
Abstract
High mobility group box 1 (HMGB1) is a ubiquitous nuclear protein that is present in almost all cells and regulates the activity of innate immune responses in both intracellular and extracellular settings. Current evidence suggests that HMGB1 plays a pivotal role in human pathological and pathophysiological processes such as the inflammatory response, immune reactions, cell migration, aging, and cell death. Sepsis is a systemic inflammatory response syndrome (SIRS) that occurs in hosts in response to microbial infections with a proven or suspected infectious etiology and is the leading cause of death in intensive care units worldwide, particularly in the aging population. Dysregulated systemic inflammation is a classic characteristic of sepsis, and suppression of HMGB1 may ameliorate inflammation and improve patient outcomes. Here, we focus on the latest breakthroughs regarding the roles of HMGB1 in sepsis and sepsis-related organ injury, the ways by which HMGB1 are released, and the signaling pathways and therapeutics associated with HMGB1. This review highlights recent advances related to HMGB1: the regulation of HMBG1 might be helpful for both basic research and drug development for the treatment of sepsis and sepsis-related organ injury.
Similar content being viewed by others


Introduction
Detecting pathogens is the first step in mounting an effective immune response to eliminate invading organisms and maintain immune homeostasis [1]. If unsuccessful, invading pathogens may enter the blood stream and trigger widespread systemic inflammation termed sepsis. Sepsis is also referred to as systemic inflammatory response syndrome (SIRS), occurs in response to microbial infections with a proven or suspected infectious etiology, and is the leading cause of death in intensive care units worldwide [2,3,4,5]. Notably, sepsis is common in the aging population and disproportionately affects patients with cancer and underlying immunosuppression [6]. The pathogenesis of sepsis is rather complex and partially attributable to dysregulated inflammatory responses propagated by innate immune cells, including macrophages, monocytes, neutrophils [7, 8], which release proinflammatory cytokines, resulting in damage to tissue and organs, metabolic acidosis, hypotension, or even death. To date, there are no effective therapeutics to cure sepsis and sepsis-related SIRS. The current therapies for sepsis in the clinic remain largely supportive and are limited to a few clinical interventions, including antibiotics, steroidal anti-inflammatory drugs, and early goal-directed therapies [9]. Unfortunately, these treatments are usually ineffective, and most patients still suffer from shock or death [10]. Moreover, some drugs have adverse effects that contribute to poor patient compliance. Accumulating evidence has demonstrated that restoring homeostasis and ameliorating dysregulated systemic inflammation are critical. Thus, a better understanding of the pathophysiological role of proinflammatory cells and cytokines in sepsis is required.
High mobility group box 1 (HMGB1) is a small protein of 215 amino acid residues composed of two tandem high mobility group (HMG) box domains (A box and B box) in the N-terminus and a continuous stretch of negatively charged residues in the C-terminus [11, 12]. HMGB1 is a ubiquitous nuclear protein present in almost all cells that regulates the activity of innate immune responses intracellularly and extracellularly [13]. HMGB1 was originally described as a nuclear DNA-binding protein. HMGB1 is properly defined as a cytokine because it stimulates proinflammatory responses in monocytes/macrophages in vivo in standardized models of systemic and local inflammation and mediates delayed endotoxin lethality in animal models of endotoxemia and sepsis [14]. Recently, a better understanding of HMGB1 has been gradually obtained. Serum levels of HMGB1 are high in patients who die of sepsis [8], whereas blocking HMGB1 preserves the ability of neutrophils to activate NADPH oxidase in patients recovering from septic shock [15]. Given the emerging roles of HMGB1 in systemic inflammation, the present review aims to discuss recent breakthroughs related to HMGB1 in sepsis and potential therapeutics.
Pathogenesis of sepsis
The range of presentations of sepsis syndrome is very wide because sepsis can originate from virtually any infecting organism. The pathogenesis of sepsis is extremely complex and involves the inflammatory response, immune dysfunction, mitochondrial dysfunction, coagulation disorders, neuroendocrine–immune network abnormalities, endoplasmic reticulum stress and autophagy.
