
Abstract
Neuronal loss is a primary factor in determining the outcome of ischemic stroke. Oridonin (Ori), a natural diterpenoid compound extracted from the Chinese herb Rabdosia rubescens, has been shown to exert anti-inflammatory and neuroregulatory effects in various models of neurological diseases. In this study we investigated whether Ori exerted a protective effect against reperfusion injury-induced neuronal loss and the underlying mechanisms. Mice were subjected to transient middle cerebral artery occlusion (tMCAO), and were injected with Ori (5, 10, 20 mg/kg, i.p.) at the beginning of reperfusion. We showed that Ori treatment rescued neuronal loss in a dose-dependent manner by specifically inhibiting caspase-9-mediated neuronal apoptosis and exerted neuroprotective effects against reperfusion injury. Furthermore, we found that Ori treatment reversed neuronal mitochondrial damage and loss after reperfusion injury. In N2a cells and primary neurons, Ori (1, 3, 6 μM) exerted similar protective effects against oxygen-glucose deprivation and reoxygenation (OGD/R)-induced injury. We then conducted an RNA-sequencing assay of the ipsilateral brain tissue of tMCAO mice, and identified receptor-interacting protein kinase-3 (RIPK3) as the most significantly changed apoptosis-associated gene. In N2a cells after OGD/R and in the ipsilateral brain region, we found that RIPK3 mediated excessive neuronal mitophagy by activating AMPK mitophagy signaling, which was inhibited by Ori or 3-MA. Using in vitro and in vivo RIPK3 knockdown models, we demonstrated that the anti-apoptotic and neuroprotective effects of Ori were RIPK3-dependent. Collectively, our results show that Ori effectively inhibits RIPK3-induced excessive mitophagy and thereby rescues the neuronal loss in the early stage of ischemic stroke.
Similar content being viewed by others

Introduction
Ischemic stroke is a major type of cerebrovascular disease, and it usually results in death or disability in aging people [1]. Despite recent developments in innovative treatment strategies, stroke remains the second leading cause of death and disability worldwide [2]. Neuronal loss plays a critical role in most important neural pathologies, especially in ischemic stroke. Importantly, the core problem in stroke is the loss of neuronal cells, which makes recovery difficult or even impossible in the late stages of the pathological process [3, 4]. Intensive treatment options applied to stroke mainly target to the re-establishment of blood flow, but the effect of thrombolytic agents is limited due to their short effective time window.
During the period of ischemic stroke pathology, the blood supply to neurons is disrupted, which promotes a cascade of pathophysiological responses resulting in neuronal loss [5]. Reportedly, two caspase-dependent apoptosis pathways are the main causes of neuronal loss after ischemic stroke [6,7,8]. The intrinsic pathway is initiated by injured mitochondria and endoplasmic reticulum (ER) stress. Mitochondrial pathway-mediated apoptosis involves the release of mitochondrial cytochrome c (Cyto-C) into the cytosol, the stimulation of caspase-3 and subsequent activation of downstream caspase-9 cascades, ultimately destroying the cell [7]. ER stress causes the accumulation of unfolded proteins in the ER, a phenomenon termed the unfolded protein response (UPR). Once the UPR is protracted or excessively active, apoptosis is induced. This type of apoptosis progression is mainly dependent on the activity of caspase-12 [8]. The extrinsic pathway is triggered by death receptors at the cell surface and is mediated by downstream caspase-8/caspase-3 hydrolytic cascades [6]. Importantly, previous studies have proven that inhibiting caspase cascade-mediated apoptosis is beneficial to ischemic stroke therapy in mice [9,10,11]. However, the specific mechanism of neuronal apoptosis after stroke has not yet been established, and there are few clinically effective drugs for ischemic stroke therapy.
Oridonin (Ori), an ent-kaurene tetracyclic diterpenoid compound, was isolated from the Chinese herb Rabdosia rubescens and has been shown to exert various biological and pharmacological effects, including antiapoptotic, anti-inflammatory and antioxidant effects, in Alzheimer’s disease and traumatic brain injury models [12,13,14,15,16]. However, whether Ori can ameliorate neuronal loss after acute ischemic stroke remains unclear. Therefore, the primary aim of the present study was to investigate the possible protective effects of Ori on neuronal loss and the mechanisms involved in an ischemic stroke model.
Materials and methods
Animals
Adult male C57BL/6 mice (20–25 g) were purchased from the Animals Resource Center of the Faculty of Medicine, Nanjing Medical University. Before the experiments, all animals were maintained in control conditions, including a 12 h light–dark cycle, free access to water and food, and suitable temperature and humidity.
All mice accepted procedures in accordance with the regulations of the Animal Protection and Use Committee of Jiangsu Association of Experimental Animals and approved by the Animal Protection and Ethics Committee of Nanjing Medical University (IACUC) of Nanjing Medical University (approval number: 2110035).
Reagents and drugs
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and penicillin–streptomycin (PS) were obtained from Gibco (Waltham, MA, USA). Ori was purchased from Targetmol (28957-04-2, Shanghai, China), dissolved in dimethyl sulfoxide (DMSO) to produce a 110 mM stock solution and then diluted to working concentrations. 3-Methyladenine (3-MA) was purchased from MedChemExpress (HY-19312, Monmouth Junction, NJ, USA), and was dissolved in dimethyl sulfoxide (DMSO) to produce a 33.25 mM stock solution and then diluted to working concentrations (5 mM).
Transient middle cerebral artery occlusion (tMCAO) in mice
The tMCAO model was established by inserting a nylon thread into the right middle cerebral artery as previously described. The MoorFLPI Full-field Laser Perfusion Imager (MoorFLPI-2, Gene&I, Beijing, China) was used to establish a transient focal cerebral ischemia and reperfusion model through local cerebral blood flow. In brief, the mice were anesthetized with 1% pentobarbitone and placed in a supine position. After skin preparation and disinfection, the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were exposed and ligated carefully. Then, a nylon monofilament (0.18 ± 0.01 mm, L1800, Guangzhou Jialing Biotechnology, Guangzhou, China) was inserted into the ICA through the ECA until reaching the origin of the middle cerebral artery. After 45 min, the nylon monofilament was pulled out from the ICA to achieve reperfusion. The mice in the sham group were subjected to a similar surgical procedure without middle cerebral artery occlusion. At the beginning of reperfusion, different concentrations of Ori (5 mg/kg, 10 mg/kg, 20 mg/kg) were intraperitoneally injected into the mice and the same amount of DMSO was applied as vehicle.
Recombinant adeno-associated virus (AAV) and infection
Recombinant AAV was generated by GeneChem Co., Ltd (Shanghai, China). Using the recombinant adeno-associated virus vector AAV-hSyn promoter-EGFP-MIR155 (MCS)-S V40 PolyA, the target sequence of the mouse Ripk3 gene, 5′-CUCCGAGGUAAAGCAUUAUTT-3′, was inserted into the MCS restriction site of the vector. After the sequencing results were accurate, the plasmid was packaged into an AAV virus of the PHP.eB serotype, and the obtained Ripk3 knockdown AAV virus was named AAV-hSyn-EGFP-MIR155 (si-Ripk3). The negative control AAV virus was AAV-hSyn-EGFP-MIR155 (si-control), the serotype was also PHP.eB, and the sequence of the negative control was 5′-TTCTCCGAACGTGTCACGT-3′. Three weeks before the preparation of tMCAO surgery (or sham-operated surgery), C57BL/6 mice were infected with 1.5 μL AAV by microinjecting into the right sensorimotor cortex (1.06 mm caudal to the bregma, 1.5 mm lateral to the midline, 0.55 mm ventral to the dura) at a flow rate of 1 μL/5 min.
