
Abstract
Dysregulation of auto-reactive T cells and autoantibody-producing B cells and excessive inflammation are responsible for the occurrence and development of autoimmune diseases. The suppression of autoreactive T cell activation and autoantibody production, as well as inhibition of inflammatory cytokine production have been utilized to ameliorate autoimmune disease symptoms. However, the existing treatment strategies are not sufficient to cure autoimmune diseases since patients can quickly suffer a relapse following the end of treatments. Pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), Nod-like receptors (NLRs), RIG-I like receptors (RLRs), C-type lectin receptors (CLRs) and various nucleic acid sensors, are expressed in both innate and adaptive immune cells and are involved in the development of autoimmune diseases. Here, we have summarized advances of PRRs signaling pathways, association between PRRs and autoimmune diseases, application of inhibitors targeting PRRs and the corresponding signaling molecules relevant to strategies targeting autoimmune diseases. This review emphasizes the roles of different PRRs in activating both innate and adaptive immunity, which can coordinate to trigger autoimmune responses. The review may also prompt the formulation of novel ideas for developing therapeutic strategies against autoimmune diseases by targeting PRRs-related signals.
Similar content being viewed by others

Background
Autoimmune diseases are generally considered uncommon, but they significantly affect the mortality and morbidity of the population. Rigorous epidemiological studies have reported that approximately 3 ~ 5% of the population suffers from autoimmune diseases [1]. In principle, the occurrence of autoimmune disease is triggered by specific recognition of self-antigens by adaptive immune cells. In this context, T cells, which recognize autopeptides presented by antigen-presenting cells (APCs), become autoreactive effector cells capable of producing inflammatory cytokines. Activated T cells also help B cells produce antibodies against self-tissues. Excessive inflammation and autoantibodies finally lead to different types of pathogenic symptoms. Currently, several classes of drugs, for example, non-steroidal anti-inflammatory drugs, glucocorticoids, immunosuppressive drugs (methotrexate, leflunomide and cyclophosphamide) and biological drugs (TNF inhibitors, IL-1 inhibitors, anti-T-cell co-stimulation fusion protein and anti-B-cell monoclonal antibody, et al.), are applied in clinic for autoimmune disease treatment. However, some treatments show limited efficacy and serious long-term side effects [2,3,4,5,6,7,8,9,10,11]. Moreover, the diseases frequently relapse in patients upon treatment removal [12,13,14,15,16,17,18]. Therefore, it is necessary to explore the complex mechanisms of autoimmunity and optimize treatment strategies.
Innate immunity is the first line to defend against variant pathogens by recognizing pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs). PAMPs, such as polysaccharides, glycolipids, lipoproteins, nucleotides and nucleic acids, are commonly expressed by various pathogens, while DAMPs are endogenous host molecules released from stressed or dying cells [19, 20]. PRRs, including Toll-like receptors (TLRs), Nod-like receptors (NLRs), RIG-I like receptors (RLRs), C type lectin receptors (CLRs) and various nucleic acid sensors, recognize PAMPs and DAMPs and become activated to initiate innate immune responses to produce proinflammatory molecules and clear various pathogens. However, overactivation of PRRs and excessive inflammation are involved in various autoimmune diseases, such as psoriasis, systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA) [21,22,23,24]. In addition, PRRs are also expressed in adaptive immune cells and trigger autoimmune responses directly by shaping adaptive immunity to a proinflammatory phenotype [25,26,27,28,29]. Therefore, targeting PRRs may be a promising strategy for treating autoimmune diseases. Indeed, a number of small molecules, peptides, antibodies, proteins, nanoparticles and drugs targeting PRRs have been investigated. Herein, we summarized the signaling pathways of PRRs, the potential roles of PRRs in activating autoimmunity, and their applications in treating autoimmune diseases.
TLRs and autoimmune diseases
TLRs and their ligands
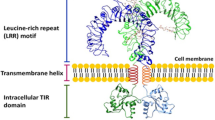
TLRs, as type I transmembrane proteins, are composed of an extracellular domain containing tandem copies of leucine-rich repeats (LRR) responsible for ligand recognition, a transmembrane domain, and an intracellular domain, also named the Toll interleukin-1 receptor homology (TIR) domain, responsible for signal transduction. TLRs can recognize 10 different PAMP types in humans and are expressed in immune cells, including dendritic cells (DCs), macrophages, natural killer (NK) cells, T and B lymphocytes and nonimmune cells such as epithelial cells, endothelial cells and fibroblasts [30]. TLR1, 2, 4, 5, 6 and 10 are mainly localized on the cell surface and are involved in the recognition of proteins and lipopeptides. Additionally, TLR3, 7, 8 and 9 are mainly localized in endosomes and are involved in the detection of nucleic acids [26, 31,32,33,34]. More specifically, TLR2 forms heterodimerizations with either TLR1 or TLR6, which respond to triacylated lipopeptides from gram-negative bacteria or diacylated lipopeptides from mycoplasma and gram-positive bacteria, respectively [34, 35]. TLR4 recognizes lipopolysaccharide (LPS) principally from the germ-negative bacterial cell wall. TLR5 recognizes flagellin, the major structural protein of bacterial flagella. In addition, TLR3 recognizes double-stranded RNA (dsRNA), TLR7 and TLR8 recognize single-stranded RNA (ssRNA), and TLR9 recognizes unmethylated CpG DNA from viruses, bacteria, and the host [34]. TLR10 is mostly expressed on the plasma membrane of monocytes. Thus far, the ligand of TLR10 remains to be illuminated. Recently, TLR10 was reported to play an anti-inflammatory role by suppressing the production of IL-6, TNF-α and IL-1β and inhibiting the activation of T cells by DCs [36, 37].
The downstream pathways of TLRs signaling
Four major adaptor proteins, myeloid differentiation primary response 88 (MyD88), MyD88 adapter like (Mal), TIR-domain containing adapter inducing interferon-β (TRIF) and TRIF-related adaptor molecule (TRAM), and two pathways, the MyD88-dependent pathway and TRIF-dependent pathway, are involved in TLR signaling (Fig. 1). Most TLRs use the MyD88-dependent pathway, except for TLR3. TLR2/1, TLR2/6 and TLR4 use MyD88 to initiate signaling in the presence of Mal, while TLRs 5, 7, 8 and 9 do not need Mal. MyD88 interacts with interleukin 1 receptor associated kinase 4 (IRAK-4), and IRAK-4 then phosphorylates IRAK-1 and IRAK-2, which in turn activate TNF receptor associated factor 6 (TRAF6). TRAF6 then activates TGF-β-activated kinase 1 (TAK1) in cooperation with TAK1-binding proteins (TAB1-3). TAK1 activates NF-κB and AP1 through the IKK and MAPK pathways, which induce the transcription of proinflammatory cytokines. In addition, TLR7, 8, and 9 trigger the IRAK-TRAF6-TRAF3-IKKα-dependent activation of IRF7, leading to transcription of type I interferons. Moreover, TLR3 signals through a TRIF-dependent pathway. TLR3 not only induces proinflammatory cytokines through TRAF6-receptor-interacting protein-1 (RIP-1)-TAK1-dependent activation of NF-κB and AP1 but also induces type I interferons by triggering TANK-binding kinase 1 (TBK1)-dependent activation of IRF3 [38,39,40,41,42,43]. After binding to LPS at the cell surface, TLR4 initially signals through the MyD88 pathway. Subsequently, receptor internalization into endosomes triggers the TRIF-dependent pathway but additionally requires TRAM [38, 44, 45].

The signaling pathways for TLRs. Cell membrane receptors, TLR2/1, TLR2/6, TLR5 and TLR4, and endosomal membrane receptors, TLR7, TLR8 and TLR9 are activated by their ligands. Then they interact with MyD88 and recruit the IRAK complex and TRAF6 to activate TAK1. TAK1 not only activates IKK complex to induce NF-κB activation, but also activates MKK to induce AP1 activation, which result in the transcription of proinflammatory cytokines (IL-1β, IL-6, IL-8 and TNF-α). Activation of TLR7, TlR8 and TLR9 trigger the IRAK-TRAF6-TRAF3-IKKα-dependent activation of IRF7, leading to transcription of type I IFNs. TLR3 signals not only through TRIF-TRAF6-RIP1-TAK1-dependent activation of NF-κB and AP1 pathways, but also through TBK1 dependent activation of IRF3 pathway. TLR4 initially signals on cell membrane through MyD88-dependent pathway. Subsequently, receptor internalization into endosomes triggers TRIF-dependent pathway, but additionally requires TRAM. The ligand and signaling pathway of TLR10 remain unclear
Relationship between TLRs and autoimmune diseases
TLRs are essential for innate immunity against infection. However, inappropriate TLR responses cause acute and chronic inflammation and further autoimmune diseases [46, 47]. For example, TLR2/1 signaling activation induces proinflammatory cytokines, such as IL-1β, TNF and IL-6, and is implicated in RA [48, 49]. TLR7 and TLR9 signaling activation-induced type I interferons are implicated in SLE [47, 50]. Moreover, some TLRs activate DCs and macrophages to produce proinflammatory cytokines and chemokines and upregulate costimulatory molecules, which in turn regulate adaptive immune cells and lead to autoimmune diseases [44, 51]. For example, TLR2 and TLR4 activation induces inflammation in the liver. The sustained increase in IL-6 and IL-12 inhibited the suppressive function of Tregs. Meanwhile, increasing IL-12 and IL-4/IL-25 promoted Th1 and Th2 responses, respectively. Thus, immune tolerance against liver tissue is disrupted, which results in autoimmune hepatitis (AIH) [52]. TLR2- or TLR4-mediated stimulation was also reported to promote DCs to produce IL-10, induce an augmented Th2 immune response, and aggravate systemic sclerosis (SSc) [53]. In addition, TLR7-mediated IL-6, IL-1β, and IL-23 production by DCs promotes the Th17 response, which results in severe experimental autoimmune uveitis (EAU) [54]. Hence, TLRs expressed on innate immune cells may promote autoimmune diseases by boosting inflammation and impacting adaptive immune responses.
In addition, some adaptive immune cells can express TLRs, which are involved in autoimmune disease [55, 56]. For example, TLR2 activation in CD4+ T cells is involved in experimental autoimmune encephalomyelitis (EAE) development by augmenting the Th17 response [27], while TLR4 activation induces EAE development by augmenting both Th1 and Th17 responses [57]. Cleonice et al. reported that the expression of TLR2, TLR4 and TLR9 was significantly higher on CD4+ and CD8+ T lymphocytes from multiple sclerosis (MS) patients than on those from healthy individuals. The expansion of different Th17 phenotypes expressing TLR2, TLR4 and TLR9 was associated with MS disease activity [58]. Furthermore, some TLRs can regulate the functions of Tregs. The activation of TLR2, TLR8 and TLR9 by their ligands can abrogate the suppressive function of Tregs [59, 60]. In contrast, the TLR5 ligand flagellin can enhance the suppressive function of Tregs by enhancing FOXP3 expression [61]. The TLR3 ligand polyI:C was reported to induce IFN-γ+Foxp3+ Tregs that have potent immunosuppressive functions and prevent food allergy (FA) development in mice [59]. Moreover, B cells were reported to recognize nucleic acids via TLR7 and TLR9 and produce autoreactive antibodies, promoting the development of SLE [62,63,64]. Thus, TLRs expressed on adaptive immune cells may promote autoimmune diseases, although there is a debate on Treg functions regulated by different TLRs.