Immune dysfunction
Immune activation in response to infection is triggered by highly conserved microbial pathogen-associated molecular patterns that are recognized by pattern-recognition receptors, including Toll-like receptors, on cells in the innate immune system. This interaction activates the release of both proinflammatory and anti-inflammatory mediators through the activation of nuclear factor κB (NF-κB) and neutrophils [6]. In addition, cytokines lead to neutrophil–endothelial cell adhesion, complement activation and clotting cascades. Sepsis was traditionally acknowledged as a systemic and proinflammatory response to infection, which subsequently leads to a cascade of immunosuppressive responses, including lymphopenia, anergy, and secondary infections [16]. Moreover, the maturation of dendritic cells (DCs) in the spleen and lymph nodes is impeded during sepsis. However, DC activation also leads to the accumulation of innate immune cells [17]. In sepsis models, impaired monocyte metabolism causes remarkable immunosuppression characterized by an extensive inhibition of metabolic processes, such as glycolysis, fatty acid oxidation, and oxidative phosphorylation [18]. Furthermore, emerging paradigms indicate that proinflammatory and immunosuppression responses might occur simultaneously, and their intensity depends on multiple factors of both the host and the pathogen, such as genetics, comorbidities, and pathogen types [19].
Mitochondrial dysfunction
Due to the limited oxygen supply and incomplete oxidative reaction, free radical production dramatically increases while the machinery of the antioxidant system, such as mitochondrial homeostasis, becomes imbalanced during sepsis. Mitochondrial dysfunction can aggravate the failure of tissue oxygen extraction, which is called cytopathic hypoxia [20]. When exposed to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPS), activated leukocytes release inflammatory cytokines and subsequently trigger the expression of NADPH oxidase [21]. In addition, inflammatory cytokines induce overproduction of reactive nitrogen species (RNS) by activating inducible nitric oxide synthase (iNOS). NO can bind ROS peroxides to form RNS, resulting in the irreversible repression of electron transfer chain (ETC) activity. During sepsis, impaired mitochondria represent a source of additional ROS production, further contributing to damage to mitochondria, including mitochondrial DNA damage and intimal damage, which ultimately triggers the apoptosis process [22]. Mitochondrial biogenesis is regulated by the AMPK/PGC-1/NRF-1/2 signaling pathway. However, the activation of AMPK and the subsequent PGC-1/NRF-1/2 pathway is disrupted by the disproportionality of the ATP/ADP ratio, thus leading to a decreased mitochondrial density after a severe sepsis episode [23, 24].
Coagulation disorders
Sepsis usually interferes with the distribution of systemic blood flow to organ systems via vasodilation and disturbances in microcirculation [25]. Either a systemic or local mismatch between oxygen delivery and tissue demand can result in severe tissue ischemia. Inflammatory regulators are also involved in the coagulopathy underlying sepsis. The coagulation manifestation can vary from fibrin deposition to massive thromboembolism in the microvascular bed, leading to a severe complication termed disseminated intravascular coagulation [25]. When the maximal activation of coagulation occurs, endogenous fibrinolysis substantially diminishes in sepsis. The release of plasminogen activators, such as tissue plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA), from vascular endothelial cell storage sites enhance the activity of plasminogen activators and the production of sub-quantitative plasmin [26]. Nevertheless, the disappearance of these effects is attributed to a continuous increase in plasminogen activator inhibitor-1 (PAI-1); a polymorphism of PAI-1 has been identified to increase the risk of septic shock caused by meningococcal infection [26].