Detection of the infarct volume
2,3,5-Triphenyltetrazolium chloride (TTC; T8877, Sigma Aldrich, MO, USA) was used to measure the infarct volume. Mice were anesthetized and sacrificed by decapitation 1 d after tMCAO. The brains were quickly removed and frozen for 15 min. Then, the brains were cut into 2-mm-thick slices. These slices were incubated in a 1% TTC solution at 37 °C for 20 min and fixed with 4% paraformaldehyde (PFA) overnight. The infarct volume was determined by subtracting the volume of noninfarcted tissue in the ipsilateral hemisphere from the volume of noninfarcted tissue in the intact contralateral hemisphere to correct for brain edema. The infarct volume was calculated by summing the infarct volumes for all slices of each brain. The infarct volume was then calculated by the ImageJ software (Loci, Madison, WI, USA).
Neurological behavior score assessment
The modified Neurological Severity Score (mNSS) was applied to assess the neurological deficits. Neurological dysfunction was tested 1 d after tMCAO. The score ranges from 0–18 points, evaluating motor, sensory, beam balance and reflex ability. Higher scores indicate the more severe neurological damage. Severity was classified as follows: 1–6 points, mild injury; 7–12 points, moderate injury; and 13–18 points, severe injury. One point was awarded to a failed specific task or to a disappeared reflex which means that higher cumulative scores indicate more severe neurological impairments in mice.
Immunofluorescence staining
The mice from different groups were perfused with PBS followed by 4% paraformaldehyde (G1101, Servicebio, Wuhan, China). After fixation with paraformaldehyde for 1 d, brain tissues were dehydrated with 30% sucrose solution. Next, brain tissues were embedded in Tissue-Tek® O.C.T compound and sliced into 15-μm-thick sections. The slices were loaded with 0.25% Triton X-100 for 10 min and incubated in 5% bovine serum albumin (BSA) at room temperature for 60 min. Subsequently, the slices were incubated with primary antibodies overnight at 4 °C. The dilutions of primary antibodies were as follows: NeuN (1:1000, ab177487, Abcam, Cambridge, MA, USA), ATPB (1:200, ab14730, Abcam, Cambridge, MA, USA), LC3 (1:200, #3868, CST, Boston, USA) and RIPK3 (1:100, #95702, CST, Boston, USA). The slices were washed three times with PBST and incubated with secondary antibodies (1:1000, Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature. Finally, the nuclei were stained with 4′, 6-diamidino-2-phenyl-indole (DAPI; Southern Biotech, Birmingham, Ala, USA). Images were captured using a fluorescence microscope (Olympus, Tokyo, Japan) and analyzed with ImageJ software.
Transmission electron microscopy
Mice were perfused with 4% paraformaldehyde 1 d after tMCAO. Then, the brains were rapidly separated, and the ischemic cerebral cortex was cut into 1-mm3 pieces. Subsequently these pieces were fixed in 2% glutaraldehyde overnight at 4 °C. After sectioning and uranium lead double staining, the samples were observed using a TEM system (JEM-1010, JEOL, Tokyo, Japan). Based on the typical mitochondrial ultrastructure, the mitochondrial damage score was used to evaluate the damage to mitochondria. Grade 0 indicated intact and normal ultrastructure of mitochondria; Grade 1 indicated that the general structure was retained, but the number of cristae was slightly reduced and there was mild swelling; Grade 2 indicated that the mitochondrial matrix basically reduced, there was a lot of swelling, and there was discontinuous membrane and cristae; Grade 3 indicated severe swelling and complete loss of cristae; and Grade 4 indicated complete destruction of the outer membrane and collapse of the mitochondrial structure.
Neuronal apoptosis detection in vivo
To detect the apoptosis of neurons in brain tissues, a TUNEL BrightGreen Apoptosis Detection Kit (A112, Vazyme, Nanjing, China) was used. After perfusion, fixation, and dehydration, the brain tissues were embedded and sliced. All procedures followed the manufacturer’s instructions. The slices were incubated with TUNEL reaction mixture at 37 °C for 1 h in the dark. Then, these slices were incubated with a NeuN antibody (1:1000, ab177487, Abcam, Cambridge, MA, USA) at 4 °C overnight. The secondary antibody, goat anti-mouse Alexa Fluor 555 (1:1000, A-31570, Thermo Fisher, MA, USA), was added to the slices for 1 h at room temperature. The nuclei were stained with DAPI. The quantification of images was analyzed with ImageJ as previously described.
N2a cell culture
Mouse neuroblastoma N2a cells were cultured in DMEM medium containing 10% FBS (10100147, Thermo Fisher, MA, USA) and 1% penicillin–streptomycin (15140163, Thermo Fisher, MA, USA). Cells were maintained in an environment with 5% CO2 at 37 °C.
Primary neuron cultures
Primary neuron cultures were prepared as previously described and were isolated from 1-day-old postnatal C57BL/6 mice. Briefly, the cerebral cortices were dissected and incubated with 0.125% trypsin/EDTA (Gibco, Grand Island, NY, USA) and 0.025% DNase I (Biofroxx, 1121MG010) for 15 min at 37 °C. After centrifugation (1000 rpm for 5 min), cell pellets were resuspended in DMEM supplemented with 10% FBS and 50 U/mL penicillin–streptomycin (Life Technologies, 15140122) and filtered through an 80-μm cell strainer. Collected cells were then seeded into 24-well plates coated with 0.01 mg/mL poly‐D‐lysine (Sigma, P6407). The medium was changed th neurobasal medium (Life Technologies, 21103-049) supplemented with 2% B27 (Life Technologies, 17504044) and 50 U/mL PS at 6 h after seeding. Neurons were maintained at 37 °C in a humidified 5% CO2 95% air atmosphere and used at 7–10 days in vitro (DIV). To prevent the growth of astrocytes, 1 μM cytosine-beta-D-arabinofuranose hydrochloride (AraC, Sigma, C1768) was added to the media and maintained for 1 d at 2 DIV. Primary neurons were used for the next experiments after 10 days of culture.
Oxygen-glucose deprivation and reoxygenation (OGD/R) and drug treatment
N2a cells
N2a cells were exposed to OGD/R conditions to mimic cerebral I/R injury as previously described. Before OGD/R, N2a cells were seeded into a 6-well plate and cultured with DMEM medium in an incubator with 95% air and 5% CO2 at 37 °C. For the OGD procedure, DMEM was replaced with glucose-free DMEM, and then the cells were placed in deoxygenated conditions (95% N2 and 5% CO2) for 6 h. When OGD was over, the cells were removed from the deoxygenated condition. A stock solution of Ori was diluted to different working concentrations (1 μM, 3 μM, 6 μM) with culture media. After replacement with these medium, the cells were cultured in a normoxic incubator.
Primary neurons
For the OGD procedure, neurobasal medium was replaced with glucose-free DMEM, and then, the cells were placed in deoxygenated conditions (95% N2 and 5% CO2) for 2 h. When OGD was over, the cells were removed from the deoxygenated condition. A stock solution of Ori was diluted to different working concentrations (6 μM) with neurobasal medium.