NLRs and autoimmune diseases
NLRs and their ligands
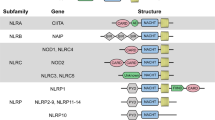
NLRs are distributed in the cytoplasm and mostly recognize invading bacteria. A total of 22 NLRs are detected in humans. All NLRs contain a C-terminal LRR, a central nucleotide-binding oligomerization domain (NOD or NACHT), and an N-terminal effector domain. The N-terminal effector domain binds with adaptor molecules and triggers downstream signals. NLRs can be divided into five subfamilies according to unique N-terminal effector domain: NLRA containing an acidic transactivation domain (AD), NLRB containing a baculovirus inhibitor of apoptosis protein repeat (BIR) domain, NLRC containing a caspase activation and recruitment domain (CARD), NLRP containing a pyrin domain (PYD) and NLRX containing an unidentified domain or mitochondria-localization sequence (MTS) [65,66,67]. The NLRA subfamily has a single member, MHC-II transcription activator (CIITA), whose primary function is to regulate the expression of MHC-II in different populations of APCs [67]. NLRB, also known as neuronal apoptosis inhibitory protein (NAIP), is expressed in the central nervous system, placental liver, spleen, lung, and peripheral blood leukocytes and exerts antiapoptotic effects [22, 68]. NLRC consists of five members (NLRC1–5). NLRC1 (NOD1) is expressed in a variety of cells, including epithelial cells, stromal cells, and endothelial cells, and recognizes g-D-glutamylmeso-diaminopimelic acid (iE-DAP), which is produced by most gram-negative and several gram-positive bacteria. NLRC2 (NOD2), mainly expressed in monocytes, DCs, macrophages, B cells and T cells, recognizes the muramyl dipeptide peptidoglycan (MDP) motifs present among both gram-positive and gram-negative bacteria and ssRNA of virus [69]. NLRC3 is expressed in myeloid cells, epithelial cells and T cells, and its ligands are largely unknown [70]. NLRC4 is expressed in epithelial cells and functions together with the NLRB protein in sensing bacterial flagellin and components of the bacterial T3SS and in forming inflammasomes [71]. NLRC5 is mostly expressed in bone marrow, lymph nodes, spleen, and mucosal surfaces, such as the lung, small intestine, colon, and uterus. It regulates MHC I transcription and expression and complements CIITA in T cell recognition [72]. The NLRP family consists of several members, of which NLRP1, NLRP3, NLRP6, NLRP7 and NLRP12 are reported to form inflammasomes in response to microbial pathogens, UV light, crystalline particles, potassium efflux and mitochondrial reactive oxygen species (ROS) production [73]. NLRP10 is widely expressed in myeloid cells, epithelial cells, and keratinocytes and plays a role in immunoregulation other than invading pathogen recognition function [74,75,76,77]. Until now, NLRX1, the only member of the NLRX family, was reported to recognize viral dsRNA in airway epithelial cells and result in barrier dysfunction in airway epithelia by inducing ROS production [67, 78].
The downstream pathways of NLRs signaling
NLRC1 and NLRC2 are in an autoinhibited monomeric state in the cytoplasm. Upon ligand recognition, NLRC1 and NLRC2 self-oligomerize and recruit receptor-interacting serine/threonine-protein kinase 2 (RIPK2) through homotypic CARD-CARD interactions. RIPK2 then recruits and activates TAK1, which induces the activation of the IKK-NF-κB and MAPK-AP1 signaling pathways, resulting in the expression of proinflammatory cytokines [79]. RIPK2 also activates the TRAF3-IKKε-IRF7 signaling pathway, resulting in the production of type I IFNs. Once NLRC2 is activated by viral ssRNA, NLRC2 oligomerizes and then interacts with the mitochondrial antiviral signaling (MAVS) protein and TRAF3 to activate IRF3. Afterward, IRF3 translocates into the nucleus and induces type I IFN expression [80]. Activated NLRC4/NAIP and NLRP members (NLRP1, 3, 6, 7 and 12) recruit apoptosis-associated speck-like protein (ASCs) containing a CARD and caspase-1 to form the basis of the inflammasome. Activated caspase-1 cleaves pro-IL-1β and pro-IL-18 into their active forms, IL-1β and IL-18. It also cleaves gasdermin D (GSDMD) and then triggers pyroptosis [65] (Fig. 2). Currently, the signaling mechanisms of NLRC3, NLRP10 and NLRX1 are unclear and need further investigation.

The signaling pathways for NLRs. Activated NLRC1 and NLRC2 respectively self-oligomerize, and then recruit RIPK2. RIPK2 not only activates TAK1 to induce IKK-NF-κB and MAPK-AP1 pathway, but also interacts with TRAF3 to induce TBK1-IKKε complex and further activates IRF7, which result in the transcription of proinflammatory cytokines (IL-1β, IL-6, IL-8 and TNF-α) and type I IFNs, respectively. Once NLRC2 is activated by ssRNA of virus, NLRC2 oligomerizes and then interacts with MAVS protein and TRAF3 to activate IRF3 pathway, which induces expression of type I IFNs. Activated NLRC4/NAIP and NLRP members (NLRP1, 3, 6, 7 and 12) recruit ASCs and caspase-1 to form the basis of the inflammasome. The activated caspase-1 not only cleaves pro-IL-1β and pro-IL-18 into IL-1β and IL-18, but also triggers pyroptosis by cleaving GSDMD. So far, the downstream signaling pathways of NLRC3, NLRP10 and NLRX1 remain to be investigated
Relationship between NLRs and autoimmune diseases
NLRs play variable roles in the development and progression of autoimmune diseases by affecting innate immunity. For example, NLRC1 was reported to be overexpressed in ulcerative colitis (UC) patients and caused excessive activation of the innate immune response in a mucosal cell line [81]. Unlike NLRC1, the loss of NLRC2 functions resulted in downregulated pathogen clearance capacity of the host and maintained lasting infection and chronic inflammation, which was associated with the development of Crohn’s disease (CD) [69, 82]. Additionally, NLRC2 polymorphisms were reported to be involved in Blau syndrome [80]. Moreover, an inherited mutation of NLRC4 in familial cold autoinflammatory syndrome (FCAS) was observed. The mutant NLRC4 activated caspase-1 and resulted in the increased secretion of IL-1β [83]. Transgenic mice that expressed mutant NLRC4 under the invariant chain promoter developed dermatitis and arthritis, accompanied by bone erosion [83]. Therefore, NLRC4 is a causative gene for FCAS and plays roles in the pathogenesis of human inflammatory diseases [83].
In addition, NLRs also influence the development of autoimmune diseases by regulating adaptive immune responses. NLRC3 is expressed both in CD4+ T cells and in DCs and limits autoimmunity by different pathways [70, 84]. Uchimura et al. revealed that NLRC3 functioned as a negative regulator affecting the proliferation of both Th1 and Th17 cells, limiting IFN-γ and TNF expression by CD4+ T cells and restricting autoimmunity by attenuating T cell signaling and metabolic pathways [70]. Moreover, NLRC3-overexpressing DCs reduced EAE progression by attenuating the antigen-presenting function of DCs via the p38 signaling pathway. NLRC3 reduced DCs’ ability to activate and polarize CD4+ T cells into Th1 and Th17 subsets [84]. Therefore, NLRC3 serves as an anti-inflammatory mediator to regulate the T cell response, either directly or indirectly. In addition, NLRC1 and NLRC2 were shown to contribute to the induction of mucosal Th1 and Th17 immune responses during infection [80]. Similarly, NLRP10 and NLRP12 have been reported to display anti-inflammatory functions or pro-inflammatory functions in different pathogen infections [74,75,76,77, 85,86,87]. However, their roles in the development of autoimmune diseases by regulating adaptive immune responses remain unknown.
RLRs and autoimmune diseases
RLRs and their ligands
RLRs localize in the cytosol in various cell types, such as myeloid cells, epithelial cells, and cells of the central nervous system [88]. RLRs are key sensors of viral infection and belong to the DExD/H (x can be any amino acid residue) box RNA helicase family. This protein family consists of three members: retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2). All RLRs contain a central DEAD box with RNA helicase activity and a C-terminal domain (CTD, also known as the regulatory or repressor domain (RD)), which are critical for RNA recognition. RIG-I and MDA5 also have two N-terminal CARD domains interacting with cascade signal molecules [89, 90]. LGP2 lacks an N-terminal CARD domain and is widely believed to regulate RIG-I and MDA5 [91]. In addition, RIG-I recognizes 5’-ppp-dsRNA, 5’-ppp-ssRNA, and blunt-ended short dsRNA (< 20 bp), while MDA-5 can recognize long dsRNA (> 1 kbp) with a blunt end, which is known to be generated by picornaviruses [91,92,93].
The downstream pathways of RLRs signaling
RIG-I and MDA5 are activated by virus-derived RNA. They then undergo lysine 63-linked polyubiquitin modification and form homotetramerization. Afterward, homotetramerization recruits MAVS and activates TRAF, which further activates the TRAF3-TBK1-IKKε-IRF3/IRF7 pathway to produce type I IFNs and activates the RIP1-IKK complex-NF-κB pathway to induce proinflammatory cytokine secretion, including IL-1β, IL-6, IL-8 and TNF-α [92,93,94] (Fig. 3).

The signaling pathways for RLRs. After RNA virus or DNA virus infection, RIG-I recognizes 5’-ppp-dsRNA, 5’-ppp-ssRNA, and short dsRNA, while MDA-5 recognizes long dsRNA. Then RIG-I and MDA5 undergo lysine 63-linked polyubiquitin modification and form homotetramerization, respectively. Afterward, the homotetramerization recruits the MAVS and activates TRAF, which further activates TRAF3-TBK1-IKKε-IRF3/IRF7 pathway to produce type I IFNs, and activates RIP1-IKK complex-NF-κB pathway to induce proinflammatory cytokines secretion, including IL-1β, IL-6, IL-8 and TNF-α
Relationship between RLRs and autoimmune diseases
Aberrant RLR activation leads to several types of autoimmune diseases, such as amyopathic dermatomyositis (ADM) [95], type 1 diabetes [96], SLE [97], Aicardi-Goutieres syndrome (AGS) [98], and Singleton-Merten syndrome (SMS) [99]. An autoantibody to MDA5 was detected in a subpopulation of ADM patients, and the level of autoantibody was strongly correlated with disease severity [99, 100]. MDA5 was also reported to be associated with type 1 diabetes [88, 101, 102]. Several polymorphisms in IFIH1, which encodes MDA5, were found to be associated with resistance to type 1 diabetes, such as T946A, E627*, I923V, R843H, IVS8 + 1, and IVS14 + 1 [103,104,105,106]. Aberrant MDA5-mediated IFN production during picornavirus infection of pancreatic cells is one of the probable pathogeneses of the onset of type 1 diabetes [88]. In addition, other mutations of MDA5 were detected in patients with SLE, AGS, and SMS, and all these diseases exhibited a type I IFN signature [107,108,109]. In addition, RLRs are involved in adaptive immune responses. It has been reported that activated RLRs markedly attenuate TLR-induced Th1 and Th17 responses by suppressing IL-12b expression in DCs and macrophages. Meanwhile, activated RLRs also reshaped the immune system toward a Th2 response, possibly by inducing the polarizing cytokine IL-4 [110]. Furthermore, RIG-I activation promoted germinal center reactions and T follicular helper cell responses, resulting in the production of long-lasting antibodies and augmented antibody affinity [111]. However, aberrant activation of RLRs causing autoimmune diseases by regulating adaptive immune responses has not been reported. Hence, RLR-mediated excessive innate immune responses, inflammatory cytokines and autoantibody production are the main reasons for RLR-associated autoimmune diseases.