Neuroendocrine–immune network abnormalities
The homeostasis involved in the interaction between the neuroendocrine and immune systems is also considered an important part of the host response during septic shock [27]. The responses of the central nervous system to sepsis depend on the following three main mechanisms: (1) circulatory inflammatory mediators connected to the central nervous system through the choroid plexus and ventricle organs; (2) the autonomic nervous system in which primary afferent nerves and sensory nerves are associated with PAMPs and enhance inflammatory cytokine activation; and (3) the activation of endothelial cells by the blood–brain barrier, leading to the release of inflammatory mediators, such as NOS metabolites [28]. Moreover, hypothalamic–pituitary–adrenal (HPA) axis dysfunction causes a reduction in the serum levels of corticotropin, adrenocorticotrophic hormone, and adrenal cortisol in sepsis, leading to adrenal insufficiency syndrome [29]. Evidence demonstrates that noradrenaline (NA) can respond to LPS by inhibiting the expression of pro-inflammatory factors, including TNF-β and IL-12, and promoting the expression of anti-inflammatory cytokines, including IL-10 [30]. In addition, the concept of the “cholinergic anti-inflammatory pathway” has been proposed in a study examining how the vagus nerves participates in the regulation of sepsis, which opens up a new direction for the treatment of sepsis [31].
Endoplasmic reticulum stress
The endoplasmic reticulum (ER) is an intracellular organelle involved in protein translocation, folding, and posttranslational modification. Notably, unfolded or misfolded proteins are accumulated in the ER during sepsis, leading to impaired homeostasis and subsequent ER stress [32]. Under ER stress, unfolded protein response sensors might switch their signals to stimulate cell death by activating unique signaling mechanisms via multiple steps, including the transcriptional activation of the CEBP homologous protein (CHOP) gene and the activation of the JNK pathway [33]. In sepsis animal models, the increased expression of glucose-regulated protein 94, CHOP, and caspase-12, all of which are markers of enhanced ER stress, can be detected in sepsis-induced multiple organ failure, including heart and liver failure [33, 34]. In addition, ER stress results in abnormal apoptosis in sepsis animals, suggesting that ER stress-mediated apoptosis represents a potential novel target for the clinical prevention of sepsis [35].
Autophagy
Autophagy plays an essential role in not only resisting external pathogens and dangerous signals but also the regulation of the natural inflammatory immune-cell response during sepsis [36]. Autophagy-deficient mice lacking the autophagy protein or ATG16L1 gene are more susceptible to LPS challenge, and the endotoxin effects in these mice are enhanced due to the inhibited immune response as a result of T-cell autophagy deficiency [37]. Moreover, in a cecal ligation and puncture (CLP)-induced mouse model of sepsis, incomplete autophagy might lead to cardiac dysfunction in sepsis, which can be restored by autophagy activation using rapamycin [19]. Therefore, autophagy exerts a protective effect in sepsis via regulation of cytokine release, the reduction in the apoptosis process, and pathogen clearance.
Inflammatory storm and sepsis
The evolvement of the inflammatory response in sepsis from the innate immune system consisting of macrophages, monocytes, neutrophils, and natural killer cells to PAMPs and DAMPs has been well described. PAMPs mainly include bacterial, fungal, and viral pathogens, such as endotoxin and 9-glucan, while DAMPs mainly include endogenous molecules released from damaged host cells. Both PAMPs and DAMPs activate the immune cascade and several epithelial cells after the recognition of receptors on the cell surface (Toll-like receptors) or in the cytosol (NOD-like receptors), activating multiple cytokines, including IL-1, IL-6, and TNF-8 [38, 39]. Inflammasomes assembled with some pattern recognition receptors are also essential for the secretion of several important cytokines, such as IL-1β and IL-18, resulting in inflammatory programmed cell death by pyroptosis [40]. In turn, proinflammatory cytokines enhance the number and activation state of innate immune cells and the expression of adhesion molecules and chemokines and promote neutrophils to release antimicrobial proteins and enzymes that form a scaffold for platelet activation [41]. In addition, tissue factor expression by blood monocytes and the release of NETS are upregulated by these increased inflammatory responses [42].