Cell viability assay
Cell viability was measured with a cell Counting Kit-8 (CCK8; A311, Vazyme, Nanjing, China). N2a cells were seeded into a 96-well plate at a density of 8000. After OGD/R and drug treatment, 10 μL of CCK8 reagent was added to each well, and the cells were incubated for 2 h at 37 °C. The absorbance was read with a microplate reader at 450 nm.
Annexin V/PI staining
An Annexin V-FITC/PI apoptosis analysis kit (abs50001, Absin, Shanghai, China) was applied to detect the apoptosis of N2a cells. According to the manufacturer’s instructions, the cells were digested with 0.25% trypsin and collected. After washing with PBS twice, the cells were resuspended in binding buffer. 5 μL of Annexin V-FITC was first added to the cells and incubated for 15 min at room temperature, and then 5 μL of PI was added. After 5 min, the apoptosis rate of the cells was examined by flow cytometry (BD FACSVerse, NJ, USA). Data were analyzed using FlowJo Software (Version 7.6.1; TreeStar, Ashland, OR, USA).
Primary neuronal apoptosis detection
TUNEL BrightRed Apoptosis (A113, Vazyme, Nanjing, China) assays were conducted to detect the apoptosis of primary neurons. Briefly, after treatment, primary neurons were fixed with 4% PFA solution, and the remaining steps were completed according to the instructions of the kit. Images were captured using a fluorescence microscope (Olympus, Tokyo, Japan) and analyzed with ImageJ software.
ΔΨm measurement and mitochondrial ROS detection
ΔΨm was measured with JC-1 (C2006, Beyotime, Shanghai, China). N2a cells were seeded into a 24-well plate. According to the protocol, cells were washed with PBS at the end of treatment. JC-1 staining solution was added to each well for 20 min at 37 °C. Then, the cells were washed twice with staining buffer and ΔΨm was examined using a fluorescence microscope. To analyze the mitochondrial ROS generation, the MitoSOX™ Red Mitochondrial Superoxide Indicator (M36008, Thermo Fisher, MA, USA) was used to detect it. After OGD/R and drug treatment, MitoSOX was added to the cells and incubated at 37 °C for 10 min in the dark. After washing with PBS twice, the cells were scanned with a microplate reader.
Cell transfection
For RIPK3 knockdown, RIPK3 siRNA (GenePharma, Shanghai, China) was added. According to the manufacturer’s instructions, RIPK3 siRNA was transfected into N2a cells using Lipofectamine 2000 (11668030, Invitrogen, CA, USA). The cells were replaced with DMEM after transfection in Opti-MEM for 6 h.
Co-immunoprecipitation
For the co-immunoprecipitation assay, N2a cells were seeded into a 12-well plate at a density of 1.0 × 105. After OGD/R and drug treatment, the cells in each well were lysed with 200 μL of immunoprecipitated lysis buffer. Ten microliters of lysate were collected as an input sample. Antibodies were added to the remaining lysates and incubated at 4 °C overnight. Subsequently, 20 μL of Protein A/G magnetic beads (HY-K0202, MCE, NJ, USA) was added and incubated at 4 °C for 3 h. After washing and boiling, the immunoprecipitates were loaded on SDS-PAGE.
RNA sequence
Total RNA was isolated using the TRIzol Reagent (Invitrogen Life Technologies), after which the concentration, quality and integrity were determined using a NanoDrop spectrophotometer (Thermo Scientific). Three micrograms of RNA were used as input material for the RNA sample preparations. Sequencing libraries were generated according to the following steps. First, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in an Illumina proprietary fragmentation buffer. First strand cDNA was synthesized using random oligonucleotides and Super Script II. Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities, and the enzymes were removed. After adenylation of the 3′ ends of the DNA fragments, Illumina PE adapter oligonucleotides were ligated to prepare for hybridization. To select cDNA fragments of the preferred 400–500 bp in length, the library fragments were purified using the AMPure XP system (Beckman Coulter, Beverly, CA, USA). DNA fragments with ligated adapter molecules on both ends were selectively enriched using Illumina PCR Primer Cocktail in a 15-cycle PCR. Products were purified (AMPure XP system) and quantified using the Agilent high sensitivity DNA assay on a Bioanalyzer 2100 system (Agilent). The sequencing library was then sequenced on a NovaSeq 6000 platform (Illumina) by Shanghai Personal Biotechnology Co. Ltd. We used HTSeq (0.9.1) statistics to compare the read count values on each gene as the original expression of the gene, and then used FPKM to standardize the expression. Then, the differential expression of genes was analyzed by DESeq (1.30.0) with the following screening conditions: expression difference multiple |log2FoldChange| >1, significant P value < 0.05. ClusterProfiler (3.4.4) software was used to carry out the enrichment analysis of the KEGG pathways of the differentially expressed genes, focusing on the significantly enriched pathways with P value < 0.05.
Western blot
Total proteins were extracted from brain tissues and N2a cells using RIPA lysis buffer containing phenylmethylsulfonyl fluoride (PMSF; ST506, Beyotime, Shanghai, China) and phosphatase inhibitor (P1081, Beyotime, Shanghai, China), and mitochondrial proteins were extracted using a Mitochondrial Extraction Kit (Beyotime, Shanghai, China). Bicinchoninic acid (BCA; E112, Vazyme, Nanjing, China) was used to measure concentrations. The volumes and contents of all samples were equalized. Next, the protein samples were electrophoresed by SDS-PAGE and transferred to PVDF membranes (IPVH00010, Merck Millipore, MA, USA). After blocking with 5% nonfat milk at room temperature for 2 h, the membranes were incubated with primary antibody at 4 °C for 12 h. On the second day, the membranes were washed with TBST and then incubated with secondary antibodies for 2 h at room temperature. After the above procedures, the images of the proteins were visualized by enhanced chemiluminescence, and the grayscale of each band was analyzed by ImageJ software.
The following primary antibodies were used: anti-Cyto C (1:1000, Santa Cruz, CA, USA), anti-Bax (1:1000, 50599-2-Ig, Proteintech, Rosemont, USA), anti-Bcl-2 (1:1000, 12789-1-AP, Proteintech, Rosemont, USA), anti-Tomm 20 (1:1000, 11802-1-AP, Proteintech, Rosemont, USA), anti-Caspase-8 (1:1000, 13423-1-AP, Proteintech, Rosemont, USA), anti-ATPB (1:1000, ab14730, Abcam, Cambridge, MA, USA), anti-Caspase12 (1:1000, ab62484, Abcam, Cambridge, MA, USA), anti-LC3 (1:1000, #3868, CST, MA, USA), anti-COX IV (1:1000, #4850, CST, MA, USA), anti-caspase-9 (1:1000, #9508, CST, MA, USA), anti-AMPK (1:1000, #2532, CST, MA, USA), anti-p-AMPK (1:1000, #2535, CST, MA, USA), anti-Parkin (1:1000, #4211, CST, MA, USA), anti-Cleaved caspase8 (1:1000, #8592, CST, MA, USA), anti-VDAC1 (1:1000, #4661, CST, MA, USA), anti-Caspase3 (1:1000, #9662, CST, MA, USA), anti-Cleaved caspase3 (1:1000, #9661, CST, MA, USA), anti-RIPK3 (1:800, sc-374639, Santa Cruz, CA, USA), anti-Pink (1:800, sc-518052, Santa Cruz, CA, USA), anti-β-actin, anti-mouse and anti-rabbit horseradish peroxidase-conjugated secondary antibodies (1:5000, Bioworld, MN, USA).
