was used to evaluate the association between the mRNA level of Hif1α, Cxcr1, and EDSS. A p-value of <0.05 was considered statistically significant.
Results
CXCR1 deficiency ameliorates EAE development
To identify the role of CXCR1 in MS, we first analyzed the mRNA expression of CXCR1 in peripheral blood leukocytes from 35 MS patients and 21 healthy donors (Table 1). The results indicated that CXCR1 was significantly upregulated in MS patients and positively correlated with the EDSS (Fig. 1A). The data suggested potential involvements of CXCR1 in the pathogenesis of MS. We analyzed and found that mRNA expression and protein level of CXCR1 in CNS was significantly increased in the later period of the EAE progression (Supplementary information, Fig. S1A, B). Given the increased mRNA of CXCR1 in MS patients and EAE mice, we aimed to gain insight into the role of CXCR1 in EAE development. Thus, we generated CXCR1-deficient mice (Supplementary information, Fig. S2), and CXCR1-deficiency showed no significant effects on the development of macrophages, DCs, B cells, CD4+ T cells, and CD8+ T cells (Supplementary information, Fig. S3). MOG35–55 was used to induce EAE, and results indicated that clinical score was significantly reduced in Cxcr1−/− mice (Fig. 1B). Pathologically, CXCR1 deficiency significantly ameliorated the leukocyte infiltration, inflammatory foci, and demyelination (Fig. 1C, D). In addition, CD4+ T cells isolated from CNS of Cxcr1−/− mice showed decreased expression of intracellular IFN-γ and IL-17A compared to those of WT mice (Fig. 1E). CD4+ T helper (Th) cells partly regulate immune response depending on the inflammation cytokine milieu [38, 39]. Therefore, we detected the inflammatory cytokines’ production in the serum of EAE mice, CXCR1 deficiency induced downregulation of IL-12p70 and IL-6, but not TGF-β1(Fig. 1F). Meanwhile, downregulated mRNA levels of Il12a and Il6 was detected in CNS of Cxcr1−/− mice compared with WT mice (Fig. 1G). It is well-known that DC plays a crucial role in CD4+ T cell differentiation via regulating the secretion of polarizing cytokines, such as IL-6, IL-12p70, and TGF-β1 [40]. We found that the expressions of IL-6 and IL-12p70, rather than TGF-β1, were significantly downregulated in splenic DC from Cxcr1−/− mice (Fig. 1H). Therefore, these data confirmed that CXCR1 was important for promoting EAE progression.

A CXCR1 mRNA expression in peripheral blood leukocytes in healthy controls (n = 21) and MS patients (n = 35) (left panel). Scatterplots showing the correlation between mRNA level of CXCR1 and EDSS in MS patients (right panel). WT and Cxcr1−/− mice were immunized with MOG35–55 peptides in CFA adjuvant and pertussis toxin to induce EAE. B Clinical scores of EAE in immunized WT and Cxcr1−/− mice (left panel), and linear regression analysis (right panel) of the recipient mice depicted (n = 6). The data are expressed as the mean ± SEM. *p < 0.05, vs. WT group (Mann–Whitney U test). C H&E staining and LFB staining of spinal cord paraffin sections from the WT and Cxcr1−/− mice 28 days after EAE induction. Scale bars, 200 μm. D Pooled data are presented from (C). E Representative flow cytometry data showing intracellular production of IFN-γ and IL-17A in CD4+ T cells from the spinal cord and brain of WT and Cxcr1−/− mice on 28 days after EAE induction. Pooled data are presented in the right panel. F ELISA analysis of cytokines (IL-12p70, IL-6, and TGF-β1) in the serum from WT and Cxcr1−/− mice 12 days after EAE induction. G mRNA levels of Il12a, Il6, and Tgfb1 in the brain from WT and Cxcr1−/− mice on days 0 and 21 after EAE induction. H ELISA analysis of IL-6, IL-12p70, and TGF-β1 in supernatant of DCs sorted from WT and Cxcr1−/− mice 12 days after EAE induction and restimulated with MOG35–55 for 48 h. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT group (one-tailed Student’s t-test). Data are representative of three independent experiments with similar results.
CXCR1 augments the production of inflammatory cytokines in DCs
The indispensable role of CD4+ T cells in the pathogenesis of EAE prompted us to investigate whether CD4+ T cell differentiation was affected by CXCR1. To analyze whether CXCR1 has a direct effect on T cell differentiation, CD4+ T cell differentiation was induced under different polarizing conditions in vitro. Flow cytometric analysis revealed that CXCR1 did not affect Treg, Th1, and Th17 cell differentiation, suggesting that CXCR1 had no obvious effect on T cell differentiation directly in vitro (Supplementary information, Fig. S4). To unequivocally demonstrate the participation of the CXCR1 in the production of inflammatory cytokines in DCs, we first sorted the DCs from the spleen of wild-type mice and Cxcr1−/− mice, then stimulated DCs with LPS. DCs from the spleen of Cxcr1−/− mice showed lower expressions of IL-6 and IL-12p70 compared with that from WT mice (Fig. 2A). Further, we established a co-culture model of DCs and naïve CD4+ T cells to investigate the ability of CXCR1 to regulate DCs in activating and polarizing CD4+ T cells into specific subsets. DCs from Cxcr1−/− and WT mice were activated with LPS and co-cultured with naïve CD4+ T cells. Analysis of intracellular IFN-γ and IL-17A expression revealed that Cxcr1-deficiency inhibited the ability of DCs to polarize CD4+ T cells differentiating into Th1 and Th17 (Fig. 2B, C) and found downregulated expressions of IFN-γ, IL-17A, IL-6, and IL-12p70 (Fig. 2D) (Supplementary information, Fig. S5). To gain insight into the direct roles of secreted cytokines (IL-6 and IL-12p70), we performed transwell experiment to test the T cell differentiation in presence of DCs with WT or Cxcr1-deficiency after LPS stimulated (Fig. 2E). The proportion of Th1 and Th17 as differentiation by the secreted cytokines from DCs into the bottom chamber significantly decreased by DCs with Cxcr1-deficiency (Fig. 2F).