Basic background of HMGB1
HMGB1
The human HMGB1 gene is located on chromosome 13 and is highly evolutionarily conserved [43]. HMGB1 is the most highly expressed of all HMG family members. The structure of HMGB1 is extremely conserved in eukaryotes among species, with 99% amino acid homology between rodents and humans [44]. HMGB1 binds DNA through its autologous structural domain to maintain the stability of the nucleosome and regulate transcription, translation, and DNA repair [45, 46]. Notably, the structural domains of HMGB1 perform different functions. Box B can bend and twine DNA, while Box A cannot [47, 48]. Blair and colleagues [49] discovered that the full-length (FL) HMGB1 bent DNA more than the individual A and B boxes. Removing the C-terminal tail of HMGB1 results in a protein that bends DNA to a greater extent than the FL protein. These data suggest that the A and B boxes simultaneously bind DNA in the absence of the C-terminal tail, but the tail modulates DNA binding and bending by one of the two HMG boxes in the FL protein.
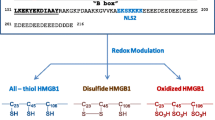
HMGB1 is both a nuclear factor and a secreted protein. HMGB1 acts as an architectural chromatin-binding factor in the cell nucleus to bind DNA and promote protein assembly on specific DNA targets. Outside the cell, HMGB1 binds to receptor for advanced glycation endproducts (RAGE) with high affinity [50]. As a classic signaling molecule, HMGB1 plays diverse biological roles in pathological and pathophysiological processes, such as inflammation, immune reactions, cell migration, differentiation, proliferation, tissue repair, angiogenesis, aging, and cell death [51]. Interestingly, the functions of HMGB1 are determined by its autologous redox state. The reduced form of HMGB1 has been implicated in chemotaxis, whereas the disulfide form of HMGB1 is pro-inflammatory [52, 53]. Physiologically, the A box and B boxes perform an internal action in the N-terminus called the reduced state, which promotes the recruitment of leukocytes and regeneration of tissues and organs [54]. However, the oxidation of HMGB1 results in the formation of intramolecular cysteine 23-cysteine 45 bonds, which enable HMGB1 to signal through TLR4-MD2 and function as a priming signal for NLRP3 inflammasome activation [52, 55]. Caspase activation targets mitochondria to produce reactive oxygen species (ROS), which are critical for the tolerance induction of apoptotic cells. ROS oxidize the potential danger signal HMGB1 released from dying cells and thereby neutralize its stimulatory activity. Apoptotic cells fail to induce tolerance and instead stimulate immune responses by scavenging or mutating target mitochondrial caspases when ROS activity is prohibited. Similarly, blocking oxidation sites in HMGB1 prevents tolerance induction by apoptotic cells [56].
Release of HMGB1
Physiologically, the levels of HMGB1 remain low in the cytoplasm and serum. However, the HMGB1 levels sharply increase under tissue injury [57]. HMGB1 is released from cells in the following two dependent ways: active secretion and passive release. Despite its clinical importance, the exact mechanism of HMGB1 release largely remains unknown. Extracellular HMGB1 performs its proinflammatory functions by binding immunostimulatory molecules, such as RNA, histone, lipopolysaccharide (LPS), and IL-1α (Fig. 1) [58,59,60].

LPS uptake into hepatocytes via TLR4 can activate the caspase-11-mediated pathway to stimulate HMGB1 mobilization from the nucleus and release from hepatocytes. Extracellular HMGB1 can deliver LPS to macrophages. Subsequently, endosomal and lysosomal rupture in macrophages leads to the release of HMGB1 and LPS into the cell cytosol and activates caspase-11 and caspase-1 signaling. Consequently, HMGB1 induces a systemic inflammatory response through TNF, IL-1, and IL-6 and triggers cell apoptosis, autophagy, and pyroptosis. LPS lipopolysaccharide, HMGB1 high mobility group box 1, TLR4 Toll-like receptors.