Statistics
GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA, USA) was used to perform all statistical tests. All the results are shown as the mean ± SD from at least three independent experiments. The experiments with only two groups were analyzed with the unpaired two-tailed Student’s t test. Single factor experiments with >2 groups were analyzed with one-way analysis of variance (ANOVA) with Dunnett’s post-hoc test. P < 0.05 was considered statistically significant, and ns represents no significance.
Results
Ori attenuates the neuronal loss and exerted neuroprotective effects following reperfusion injury
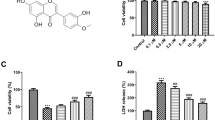
To determine the protective effects of Ori (S Fig. 1a) on neurons following reperfusion injury, we first investigated changes in cell availability and numbers of N2a cultures and primary neurons after OGD/R. As shown in Fig. 1a, Ori effectively promoted the viability of N2a cells after OGD/R in a concentration-dependent manner, as indicated by the CCK-8 assay. Moreover, Ori attenuated neuronal loss after OGD/R in a concentration-dependent manner in N2a cell cultures (Fig. 1b, c). Importantly, we also found that 6 μM Ori could reverse the loss of primary neurons after OGD/R (Fig. 1d, e). These results indicated that Ori could protect neurons against reperfusion injury.

a A CCK-8 assay was performed to detect the viability of N2a cells after OGD/R (n = 8). b, c The number of N2a cells was observed after OGD/R by brightfield microscopy. Representative images of different groups are shown (n = 4). d, e The number of primary neurons was observed after OGD/R by MAP2 staining. Representative images of different groups are shown (n = 5). f, g The apoptotic rate of N2a cells was analyzed by FACS analysis after OGD/R. Representative images of different groups are shown (n = 4). h, i The infarct volume was determined by conducting TTC staining at 1 d after reperfusion. Representative TTC-stained brain slices of different groups are shown (n = 6). j mNSS scores of the mice measured at 1 d after reperfusion. Higher scores indicate greater deficits (n = 6). k, l Nissl staining was used to measure the number of neurons in the ipsilateral brain region at 1 d after reperfusion. Representative images of different groups are shown (n = 4). m, n TUNEL and NeuN co-staining was performed to assess neuronal apoptosis at 1 d after reperfusion. Representative images from different groups are shown (n = 6). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Studies have implied that apoptosis principally contributes to neuronal loss after reperfusion injury, and therefore, inhibiting apoptosis can reduce neuronal loss and promote recovery after ischemic stroke [17, 18]. Therefore, we next examined whether Ori inhibited the neuronal apoptosis induced by reperfusion injury both in the N2a cell line and in primary neurons. As shown in Fig. 1f, g, the flow cytometry results showed that Ori decreased the apoptotic rate of N2a cell cultures in a concentration-dependent manner after OGD/R. Additionally, 6 μM Ori also significantly inhibited OGD/R-induced apoptosis in primary neurons (S Fig. 1b, c), as indicated by the TUNEL assay. Because of the close relationship between neuronal loss in the ipsilateral brain and recovery after ischemic stroke, we examined the protective effects of Ori in vivo. The operational steps and time points of the animal experiments are described in S Fig. 1d, the tMCAO surgery was performed in mice (S Fig. 1e), while blood flow was monitored with a laser Doppler Blood Flow Meter (S Fig. 1f). Serial doses of Ori were applied to treat tMCAO mice, and changes in infarct volume were determined. The results showed that Ori significantly reduced the infarct volume (Fig. 1h, i) and restored neurological deficits (Fig. 1j) in a dose-dependent manner. Considering the best therapeutic effects of Ori at a dose of 20 mg/kg on tMCAO mice, the following in vivo studies were conducted using this dose. In addition, the application of Ori increased the number of Nissl+ neurons (Fig. 1k, l) and decreased the number of NeuN+TUNEL+ apoptotic neurons (Fig. 1m, n) in the ipsilateral brain region. Accordingly, the results above confirmed that Ori inhibited apoptosis, rescued neuronal loss after reperfusion injury, and promoted the recovery of tMCAO mice.
Ori inhibits caspase-9-dependent neuronal apoptosis following reperfusion injury
Caspase-9-, caspase-8- and caspase-12-mediated signaling pathways are the major apoptosis pathways that contribute to neuronal loss after reperfusion injury [6,7,8]. Therefore, to identify the specific anti-apoptotic pathway activated by Ori, we observed changes in the cleavage rates of caspase-9, caspase-8, and caspase-12 after treatment with Ori both in N2a cells and in vivo. We found that Ori effectively decreased the expression level of cleaved caspase-9 in N2a cell cultures (Fig. 2a, b) in a concentration-dependent manner after OGD/R, but no significant changes in the levels of cleaved caspase-8 expression (Fig. 2a, c) or cleaved caspase-12 expression (Fig. 2a, d) were observed. Notably, we also found that Ori reduced the expression level of cleaved caspase-9 (Fig. 2e, f) but did not change that of cleaved caspase-8 (Fig. 2e, g) or that of cleaved caspase-12 (Fig. 2e, h) in vivo.

a–d Western blotting was used to analyze the protein expression levels of caspase-9, caspase-8, caspase-12 and their cleaved bodies in N2a cells after OGD/R. e–h Western blotting was used to analyze the protein expression levels of caspase-9, caspase-8, caspase-12 and their cleaved bodies in the ipsilateral brain region at 1 d after reperfusion (n = 3). i–n The expression levels of Bax, Bcl-2, mitochondrial Cyto-C, cytoplasmic Cyto-C, caspase-3 and cleaved caspase-3 were measured by Western blot analysis of N2a cells after OGD/R. o–t The expression levels of Bax, Bcl-2, mitochondrial Cyto-C, cytoplasmic Cyto-C, caspase-3 and cleaved caspase-3 were measured by Western blot analysis in the ipsilateral brain region at 1 d after reperfusion (n = 3). Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The release of mitochondrial cytochrome c (Cyto-C) leads to caspase-9 and caspase-3 activation, which ultimately leads to apoptosis [19]. In addition, the pro-apoptotic protein Bax promotes Cyto-C release from mitochondria to the cytosol upon death signaling, whereas the anti-apoptotic protein Bcl-2 inhibits Cyto-C release [20, 21]. Therefore, we next focused on changes in these molecules in the caspase-9-mediated pro-apoptotic signaling pathway after treatment with Ori. We found that Ori significantly decreased the expression of Bax (Fig. 2i, j) and increased the expression of Bcl-2 (Fig. 2i, k) in a concentration-dependent manner after OGD/R in N2a cell cultures. Furthermore, treatment with Ori increased the expression of mitochondrial Cyto-C (Fig. 2i, l) and decreased that in the cytosol (Fig. 2i, m), which indicated that Ori prevented Cyto-C release from mitochondria to the cytosol after OGD/R in N2a cell cultures. In addition, Ori effectively decreased the level of cleaved caspase-3 (Fig. 2i, n) in a concentration-dependent manner after OGD/R in N2a cell cultures. Similarly, treatment with Ori in vivo reduced the expression of Bax (Fig. 2o, p), increased the expression of Bcl-2 (Fig. 2o, q), increased the expression of mitochondrial Cyto-C (Fig. 2o, r), decreased the levels in cytosol (Fig. 2o, s), and reduced the caspase-3 cleavage rate (Fig. 2o, t). These data confirmed that Ori inhibited the apoptosis of neurons by downregulating the caspase-9-dependent apoptotic pathway.