A DCs sorted from WT and Cxcr1−/− mice and treated with LPS (100 ng/ml) for 24 h. IL-6 and IL-12p70 in DCs supernatant were examined by ELISA. B Representative flow cytometry data showing intracellular production of IFN-γ and IL-17A in CD4+ T cells from DC-T cell coculture system in vitro. C Pooled data are presented from (B). D ELISA detection of IFN-γ and IL-17A in supernatants of cocultured DC-T cells. E DCs from WT or Cxcr1−/− mice were stimulated with LPS for 24 h and cultured in a transwell to naïve CD4+ T cell differentiation. F Representative flow cytometry data showing intracellular production of IFN-γ and IL-17A in CD4+ T cells. G, H The percentage of positive cells and protein levels of IL-17A (G) and IFN-γ (H) in cocultured DC-T cells or their supplemented. Pooled data are presented in the right panel. I Schematic diagram of cell adoptive transfer. J Representative flow cytometry data showing intracellular production of IFN-γ and IL-17A in CD4+ T cells 7 days after transfer. Pooled data are presented in the right panel. Data are mean ± SEM (n = 5–6). *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT or indicated group. (one-tailed Student’s t-test). Data are representative of three independent experiments with similar results.
To determine whether the impaired IL-6 and IL-12p70 production in Cxcr1−/− DCs was related to the regulation of T cell differentiation, we applied the co-culture model of DCs and naïve CD4+ T cells and showed that additional supplementation of IL-6 and IL-12p70 significantly reversed CXCR1 deficiency-induced Th17 and Th1 differentiation impairment, respectively (Fig. 2G, H). To further evaluate the effects of CXCR1 in DCs required on CD4+ T cell differentiation in vivo, splenic DCs obtained from WT and Cxcr1−/− mice were primed with LPS and OVA and then injected into two groups of recipient mice that previously received naïve GFP OT-II T cells (Fig. 2I). A significant decrease of Th17 and Th1 differentiation was observed in mice receiving DCs with CXCR1 deficiency (Fig. 2J). Together, these data emphasized that CXCR1 is a prerequisite to produce proinflammatory cytokines by DCs and mediate T cell differentiation.
CXCR1 expression in DCs accelerates EAE progression
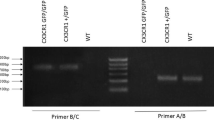
To further study the role of CXCR1 in DCs during EAE, we first assessed the expression of CXCR1 in DCs from WT mice with or without EAE. Early in the pathogenesis of EAE, DCs in the spleen were isolated from mice, and the mRNA level of Cxcr1 by quantitative real-time PCR. Compared with the WT mice without EAE, we observed the upregulated level of Cxcr1 mRNA level in WT mice with EAE (Supplementary information, Fig. S6). Thus, to determine whether CXCR1 in DCs has an important role in the regulation of EAE pathogenesis, we crossbred Itgaxcre transgenic mice to mice with CXCR1 loci flanked by loxp sequences (CXCR1fl/fl) to generate CXCR1fl/fl Itgaxcre (termed DC (Cxcr1−/−) mice), in which CXCR1 was conditional knockout in DCs (Supplementary information, Fig. S7A, B). The percentage and absolute number of DCs, macrophages, B cells, CD4+ T cells, and CD8+ T cells were not altered upon CXCR1 deficiency (Supplementary information, Fig. S7C, D). Importantly, the mRNA level of Cxcr1 in the brain of DC (Cxcr1−/−) mice with EAE was significantly lesser than that observed in WT mice with EAE but similar to that seen in the WT mice (Supplementary information, Fig. S8).
As expected, DC (Cxcr1−/−) mice showed lower disease scores than DC (WT) group after immunization MOG35–55 (Fig. 3A). The expression of IFN-γ+ CD4+ T and IL-17A+ CD4+ T cells in CNS of DC (Cxcr1−/−) mice was lower compared to that in DC (WT) mice (Fig. 3B). Associated pathologies (leukocyte infiltration, inflammatory foci, and demyelination) were dramatically ameliorated in DC (Cxcr1−/−) mice (Fig. 3C, D). We further isolated serum from DC (Cxcr1−/−) and DC (WT) mice for 12 days in the EAE model, DCs specific CXCR1-deficiency downregulated the expression of IL-12p70 and IL-6, concomitant with no difference expression of TGF-β1 (Fig. 3E). QPCR analysis revealed that Il6 mRNA level was downregulated significantly in spinal cord of mice with CXCR1-deficiency in DCs, whereas the expression of Il12a and Tgfb1 remained unchanged (Fig. 3F). These results suggested that CXCR1 deficiency in DCs restricted the progression of EAE.