Active secretion
Evidence suggests that the HMGB1 protein is secreted by monocytes via a nonclassical, vesicle-mediated secretory pathway [61, 62]. Mechanistically, the activation of monocytes contributes to the redistribution of HMGB1 from the nucleus to cytoplasmic organelles that display the ultrastructural features of endolysosomes. Then, HMGB1 secretion is induced by stimuli triggering lysosomal exocytosis [63]. HMGB1 actively shuttles between the cytoplasm and nucleus, which is the first step of active secretion in almost all cells. Furthermore, the process of HMGB1 translocation from the nucleus to the cytoplasm significantly requires posttranslational modifications, such as phosphorylation and acetylation (Table 1). Nevertheless, the upstream pathway that phosphorylates HMGB1 is not fully understood [64]. Deng et al. [65] reported that calmodulin-dependent protein kinase (CaMK) IV may be involved in HMGB1 phosphorylation during the release of HMGB1 from LPS-stimulated macrophages. In monocytes and macrophages, HMGB1 is extensively acetylated by PCAF, CBP and p300 upon activation with LPS. In addition, increased hyperacetylation of HMGB1 in resting macrophages results in its relocalization to the cytosol. Then, cytosolic HMGB1 is concentrated by default in secretory lysosomes and subsequently secreted when monocytic cells receive an appropriate second signal [66]. More importantly, Deng et al. [67] recently found that hepatocytes rather than myeloid immune cells are the dominant source of systemic HMGB1 release during endotoxemia, which is dependent on the activation of caspase-11 to eventually induce gasdermin D-mediated pyroptosis of immune cells [68].
Moreover, interferon (IFN)-γ induces HMGB1 release partially via a tumor necrosis factor (TNF)-dependent mechanism. Rendon-Mitchell and colleagues [69] discovered that IFN-γ, which is an immunoregulatory cytokine known to mediate the innate immune response, dose-dependently induces the release of HMGB1, TNF, and nitric oxide. The pharmacological suppression of TNF with neutralizing antibodies or genetic knockout of TNF expression partially restrains IFN-γ-mediated HMGB1 release. AG490, which is a specific inhibitor of Janus kinase (JAK) 2 of the IFN-γ pathway, dose-dependently attenuates IFN-γ-induced HMGB1 release. IFN-β, IFN-γ, and LPS [70,71,72,73], which are early proinflammatory cytokines, activate the JAK/signal transducer and activator of transcription (STAT) 1 pathway, which, in turn, promotes the hyperacetylation of HMGB1. Then, HMGB1 is translocated from the nucleus to the cytoplasm and is subsequently released [74]. Notably, sirtuin1 (SIRT1) directly interacts with HMGB1 via its N-terminal lysine residues (28–30) and thereby suppresses HMGB1 release to improve survival in an experimental model of sepsis. In contrast, inflammatory stimuli, such as LPS and TNF-α, promote HMGB1 release by inducing its dissociation from SIRT1 to increase its acetylation, thereby increasing the association between HMGB1 and chromosome region maintenance 1, contributing to HMGB1 translocation [75]. In addition, platelet-derived HMGB1 is released and is involved in inflammation and thrombosis. Activated platelets release large amounts of HMGB1, which is transferred to the cytomembrane when blood vessels are injured [76, 77].