Ori restores neuronal mitochondrial function following reperfusion injury
Damaged mitochondria play a central role in the initiation and activation of caspase-9-mediated apoptosis [22, 23]. Mitochondrial dysfunction during cell apoptosis is mainly characterized by increased production of ROS (reactive oxygen species), a decline in MMP (mitochondrial membrane potential) and the loss of mitochondria. Thus, our next analyses were directed to determine whether the anti-apoptotic effect of Ori was attributable to the reversal of mitochondrial damage. ROS are the major factors involved in mitochondrial damage during reperfusion injury [24], and we found that Ori decreased overloaded mitochondrial ROS levels after OGD/R in a concentration-dependent manner in N2a cell cultures (Fig. 3a, b). Intact MMP is necessary for selective elimination of dysfunctional mitochondria to maintain mitochondrial homeostasis [25]. Ori treatment increased the ratio of JC-1 aggregate/monomer (Fig. 3c, d) after OGD/R in N2a cell cultures, which indicated that Ori reversed the decline in MMP. In addition, the expression levels of the mitochondrial markers ATPB (ATPase beta subunit gene), Tomm 20 (translocase of outer mitochondrial membrane 20) and VDAC (voltage-dependent anion channel) were evaluated as indicators of mitochondrial quality and activity. We found that Ori elevated the expression of ATPB (Fig. 3e, f), Tomm 20 (Fig. 3e, g) and VDAC (Fig. 3e, h) in a concentration-dependent manner in N2a cell cultures.

a, b The production of ROS in mitochondria in N2a cells after OGD/R was measured by MitoSOX staining. Representative images of different groups are shown (n = 5). c, d Mitochondrial membrane potential was assessed by JC-1 staining. Representative images of different groups are shown (n = 5). e–h Western blotting was performed to analyze the protein expression levels of ATPB, Tomm20, and VDAC, three mitochondrial marker proteins, in N2a cells after OGD/R. i–k Electron micrographs were obtained to observe the number of mitochondria. Histogram showing mitochondrial injury in the brain at 1 d after reperfusion. Representative images of different groups are shown (n = 5). l–o Western blotting was performed to analyze the protein expression levels of ATPB, Tomm20, and VDAC in the ipsilateral brain region at 1 d after reperfusion (n = 3). Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In vivo, we performed electron microscopy to evaluate the changes in mitochondrial quantity and morphology after treatment with Ori. As shown in Fig. 3i, j, Ori effectively reversed the decrease in the number of mitochondria. Morphologically, the mitochondria were severely swollen, the matrix was basically reduced, and the number of cristae was profoundly decreased after reperfusion injury, but treatment with Ori effectively restored these changes (Fig. 3i). Moreover, the damage scores of mitochondria were used to quantify these changes, and we found that Ori significantly reduced the mitochondrial swelling score (Fig. 3k). In line with the data presented in Fig. 3e–h, Ori also increased the expression of the mitochondrial membrane protein ATPB (Fig. 3l, m), Tomm 20 (Fig. 3l, n) and VDAC (Fig. 3l, o) after reperfusion injury in vivo. Taken together, these results indicated that Ori effectively reversed mitochondrial damage when neuronal apoptosis was inhibited after reperfusion injury both in vitro and in vivo.
Ori prevents neuronal mitochondrial apoptosis and showed neuroprotective effects in a RIPK3-dependent manner
To investigate the specific apoptosis regulation mechanism of Ori, we performed an RNA-sequencing assay on the ipsilateral brain tissue of the sham, tMCAO + veh and tMCAO + Ori mice. A PCA plot confirmed that there was no overlap between the 3 groups (Fig. 4a); therefore, the 3 groups analyzed were transcriptionally distinct. A Venn plot showed the number of genes with changed expression (Fig. 4b), and we distinguished the genes regulating apoptosis from the KEGG pathway database from the sham and tMCAO + veh groups (Fig. 4c). We found that Ori significantly downregulated the RNA expression of RIPK3 in tMCAO mice (Fig. 4d). To examine whether the protective effects of Ori on the mitochondrial apoptosis and mitochondrial function of neurons were RIPK3 dependent, we observed the effects of Ori after downregulating the expression of RIPK3 in N2a cell cultures. The efficiency of RIPK3 expression interference was verified by Western blot analysis (S Fig. 2a). Then, we observed the effects of Ori on the neuronal apoptosis after knocking down RIPK3 expression. The results showed that downregulation of RIPK3 expression decreased the expression of cleaved caspase-9 (Fig. 4e, f) and inhibited the apoptosis of neurons (Fig. 4g, h). However, Ori treatment did not further change the expression of cleaved caspase-9 (Fig. 4e, f) or inhibit the apoptosis of neurons (Fig. 4g, h) after OGD/R. In addition, to further validate whether the protective effects of Ori were RIPK3 dependent in vivo, a neuron-specific RIPK3 knockdown mouse model was generated by stereotaxic injection of neuron-specific adeno-associated virus. As shown in Fig. 4i, j, AAV-hSyn-EGFP-MIR155 (AAV-siRIPK3) or AAV-Syn-GFP (AAV-sicon) was delivered into the perspective penumbra by microinjection. RIPK3 immunofluorescence on neurons was measured in the injected area followed by tMCAO, and although RIPK3 was robustly increased on neurons in the injected area followed by tMCAO (Fig. 4k, l), RIPK3 expression on neurons was significantly reduced in mice receiving the AAV-si-RIPK3 injection at 1 d after tMCAO (Fig. 4m, n). Neuronal RIPK3 knockdown and Ori showed comparable efficiency in anti-apoptosis, and importantly, but Ori did not further inhibit neuronal apoptosis upon neuronal RIPK3 knockdown (Fig. 4o, p). Moreover, neuronal RIPK3 knockdown and Ori treatments showed comparable effects on reducing infarct volume (Fig. 4q, r) and alleviating neurological defects (Fig. 4s), and Ori did not further show any significant additive protection upon neuronal RIPK3 knockdown (Fig. 4q–s).

a The PCA plot showed that the three groups (sham, tMCAO + veh, tMCAO + Ori) analyzed were transcriptionally distinct. b Venn diagram showing the number of genes with changed expression (expression difference multiple |log2FoldChange| > 1, significant P value < 0.05). c Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses revealed the potential roles of differentially expressed genes. d Heatmap representation of transcriptome array data for the expression levels of apoptosis-associated regulators from the sham, tMCAO + veh, and tMCAO + Ori datasets. e, f Western blot analysis of caspase-9 and cleaved caspase-9 expression after RIPK3 knockdown in OGD/R injury. g, h The apoptotic rate of N2a cells was analyzed by FACS analysis after knockdown of RIPK3 in OGD/R injury. i Construction of adeno-associated virus that specifically knocked down neuronal RIPK3. j Adeno-associated virus was assessed by detecting GFP expression in vivo. k, l The colocalization of RIPK3 and neurons (NeuN+) was assessed by immunofluorescence at 1 d after reperfusion (n = 3). m, n Neuronal RIPK3 from different groups was assessed by immunofluorescence at 1 d after reperfusion (n = 3). o, p A TUNEL assay was conducted to assess the apoptosis of neurons in different groups (n = 4). q, r The infarct volume was determined by TTC staining at 1 d after reperfusion. Representative TTC-stained brain slices of different groups are shown (n = 5). s mNSS scores of the mice measured 1 d after reperfusion (n = 5). Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ns represents no significance.