WT or DC conditional knockout mice (DC (WT) or DC (Cxcr1−/−)) were immunized with MOG35–55 peptides in CFA adjuvant and pertussis toxin to induce EAE. A Mean clinical scores of EAE in DC (WT) and DC (Cxcr1−/−) mice (left panel), and linear regression analysis (right panel) of the recipient mice depicted (n = 5). The data are expressed as the mean ± SEM. *p < 0.05, vs. DC (WT) group (Mann–Whitney U test). B Representative flow cytometry data showing intracellular production of IFN-γ and IL-17A in CD4+ T cells from the spinal cord and brain of DC (WT) and DC (Cxcr1−/−) mice 28 days after EAE induction. Pooled data are presented in the right panel. C H&E staining and LFB staining (D) of spinal cord paraffin sections from the DC (WT) and DC (Cxcr1−/−) mice 28 days after EAE induction. Scale bars, 200 μm. Pooled data are presented in the right panel. E IL-12p70, IL-6, and TGF-β1 levels in the serum of DC (WT) and DC (Cxcr1−/−) mice 12 days after EAE induction were detected by ELISA. F Expression of Il12a, Il6, and Tgfb1 mRNA in the spinal cord and brain from the DC (WT) and DC (Cxcr1−/−) mice 28 days after EAE induction. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. DC(WT) group (one-tailed Student’s t-test).
CXCL5/CXCR1 axis is required for aggravating the severity of EAE
To unequivocally demonstrate the participation of CXCL5 in disease progression, we first analyzed the expression of CXCL5 in serum and the mRNA level of Cxcl5 in the brain of the EAE model. Of note, we found a significant upregulation of CXCL5 expression in serum in the early period of EAE progression and Cxcl5 mRNA in the brain in the later period of EAE progression (Supplementary information, Fig. S9A, B). An increased expression of CXCL5 both in protein and mRNA suggests that CXCL5 at least participates in the progression of EAE. Next, we neutralized CXCL5 to analyze the function of CXCL5 in EAE pathogenesis. C57BL/6 mice were induced EAE animal model by MOG35–55 and were treated at day 0/4/8/12/16 post-immunization with intraperitoneal injection of neutralizing anti-CXCL5 antibody or control IgG (Fig. 4A). After anti-CXCL5 antibody treatment, lower clinical scores were observed in mice received 1.6 μg or 5 μg anti-CXCL5 antibody compared to mice injected with IgG (Fig. 4B), with lower associated pathology (leukocyte infiltration, inflammatory foci, and demyelination) (Fig. 4C–E). CD4+ T cells isolated from CNS were quantified, and decreased percentages of CD4+ T cells were found in CXCL5 neutralized mice compared to those in IgG-treated mice (Fig. 4F). The number of Th1 and Th17 cells in the CNS showed a significant decrease in 5 μg anti-CXCL5 antibody-treated mice (Fig. 4G, H). To determine whether CXCL5 is a receptor agonist for CXCR1 to mobilize the production of inflammatory cytokines in DCs, we first measured the expression of CXCL5 in vitro. We observed the high level of CXCL5 in DCs with LPS stimulation was consistent with DCs co-cultured with naïve CD4+ T cells (Supplementary information, Fig. S9C, D). We next assessed the impact of CXCL5 in DC (Cxcr1−/−) mice during the progression of EAE. Both DC (Cxcr1−/−) mice and DC (Cxcr1−/−) mice with anti-CXCL5 antibody treatment reduced the clinical score of EAE. Meanwhile, there were no significant different between DC (Cxcr1−/−) mice and DC (Cxcr1−/−) mice with anti-CXCL5 antibody treatment (Fig. 4I, J). Overall, the data demonstrated that CXCL5 aggressively participates in the progression of EAE through CXCR1.

A Schematic diagram of EAE mice treated with CXCL5 neutralizing antibody (anti-CXCL5) or isotype control (IgG). B Clinical scores (left panel) and linear regression analysis (right panel) of the recipient mice. The data are expressed as the mean ± SEM (n = 7). **p < 0.01, ***p < 0.001 vs. IgG group (Mann–Whitney U test). C LFB staining and D H&E staining of spinal cord paraffin sections from the IgG and anti-CXCL5 (5 μg) treated mice 28 days after EAE induction. Pooled data are presented in the right panel. E Pooled data are presented from (C, D). F Frequencies of CD4+ T cells and G expression of IFN-γ and IL-17A from the spinal cord and brain 28 days after EAE induction. H Pooled data are presented from (G). *p < 0.05, **p < 0.01, ***p < 0.001 vs. IgG group (one-tailed Student’s t-test). I Schematic diagram of DC (WT) or DC (Cxcr1−/−) mice that were induced EAE and treated with 1.6 μg anti-CXCL5 or IgG. J Clinical scores (left panel) and linear regression analysis (right panel) of the recipient mice. The data are expressed as the mean ± SEM (n = 7). **p < 0.01, ***p < 0.001 vs. IgG group (Mann–Whitney U test).