Passive release
HMGB1 can be released passively under a severe inflammatory response or lytic cell death, including necrosis, pyroptosis, and necroptosis, during sepsis and trauma. HMGB1 is loosely bound to chromatin in the interphase and mitotic cells and rapidly leaks into the medium when the membrane integrity is impaired in permeabilized or lytic cells [44]. HMGB1 knockout necrotic cells have a greatly reduced ability to promote inflammation, suggesting that the release of HMGB1 transmits the demise signal of a cell to adjacent cells. Apoptotic cells do not release HMGB1 even after undergoing secondary necrosis and partial autolysis and, thus, fail to promote inflammation, even if not cleared promptly by phagocytic cells. In apoptotic cells, HMGB1 is firmly bound to chromatin because of the generalized hypoacetylation of histone and is released into the extracellular medium if chromatin deacetylation is prevented [50]. These findings are consistent with evidence suggesting that most HMGB1 released passively is non-acetylated HMGB1 still located within the nucleus [78]. Therefore, the non-acetylated form of HMGB1 has been utilized in investigations to clarify the proportion of HMGB1 passively released compared to its active release from cells in the inflammatory response. Moreover, HMGB1 has been observed to be in its absolute reductive form during passive release, which obviously affects the function and pro-inflammatory potential of HMGB1 [79]. Lipid peroxidation and aerobic glycolysis have been implicated in promoting HMGB1 release via inflammasome activation in sepsis [80, 81]. However, whether these metabolic stresses trigger the passive release of HMGB1 require further clarification. Altogether, the identification of the detailed mechanisms underlying the active and passive release of HMGB1 may provide optimal and effective therapeutics designed to suppress either lytic cell death or active HMGB1 release.
HMGB1 and the inflammatory storm
Disulfide HMGB1 has the most potent inflammatory effects and acts as a DAMP when released from cells in this form. HMGB1 can trigger massive inflammatory cytokine production from immune cells through multiple signaling pathways involving cell surface pattern recognition receptors, such as TLR4 and RAGE.
Toll-like receptors
Innate immune receptors for pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) orchestrate inflammatory responses to infection and sterile injury. Secreted by activated immunocytes or passively released from dying cells, HMGB1 is subjected to redox modification that distinctly influences its extracellular functions [55]. Toll-like receptors (TLRs) belong to an evolutionarily conserved type I transmembrane superfamily containing an extracellular leucine-rich repeat (LRR) and a cytoplasmic Toll/IL-1 receptor (TIR) domain [82,83,84,85,86]. Surface plasmon resonance studies indicate that HMGB1 specifically binds TLR4 and that this binding requires a cysteine at position 106. A wholly synthetic 20-mer peptide containing cysteine 106 within the cytokine-stimulating B box mediates TLR4-induced activation of macrophage TNF release. The inhibition of TLR4 binding with neutralizing anti-HMGB1 mAbs or by mutating cysteine 106 prevents the activation of HMGB1 [87]. In addition, Yang and colleagues [55] demonstrated that the extracellular TLR4 adaptor myeloid differentiation factor 2 (MD-2) specifically binds the cytokine-inducing disulfide isoform of HMGB1. These results suggest a requirement for HMGB1-dependent TLR4 signaling in MD-2-deficient mice and MD-2-silenced macrophages. P5779 (FSSE), which is a tetramer peptide for a specific MD-2 antagonist, prevents MD-2-HMGB1 interaction and TLR4 signaling. Mechanistically, P5779 does not interfere with LPS-induced cytokine/chemokine production, thus preserving PAMP-mediated TLR4–MD-2 responses. Furthermore, P5779 protects mice against sepsis. These findings reveal a novel mechanism by which innate systems selectively recognize specific HMGB1 isoforms [55]. Park and colleagues used fluorescence resonance energy transfer (FRET) and immunoprecipitation to directly investigate the cell surface interactions between HMGB1 and TLR2 or TLR4. Fluorescence resonance energy transfer images in RAW264.7 macrophages identified an association between HMGB1 and TLR2 or TLR4. Transient transfections of human embryonic kidney-293 cells showed that HMGB1 induced cellular activation and NF-κB-dependent transcription through TLR2 or TLR4. Coimmunoprecipitation also revealed an interaction between HMGB1 and TLR2 or TLR4. Moreover, during the release process of HMGB1 from hepatocytes, enhanced TLR4 signaling promoted both HMGB1 release from hepatocytes and LPS delivery to the cytosol of hepatocytes [67]. These studies provide the first direct evidence suggesting that HMGB1 interacts with both TLR2 and TLR4 to activate inflammatory responses [88].