Overall, these results confirmed that RIPK3 was required for oridonin-induced anti-apoptosis and neuroprotective effects after ischemic stroke.
RIPK3-mediated mitophagy is required for oridonin-induced anti-apoptotic effects
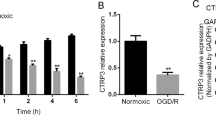
It has been reported that RIPK3 overexpression is closely associated with the mitochondrial apoptosis and that RIPK3 interacts with AMPK to activate early autophagy [26,27,28]. Importantly, increasing evidence has demonstrated that overactivated mitophagy at the early stage contributes to mitochondrial damage and apoptosis after ischemic stroke [29,30,31]. These reports and our results indicated that oridonin might protect neurons against apoptosis by regulating the RIPK3-mediated mitophagy. We quantified the dynamic changes in the RIPK3/AMPK-mediated mitochondrial LC3 signaling pathway within 6 h to 7 d after the induction of ischemic stroke pathogenesis. The results showed that the expression of RIPK3 (Fig. 5a, b), p-AMPK (Fig. 5c, d) and mitochondrial LC3 (Fig. 5e, f) synchronously increased within 6 h and reached a marked peak 1 d after reperfusion injury. Additionally, double immunofluorescence experiments were conducted to further observe the dynamic change in mitochondrial LC3, and the results confirmed its significant increases at 12 h and 1 d after reperfusion (Fig. 5g, h). Moreover, we confirmed the interaction of RIPK3 and AMPK after OGD/R by performing co-IP (Fig. 5i). In addition, we identified the cell types that express RIPK3 in the brain, and we found that RIPK3 was mainly expressed on NeuN+ neurons (Fig. 4i) and was expressed at low levels on Iba1+ microglia (S Fig. 3a) and GFAP+ astrocytes (S Fig. 3b) in brain slices from sham and tMCAO mice. These results indicated that the RIPK3-dependent mitophagy signaling pathway in neurons was activated in the hyperacute phase after reperfusion.

a–f The expression levels of the proteins RIPK3, p-AMPK and mitochondrial LC3 at different time points of reperfusion were measured by Western blot in the ipsilateral brain region (n = 4). g, h The level of mitochondrial LC3 was assessed by conducting double fluorescence at 12 h and 1 d after reperfusion (n = 4). i RIPK3 and AMPK interaction was assessed by coimmunoprecipitation experiments (n = 3). Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Subsequently, we sought to explore the impossible effects of Ori on the RIPK3-mediated mitophagy signaling pathway. We found that Ori treatment decreased the expression of RIPK3 (Fig. 6a, b), p-AMPK (Fig. 6c, d), mitochondrial Pink and Parkin (Fig. 6e, f) and increased the expression of p62 (Fig. 6g, h) in N2a cell cultures after OGD/R in a concentration-dependent manner. Similarly, the results in primary neurons also showed that oridonin decreased the expression of RIPK3 after OGD/R (S Fig. 4a). In vivo, the expression of RIPK3 at 12 h (Fig. 6i, j) and 1 d (Fig. 6k, l) after reperfusion was both significantly reduced after treatment with oridonin, and our double immunofluorescence experiments in vivo confirmed the results at 1 d again (Fig. 6m, n). Moreover, Ori treatment reduced the expression of p-AMPK (Fig. 6o, p), mitochondrial Pink and Parkin (Fig. 6q, r) and increased the expression of p62 (Fig. 6s, t) in vivo. Next, we tested whether Ori inhibited mitophagy after reperfusion injury. The results showed that Ori decreased the expression of mitochondrial LC3 (Fig. 7a, b) and the colocalization of LC3 puncta and Mitotracker after OGD/R in N2a cell cultures in a concentration-dependent manner (Fig. 7c, d). Additionally, Ori greatly inhibited OGD/R-induced mitophagy in primary neurons (S Fig. 5a). In vivo, we observed that fewer mitochondria in the ipsilateral brain region were enclosed by autophagosomes after treatment with Ori, as determined through electron microscopy (Fig. 7e, f). Similarly, Ori effectively reversed the elevated expression of mitochondrial LC3 at 12 h (Fig. 7g, h) and 1 d (Fig. 7i, j) after reperfusion, and confocal images directly revealed that Ori reduced the accumulation of LC3-positive puncta with the mitochondrial marker ATPB at 1 d after reperfusion in the ipsilateral brain region (Fig. 7k, l). The results above confirmed that Ori inhibited the RIPK3-mediated overactivated mitophagy after reperfusion injury. Then, we questioned whether the inhibitory effects of Ori on mitophagy of oridonin were RIPK3 dependent. Therefore, we observed the effects of Ori on the RIPK3-mediated mitophagy signaling pathway after knocking down RIPK3 expression in N2a cells. As shown in Fig. 7m, n, RIPK3 knockdown reduced the expression of RIPK3 and p-AMPK, but Ori did not change these levels after OGD/R. Notably, RIPK3 knockdown reduced reperfusion-induced LC3 puncta accumulation around mitochondria, but Ori treatment did not change this accumulation level (Fig. 7o, p). Taken together, these results confirmed that Ori prevented mitophagy mainly by inhibiting RIPK3-mediated mitophagy signaling pathway activation. Finally, we explored the contribution of mitophagy inhibition by Ori to its anti-apoptosis effect. To address this question, autophagy inhibition using 3-MA was conducted in N2a cell culture. The results showed that Ori and inhibiting autophagy via 3-MA significantly protected N2a cells from OGD/R-induced apoptosis, but Ori did not further show any significant anti-apoptotic effect upon inhibiting autophagy (Fig. 7q, r). Accordingly, RIPK3-mediated mitophagy was required for oridonin-induced anti-apoptotic effects.

a–d Western blotting was used to analyze the protein expression levels of RIPK3 and p-AMPK in N2a cells after OGD/R. e, f Western blotting was used to analyze the mitochondrial protein expression levels of Pink and Parkin in N2a cells after OGD/R. g, h Western blotting was used to analyze the protein expression level of P62 in N2a cells after OGD/R. i–l Western blotting was used to analyze the protein expression levels of RIPK3 in the ipsilateral brain region at 12 h and 1 d after reperfusion (n = 3). m, n The co-staining of RIPK3 and NeuN was assessed. Representative images from different groups are shown (n = 4). o, p Western blotting was used to analyze the protein expression levels of p-AMPK and AMPK in the ipsilateral brain region (n = 3). q, r Western blotting was used to analyze the mitochondrial protein expression levels of Pink and Parkin in the ipsilateral brain region (n = 3). s, t Western blotting was used to analyze the protein expression level of p62 in the ipsilateral brain region (n = 3). Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.

a, b Western blotting was performed to analyze the expression level of mitochondrial LC3 in N2a cells after OGD/R. c, d Laser confocal microscopy was performed to observe mitophagy in N2a cells. Representative images of different groups are shown (n = 9). e, f Electron micrographs were obtained to observe mitophagy in neurons. Representative images of different groups are shown (n = 5). g–j Western blotting was performed to analyze the expression level of mitochondrial LC3 in the ipsilateral brain region at 12 h and 1 d after reperfusion (n = 3). k, l The colocalization of ATPB (a special mitochondrial marker) and LC3 was observed and assessed at 1 d after reperfusion. Representative images of different groups are shown (n = 5). m, n Western blot analysis of RIPK3, p-AMPK, and AMPK expression after knockdown of RIPK3 in OGD/R injury in N2a cells (n = 3). o, p Laser confocal microscopy was conducted to observe mitophagy after RIPK3 knockdown in OGD/R injury. Representative images from different groups are shown (n = 9). q, r The apoptotic rate of N2a cells was analyzed by FACS analysis after different treatments. Representative images of different groups are shown (n = 4). Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns represents no significance.