HIF-1α is necessary for CXCL5/CXCR1 axis-mediated proinflammatory cytokine production in DCs
To further evaluate the effect of CXCL5 in DCs-mediated proinflammatory cytokine production, we first sorted the DCs from the spleens of wild-type and then stimulated DCs with LPS under CXCL5 neutralization antibody and found that CXCL5 neutralization led to significantly decreased IL-6 and IL-12p70 production by DCs (Fig. 5A). To determine specific molecular pathways, DCs were isolated from Cxcr1−/− and WT mice to for further detection of phosphorylation on extracellular signal-regulated kinases (ERK), protein kinase B (AKT), S6, p38, and c-Jun N-terminal kinase (JNK) after stimulated with LPS. Cxcr1 deficiency leads to significantly reduced phosphorylation of ERK, not the phosphorylation of AKT, S6, p38, and JNK (Supplementary information, Fig. S10A). HIF-1α has been reported as being activated by phosphorylated ERK and implicated in controlling cytokine production such as IL-6, and IL-12p70 [41, 42], we investigated the effect of inhibiting ERK phosphorylation on activation of HIF-1α in DCs. FACS and Immunoblot analysis showed that ERK inhibitor (U0126) treatment significantly decreased the expression of HIF-1α in DCs (Supplementary information, Fig. S10B). To further investigate if HIF-1α regulates the IL-6 and IL-12p70 production by DCs, we inhibited HIF-1α with a hypoxic inhibitor (KC7F2) and found KC7F2 treatment led to significantly decreased production of IL-6 and IL-12p70 in DCs (Supplementary information, Fig. S11). Then we interrogated whether HIF-1α expression was affected by CXCL5/CXCR1 axis in DCs and found CXCL5 neutralization and Cxcr1 deficiency both greatly reduced the protein level of HIF-1α in DCs (Fig. 5B, C) (Supplementary information, Fig. S12). We next tested the protein level of HIF-1α during the progression of EAE, spinal cords were isolated from WT and Cxcr1−/− mice at 0 day and 26 days after EAE induction; the data showed that Cxcr1 deficiency leads to prominently decreased HIF-1α expression at 26 days after EAE induction (Fig. 5D), consistent with the tendency of HIF-1α in DC (Cxcr1−/−) mice (Fig. 5E). We further analyzed whether HIF-1α was required to mediate the regulation function of CXCR1 on IL-6 and IL-12p70 production in DCs, results from cytokine detection revealed that HIF-1α agonist (CoCl2) significantly retuned decreased expressions of IL-6 and IL-12p70 caused by CXCR1 deficiency in DCs (Fig. 5F). These results demonstrated that CXCL5/CXCR1 positively regulates inflammation secretion function of DCs via HIF-1α.

DCs were sorted from spleens of WT mice and treated with LPS (100 ng/ml) for 24 h in the presence of anti-CXCL5 antibody or IgG isotype control. A Concentrations of IL-6 and IL-12p70 in culture supernatants of DCs were detected by ELISA. B Flow cytometry analyzed the expression of HIF-1α (left), Pooled data are presented from (right). C DCs were sorted from spleens of WT or Cxcr1−/− mice and stimulated by LPS or not for 24 h. The expression of HIF-1α was examined by Immunoblot analysis. D, E Four different groups of mice (WT, Cxcr1−/−, DC(WT) and DC(Cxcr1−/−)) were immunized with MOG 35–55 peptide in CFA adjuvant and pertussis toxin to induce EAE for 28 days. The spinal cord and brain lysates were probed for HIF-1α protein level by Immunoblot analysis. F Purified DCs were sorted from spleens of WT or Cxcr1−/− mice and treated for 24 h with LPS (100 ng/ml) in the presence or absence of the HIF-1α agonist (CoCl2, 200 μM). Concentrations of IL-6 and IL-12p70 in supernatants were detected by ELISA. G Purified DCs were treated for 24 h with LPS (100 ng/ml) in the presence or absence of the HIF-1α agonist (CoCl2, 200 μM) or inhibitor (KC7F2, 20 μM) and concentrations of CXCL5 in supernatants were detected by ELISA. I Schematic diagram of two HREs in the CXCL5 promoter. H, J Luciferase assay to analyze the function of HIF-1α in regulation of CXCL5 promoter activity. Luciferase activity was calculated as the ratio of firefly/Renilla luciferase activity. K ChIP assay demonstrating the direct binding of HIF-1α to CXCL5 promoter. L IL-6 or IL-12p70 production by human DCs stimulated with LPS (100 ng/ml) alone or combined with indicated conditions. M HIF1A mRNA level in peripheral blood leukocytes from MS patients (n = 20) or control healthy donors (n = 18). N Scatterplots showing the correlation between mRNA level of HIF1A and EDSS. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT or indicated group (one-tailed Student’s t-test). Data are representative of three independent experiments with similar results.
CXCL5/CXCR1/HIF-1α feedback loop positively regulates IL-6 and IL-12p70 secretion in DCs
It is known that HIF-1α is a transcription factor with the ability to regulate its own upstream activators [43, 44]. We, therefore, explored the possibility that HIF-1α upregulates CXCL5 expression, thereby forming a positive feedback loop to control inflammatory cytokines production in DCs. Interestingly we really found that the production of CXCL5 in DCs was greatly enhanced upon HIF-1α activator treatment, while significantly decreased by HIF-1α inhibitor treatment (Fig. 5G). We speculate that HIF-1α may regulate CXCL5 transcription by direct binding to its promoter region, data from luciferase reporter assay showed that luciferase activity of the Cxcl5 promoter can be significantly enhanced by overexpression of HIF-1α and 1β (Fig. 5H). Combined with the fact that two putative hypoxia response elements (HREs) containing the consensus sequence (A/G)CGTG were found within the promoter of mouse CXCL5 by using the JASPAR database (Fig. 5I), while the enhanced luciferase activity by overexpression of HIF was greatly impaired when we mutated the HIF binding sites in the promoter region of Cxcl5 (Fig. 5J). Moreover, data from chromatin immunoprecipitation (ChIP) analyses further confirmed the direct binding of HIF-1α onto the promoter region of Cxcl5 (Fig. 5K). These observations confirmed that HIF-1α is a direct transcriptional factor for CXCL5.