CLRs and autoimmune diseases
CLRs and their ligands
CLRs are characterized by containing at least one C-type lectin-like domain (CTLD) to bind carbohydrates, which is also named the carbohydrate recognition domain (CRD). CLRs are mainly expressed by myeloid cells and play important functions in the antifungal immune response [112]. CLRs can be divided into three subgroups based on their intracellular signaling motifs: CLRs with immunoreceptor tyrosine-based activation motif (ITAM)-like domains (also named hem-ITAM), for example, DC-associated C-type lectin-1 (Dectin-1); CLRs associated with ITAM containing the Fc receptor γ chain (FcR-γ), for example, Dectin-2, Dectin-3, and Mincle; and CLRs containing nonimmunoreceptor tyrosine-based motifs, for example, DC-specific ICAM3-grabbing nonintegrin (DC-SIGN) [113]. Dectin-1 recognizes β-glucans, while Dectin-2, Dectin-3, Mincle and DC-SIGN can recognize mannans and mannoproteins [113, 114].
The downstream pathways of CLRs signaling
Upon recognizing ligands, Dectin-1 homodimerizes and transduces intracellular signaling directly through hem-ITAM within its cytoplasmic tails, while Dectin-2 and Mincle heterodimerize with Dectin-3 and transduce intracellular signaling through FcR-γ containing ITAM [115]. CLR activation induces the tyrosine phosphorylation of hem-ITAM or ITAM by Src family kinases, leading to the recruitment and activation of Syk kinase. Subsequently, the kinase initiates caspase activation, recruits the complex of domain-containing protein 9 (CARD9)/B-cell lymphoma/leukemia 10 (BCL-10)/mucosa-associated lymphoid tissue 1 (MALT1), activates the canonical NF-κB signaling pathway and induces proinflammatory molecule production [116]. In addition, Dectin-1 activation also induces inflammatory cytokines by five other pathways: (i) Dectin-1-Syk kinase signaling initiates ROS production, which can mediate NLRP3 inflammasome formation and maturation of IL-1β and IL-18 [112]; (ii) Dectin-1-Syk kinase signaling is also involved in the formation of noncanonical caspase-8 inflammasome, which is responsible for active IL-1β production [117]; (iii) Dectin-1-Syk kinase signaling initiates nuclear factor of activated T cells (NFAT) activation in a calcineurin-dependent fashion, which integrates with NF-κB signaling [112]; (IV) Dectin-1 signaling pathway induces Syk-dependent activation of Ras-GRF1, which recruits H-Ras via CARD9 and ultimately leads to extracellular signal-regulated protein kinase (ERK) activation [112, 118]; (V) Dectin-1 transduces signals through the Raf-1-dependent pathway, as does DC-SIGN. Dectin-1 and DC-SIGN activate Ras, induce Raf-1-mediated noncanonical NF-κB signaling activation and subsequently promote proinflammatory molecule expression [119] (Fig. 4).

The signaling pathways for CLRs. Dectin-1, Dectin-2/Dectin3 and Mincle/Dectin-3 are activated by their ligands and then activate Syk. Afterward, Syk initiates caspase activation and recruits CARD9/BCL10/MALT1 complex to induce the expression of canonical NF-κB dependent proinflammatory molecules. Moreover, Dectin-1-Syk kinase signaling (i) initiates ROS production, which can mediate NLRP3 inflammasome formation and maturation of IL-1β and IL-18; (ii) promotes noncanonical caspase-8 inflammasome and then actives IL-1β production; (iii) initiates NFAT activation; (iv) mediates Ras-GRF1/H-Ras/CARD9 complex formation to induce ERK activation. In addition, Dectin-1 and DC-SIGN activate Ras, then induce Raf-1 mediated non-canonical NF-κB activation and subsequently promote proinflammatory molecule expression
Relationship between CLRs and autoimmune diseases
Excessive activation of CLRs can facilitate the development of autoimmune diseases by regulating innate and adaptive immunity [120,121,122]. Blockade of Dectin-1 can prevent SKG arthritis triggered by β-glucans by inhibiting the activation of synovial cells, including synovial macrophages/DCs and granulocytes [123]. Consistently, Dectin-1-deficient mice are resistant to both dextran sodium sulfate (DSS)- and CD45RBhigh naïve CD4 T cell-induced colitis because of an increase in Treg cells [124]. Conversely, Dectin-1-activated DCs promote the Treg response to pancreatic β-cell antigen and prevent type 1 diabetes by producing large amounts of the immune regulatory cytokines IL-2, IL-10 and TGF-β1 [125]. These studies suggest that Dectin-1 plays different roles in different types of autoimmune diseases. Dectin-2 and Mincle activation were reported to induce EAU through the CARD9 signaling axis and IL-17 production [126, 127]. In addition, the expression of Dectin3 and Mincle on myeloid cells in the central nervous system is crucial for T cell recruitment and reactivation into a pathogenic Th17 phenotype, and their lower expression is associated with a drastic reduction in EAE incidence [128]. In addition, the activation of Mincle-Syk signaling aggravates intestinal inflammation by inducing macrophage pyroptosis and triggers Crohn's disease [129].
Other sensors and autoimmune diseases
Nucleic acid sensing PRRs, including DNA sensors and RNA sensors, can trigger an immune response and are associated with autoimmune diseases. Apart from TLRs, RLRs and CLRs, there are several nucleic acid sensing PRRs, such as cyclic GMP-AMP (cGAMP) synthase (cGAS), stimulator of interferon genes (STING), AIM-like receptors (ALRs) and non-RLRs DExD/H-box family of helicases (DEAH-box helicases (DHX) family and DEAD-box helicases (DDX) family) [130].
c-GAS-STING
cGAS, as a DNA sensor, recognizes cytoplasmic DNA of bacteria or viruses, catalyzes the synthesis of cGAMP, and then activates STING. STING recruits and activates TBK1, which in turn phosphorylates STING. Phosphorylated and activated STING recruits and licenses IRF3 for phosphorylation by TBK1. Finally, phosphorylated IRF3 enters the nucleus and induces IFN-β, CCL2 and CCL20 production [130]. The cGAS-STING pathway mediates protective immune defense against infection by a large variety of DNA-containing pathogens [131]. However, aberrant activation of the cGAS-STING pathway by self-DNA can also lead to autoimmune diseases, such as AGS [132]. Some molecules that can inhibit the cGAS-STING signaling pathway have been shown to alleviate autoimmune disease, as reviewed by Zhou et al. [133], supporting that cGAS-STING is one of the inducers of the development of autoimmune diseases.
ALRs
ALRs consist of 4 proteins in humans, including gamma-interferon-inducible protein 16 (IFI16), absent in melanoma 2 (AIM2), myeloid cell nuclear differentiation antigen (MNDA) and interferon-inducible protein X (IFIX) [19]. After recognizing exogenous and endogenous DNA in the cytoplasm or nucleus, ALRs bind to ASCs and recruit pro-caspase-1 to form inflammasomes. Activated caspase-1 not only promotes the maturation of IL-1β and IL-18 but also promotes cell pyroptosis [130, 134]. In addition, some ALRs can also induce IFN-β production [134]. ALR activation is important for pathogen clearance under physiological conditions. However, aberrant ALR signaling can cause autoimmunity by producing excessive proinflammatory molecules and promoting autoreactive T and B cell responses [134, 135]. Until now, IFI16 and AIM2 have been reported to be involved in the pathogenesis of SLE and RA [134,135,136,137].
Non-RLRs DExD/H-box family of helicases
The non-RLR DExD/H-box family of helicases, such as DHX9, DHX36, DDX41 and DDX3, has proinflammatory effects. DHX9 and DHX36 are known to interact with unmethylated CpG-DNA and then activate downstream NF-κB and IRF7 signaling [138, 139]. DDX41 recognizes ds-DNA or cGAMP and then interacts with STING and triggers TBK1 to activate downstream IRF3 signaling [138]. DDX3, as an RNA sensor, induces antiviral immunity in DCs [140]. However, the role of the non-RLR DExD/H-box family of helicases in the development of autoimmune diseases has not been reported and needs to be explored.
Targeting innate immune sensors in the treatment of autoimmune diseases
Recently, an increasing number of inhibitors suppressing innate immune molecule responses have been studied, some of which have been applied to the treatment of autoimmune diseases. Here, we reviewed the research progress of these inhibitors according to their targets at three levels, namely, targeting receptors, signal transduction and terminal inflammatory molecules. The characteristics of inhibitors and their applications in different autoimmune diseases are summarized in Table 1.
Inhibitors targeting innate receptors
Inhibitors targeting TLRs
TLR antagonists, including small molecules, oligonucleotides, peptides, antibodies, proteins, nanoparticle inhibitors and drugs, have been developed for blocking ligand recognition or receptors dimerization, of which eight antagonists have been applied in the clinic or studied in clinical trials (Table 1). Three antimalarial drugs, chloroquine, hydroxychloroquine, and quinacrine, act as antagonists of TLR7, 8, 9 and have been used to treat SLE and RA. These drugs inhibit endosomal acidification and directly interact with nucleic acids to prevent their binding to endosomal TLRs [46, 141]. Clinical observations showed that these drugs could alleviate disease, prevent disease relapse, reduce disease complications and promote the prognosis of patients [142]. However, various adverse effects including gastrointestinal effects, myopathy, cardiotoxic effects, and retinopathy limit their clinical applications [142]. CpG-52364 (a quinacrine derivative) was more effective and safer compared to hydroxychloroquine in an animal study, and its Phase I clinical trial in SLE patients was completed [143]. Two TLR antagonists have finished phase II clinical trials in psoriasis patients. The results showed that the Psoriasis Area Severity Index score was decreased in psoriasis patients treated with immune modulatory oligonucleotide- (IMO-) 3100, an antagonist of TLR7 and TLR9 [141]. IMO-8400, an oligonucleotide-based antagonist of TLR7,8,9, was reported to be safe and effective in the treatment of plaque psoriasis patients, only with mild adverse effects [144]. Moreover, several clinical trials to evaluate the effects of NI-0101 (a humanized monoclonal antibody that interferes with TLR4 dimerization) and baclofen (a TLR3 and TLR4 signaling small molecule inhibitor) are in the progress in RA patients and MS patients [145,146,147].
In addition, some TLR inhibitors have been verified to be effective in the treatment of autoimmune diseases in mouse models. For example, ( +)-naltrexone [( +)-NTX], an antagonist of TLR2 and TLR4, was effective at treating MS-related memory deficits with both lower- and higher-dose in EAE mice but failed to alleviate EAE-induced motor deficits [148]. TLR7/9 antagonists, compound 29 and IRS-954 (immunoregulatory DNA sequence-954), showed therapeutic efficacy in a preclinical murine model of psoriasis and lupus, respectively [149, 150]. Wang et al. found that total coumarins from Urtica dentata Hand could prevent murine autoimmune diabetes by suppressing the TLR4 signaling pathways in DCs [151]. In addition, drug-coated nanoparticles tend to accumulate in inflammatory joints and thus possess augmented effectiveness. Opuntiol-coated silver and gold nanoparticles (OP-AgNPs and OP-AuNPs) were reported to prevent disease progression in complete Freund's adjuvant (CFA)-induced arthritic rats by suppressing the expression of TLR2 and TLR4 [152]. Therefore, PRR inhibitors coated nanoparticles with increasing bioavailability, stability and targeted drug delivery capacity are promising strategies for autoimmune diseases.