Discussion
Neuronal loss after reperfusion injury contributes to cerebral injury and makes recovery difficult or impossible in the later stages of disease progression [3, 4]. However, many studies have concluded that different pathways, such as apoptosis, oxidative stress, neuroinflammation and excitotoxicity pathways, affect neuronal loss in the acute phase of stroke [17, 32]. Unfortunately, there is still no effective drug to rescue neuronal loss in stroke patients. In the current study, we provide evidence to explain the core role played by RIPK3/AMPK-mediated mitophagy in the pathogenesis and disease progression of cerebral I/R injury. Moreover, we found that Ori, an ent-kaurene tetracyclic diterpenoid compound, inhibited the caspase-9-mediated apoptosis of neurons by attenuating RIPK3/AMPK-mediated mitophagy and thus accelerated the recovery of tMCAO mice (Fig. 8).

Under ischemia, RIPK3 was activated. Activated RIPK3 interacts with AMPK to phosphorylate AMPK, and phosphorylated AMPK results in excessive mitophagy, leading to the activation of the caspase-9-dependent apoptotic pathway. Ori reduces the phosphorylation of AMPK by inhibiting RIPK3, thereby alleviating excessive mitophagy and preventing neuronal caspase-9-dependent apoptosis.
Mounting evidence implies that Ori affects many biological activities and exerts effects on different animal models. We have previously reported that treatment with Ori attenuated neuroinflammation and improved cognitive behavior deficits [14, 16]. In addition, other studies have shown that Ori exerts protective effects on reperfusion injury, such as acute kidney injury and brain ischemic stroke, in animal models by reducing oxidative stress and inflammation [33, 34]. However, no focus on the direct protective effect of Ori on neurons has been reported. According to our results, Ori significantly reduced neuronal loss and inhibited neuronal apoptosis after reperfusion injury both in vivo and in vitro. Moreover, our results revealed the specific anti-apoptotic pathway affected by Ori. The activated mitochondrial apoptotic pathway, characterized by the release of mitochondrial Cyto-c into the cytosol, increases cleaved caspase-3 and caspase-9 levels and the Bax/Bcl-2 ratio [19,20,21]. Our findings showed that Ori effectively reversed the aforementioned changes and confirmed that Ori inhibited caspase-9-associated mitochondrial apoptosis.
After ischemic stroke, the dynamic balance maintained by mitochondria is disrupted, which leads to Cyto-c release and caspase-9-associated apoptosis [35, 36]. Thus, mitochondrial dysfunction plays an important role in the activation of caspase-9-associated apoptosis [37, 38]. Our results demonstrated that Ori profoundly reversed mitochondrial dysfunction after reperfusion injury by decreasing the level of ROS, promoting MMP and increasing the expression of mitochondrial membrane proteins. Mitophagy plays a significant role in mitochondrial dysfunction, and elimination of abnormal mitochondria through mitophagy is essential for maintaining neuronal function in ischemic stroke [39, 40]. However, whether mitophagy is beneficial or harmful in ischemic stroke has been unclear until now. Some studies have shown that activating mitophagy to clear excessively aggregated and damaged mitochondria can reduce neuronal damage caused by cerebral I/R injury [7, 41, 42]; however, other studies have shown that inhibiting excessive mitophagy can play a protective role in cerebral I/R injury [43,44,45]. In our study, we provided evidence that Ori reduced the mitochondrial apoptosis of neurons by inhibiting the overactivated mitophagy after I/R injury.
In addition, our RNA-sequencing results clearly identified the most significantly downregulated apoptosis-associated gene, RIPK3, which plays an important role in autophagy/mitophagy and cell apoptosis. Afterward, we found that neuronal RIPK3 knockdown and Ori showed comparable efficiency in anti-apoptosis and neuroprotection, and very importantly, Ori did not show any significant additive protection upon neuronal RIPK3 knockdown both in vivo and in vitro. This result confirms that RIPK3 signaling is required for Ori to prevent neuronal mitochondrial apoptosis and exerts neuroprotective effects. Reportedly, RIPK3 inhibition reduced mitochondria-initiated oxidative damage, sustained mitochondrial dynamics and inhibited cell death [29,30,31]. Therefore, our findings and previous reports together suggest that RIPK3 is an important target for protecting neurons after I/R injury. Studies have suggested that RIPK3 interacts with AMPK to initiate AMPK-mediated autophagy and induce cell death [26,27,28, 46]. In our study, RIPK3/AMPK-mediated mitophagy signaling was indeed activated at an early stage of disease progression and peaked at 1 h after I/R injury. We also provided evidence that Ori reduced the activation of mitophagy, inhibited RIPK3/AMPK mitophagy signaling and decreased caspase-9 mitochondrial apoptosis in neurons. Importantly, both the blockage of RIPK3 via siRNA and mitophagy via 3-MA and treatment with Ori showed comparable anti-apoptosis effects, which confirms that the inhibition of RIPK3-mitophagy is required for Ori’s neuroprotective effect after ischemic stroke.
In conclusion, our findings provide evidence showing that Ori rescued neuronal loss by inhibiting caspase-9-dependent neuronal apoptosis in a RIPK3-dependent manner. By inhibiting the expression of RIPK3, Ori suppressed AMPK-mediated neuronal excessive mitophagy and improved mitochondrial function. These results established the beneficial effects of Ori and its special mechanism of action, suggesting that it is a novel therapeutic approach/target of stroke in humans.
References
Yousufuddin M, Young N. Aging and ischemic stroke. Aging. 2019;11:2542–4.
Feske SK. Ischemic stroke. Am J Med. 2021;134:1457–64.
Kim Y, Lee S, Zhang H, Lee S, Kim H, Kim Y, et al. CLEC14A deficiency exacerbates neuronal loss by increasing blood-brain barrier permeability and inflammation. J Neuroinflammation. 2020;17:48.
Brown GC. Neuronal loss after stroke due to microglial phagocytosis of stressed neurons. Int J Mol Sci. 2021;22:13442.
Koronowski KB, Perez-Pinzon MA. Sirt1 in cerebral ischemia. Brain Circ. 2015;1:69–78.
Muhammad IF, Borne Y, Melander O, Orho-Melander M, Nilsson J, Soderholm M, et al. FADD (Fas-Associated Protein With Death Domain), caspase-3, and caspase-8 and incidence of ischemic stroke. Stroke. 2018;49:2224–6.
Cai Y, Yang E, Yao X, Zhang X, Wang Q, Wang Y, et al. FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 2021;38:101792.
Gong L, Tang Y, An R, Lin M, Chen L, Du J. RTN1-C mediates cerebral ischemia/reperfusion injury via ER stress and mitochondria-associated apoptosis pathways. Cell Death Dis. 2017;8:e3080.
Shabanzadeh AP, D’Onofrio PM, Monnier PP, Koeberle PD. Targeting caspase-6 and caspase-8 to promote neuronal survival following ischemic stroke. Cell Death Dis. 2015;6:e1967.
Dai Q, Han S, Liu T, Zheng J, Liu C, Li J, et al. IL-17A Neutralization improves the neurological outcome of mice with Ischemic Stroke and inhibits caspase-12-dependent apoptosis. Front Aging Neurosci. 2020;12:274.