Further, we confirmed the roles of CXCR1 and HIF-1α regulating of IL-6 and IL-12p70 secretion in human DCs. We found that inhibition of any of CXCR1 or HIF-1α can lead to a significant reduction of IL-6 and IL-12p70 production in human DCs (Fig. 5L). Moreover, we also examined the mRNA level of HIF1A in peripheral blood leukocytes of healthy control and MS patients and found a significant increase of HIF1A in MS patients (Fig. 5M) and a positive correlation between HIF1A expression level and EDSS scores (Fig. 5N). These results confirmed that CXCR1 governs IL-6 and IL-12p70 production via increasing CXCL5/CXCR1/HIF-1α positive feedback loop in DCs.
Deletion of CXCR1 prevents LPS-induced ARDS
As DCs-mediated IL-6 production also plays a pivotal role in the pathogenesis of LPS-induced ARDS [45, 46], we further examined whether the CXCL5/CXCR1 axis is necessary for IL-6 production in LPS-induced ARDS. Compared with the WT mice, the level of CXCL5 in serum and BALF increased when stimulated with LPS (Supplementary information, Fig. S13A, B). Thus, we further examined whether CXCR1 deficiency could ameliorate ARDS. By i.p. injected with a single bolus of 10 mg/kg LPS, the mouse ARDS model was induced, and a 6-day survival rate was calculated. The results indicated that Cxcr1−/− mice showed a significantly higher survival rate compared to WT mice (Fig. 6A). Histological assessment of lung sections showed that CXCR1 deficiency prevented septa thickening and inhibited infiltration of inflammatory cells into the bronchioles (Fig. 6B). Importantly, Cxcr1−/− mice showed a strongly decreased total CD4+ T cells and IFN-γ+ and IL-17A+ percentage in CD4+ T cells (Fig. 6C, D), and accompanied by decreased IL-6 and CXCL5 in BALF (Fig. 6E). Consistently, mice lacking CXCR1 were protected from LPS-induced ARDS by exhibiting reduced CD4+ T cell percentage (Fig. 6F), the proportion of IFN-γ+ and IL-17A+ (Fig. 6G) and the mRNA levels of Il6, Il12a, Cxcl5, and Hif1α in lung tissue (Fig. 6H). Flow cytometry analysis and western blot suggested that CXCR1 deficiency resulted in the loss of HIF-1α protein level in lung tissue (Supplementary information, Fig. S13C, D).

A Percent survival of WT and Cxcr1−/− mice after LPS (10 mg/kg) intraperitoneal compared with saline controls. (n = 8, Kaplan–Meier survival curves/Mantel–Cox analysis). B H&E staining images of lung tissue from WT and Cxcr1−/− mice after saline or 10 mg/kg LPS challenge. Tissue was collected 24 h after administration. C–E Bronchoalveolar lavage fluid (BALF) was obtained after WT and Cxcr1−/− mice 24 h intraperitoneal injection of LPS (10 mg/kg). Representative flow cytometry data showing frequencies of CD4+ T cells (C) IFN-γ+ and IL-17A+ (D). Pooled data are presented in the right panel. E ELISA analysis of IL-6, IL-12p70, and CXCL5 concentrations in BALF. F, G Immune cells were sorted from the lung tissue, and the percentage of CD4+ T cells (F), IFN-γ, and IL-17A in CD4+ T (G) were analyzed by flow cytometry. Pooled data are presented in the right panel. H Quantitation of Il6, Il12a, Cxcl5, and Hif1α mRNA level in lung tissue of WT and Cxcr1−/− mice stimulated with LPS for 24 h. Data are mean ± SEM (n = 5–6 in B–E, G, H). *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT group (one-tailed Student’s t-test). Data are representative of three independent experiments with similar results.
Next, we tested whether CXCR1 deficiency in DCs can ameliorate LPS-induced ARDS. As expected, DC(Cxcr1−/−) mice exhibited higher survival rate than DC(WT) mice after LPS i.p. injection (Fig. 7A). Histological pathogenesis of lung sections, cytokine production, IFN-γ+ and IL-17A+ percentage of CD4+ T cells in BALF were all significantly alleviated in mice with conditional deletion of Cxcr1 in DCs (Fig. 7B–E). In agreement with the data from Cxcr1−/− mice, DC(Cxcr1−/−) mice showed reduced CD4+ IFN-γ+ T and CD4+ IL-17A+ T cell percentage (Fig. 7F, G) and the decreased mRNA levels of Il6, Il12a, and Hif1α in lung tissue compared with the DC(WT) mice (Fig. 7H). We concluded that CXCR1 deficiency can ameliorate LPS-induced ARDS, and the CXCL5/CXCR1/HIF-1α positive feedback loop also inhibits the production of inflammatory cytokines in LPS-ARDS.