Inhibitors targeting NLRs
Overactivation of the NLRP3-containing inflammasome has been widely associated with various autoimmune diseases, including SLE, RA, SSc, IBD, MS and autoimmune thyroiditis (AIT) [153,154,155,156,157,158,159]. Thus, targeting NLRP3 is a promising strategy to treat autoimmune diseases. Indeed, Tofacitinib, a Janus kinase (JAK) inhibitor, was recently reported to alleviate collagen-induced arthritis (CIA) by inhibiting the NLRP3 inflammasome [160]. RRx-001, a well-tolerated anticancer drug undergoing phase III clinical trials, was reported to specifically inhibit the activation of the NLRP3 inflammasome and attenuate the symptoms of dextran sulfate sodium (DSS)-induced colitis and EAE in mice [161]. CY-09, which can inhibit NLRP3 ATPase activity, was shown to have remarkable therapeutic effects in mouse models of CAPS and type 2 diabetes [162]. In addition, OLT1177, thiolutin and MCC950 significantly ameliorated the clinical signs of EAE mice by specifically targeting NLRP3 inflammasome [160, 161, 163]. Although a large number of mouse studies have shown that NLRP3 inhibitors are effective in the treatment of autoimmune diseases, the clinical use of NLRP3 inhibitors has not been reported.
Inhibitors targeting RLRs
MDA5 is involved in several autoimmune diseases, including type 1 diabetes, MS, psoriasis and SLE [96, 164,165,166]. There is no report of inhibitors targeting MDA5. However, some MDA5-binding proteins could be exploited as therapeutic targets by blocking their interactions. For example, ARL5B was found to prevent the interaction between MDA5 and dsRNA by binding to MDA5 [167]. DNAJB1 disrupted MDA5 multimer formation after binding to MDA5, resulting in the suppression of type I IFN production [96]. In addition, ubiquitin-specific protease 3 (USP3), a deubiquitinase, was reported to bind to MDA5, remove K63-polyubiquitin chains on the N-terminal CARDs, and then inhibit MDA5 activity [168]. The treatment effect of small molecules targeting MDA5 binding proteins needs further study.
Inhibitors targeting CLRs
Dysregulation of CLRs is associated with the development of autoimmune diseases, allergies, and cancer [121]. Although several agonists of CLRs for cancer treatment have been applied in the clinic [169], targeting CLRs in the treatment of autoimmune diseases are still in the early experimental stage. For example, laminarin, a Dectin-1 antagonist, was reported to suppress the development of DSS-induced colitis by inducing Treg cells in mice [124].
Inhibitors targeting signal transduction molecules
Inhibitors targeting TLR signal transduction molecules
Several molecules, including MyD88, IRAK4, TRAF6, TAK1, TRIF, TBK1 and NF-κB, are involved in TLR-associated signal transduction. Inhibitors targeting these molecules have been explored in the treatment of autoimmune diseases. However, only three IRAK4 inhibitors have entered clinical trials. The results of a 12-week phase II clinical trial showed that activated RA patients treated with once daily doses of PF-06650833 (Pfizer) exhibited reduced disease activity scores and an improved rate of ACR50 responses compared to the placebo control group. Only 6.4% (12/187) of participants dropped off the study due to adverse effects [170]. BAY1834845 (Bayer) and BAY1830839 (Bayer) have completed phase I clinical trials in healthy volunteers, and phase I/II clinical trials are under way [171].
Most inhibitors are at the interface between basic and clinical research, using mouse models to explore their treatment efficiency. For example, the MyD88 function inhibitors, c(MyD4–4) and TJ-M2010–6, were reported to alleviate the severity of EAE and reduce the onset of autoimmune diabetes in experimental NOD mice, respectively [172, 173]. IRAK4 inhibitors, HS-243, ND-2158 and ND-2110, were shown to suppress LPS-induced proinflammatory cytokines and relieve the symptoms in rheumatoid arthritis fibroblast-like synoviocytes (RAFLSs) and CIA mouse models [174,175,176]. Two NF-κB inhibitors, polyphyllin and taraxasterol, could block the production of proinflammatory effectors obviously in CIA mice [177, 178]. Furthermore, TAK1 inhibitors, takinib and oxozeaenol, were reported to alleviate the severity of CIA in mouse model [179] and reduce the onset of autoimmune diabetes in NOD mice [180]. Remarkably, siRNAs, which serve as treatments for autoimmune disease, were applied in mouse models. Adenoviral-mediated siRNA against TAK1 treatment helped to control joint inflammation by suppressing JNK activation and the expression of proinflammatory cytokines in CIA mice [181]. Another study performed by Wang et al. showed that siRNAs targeting the TRIF gene alleviated the severity of EAE in a mouse model by reducing IFN-γ and IL-2 levels [182]. Therefore, further studies are required to accelerate the clinical application of inhibitors targeting TLR signal transduction molecules.
Inhibitors targeting NLR signal transduction molecules
RIPK2, which mediate proinflammatory signaling from NOD1 and NOD2, is an emerging therapeutic target in autoimmune diseases [183,184,185]. A selective RIPK2 kinase inhibitor, WEHI-345, was reported to delay RIPK2 ubiquitylation, prevent cytokine production and ameliorate EAE in mice [186]. Remarkably, four RIPK2 inhibitors (ponatinib, regorafenib, gefitinib and erlotinib), which are approved by the US Food and Drug Administration, are used clinically against various forms of cancer [183, 187, 188]. Considering the anti-inflammatory effect of these drugs by inhibiting NLR signal transduction, their therapeutic potential in autoimmune diseases is worth further investigation.
Inhibitors targeting RLR and CLR signal transduction molecules
Reports related to inhibitors targeting RLR signal transduction molecules are rare. E3 ubiquitin ligases, RNF125, A20, MARCH5, SMURF2, and AIP4, are implicated in inhibiting RLR signaling by targeting MAVS [189], which could be exploited as therapeutic targets for the treatment of autoimmune disease. Syk kinase is a key signal transduction molecule of the CLR signaling pathway. It has been reported that R788, a selective Syk inhibitor, obviously delays spontaneous diabetes onset in NOD mice [190] and prevents and suppresses skin injury in lupus MRL/lpr mice [191]. Furthermore, R788 was reported to be effective in RA patients in a randomized clinical phase II trial [192], but unfortunately, the clinical use of R788 was hampered due to obvious side effects [193].
Inhibitors targeting terminal proinflammatory cytokines
Most innate immune molecules function by producing proinflammatory cytokines. Thus, inhibiting terminal proinflammatory cytokines is considered an effective and rapid treatment strategy for autoimmune diseases. To date, nine proinflammatory cytokine inhibitors targeting TNF, IL-6R and IL-1 have been used in the clinical treatment of RA [194,195,196,197,198,199,200] (Table 1). Six IL-6 blockers for SLE treatments and one IL-18 inhibitor for patients with NLRC4 and XIAP deficiency (monogenic IL-18 associated autoinflammatory conditions) are in phase II/III clinical trials (Table 1) [6, 198, 201,202,203,204,205]. Although the administration of the proinflammatory cytokine blockade increased therapeutic efficacy on autoimmune diseases, the serious side effects, for example, increased risk of infection, malignancy and induction of multiple sclerosis, lupus, psoriasis, or heart failure, and the expensive costs limited their clinical applications [206,207,208]. Therefore, it is of great significance to explore safer, more effective and more economical proinflammatory cytokines blockade strategies for autoimmune diseases.
Conclusions
PRRs are capable of activating the inflammatory pathway in innate immune cells and collaborating with antigen receptors to enhance activation in adaptive immune cells. Both synergistically and individually contribute to the initiation and/or development of autoimmune diseases. So far, a total of 30 inhibitors that inhibit innate immune molecular responses are in clinical use or undergoing clinical trials, several of which have the obvious advantages of better clinical efficacy. However, the serious side effects and the expensive costs are still considerable problems to be solved. In addition, many small molecule inhibitors which work well in animal models of autoimmune diseases are ideal candidates for further study. Therefore, PRRs might be considered potent targets for manipulating the kinetic status of the autoimmune response. In the future, it is worth exploring more molecules and related pathways that are critical for both innate and adaptive immunity. It should also evaluate the efficacy of PRR inhibitors combined with conventional drugs for anti-autoimmune diseases.
Availability of data and materials
Not applicable.
Abbreviations
- PRRs:
-
Pattern recognition receptors
- TLRs:
-
Toll-like receptors
- NLRs:
-
Nod-like receptors
- RLRs:
-
RIG-I like receptors
- CLRs:
-
C-type lectin receptors
- APCs:
-
Antigen-presenting cells
- PAMPs:
-
Pathogen-associated molecular patterns
- DAMPs:
-
Danger-associated molecular patterns
- SLE:
-
Systemic lupus erythematosus
- RA:
-
Rheumatoid arthritis
- LRR:
-
Leucine-rich repeats
- TIR:
-
Toll interleukin-1 receptor homology
- DCs:
-
Dendritic cells
- NK:
-
Natural killer
- LPS:
-
Lipopolysaccharide
- dsRNA:
-
Double-stranded RNA
- ssRNA:
-
Single-stranded RNA
- MyD88:
-
Myeloid differentiation primary response 88
- Mal:
-
MyD88 adapter like
- TRIF:
-
TIR-domain containing adapter inducing interferon-β
- TRAM:
-
TRIF-related adaptor molecule
- IRAK-4:
-
Interleukin 1 receptor associated kinase 4
- TRAF6:
-
TNF receptor associated factor 6
- TAK1:
-
TGF-β-activated kinase 1
- TAB:
-
TAK1-binding proteins
- RIP-1:
-
Receptor-interacting protein-1
- TBK1:
-
TANK-binding kinase 1
- AIH:
-
Autoimmune hepatitis
- SSc:
-
Systemic sclerosis
- EAU:
-
Experimental autoimmune uveitis
- EAE:
-
Experimental autoimmune encephalomyelitis
- MS:
-
Multiple sclerosis
- FA:
-
Food allergy
- NOD:
-
Nucleotide-binding oligomerization domain
- AD:
-
Acidic transactivation domain
- BIR:
-
Baculovirus inhibitor of apoptosis protein repeat domain
- CARD:
-
Caspase activation and recruitment domain
- PYD:
-
Pyrin domain
- MTS:
-
Mitochondria-localization sequence
- CIITA:
-
MHC-II transcription activator
- NAIP:
-
Neuronal apoptosis inhibitory protein
- MDP:
-
Muramyl dipeptide peptidoglycan
- ROS:
-
Reactive oxygen species
- RIPK2:
-
Receptor-interacting serine/threonine-protein kinase 2
- MAVS:
-
Mitochondrial antiviral signaling
- ASCs:
-
Apoptosis-associated speck-like protein
- UC:
-
Ulcerative colitis
- CD:
-
Crohn’s disease
- FCAS:
-
Familial cold autoinflammatory syndrome
- RIG-I:
-
Retinoic acid-inducible gene I
- MDA5:
-
Melanoma differentiation-associated gene 5
- LGP2:
-
Laboratory of genetics and physiology 2
- CTD:
-
C-terminal domain
- RD:
-
Repressor domain
- AGS:
-
Aicardi-Goutieres syndrome
- SMS:
-
Singleton-Merten syndrome
- CTLD:
-
C-type lectin-like domain
- CRD:
-
Carbohydrate recognition domain
- ITAM:
-
Immunoreceptor tyrosine-based activation motif
- Dectin-1:
-
DC-associated C-type lectin-1
- FcR-γ:
-
Fc receptor γ chain
- DC-SIGN:
-
DC-specific ICAM3-grabbing nonintegrin
- CARD9:
-
Caspase activation, recruits the complex of domain-containing protein 9
- BCL-10:
-
B-cell lymphoma/leukemia 10
- MALT1:
-
Mucosa-associated lymphoid tissue 1
- NFAT:
-
Nuclear factor of activated T cells
- ERK:
-
Extracellular signal-regulated protein kinase
- cGAMP:
-
Cyclic GMP-AMP
- cGAS:
-
Cyclic GMP-AMP synthase
- STING:
-
Stimulator of interferon genes
- ALRs:
-
AIM-like receptors
- IFI16:
-
Gamma-interferon-inducible protein 16
- AIM2:
-
Absent in melanoma 2
- MNDA:
-
Myeloid cell nuclear differentiation antigen
- IFIX:
-
Interferon-inducible protein X
- IMO:
-
Immune modulatory oligonucleotide
- IRS-954:
-
Immunoregulatory DNA sequence-954
- CFA:
-
Complete Freund's adjuvant
- AIT:
-
Autoimmune thyroiditis
- JAK:
-
Janus kinase
- CIA:
-
Collagen-induced arthritis
- DSS:
-
Dextran sulfate sodium
- USP3:
-
Ubiquitin-specific protease 3
References
Wang L, Wang FS, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med. 2015;278(4):369–95.