Wu M, Zhang H, Kai J, Zhu F, Dong J, Xu Z, et al. Rapamycin prevents cerebral stroke by modulating apoptosis and autophagy in penumbra in rats. Ann Clin Transl Neurol. 2018;5:138–46.
Li X, Zhang CT, Ma W, Xie X, Huang Q. Oridonin: a review of its pharmacology, pharmacokinetics and toxicity. Front Pharmacol. 2021;12:645824.
Yang H, Lv H, Li H, Ci X, Peng L. Oridonin protects LPS-induced acute lung injury by modulating Nrf2-mediated oxidative stress and Nrf2-independent NLRP3 and NF-kappaB pathways. Cell Commun Signal. 2019;17:62.
Zhang ZY, Daniels R, Schluesener HJ. Oridonin ameliorates neuropathological changes and behavioural deficits in a mouse model of cerebral amyloidosis. J Cell Mol Med. 2013;17:1566–76.
Yan C, Yan H, Mao J, Liu Y, Xu L, Zhao H, et al. Neuroprotective effect of Oridonin on traumatic brain injury via inhibiting NLRP3 inflammasome in experimental mice. Front Neurosci. 2020;14:557170.
Zhao XJ, Zhu HY, Wang XL, Lu XW, Pan CL, Xu L, et al. Oridonin Ameliorates traumatic brain injury-induced neurological damage by improving mitochondrial function and antioxidant capacity and suppressing neuroinflammation through the Nrf2 pathway. J Neurotrauma. 2022;39:530–43.
Sekerdag E, Solaroglu I, Gursoy-Ozdemir Y. Cell death mechanisms in stroke and novel molecular and cellular treatment options. Curr Neuropharmacol. 2018;16:1396–415.
Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC. Neuronal cell death. Physiol Rev. 2018;98:813–80.
Arama E, Bader M, Srivastava M, Bergmann A, Steller H. The two Drosophila cytochrome C proteins can function in both respiration and caspase activation. EMBO J. 2006;25:232–43.
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–32.
Khan A, Ali T, Rehman SU, Khan MS, Alam SI, Ikram M, et al. Neuroprotective effect of Quercetin against the detrimental effects of LPS in the adult mouse brain. Front Pharmacol. 2018;9:1383.
Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. 2021;184:2896–910. e13.
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–6.
Wu M, Gu X, Ma Z. Mitochondrial quality control in cerebral ischemia-reperfusion injury. Mol Neurobiol. 2021;58:5253–71.
Li H, Ham A, Ma TC, Kuo SH, Kanter E, Kim D, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy. 2019;15:113–30.
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507.
Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347–53.
Wu W, Wang X, Sun Y, Berleth N, Deitersen J, Schlutermann D, et al. TNF-induced necroptosis initiates early autophagy events via RIPK3-dependent AMPK activation, but inhibits late autophagy. Autophagy. 2021;17:3992–4009.
Deng Z, Ou H, Ren F, Guan Y, Huan Y, Cai H, et al. LncRNA SNHG14 promotes OGD/R-induced neuron injury by inducing excessive mitophagy via miR-182-5p/BINP3 axis in HT22 mouse hippocampal neuronal cells. Biol Res. 2020;53:38.
Shi RY, Zhu SH, Li V, Gibson SB, Xu XS, Kong JM. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther. 2014;20:1045–55.
Lan R, Wu JT, Wu T, Ma YZ, Wang BQ, Zheng HZ, et al. Mitophagy is activated in brain damage induced by cerebral ischemia and reperfusion via the PINK1/Parkin/p62 signalling pathway. Brain Res Bull. 2018;142:63–77.
Takano T, Oberheim N, Cotrina ML, Nedergaard M. Astrocytes and ischemic injury. Stroke. 2009;40:S8–12.
Li L, Cheng SQ, Guo W, Cai ZY, Sun YQ, Huang XX, et al. Oridonin prevents oxidative stress-induced endothelial injury via promoting Nrf-2 pathway in ischaemic stroke. J Cell Mol Med. 2021;25:9753–66.
Tan RZ, Yan Y, Yu Y, Diao H, Zhong X, Lin X, et al. Renoprotective effect of Oridonin in a mouse model of acute kidney injury via suppression of macrophage involved inflammation. Biol Pharm Bull. 2021;44:714–23.
Yorns WR Jr., Hardison HH. Mitochondrial dysfunction in migraine. Semin Pediatr Neurol. 2013;20:188–93.
Hoehn B, Yenari MA, Sapolsky RM, Steinberg GK. Glutathione peroxidase overexpression inhibits cytochrome C release and proapoptotic mediators to protect neurons from experimental stroke. Stroke. 2003;34:2489–94.
Nhu NT, Li Q, Liu Y, Xu J, Xiao SY, Lee SD. Effects of Mdivi-1 on neural mitochondrial dysfunction and mitochondria-mediated apoptosis in Ischemia-Reperfusion Injury after stroke: a systematic review of preclinical studies. Front Mol Neurosci. 2021;14:778569.
Dubois-Dauphin M, Pfister Y, Vallet PG, Savioz A. Prevention of apoptotic neuronal death by controlling procaspases? A point of view. Brain Res Brain Res Rev. 2001;36:196–203.
Shao Z, Dou S, Zhu J, Wang H, Xu D, Wang C, et al. The role of mitophagy in ischemic stroke. Front Neurol. 2020;11:608610.
Yang JL, Mukda S, Chen SD. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018;16:263–75.
Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu J, et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy. 2017;13:1754–66.
Shen Z, Zheng Y, Wu J, Chen Y, Wu X, Zhou Y, et al. PARK2-dependent mitophagy induced by acidic postconditioning protects against focal cerebral ischemia and extends the reperfusion window. Autophagy. 2017;13:473–85.
Zhang Y, He Y, Wu M, Chen H, Zhang L, Yang D, et al. Rehmapicroside ameliorates cerebral ischemia-reperfusion injury via attenuating peroxynitrite-mediated mitophagy activation. Free Radic Biol Med. 2020;160:526–39.
Feng J, Chen X, Lu S, Li W, Yang D, Su W, et al. Naringin attenuates Cerebral Ischemia-Reperfusion Injury through inhibiting Peroxynitrite-mediated mitophagy activation. Mol Neurobiol. 2018;55:9029–42.
Zhang Z, Yu J. NR4A1 Promotes cerebral ischemia reperfusion injury by repressing Mfn2-mediated mitophagy and inactivating the MAPK-ERK-CREB signaling pathway. Neurochem Res. 2018;43:1963–77.
Naito MG, Xu D, Amin P, Lee J, Wang H, Li W, et al. Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc Natl Acad Sci USA. 2020;117:4959–70.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grants 82071209 and 81771171 to ZYZ).
Author information
Authors and Affiliations
Contributions
ZYZ and LX designed the experiments, obtained resources, and acquired funding. ZYZ conducted the experiments with assistance from LL and LX. LL, JJS, HWZ, MXZ, HYZ and WG collected the data. WG, CLP and XL contributed to the statistical analysis. LL, JJS, and ZYZ analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, L., Song, Jj., Zhang, Mx. et al. Oridonin ameliorates caspase-9-mediated brain neuronal apoptosis in mouse with ischemic stroke by inhibiting RIPK3-mediated mitophagy. Acta Pharmacol Sin 44, 726–740 (2023). https://doi.org/10.1038/s41401-022-00995-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00995-3
- Springer Nature Singapore Pte Ltd.