A Percent survival of DC(WT) and DC(Cxcr1−/−) mice after LPS (10 mg/kg) intraperitoneal compared with saline controls. (n = 8, Kaplan–Meier survival curves/Mantel–Cox analysis). B H&E staining images of lung tissue from DC(WT) and DC(Cxcr1−/−) mice after saline or 10 mg/kg LPS challenge. Tissue was collected 24 h after administration. C–E Bronchoalveolar lavage fluid (BALF) was obtained after DC(WT) and DC(Cxcr1−/−) mice 24 h intraperitoneal injection of LPS (10 mg/kg). Representative flow cytometry data showing frequencies of CD4+ T cells (C) IFN-γ+ and IL-17A+ (D). Pooled data are presented in the right panel. E ELISA analysis of IL-6, IL-12p70, and CXCL5 concentrations in BALF. F, G Immune cells were sorted from the lung tissue, and the percentage of CD4+ T cells (F) and the proportion of IFN-γ and IL-17A in CD4+ T cells (G) were analyzed by flow cytometry. Pooled data are presented in the right panel. H Quantitation of Il6, Il12a, and Hif1α mRNA levels in lung tissue of DC(WT) and DC(Cxcr1−/−) mice stimulated with LPS for 24 h. Data are mean ± SEM (n = 5–6 in B–E, G, H). *p < 0.05, **p < 0.01, ***p < 0.001 vs. DC (WT) group (one-tailed Student’s t-test). Data are representative of three independent experiments with similar results.
Discussion
DCs are critical to driving the development of a myriad of autoimmune and inflammatory diseases because of their antigen-presenting function and high expression of anti-inflammation and pro-inflammation cytokines [47, 48]. It is, therefore, important to understand the mechanisms controlling cytokines secretion in DCs. In this study, we identified a mechanism that positively regulates DCs function through CXCR1 and its CXCL5 in mice. We showed that CXCR1 strengthened the pro-inflammation cytokines production of DCs and further accelerated inflammation and autoimmunity disease progression via a positive feedback loop of CXCL5/CXCR1/HIF-1α (Fig. 8).

LPS or virus infection upregulates the level of HIF-1α via CXCR1 and Erk signaling pathways in DCs. The increasing level of HIF-1α binds to the CXCL5 promoter and causes an upregulation of CXCL5 expression, then forming a CXCL5/CXCR1/HIF-1α positive feedback loop. This feedback loop promotes IL-6 and IL-12p70 expression, ultimately leading to inflammation damage in the EAE and ARDS mice model. LPS lipopolysaccharide, DC dendritic cells, ARDS acute respiratory distress syndrome.
Previous studies have shown that IL-6 and IL-12p70 secreted by DCs induce naïve CD4+ T cells to Th17 and Th1 differentiation. Most studies about the function of CXCR1 were focused on neutrophils and DCs, such as degranulation and migration, while its influence on the pro-inflammation production of DCs has not been fully understood [49, 50]. In this study, we found that CXCR1 in DCs promoted the expression of IL-6 and IL-12p70; however, there was no change in the expression of TGF-β1 in vitro and in vivo. This suggested that CXCR1 in DCs specifically regulated IL-6 and IL-12p70. Consistent with these findings, neutralized CXCL5, the expression of IL-6 and IL-12p70 also decreased. Multiple mechanisms may contribute to the inflammation cytokine secretion of DCs during autoimmunity, and mitogen-activated protein kinase (MAPK) has been shown to play a significant role in positive modulation of the inflammation cytokine secretion [51, 52]. Indeed, we found that CXCR1 altered ERK phosphorylation but not p38, PI3K-AKT, or JNK signaling pathways in DCs.
Previous studies have revealed that CXCR1 and CXCR2 receptors share 76% sequence homology with each other and interact with ELR+ CXCL chemokines playing high significance in many inflammatory diseases [49]. However, differences in the affinities of CXCR1 and CXCR2 towards CXCL chemokines monomer versus dimer may lead to different signaling outcomes, such as phospholipase D (PLD) activation and superoxide anion production in neutrophils is exclusively mediated by CXCR1, but not CXCR2 [53]. In autoimmune diseases, CXCL3-induced migration becomes exclusively CXCR1-dependent, which underlines the significance of CXCR1 in the pathogenesis of airway remodeling associated with asthma [54]. Our data indicated that CXCR1 mRNA in the peripheral white blood cells of MS patients increased more than compared with healthy controls. Further investigation is therefore warranted to understand how CXCR1 contributes to disease progression during inflammation and autoimmunity.
As a master transcription factor, HIF-1α is situated at the convergence of multiple oncogenic and tumor suppressor pathways, including MAPK/ERK pathways [42]. A previous study found that HIF-1α promoted the production of inflammatory cytokines from macrophages, including IL-6, IL-12p70, and IL-1 in LPS-induced sepsis. This result was in congruence with our results that showed inhibiting HIF-1α could downregulate IL-6 and IL-12p70 in LPS-induced DCs. We also discovered that HIF-1α promoted CXCL5 expression by directly activating CXCL5 transcription. Meanwhile, our study went one step forward by revealing that once HIF-1α was triggered by CXCR1, the transcription of CXCL5 continued to elevate, suggesting that a positive feedback loop containing CXCR1 and HIF-1α is involved in the transcription of CXCL5.
Conclusion
Collectively, this study demonstrated that pro-inflammation secretion of DCs driven by the CXCR1 played an inflammation-promoting role in inflammation and autoimmunity diseases, and a mechanism was further revealed whereby CXCR1 promoted IL-6 and IL-12p70 production via CXCL5/CXCR1/HIF-1α positively feedback loop. Given the important role of CXCR1 in LPS-infected ARDS development and progression, we propose that CXCR1 may be a future therapeutic approach in such diseases.
Data availability
All datasets generated and analyzed during this study are included in this published article and its Supplementary Information files. Additional data are available from the corresponding author upon reasonable request.