Bays AM, Gardner G. Pharmacologic therapies for rheumatologic and autoimmune conditions. Med Clin North Am. 2016;100(4):719–31.
Thakur S, Riyaz B, Patil A, Kaur A, Kapoor B, Mishra V. Novel drug delivery systems for NSAIDs in management of rheumatoid arthritis: an overview. Biomed Pharmacother. 2018;106:1011–23.
Seeliger B, Prasse A. Immunomodulation in autoimmune interstitial lung disease. Respiration. 2020;99(10):819–29.
Lascano AM, Lalive PH. Update in immunosuppressive therapy of myasthenia gravis. Autoimmun Rev. 2021;20(1): 102712.
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38.
Salomon BL. Insights into the biology and therapeutic implications of TNF and regulatory T cells. Nat Rev Rheumatol. 2021;17(8):487–504.
Shu SA, Wang J, Tao MH, Leung PS. Gene therapy for autoimmune disease. Clin Rev Allergy Immunol. 2015;49(2):163–76.
Esensten JH, Muller YD, Bluestone JA, Tang Q. Regulatory T-cell therapy for autoimmune and autoinflammatory diseases: the next frontier. J Allergy Clin Immunol. 2018;142(6):1710–8.
Ryba-Stanisławowska M, Sakowska J, Zieliński M, Ławrynowicz U, Trzonkowski P. Regulatory T cells: the future of autoimmune disease treatment. Expert Rev Clin Immunol. 2019;15(7):777–89.
Wahren-Herlenius M, Dörner T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet. 2013;382(9894):819–31.
Hasselbalch HC. B-cell depletion with rituximab-a targeted therapy for Graves’ disease and autoimmune thyroiditis. Immunol Lett. 2003;88(1):85–6.
Edwards JC, Cambridge G. Prospects for B-cell-targeted therapy in autoimmune disease. Rheumatol. 2005;44(2):151–6.
Mease PJ. B cell-targeted therapy in autoimmune disease: rationale, mechanisms, and clinical application. J Rheumatol. 2008;35(7):1245–55.
Barnas JL, Looney RJ, Anolik JH. B cell targeted therapies in autoimmune disease. Curr Opin Immunol. 2019;61:92–9.
Ulivieri C, Baldari CT. T-cell-based immunotherapy of autoimmune diseases. Expert Rev Vaccines. 2013;12(3):297–310.
Chen Y, Sun J, Liu H, Yin G, Xie Q. Immunotherapy deriving from CAR-T cell treatment in autoimmune diseases. J Immunol Res. 2019;2019:5727516.
Boehncke WH, Brembilla NC. Autoreactive T-Lymphocytes in inflammatory skin diseases. Front Immunol. 2019;10:1198.
Mullen LM, Chamberlain G, Sacre S. Pattern recognition receptors as potential therapeutic targets in inflammatory rheumatic disease. Arthritis Res Ther. 2015;17(1):122.
Riera Romo M, Pérez-Martínez D, Castillo FC. Innate immunity in vertebrates: an overview. Immunology. 2016;148(2):125–39.
Sener AG, Afsar I. Infection and autoimmune disease. Rheumatol Int. 2012;32(11):3331–8.
Zhu G, Xu Y, Cen X, Nandakumar KS, Liu S, Cheng K. Targeting pattern-recognition receptors to discover new small molecule immune modulators. Eur J Med Chem. 2018;144:82–92.
de Koning HD, Simon A, Zeeuwen PL, Schalkwijk J. Pattern recognition receptors in immune disorders affecting the skin. J Innate Immun. 2012;4(3):225–40.
O’Reilly S. Pound the alarm: danger signals in rheumatic diseases. Clin Sci. 2015;128(5):297–305.
Sun L, Liu W, Zhang LJ. The role of Toll-Like receptors in skin host defense, psoriasis, and atopic dermatitis. J Immunol Res. 2019;2019:1824624.
Hua Z, Hou B. TLR signaling in B-cell development and activation. Cell Mol Immunol. 2013;10(2):103–6.
Mills KH. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol. 2011;11(12):807–22.
Koh YT, Scatizzi JC, Gahan JD, Lawson BR, Baccala R, Pollard KM, Beutler BA, Theofilopoulos AN, Kono DH. Role of nucleic acid-sensing TLRs in diverse autoantibody specificities and anti-nuclear antibody-producing B cells. J Immunol. 2013;190(10):4982–90.
Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, Ma L, Yang XO, Nurieva RI, Tian Q, Dong C. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. 2010;32(5):692–702.
Murgueitio MS, Rakers C, Frank A, Wolber G. Balancing inflammation: computational design of small-molecule toll-like receptor modulators. Trends Pharmacol Sci. 2017;38(2):155–68.
Yang X, Cheng Y, Li C. The role of TLRs in cervical cancer with HPV infection: a review. Signal Transduct Target Ther. 2017;2:17055.
Patidar A, Selvaraj S, Sarode A, Chauhan P, Chattopadhyay D, Saha B. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine. 2018;104:114–23.
Bagheri M, Zahmatkesh A. Evolution and species-specific conservation of toll-like receptors in terrestrial vertebrates. Int Rev Immunol. 2018;37(5):217–28.
Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol. 2015. https://doi.org/10.1002/0471142735.im1412s109.
Lu BL, Williams GM, Brimble MA. TLR2 agonists and their structure-activity relationships. Org Biomol Chem. 2020;18(27):5073–94.
Hess NJ, Felicelli C, Grage J, Tapping RI. TLR10 suppresses the activation and differentiation of monocytes with effects on DC-mediated adaptive immune responses. J Leukoc Biol. 2017;101(5):1245–52.
Fore F, Indriputri C, Mamutse J, Nugraha J. TLR10 and Its unique anti-inflammatory properties and potential use as a target in therapeutics. Immune Netw. 2020;20(3): e21.
Ullah MO, Sweet MJ, Mansell A, Kellie S, Kobe B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol. 2016;100(1):27–45.
Balka KR, De Nardo D. Understanding early TLR signaling through the Myddosome. J Leukoc Biol. 2019;105(2):339–51.
Hu W, Jain A, Gao Y, Dozmorov IM, Mandraju R, Wakeland EK, Pasare C. Differential outcome of TRIF-mediated signaling in TLR4 and TLR3 induced DC maturation. Proc Natl Acad Sci U S A. 2015;112(45):13994–9.
Uematsu S, Akira S. Toll-like receptors and Type I interferons. J Biol Chem. 2007;282(21):15319–23.
Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, Takeuchi O, Akira S. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5(10):1061–8.
Sakaniwa K, Shimizu T. Targeting the innate immune receptor TLR8 using small-molecule agents. Acta Crystallogr D Struct Biol. 2020;76(Pt 7):621–9.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–84.
Rahimifard M, Maqbool F, Moeini-Nodeh S, Niaz K, Abdollahi M, Braidy N, Nabavi SM, Nabavi SF. Targeting the TLR4 signaling pathway by polyphenols: a novel therapeutic strategy for neuroinflammation. Ageing Res Rev. 2017;36:11–9.
Chen JQ, Szodoray P, Zeher M. Toll-Like receptor pathways in autoimmune diseases. Clin Rev Allergy Immunol. 2016;50(1):1–17.
Hamerman JA, Pottle J, Ni M, He Y, Zhang ZY, Buckner JH. Negative regulation of TLR signaling in myeloid cells–implications for autoimmune diseases. Immunol Rev. 2016;269(1):212–27.
Thwaites RS, Unterberger S, Chamberlain G, Walker-Bone K, Davies KA, Sacre S. TLR1/2 and 5 induce elevated cytokine levels from rheumatoid arthritis monocytes independent of ACPA or RF autoantibody status. Rheumatol. 2020;59(11):3533–9.
Thwaites RS, Unterberger S, Chamberlain G, Gray H, Jordan K, Davies KA, Harrison NA, Sacre S. Expression of sterile-α and armadillo motif in rheumatoid arthritis monocytes correlates with TLR2 induced IL-1β and disease activity. Rheumatology. 2021;60:5843–53.
Elkon KB, Wiedeman A. Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol. 2012;24(5):499–505.
Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OBJ. Toll-like receptor signaling drives btk-mediated autoimmune disease. Front Immunol. 2019;10:95.
Chi G, Feng XX, Ru YX, Xiong T, Gao Y, Wang H, Luo ZL, Mo R, Guo F, He YP, Zhang GM, Tian DA, Feng ZH. TLR2/4 ligand-amplified liver inflammation promotes initiation of autoimmune hepatitis due to sustained IL-6/IL-12/IL-4/IL-25 expression. Mol Immunol. 2018;99:171–81.
van Bon L, Popa C, Huijbens R, Vonk M, York M, Simms R, Hesselstrand R, Wuttge DM, Lafyatis R, Radstake TR. Distinct evolution of TLR-mediated dendritic cell cytokine secretion in patients with limited and diffuse cutaneous systemic sclerosis. Ann Rheum Dis. 2010;69(8):1539–47.
Xiao Q, Li X, Sun D, Yi H, Lu X, Nian H. TLR7 Engagement on Dendritic Cells Enhances Autoreactive Th17 Responses via Activation of ERK. J Immunol. 2016;197(10):3820–30.
Xu D, Liu H, Komai-Koma M. Direct and indirect role of Toll-like receptors in T cell mediated immunity. Cell Mol Immunol. 2004;1(4):239–46.
Rahman AH, Taylor DK, Turka LA. The contribution of direct TLR signaling to T cell responses. Immunol Res. 2009;45(1):25–36.
Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci USA. 2012;109(32):13064–9.
Ferreira TB, Hygino J, Wing AC, Kasahara TM, Sacramento PM, Camargo S, Rueda F, Alves-Leon SV, Alvarenga R, Vasconcelos CC, Agrawal A, Gupta S, Bento CAM. Different interleukin-17-secreting Toll-like receptor(+) T-cell subsets are associated with disease activity in multiple sclerosis. Immunology. 2018;154(2):239–52.
Aksoy E. TLRs toll for Tregs. J Leukoc Biol. 2019;106(6):1193–5.
Chiffoleau E, Heslan JM, Heslan M, Louvet C, Condamine T, Cuturi MC. TLR9 ligand enhances proliferation of rat CD4+ T cell and modulates suppressive activity mediated by CD4+ CD25+ T cell. Int Immunol. 2007;19(2):193–201.
Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175(12):8051–9.
Suthers AN, Sarantopoulos S. TLR7/TLR9- and B Cell receptor-signaling crosstalk: promotion of potentially dangerous B cells. Front Immunol. 2017;8:775.
Fillatreau S, Manfroi B, Dörner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17(2):98–108.
Capolunghi F, Rosado MM, Cascioli S, Girolami E, Bordasco S, Vivarelli M, Ruggiero B, Cortis E, Insalaco A, Fantò N, et al. Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology. 2010;49(12):2281–9.
Yang X, Lin G, Han Z, Chai J. Structural Biology of NOD-Like Receptors. Adv Exp Med Biol. 2019;1172:119–41.
Lechtenberg BC, Mace PD, Riedl SJ. Structural mechanisms in NLR inflammasome signaling. Curr Opin Struct Biol. 2014;29:17–25.
Chen L, Cao SQ, Lin ZM, He SJ, Zuo JP. NOD-like receptors in autoimmune diseases. Acta Pharmacol Sin. 2021;42:1742–56.
Davoodi J, Ghahremani MH, Es-Haghi A, Mohammad-Gholi A, Mackenzie A. Neuronal apoptosis inhibitory protein, NAIP, is an inhibitor of procaspase-9. Int J Biochem Cell Biol. 2010;42(6):958–64.
Caruso R, Warner N, Inohara N, Núñez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41(6):898–908.
Uchimura T, Oyama Y, Deng M, Guo H, Wilson JE, Rampanelli E, Cook KD, Misumi I, Tan X, Chen L, et al. The Innate Immune Sensor NLRC3 Acts as a Rheostat that Fine-Tunes T Cell Responses in Infection and Autoimmunity. Immunity. 2018;49(6):1049-1061.e1046.
Li X, Deng M, Petrucelli AS, Zhu C, Mo J, Zhang L, Tam JW, Ariel P, Zhao B, Zhang S, et al. Viral DNA Binding to NLRC3, an inhibitory nucleic acid sensor, unleashes STING, a cyclic dinucleotide receptor that activates type I interferon. Immunity. 2019;50(3):591-599.e596.
Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol. 2012;12(12):813–20.
Meunier E, Broz P. Evolutionary convergence and divergence in NLR function and structure. Trends Immunol. 2017;38(10):744–57.
Clay GM, Valadares DG, Graff JW, Ulland TK, Davis RE, Scorza BM, Zhanbolat BS, Chen Y, Sutterwala FS, Wilson ME. An Anti-Inflammatory Role for NLRP10 in Murine Cutaneous Leishmaniasis. J Immunol. 2017;199(8):2823–33.
Lautz K, Damm A, Menning M, Wenger J, Adam AC, Zigrino P, Kremmer E, Kufer TA. NLRP10 enhances Shigella-induced pro-inflammatory responses. Cell Microbiol. 2012;14(10):1568–83.
Mirza N, Sowa AS, Lautz K, Kufer TA. NLRP10 Affects the Stability of Abin-1 To Control Inflammatory Responses. J Immunol. 2019;202(1):218–27.
Vacca M, Böhme J, Zambetti LP, Khameneh HJ, Paleja BS, Laudisi F, Ho AWS, Neo K, Leong KWK, Marzuki M, et al. NLRP10 enhances CD4(+) T-cell-mediated IFNγ response via regulation of dendritic cell-derived IL-12 release. Front Immunol. 2017;8:1462.
Unger BL, Ganesan S, Comstock AT, Faris AN, Hershenson MB, Sajjan US. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J Virol. 2014;88(7):3705–18.
Lu Y, Zheng Y, Coyaud É, Zhang C, Selvabaskaran A, Yu Y, Xu Z, Weng X, Chen JS, Meng Y, et al. Palmitoylation of NOD1 and NOD2 is required for bacterial sensing. Science. 2019;366(6464):460–7.
Mukherjee T, Hovingh ES, Foerster EG, Abdel-Nour M, Philpott DJ, Girardin SE. NOD1 and NOD2 in inflammation, immunity and disease. Arch Biochem Biophys. 2019;670:69–81.
McKernan DP. Pattern recognition receptors as potential drug targets in inflammatory disorders. Adv Protein Chem Struct Biol. 2020;119:65–109.
Heim VJ, Stafford CA, Nachbur U. NOD Signaling and Cell Death. Front Cell Dev Biol. 2019;7:208.
Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med. 2014;211(12):2385–96.
Fu Y, Zhan X, Wang Y, Jiang X, Liu M, Yang Y, Huang Y, Du X, Zhong XP, Li L, et al. NLRC3 expression in dendritic cells attenuates CD4(+) T cell response and autoimmunity. Embo j. 2019;38(16): e101397.
Tuladhar S, Kanneganti TD. NLRP12 in innate immunity and inflammation. Mol Aspects Med. 2020;76:100887.
Normand S, Waldschmitt N, Neerincx A, Martinez-Torres RJ, Chauvin C, Couturier-Maillard A, Boulard O, Cobret L, Awad F, Huot L, et al. Proteasomal degradation of NOD2 by NLRP12 in monocytes promotes bacterial tolerance and colonization by enteropathogens. Nat Commun. 2018;9(1):5338.
Chen L, Wilson JE, Koenigsknecht MJ, Chou WC, Montgomery SA, Truax AD, Brickey WJ, Packey CD, Maharshak N, Matsushima GK, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol. 2017;18(5):541–51.
Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity. 2011;34(5):680–92.
Fan X, Jin T. Structures of RIG-I-Like Receptors and Insights into Viral RNA Sensing. Adv Exp Med Biol. 2019;1172:157–88.
Brisse M, Ly H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front Immunol. 2019;10:1586.
Li XL, Ezelle HJ, Hsi TY, Hassel BA. A central role for RNA in the induction and biological activities of type 1 interferons. Wiley Interdiscip Rev RNA. 2011;2(1):58–78.
Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol. 2015;32:48–53.
Zhao Y, Karijolich J. Know thyself: RIG-I-like receptor sensing of DNA virus infection. J Virol. 2019;93(23):e01085-e1119.
Singh H, Koury J, Kaul M. Innate immune sensing of viruses and its consequences for the central nervous system. Viruses. 2021;13(2):170.
Sontheimer RD. MDA5 autoantibody-another indicator of clinical diversity in dermatomyositis. Ann Transl Med. 2017;5(7):160.
Blum SI, Tse HM. Innate Viral Sensor MDA5 and coxsackievirus interplay in type 1 diabetes development. Microorganisms. 2020;8(7):993.
Munroe ME, Pezant N, Brown MA, Fife DA, Guthridge JM, Kelly JA, Wiley G, Gaffney PM, James JA, Montgomery CG. Association of IFIH1 and pro-inflammatory mediators: Potential new clues in SLE-associated pathogenesis. PLoS ONE. 2017;12(2): e0171193.
Xiao W, Feng J, Long H, Xiao B, Luo ZH. Case report: aicardi-goutières syndrome and singleton-merten syndrome caused by a gain-of-function mutation in IFIH1. Front Genet. 2021;12:660953.
Kato H, Fujita T. RIG-I-like receptors and autoimmune diseases. Curr Opin Immunol. 2015;37:40–5.
Kurasawa K, Arai S, Namiki Y, Tanaka A, Takamura Y, Owada T, Arima M, Maezawa R. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57(12):2114–9.
Soda N, Sakai N, Kato H, Takami M, Fujita T. Singleton-Merten Syndrome-like Skeletal Abnormalities in Mice with Constitutively Activated MDA5. J Immunol. 2019;203(5):1356–68.
Sun W, Wang H, Qi CF, Wu J, Scott B, Bolland S. Antiviral Adaptor MAVS promotes murine lupus with a B Cell Autonomous Role. Front Immunol. 2019;10:2452.
Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu-Tirgoviste C, Widmer B, Dunger DB, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38(6):617–9.
Concannon P, Onengut-Gumuscu S, Todd JA, Smyth DJ, Pociot F, Bergholdt R, Akolkar B, Erlich HA, Hilner JE, Julier C, et al. A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes. 2008;57(10):2858–61.
Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324(5925):387–9.
Liu S, Wang H, Jin Y, Podolsky R, Reddy MV, Pedersen J, Bode B, Reed J, Steed D, Anderson S, et al. IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet. 2009;18(2):358–65.
Okude H, Ori D, Kawai T. Signaling through nucleic acid sensors and their roles in inflammatory diseases. Front Immunol. 2020;11:625833.
Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M, Minowa O, Yoshida A, Deguchi K, Sato H, et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity. 2014;40(2):199–212.
Robinson T, Kariuki SN, Franek BS, Kumabe M, Kumar AA, Badaracco M, Mikolaitis RA, Guerrero G, Utset TO, Drevlow BE, et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. J Immunol. 2011;187(3):1298–303.
Negishi H, Yanai H, Nakajima A, Koshiba R, Atarashi K, Matsuda A, Matsuki K, Miki S, Doi T, Aderem A, et al. Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat Immunol. 2012;13(7):659–66.
Kulkarni RR, Rasheed MA, Bhaumik SK, Ranjan P, Cao W, Davis C, Marisetti K, Thomas S, Gangappa S, Sambhara S, et al. Activation of the RIG-I pathway during influenza vaccination enhances the germinal center reaction, promotes T follicular helper cell induction, and provides a dose-sparing effect and protective immunity. J Virol. 2014;88(24):13990–4001.
Tang J, Lin G, Langdon WY, Tao L, Zhang J. Regulation of C-type lectin receptor-mediated antifungal immunity. Front Immunol. 2018;9:123.
Shiokawa M, Yamasaki S, Saijo S. C-type lectin receptors in anti-fungal immunity. Curr Opin Microbiol. 2017;40:123–30.
Plato A, Hardison SE, Brown GD. Pattern recognition receptors in antifungal immunity. Semin Immunopathol. 2015;37(2):97–106.
Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9(7):465–79.
Speakman EA, Dambuza IM, Salazar F, Brown GD. T Cell Antifungal Immunity and the Role of C-Type Lectin Receptors. Trends Immunol. 2020;41(1):61–76.
Geijtenbeek TB, Gringhuis SI. C-type lectin receptors in the control of T helper cell differentiation. Nat Rev Immunol. 2016;16(7):433–48.
Höft MA, Hoving JC, Brown GD. Signaling C-Type Lectin Receptors in Antifungal Immunity. Curr Top Microbiol Immunol. 2020;429:63–101.
Sancho D, Reis e Sousa C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol. 2012;30:491–529.
Brown GD, Willment JA, Whitehead L. C-type lectins in immunity and homeostasis. Nat Rev Immunol. 2018;18(6):374–89.