References
Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. 2016;15:551–67.
Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol. 2019;37:238–51.
Wang L, Wang FS, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med. 2015;278:369–95.
Zarrin AA, Bao K, Lupardus P, Vucic D. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discov. 2021;20:39–63.
Spinelli FR, Conti F, Gadina M. HiJAKing SARS-CoV-2? The potential role of JAK inhibitors in the management of COVID-19. Sci Immunol. 2020;5:eabc5367.
Murakami M, Harada M, Kamimura D, Ogura H, Okuyama Y, Kumai N, et al. Disease-association analysis of an inflammation-related feedback loop. Cell Rep. 2013;3:946–59.
Tait Wojno ED, Hunter CA, Stumhofer JS. The immunobiology of the interleukin-12 family: room for discovery. Immunity. 2019;50:851–70.
Hirano T. IL-6 in inflammation, autoimmunity and cancer. Int Immunol. 2021;33:127–48.
Liu J, Zhang X, Cheng Y, Cao X. Dendritic cell migration in inflammation and immunity. Cell Mol Immunol. 2021;18:2461–71.
Colorado LH, Beecher L, Pritchard N, Al Rashah K, Dehghani C, Russell A, et al. Corneal dendritic cell dynamics are associated with clinical factors in type 1 diabetes. J Clin Med. 2022;11:2611.
Fu Y, Zhan X, Wang Y, Jiang X, Liu M, Yang Y, et al. NLRC3 expression in dendritic cells attenuates CD4(+) T cell response and autoimmunity. EMBO J. 2019;38:e101397.
Liu J, Zhang H, Su Y, Zhang B. Application and prospect of targeting innate immune sensors in the treatment of autoimmune diseases. Cell Biosci. 2022;12:68.
Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. 2013;38:336–48.
Zhang Q, Zhao K, Shen QC, Han YM, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–93.
Yousif AS, Ronsard L, Shah P, Omatsu T, Sangesland M, Moreno TB, et al. The persistence of interleukin-6 is regulated by a blood buffer system derived from dendritic cells. Immunity. 2021;54:235–e235.
Tokumasa N, Suto A, Kagarni SI, Furuta S, Hirose K, Watanabe N, et al. Expression of Tyk2 in dendritic cells is required for IL-12, IL-23, and IFN-gamma production and the induction of Th1 cell differentiation. Blood. 2007;110:553–60.
Kumar V. How could we forget immunometabolism in SARS-CoV2 infection or COVID-19? Int Rev Immunol. 2021;40:72–107.
Law HKW, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood. 2005;106:2366–74.
Comabella M, Montalban X, Munz C, Lunemann JD. Targeting dendritic cells to treat multiple sclerosis. Nat Rev Neurol. 2010;6:499–507.
Gao W, Liu H, Yuan J, Wu C, Huang D, Ma Y, et al. Exosomes derived from mature dendritic cells increase endothelial inflammation and atherosclerosis via membrane TNF-alpha mediated NF-kappaB pathway. J Cell Mol Med. 2016;20:2318–27.
Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–84.
Bachelerie F, Graham GJ, Locati M, Mantovani A, Murphy PM, Nibbs R, et al. An atypical addition to the chemokine receptor nomenclature: IUPHAR Review 15. Br J Pharm. 2015;172:3945–9.
Sozzani S, Vermi W, Del Prete A, Facchetti F. Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol. 2010;31:270–7.
Lukacs-Kornek V, Engel D, Tacke F, Kurts C. The role of chemokines and their receptors in dendritic cell biology. Front Biosci. 2008;13:2238–52.
Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702.
Tiberio L, Del Prete A, Schioppa T, Sozio F, Bosisio D, Sozzani S. Chemokine and chemotactic signals in dendritic cell migration. Cell Mol Immunol. 2018;15:346–52.
Swamydas M, Gao JL, Break TJ, Johnson MD, Jaeger M, Rodriguez CA, et al. CXCR1-mediated neutrophil degranulation and fungal killing promote Candida clearance and host survival. Sci Transl Med. 2016;8:322ra310.
Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics. 2017;7:1543–88.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33:1444–52.
Wang Y, Hao N, Lin H, Wang T, Xie J, Yuan Y. Down-regulation of CASK in glucotoxicity-induced insulin dysfunction in pancreatic beta cells. Acta Biochim Biophys Sin. 2018;50:281–7.
Yang C, Lai W, Zhou J, Zheng X, Cai Y, Yang W, et al. Betaine ameliorates experimental autoimmune encephalomyelitis by inhibiting dendritic cell-derived IL-6 production and Th17 differentiation. J Immunol. 2018;200:1316–24.
Xian H, Liu Y, Rundberg Nilsson A, Gatchalian R, Crother TR, Tourtellotte WG, et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity. 2021;54:1463–77.e11.
Pedrazza L, Cunha AA, Luft C, Nunes NK, Schimitz F, Gassen RB, et al. Mesenchymal stem cells improves survival in LPS-induced acute lung injury acting through inhibition of NETs formation. J Cell Physiol. 2017;232:3552–64.
Osterholzer JJ, Curtis JL, Polak T, Ames T, Chen GH, McDonald R, et al. CCR2 mediates conventional dendritic cell recruitment and the formation of bronchovascular mononuclear cell infiltrates in the lungs of mice infected with Cryptococcus neoformans. J Immunol. 2008;181:610–20.