Tone K, Stappers MHT, Willment JA, Brown GD. C-type lectin receptors of the Dectin-1 cluster: physiological roles and involvement in disease. Eur J Immunol. 2019;49(12):2127–33.
Ilarregui JM, Kooij G, Rodríguez E, van der Pol SMA, Koning N, Kalay H, van der Horst JC, van Vliet SJ, García-Vallejo JJ, de Vries HE, et al. Macrophage galactose-type lectin (MGL) is induced on M2 microglia and participates in the resolution phase of autoimmune neuroinflammation. J Neuroinflammation. 2019;16(1):130.
Yoshitomi H, Sakaguchi N, Kobayashi K, Brown GD, Tagami T, Sakihama T, Hirota K, Tanaka S, Nomura T, Miki I, et al. A role for fungal {beta}-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J Exp Med. 2005;201(6):949–60.
Tang C, Kamiya T, Liu Y, Kadoki M, Kakuta S, Oshima K, Hattori M, Takeshita K, Kanai T, Saijo S, et al. Inhibition of Dectin-1 signaling ameliorates colitis by inducing lactobacillus-mediated regulatory T cell expansion in the intestine. Cell Host Microbe. 2015;18(2):183–97.
Karumuthil-Melethil S, Sofi MH, Gudi R, Johnson BM, Perez N, Vasu C. TLR2- and Dectin 1-associated innate immune response modulates T-cell response to pancreatic β-cell antigen and prevents type 1 diabetes. Diabetes. 2015;64(4):1341–57.
Brown BR, Lee EJ, Snow PE, Vance EE, Iwakura Y, Ohno N, Miura N, Lin X, Brown GD, Wells CA, et al. Fungal-derived cues promote ocular autoimmunity through a Dectin-2/Card9-mediated mechanism. Clin Exp Immunol. 2017;190(3):293–303.
Lee EJ, Brown BR, Vance EE, Snow PE, Silver PB, Heinrichs D, Lin X, Iwakura Y, Wells CA, Caspi RR, et al. Mincle activation and the syk/card9 signaling axis are central to the development of autoimmune disease of the eye. J Immunol. 2016;196(7):3148–58.
N’Diaye M, Brauner S, Flytzani S, Kular L, Warnecke A, Adzemovic MZ, Piket E, Min JH, Edwards W, Mela F, et al. C-type lectin receptors Mcl and Mincle control development of multiple sclerosis-like neuroinflammation. J Clin Invest. 2020;130(2):838–52.
Gong W, Zheng T, Kun G, Miao F, Xie H, Weijie L, Tang Q, Hong Z, Ren H, Gu G, et al. Mincle/Syk signaling promotes intestinal mucosal inflammation through induction of macrophage pyroptosis in Crohn’s disease. J Crohns Colitis. 2020;14:1734–47.
He L, Chen Y, Wu Y, Xu Y, Zhang Z, Liu Z. Nucleic acid sensing pattern recognition receptors in the development of colorectal cancer and colitis. Cell Mol Life Sci. 2017;74(13):2395–411.
Zhang X, Bai XC, Chen ZJ. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity. 2020;53(1):43–53.
Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–9.
Zhou R, Xie X, Li X, Qin Z, Wei C, Liu J, Luo Y. The triggers of the cGAS-STING pathway and the connection with inflammatory and autoimmune diseases. Infect Genet Evol. 2020;77: 104094.
Caneparo V, Landolfo S, Gariglio M, De Andrea M. The Absent in Melanoma 2-Like Receptor IFN-Inducible Protein 16 as an Inflammasome Regulator in Systemic Lupus Erythematosus: The Dark Side of Sensing Microbes. Front Immunol. 2018;9:1180.
Bawadekar M, De Andrea M, Gariglio M, Landolfo S. Mislocalization of the interferon inducible protein IFI16 by environmental insults: implications in autoimmunity. Cytokine Growth Factor Rev. 2015;26(2):213–9.
Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol. 2016;46(2):269–80.
Ciążyńska M, Olejniczak-Staruch I, Sobolewska-Sztychny D, Narbutt J, Skibińska M, Lesiak A. The role of NLRP1, NLRP3, and AIM2 inflammasomes in psoriasis: review. Int J Mol Sci. 2021;22(11):5898.
Briard B, Place DE, Kanneganti TD. DNA sensing in the innate immune response. Physiology. 2020;35(2):112–24.
Kim T, Pazhoor S, Bao M, Zhang Z, Hanabuchi S, Facchinetti V, Bover L, Plumas J, Chaperot L, Qin J, et al. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc Natl Acad Sci USA. 2010;107(34):15181–6.
Stunnenberg M, Geijtenbeek TBH, Gringhuis SI. DDX3 in HIV-1 infection and sensing: a paradox. Cytokine Growth Factor Rev. 2018;40:32–9.
Lai CY, Su YW, Lin KI, Hsu LC, Chuang TH. Natural Modulators of Endosomal Toll-Like Receptor-Mediated Psoriatic Skin Inflammation. J Immunol Res. 2017;2017:7807313.
Schrezenmeier E, Dörner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol. 2020;16(3):155–66.
Gao W, Xiong Y, Li Q, Yang H. Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: a journey from molecular to nano therapeutics. Front Physiol. 2017;8:508.
Balak DM, van Doorn MB, Arbeit RD, Rijneveld R, Klaassen E, Sullivan T, Brevard J, Thio HB, Prens EP, Burggraaf J, et al. IMO-8400, a toll-like receptor 7, 8, and 9 antagonist, demonstrates clinical activity in a phase 2a, randomized, placebo-controlled trial in patients with moderate-to-severe plaque psoriasis. Clin Immunol. 2017;174:63–72.
Ain QU, Batool M, Choi S. TLR4-targeting therapeutics: structural basis and computer-aided drug discovery approaches. Molecules. 2020;25(3):627.
Crowley T, Fitzpatrick JM, Kuijper T, Cryan JF, O’Toole O, O’Leary OF, Downer EJ. Modulation of TLR3/TLR4 inflammatory signaling by the GABAB receptor agonist baclofen in glia and immune cells: relevance to therapeutic effects in multiple sclerosis. Front Cell Neurosci. 2015;9:284.
Federico S, Pozzetti L, Papa A, Carullo G, Gemma S, Butini S, Campiani G, Relitti N. Modulation of the Innate Immune Response by Targeting Toll-like Receptors: A Perspective on Their Agonists and Antagonists. J Med Chem. 2020;63(22):13466–513.
Kwilasz AJ, Todd LS, Duran-Malle JC, Schrama AEW, Mitten EH, Larson TA, Clements MA, Harris KM, Litwiler ST, Wang X, et al. Experimental autoimmune encephalopathy (EAE)-induced hippocampal neuroinflammation and memory deficits are prevented with the non-opioid TLR2/TLR4 antagonist (+)-naltrexone. Behav Brain Res. 2020;396:112896.
Kundu B, Raychaudhuri D, Mukherjee A, Sinha BP, Sarkar D, Bandopadhyay P, Pal S, Das N, Dey D, Ramarao K, et al. Systematic optimization of potent and orally bioavailable purine scaffold as a dual inhibitor of toll-like receptors 7 and 9. J Med Chem. 2021;64(13):9279–301.
Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37(12):3582–6.
Wang J, Lu J, Lan Y, Zhou H, Li W, Xiang M. Total coumarins from Urtica dentata Hand prevent murine autoimmune diabetes via suppression of the TLR4-signaling pathways. J Ethnopharmacol. 2013;146(1):379–92.
Roome T, Aziz S, Razzak A, Aslam Z, Lubna, Jamali KS, Sikandar B, Fatima T, Abidi L, Imran M et al. Opuntioside, opuntiol and its metallic nanoparticles attenuate adjuvant-induced arthritis: Novel suppressors of Toll-like receptors -2 and -4. Biomed Pharmacother. 2019; 112: 108624.
Guo Q, Wu Y, Hou Y, Liu Y, Liu T, Zhang H, Fan C, Guan H, Li Y, Shan Z, et al. Cytokine Secretion and Pyroptosis of Thyroid Follicular Cells Mediated by Enhanced NLRP3, NLRP1, NLRC4, and AIM2 Inflammasomes Are Associated With Autoimmune Thyroiditis. Front Immunol. 2018;9:1197.
Soares JL, Oliveira EM, Pontillo A. Variants in NLRP3 and NLRC4 inflammasome associate with susceptibility and severity of multiple sclerosis. Mult Scler Relat Disord. 2019;29:26–34.
Shen HH, Yang YX, Meng X, Luo XY, Li XM, Shuai ZW, Ye DQ, Pan HF. NLRP3: a promising therapeutic target for autoimmune diseases. Autoimmun Rev. 2018;17(7):694–702.
Li Z, Guo J, Bi L. Role of the NLRP3 inflammasome in autoimmune diseases. Biomed Pharmacother. 2020;130: 110542.
Guo C, Fu R, Wang S, Huang Y, Li X, Zhou M, Zhao J, Yang N. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol. 2018;194(2):231–43.
Henderson J, Bhattacharyya S, Varga J, O’Reilly S. Targeting TLRs and the inflammasome in systemic sclerosis. Pharmacol Ther. 2018;192:163–9.
Chen X, Liu G, Yuan Y, Wu G, Wang S, Yuan L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. 2019;10(12):906.
Yang X, Zhan N, Jin Y, Ling H, Xiao C, Xie Z, Zhong H, Yu X, Tang R, Ma J, et al. Tofacitinib restores the balance of γδTreg/γδT17 cells in rheumatoid arthritis by inhibiting the NLRP3 inflammasome. Theranostics. 2021;11(3):1446–57.
Chen Y, He H, Lin B, Chen Y, Deng X, Jiang W, Zhou R. RRx-001 ameliorates inflammatory diseases by acting as a potent covalent NLRP3 inhibitor. Cell Mol Immunol. 2021;18(6):1425–36.
Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, Wang A, Tao J, Wang C, Liu Q, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214(11):3219–38.
Sánchez-Fernández A, Skouras DB, Dinarello CA, López-Vales R. OLT1177 (Dapansutrile), a Selective NLRP3 Inflammasome Inhibitor, Ameliorates Experimental Autoimmune Encephalomyelitis Pathogenesis. Front Immunol. 2019;10:2578.
Sadler AJ. The role of MDA5 in the development of autoimmune disease. J Leukoc Biol. 2018;103(2):185–92.
Oliveira L, Sinicato NA, Postal M, Appenzeller S, Niewold TB. Dysregulation of antiviral helicase pathways in systemic lupus erythematosus. Front Genet. 2014;5:418.
Zahn S, Barchet W, Rehkämper C, Hornung T, Bieber T, Tüting T, Wenzel J. Enhanced skin expression of melanoma differentiation-associated gene 5 (MDA5) in dermatomyositis and related autoimmune diseases. J Am Acad Dermatol. 2011;64(5):988–9.
Kitai Y, Takeuchi O, Kawasaki T, Ori D, Sueyoshi T, Murase M, Akira S, Kawai T. Negative regulation of melanoma differentiation-associated gene 5 (MDA5)-dependent antiviral innate immune responses by Arf-like protein 5B. J Biol Chem. 2015;290(2):1269–80.
Cui J, Song Y, Li Y, Zhu Q, Tan P, Qin Y, Wang HY, Wang RF. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 2014;24(4):400–16.
Chiffoleau E. C-Type Lectin-Like Receptors As Emerging Orchestrators of Sterile Inflammation Represent Potential Therapeutic Targets. Front Immunol. 2018;9:227.
Danto SISN, Singh RSP, et al. Efficacy and Safety of the Selective Interleukin-1 Receptor Associated Kinase 4 Inhibitor, PF-06650833, in patients with active rheumatoid arthritis and inadequate response to methotrexate. Am College Rheumatol Annual Meet. 2019;73:2206–18.
Wiese MD, Manning-Bennett AT, Abuhelwa AY. Investigational IRAK-4 inhibitors for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs. 2020;29(5):475–82.
Dishon S, Schumacher-Klinger A, Gilon C, Hoffman A, Nussbaum G. Myristoylation Confers Oral Bioavailability and Improves the Bioactivity of c(MyD 4–4), a Cyclic Peptide Inhibitor of MyD88. Mol Pharm. 2019;16(4):1516–22.
Shiratori E, Itoh M, Tohda S. MYD88 Inhibitor ST2825 Suppresses the Growth of Lymphoma and Leukaemia Cells. Anticancer Res. 2017;37(11):6203–9.
De Nardo D, Balka KR, Cardona Gloria Y, Rao VR, Latz E, Masters SL. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J Biol Chem. 2018;293(39):15195–207.
Kelly PN, Romero DL, Yang Y, Shaffer AL 3rd, Chaudhary D, Robinson S, Miao W, Rui L, Westlin WF, Kapeller R, et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med. 2015;212(13):2189–201.
Giménez N, Schulz R, Higashi M, Aymerich M, Villamor N, Delgado J, Juan M, López-Guerra M, Campo E, Rosich L, et al. Targeting IRAK4 disrupts inflammatory pathways and delays tumor development in chronic lymphocytic leukemia. Leukemia. 2020;34(1):100–14.
Wang Q, Zhou X, Zhao Y, Xiao J, Lu Y, Shi Q, Wang Y, Wang H, Liang Q. Polyphyllin I Ameliorates Collagen-Induced Arthritis by Suppressing the Inflammation Response in Macrophages Through the NF-κB Pathway. Front Immunol. 2018;9:2091.
Chen J, Wu W, Zhang M, Chen C. Taraxasterol suppresses inflammation in IL-1β-induced rheumatoid arthritis fibroblast-like synoviocytes and rheumatoid arthritis progression in mice. Int Immunopharmacol. 2019;70:274–83.
Scarneo SA, Eibschutz LS, Bendele PJ, Yang KW, Totzke J, Hughes P, Fox DA, Haystead TAJ. Pharmacological inhibition of TAK1, with the selective inhibitor takinib, alleviates clinical manifestation of arthritis in CIA mice. Arthritis Res Ther. 2019;21(1):292.
Cao H, Lu J, Du J, Xia F, Wei S, Liu X, Liu T, Liu Y, Xiang M. TAK1 inhibition prevents the development of autoimmune diabetes in NOD mice. Sci Rep. 2015;5:14593.
Luo X, Chen Y, Lv G, Zhou Z, Chen J, Mo X, Xie J. Adenovirus-Mediated Small Interfering RNA Targeting TAK1 Ameliorates Joint Inflammation with Collagen-Induced Arthritis in Mice. Inflammation. 2017;40(3):894–903.
Wang X, Zheng X, Ma C, Zhao L. Role of TRIF Small Interference RNA (siRNA) in Chronic Experimental Allergic Encephalomyelitis (EAE). Med Sci Monit. 2015;21:2583–7.
Canning P, Ruan Q, Schwerd T, Hrdinka M, Maki JL, Saleh D, Suebsuwong C, Ray S, Brennan PE, Cuny GD, et al. Inflammatory Signaling by NOD-RIPK2 Is Inhibited by Clinically Relevant Type II Kinase Inhibitors. Chem Biol. 2015;22(9):1174–84.
Topal Y, Gyrd-Hansen M. RIPK2 NODs to XIAP and IBD. Semin Cell Dev Biol. 2021;109:144–50.
Shaw PJ, Barr MJ, Lukens JR, McGargill MA, Chi H, Mak TW, Kanneganti TD. Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity. 2011;34(1):75–84.
Nachbur U, Stafford CA, Bankovacki A, Zhan Y, Lindqvist LM, Fiil BK, Khakham Y, Ko HJ, Sandow JJ, Falk H, et al. A RIPK2 inhibitor delays NOD signalling events yet prevents inflammatory cytokine production. Nat Commun. 2015;6:6442.
Jun JC, Cominelli F, Abbott DW. RIP2 activity in inflammatory disease and implications for novel therapeutics. J Leukoc Biol. 2013;94(5):927–32.
Tigno-Aranjuez JT, Asara JM, Abbott DW. Inhibition of RIP2’s tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. 2010;24(23):2666–77.
Quicke KM, Diamond MS, Suthar MS. Negative regulators of the RIG-I-like receptor signaling pathway. Eur J Immunol. 2017;47(4):615–28.
Colonna L, Catalano G, Chew C, D’Agati V, Thomas JW, Wong FS, Schmitz J, Masuda ES, Reizis B, Tarakhovsky A, et al. Therapeutic targeting of Syk in autoimmune diabetes. J Immunol. 2010;185(3):1532–43.
Deng GM, Liu L, Bahjat FR, Pine PR, Tsokos GC. Suppression of skin and kidney disease by inhibition of spleen tyrosine kinase in lupus-prone mice. Arthritis Rheum. 2010;62(7):2086–92.
Genovese MC, Kavanaugh A, Weinblatt ME, Peterfy C, DiCarlo J, White ML, O’Brien M, Grossbard EB, Magilavy DB. An oral Syk kinase inhibitor in the treatment of rheumatoid arthritis: a three-month randomized, placebo-controlled, phase II study in patients with active rheumatoid arthritis that did not respond to biologic agents. Arthritis Rheum. 2011;63(2):337–45.
Deng GM, Kyttaris VC, Tsokos GC. Targeting Syk in Autoimmune Rheumatic Diseases. Front Immunol. 2016;7:78.
Tak PP. Effects of infliximab treatment on rheumatoid synovial tissue. J Rheumatol Suppl. 2005; 74: 31–34.
Catrina AI, Trollmo C, af Klint E, Engstrom M, Lampa J, Hermansson Y, Klareskog L, Ulfgren AK. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum. 2005;52(1):61–72.
Vigna-Pérez M, Abud-Mendoza C, Portillo-Salazar H, Alvarado-Sánchez B, Cuevas-Orta E, Moreno-Valdés R, Baranda L, Paredes-Saharopulos O, González-Amaro R. Immune effects of therapy with Adalimumab in patients with rheumatoid arthritis. Clin Exp Immunol. 2005;141(2):372–80.
Mihara M, Kasutani K, Okazaki M, Nakamura A, Kawai S, Sugimoto M, Matsumoto Y, Ohsugi Y. Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int Immunopharmacol. 2005;5(12):1731–40.
Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–52.
Cunnane G, Madigan A, Murphy E, FitzGerald O, Bresnihan B. The effects of treatment with interleukin-1 receptor antagonist on the inflamed synovial membrane in rheumatoid arthritis. Rheumatology. 2001;40(1):62–9.
Bresnihan B, Newmark R, Robbins S, Genant HK. Effects of anakinra monotherapy on joint damage in patients with rheumatoid arthritis. Extension of a 24-week randomized, placebo-controlled trial. J Rheumatol. 2004;31(6):1103–11.
Xia L, Lu J, Xiao W. Blockage of TNF-α by infliximab reduces CCL2 and CCR2 levels in patients with rheumatoid arthritis. J Investig Med. 2011;59(6):961–3.
Silva LC, Ortigosa LC, Benard G. Anti-TNF-α agents in the treatment of immune-mediated inflammatory diseases: mechanisms of action and pitfalls. Immunotherapy. 2010;2(6):817–33.
Kim GW, Lee NR, Pi RH, Lim YS, Lee YM, Lee JM, Jeong HS, Chung SH. IL-6 inhibitors for treatment of rheumatoid arthritis: past, present, and future. Arch Pharm Res. 2015;38(5):575–84.
Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, Davis JC Jr, Kennedy WP. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis. 2016;75(1):196–202.
Hausmann JS. Targeting cytokines to treat autoinflammatory diseases. Clin Immunol. 2019;206:23–32.
Bouman CA, van Herwaarden N, van den Hoogen FH, Fransen J, van Vollenhoven RF, Bijlsma JW, Maas AV, den Broeder AA. Long-term outcomes after disease activity-guided dose reduction of TNF inhibition in rheumatoid arthritis: 3-year data of the DRESS study-a randomised controlled pragmatic non-inferiority strategy trial. Ann Rheum Dis. 2017;76(10):1716–22.
Kirou KA, Gkrouzman E. Anti-interferon alpha treatment in SLE. Clin Immunol. 2013;148(3):303–12.
Borchers AT, Leibushor N, Cheema GS, Naguwa SM, Gershwin ME. Immune-mediated adverse effects of biologicals used in the treatment of rheumatic diseases. J Autoimmun. 2011;37(4):273–88.
Suárez-Fariñas M, Arbeit R, Jiang W, Ortenzio FS, Sullivan T, Krueger JG. Suppression of molecular inflammatory pathways by Toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS ONE. 2013;8(12): e84634.
Clemente JC, Manasson J, Scher JU. The role of the gut microbiome in systemic inflammatory disease. BMJ. 2018;360: j5145.
Smolen JS, Weinblatt ME, Sheng S, Zhuang Y, Hsu B. Sirukumab, a human anti-interleukin-6 monoclonal antibody: a randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2014;73(9):1616–25.
Finch DK, Sleeman MA, Moisan J, Ferraro F, Botterell S, Campbell J, Cochrane D, Cruwys S, England E, Lane S, et al. Whole-molecule antibody engineering: generation of a high-affinity anti-IL-6 antibody with extended pharmacokinetics. J Mol Biol. 2011;411(4):791–807.
Mease P, Strand V, Shalamberidze L, Dimic A, Raskina T, Xu LA, Liu Y, Smith J. A phase II, double-blind, randomised, placebo-controlled study of BMS945429 (ALD518) in patients with rheumatoid arthritis with an inadequate response to methotrexate. Ann Rheum Dis. 2012;71(7):1183–9.
Shaw S, Bourne T, Meier C, Carrington B, Gelinas R, Henry A, Popplewell A, Adams R, Baker T, Rapecki S, et al. Discovery and characterization of olokizumab: a humanized antibody targeting interleukin-6 and neutralizing gp130-signaling. MAbs. 2014;6(3):774–82.
Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, Drappa J, Wang L, Greth W. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(11):1909–16.
Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, Illei GG, Drappa J, Wang L, Yoo S. Anifrolumab, an anti-interferon-α receptor monoclonal antibody, moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 2017;69(2):376–86.
Acknowledgements
Not applicable.
Funding
This work was supported by Major International (Regional) Joint Research Project grants 81820108017 (to B.Z.), the National Natural Science Foundation of China grants 81771673 (to B.Z.), Young Talent Program of Xi'an Jiaotong University grants YX1J005 (to B.Z.) and National young talent program (to B.Z.).
Author information
Authors and Affiliations
Contributions
JL, HZ and YS wrote the manuscript. BZ generated the idea and design the manuscript structure. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, J., Zhang, H., Su, Y. et al. Application and prospect of targeting innate immune sensors in the treatment of autoimmune diseases. Cell Biosci 12, 68 (2022). https://doi.org/10.1186/s13578-022-00810-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-022-00810-w