Wang C, Yang J, Xie L, Saimaier K, Zhuang W, Han M, et al. Methyl butyrate alleviates experimental autoimmune encephalomyelitis and regulates the balance of effector T cells and regulatory T cells. Inflammation. 2022;45:977–91.
Zhou J, Lai W, Yang W, Pan J, Shen H, Cai Y, et al. BLT1 in dendritic cells promotes Th1/Th17 differentiation and its deficiency ameliorates TNBS-induced colitis. Cell Mol Immunol. 2018;15:1047–56.
Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10:1252–9.
Bluestone JA, Mackay CR, O’Shea JJ, Stockinger B. The functional plasticity of T cell subsets. Nat Rev Immunol. 2009;9:811–6.
Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol. 2018;15:458–69.
Ohno Y, Kitamura H, Takahashi N, Ohtake J, Kaneumi S, Sumida K, et al. IL-6 down-regulates HLA class II expression and IL-12 production of human dendritic cells to impair activation of antigen-specific CD4(+) T cells. Cancer Immunol Immunother. 2016;65:193–204.
Groneberg M, Hoenow S, Marggraff C, Fehling H, Metwally NG, Hansen C, et al. HIF-1alpha modulates sex-specific Th17/Treg responses during hepatic amoebiasis. J Hepatol. 2022;76:160–73.
Wan J, Wu W. Hyperthermia induced HIF-1a expression of lung cancer through AKT and ERK signaling pathways. J Exp Clin Cancer Res. 2016;35:119.
Rezaeian AH, Li CF, Wu CY, Zhang X, Delacerda J, You MJ, et al. A hypoxia-responsive TRAF6-ATM-H2AX signalling axis promotes HIF1alpha activation, tumorigenesis and metastasis. Nat Cell Biol. 2017;19:38–51.
Piotrowski WJ, Kiszalkiewicz J, Pastuszak-Lewandoska D, Gorski P, Antczak A, Migdalska-Sek M, et al. Expression of HIF-1A/VEGF/ING-4 axis in pulmonary sarcoidosis. Adv Exp Med Biol. 2015;866:61–69.
Kormann MS, Hector A, Marcos V, Mays LE, Kappler M, Illig T, et al. CXCR1 and CXCR2 haplotypes synergistically modulate cystic fibrosis lung disease. Eur Respir J. 2012;39:1385–90.
Jose R, Williams A, Sulikowski M, Brealey D, Brown J, Chambers R. Regulation of neutrophilic inflammation in lung injury induced by community-acquired pneumonia. Lancet. 2015;385:S52.
Hussain A, Rafeeq H, Munir N, Jabeen Z, Afsheen N, Rehman KU, et al. Dendritic cell-targeted therapies to treat neurological disorders. Mol Neurobiol. 2022;59:603–19.
Galicia G, Gommerman JL. Plasmacytoid dendritic cells and autoimmune inflammation. Biol Chem. 2014;395:335–46.
Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52:856–71.e8.
Feijoo E, Alfaro C, Mazzolini G, Serra P, Penuelas I, Arina A, et al. Dendritic cells delivered inside human carcinomas are sequestered by interleukin-8. Int J Cancer. 2005;116:275–81.
Goel C, Kalra N, Dwarakanath BS, Gaur SN, Arora N. Per a 10 protease activity modulates CD40 expression on dendritic cell surface by nuclear factor-kappaB pathway. Clin Exp Immunol. 2015;180:341–51.
Xu J, Qi Y, Xu WH, Liu Y, Qiu L, Wang KQ, et al. Matrine derivate MASM suppresses LPS-induced phenotypic and functional maturation of murine bone marrow-derived dendritic cells. Int Immunopharmacol. 2016;36:59–66.
Joseph PR, Rajarathnam K. Solution NMR characterization of WT CXCL8 monomer and dimer binding to CXCR1 N-terminal domain. Protein Sci. 2015;24:81–92.
Haurogne K, Pavlovic M, Rogniaux H, Bach JM, Lieubeau B. Type 1 diabetes prone NOD mice have diminished Cxcr1 mRNA expression in polymorphonuclear neutrophils and CD4+T lymphocytes. PloS ONE. 2015;10:e0134365.
Acknowledgements
We thank Professor Zhongjun Dong, Tsinghua University (Beijing, China), for assistance with the OT-II C57BL/6J mice and Professor Jiuhong Kang, Tongji University (Shanghai, China) for assistance with the HEK293T cell line. Graphical image was drawn by Figdraw.
Funding
This work was supported by funds from the National Natural Science Foundation of China (31871404, 32070768, and 32270754).
Author information
Authors and Affiliations
Contributions
WZ, JZ, LZ, and CD designed the research. WZ and JZ performed research and wrote the paper. All authors contributed to the experimental design. JZ, JL, GL, and LX helped establish EAE and ARDS models. CS, SH, QH, and CW performed all experiments on cell lines and mice. JL, GL, RZ, CW, and KS analyzed data. All authors reviewed the paper and discussed the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Patient samples were collected with informed consent and in accordance with the Helsinki Declaration upon approval from the Ethics Committee of Tongji University. All animal experiments were performed with the approval of the Animal Care Committee of Tongji University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Yufang Shi
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhuang, W., Zhou, J., Zhong, L. et al. CXCR1 drives the pathogenesis of EAE and ARDS via boosting dendritic cells-dependent inflammation. Cell Death Dis 14, 608 (2023). https://doi.org/10.1038/s41419-023-06126-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-06126-y
- Springer Nature Limited