RAGE
RAGE was first described as a signal transduction receptor for advanced glycation endproducts (AGE) [89,90,91]. Kokkola and colleagues [92] found that macrophages from RAGE–/– mice produced significantly lower levels of TNF, IL-1β, and IL-6 in response to HMGB1 stimulation. Under physiological conditions, RAGE mediates interactions with ligands that are distinct from AGE. Cultured embryonic rat neurons, which express RAGE, dose-dependently bind 125I-amphoterin, which is restrained by blocking RAGE using antibodies against the receptor or excess soluble receptor (sRAGE) [93]. Recent evidence has revealed that HMGB1 is sensitive to cleavage by caspase-1 activation, resulting in the production of a fragment within its N-terminal DNA binding domain (A-box) that reverses apoptosis-induced tolerance by binding RAGE. In a double-hit mouse model of sepsis initially injected intraperitoneally with a low dose of LPS (400 mg/kg) for 6 h and subsequently challenged with a high dose of LPS (10 mg/kg), LeBlanc et al. [94] discovered that tolerance to secondary infection and its associated mortality were effectively reversed by the active immunization of dendritic cells treated with HMGB1 or the A-box fragment but not a form of HMGB1 that cannot be cleaved. In addition, HMGB1 could initiate endocytosis and induce a cascade of molecular events, including cathepsin B release from ruptured lysosomes, which triggers caspase-1 activation and macrophage pyroptosis through RAGE-dependent signaling. Furthermore, the pyroptosis induced by HMGB1 has also been observed in vivo during endotoxemia, highlighting the pathophysiological significance of HMGB1 in pyroptosis during the development of inflammation [95]. Notably, Deng et al. [67] showed that HMGB1 could significantly mediate caspase-11-dependent pyroptosis and lethality in sepsis by delivering extracellular LPS to the cytosol of macrophages and endothelial cells [96]. The important mode in this process is the RAGE-mediated internalization of HMGB1-LPS complexes. Taken together, these findings represent a novel link between RAGE and HMGB1 delivery and release with potential therapeutic implications for inflammatory diseases (Fig. 1).
Roles of HMGB1 in sepsis and sepsis-related organ injury
Sepsis-related lung injury
Acute lung injury (ALI) is closely correlated with sepsis in the intensive care unit setting [97,98,99,100]. Sepsis-induced ALI has been reported to have a higher fatality rate than other pathogens that trigger ALI [101]. Lan and colleagues [102] reported that salidroside ameliorates sepsis-induced ALI and mortality in mice by downregulating the SIRT1/HMGB1 pathway. Salidroside, which is a substance isolated from Rhodiola rosea, possesses antioxidant and anti-inflammatory properties. Salidroside suppresses the inflammatory responses and HMGB1 production and reverses the reduced expression of the SIRT1 protein in bacterial LPS-treated macrophages and mice. Moreover, salidroside alleviates sepsis-induced lipid peroxidation, histopathological changes and mortality in a lung edema model and improves the lung PaO2/FiO2 ratio in CLP-induced septic mice. Sepsis decreases SIRT1 protein expression in the lungs of CLP-induced septic mice. However, salidroside significantly upregulates SIRT1 expression and inhibits inflammatory responses in CLP-induced septic mouse lungs [102]. The injection of Xuebijing, which is a traditional Chinese medicine, has been reported to be a promising approach in the treatment of sepsis in China. Xuebijing reduces morphological destruction, neutrophil infiltration and the lung wet/dry weight ratio in the alveolar space, which improves mortality in cecal ligation and puncture (CLP)-induced lung injury. Moreover, Xuebijing treatment downregulates HMGB1 and RAGE expression, neutrophil counts and production of IL-1, IL-6, and TNF-α in bronchoalveolar lavage fluid. These results indicate that Xuebijing reduces mortality in mice with CLP-induced lung injury during sepsis by inhibiting the proinflammatory cytokine secretion mediated by the HMGB1/RAGE axis [103]. The rats in the CLP + ulinastatin group exhibited higher 7-d survival rates, less lung injury, and reduced HMGB1 expression in the lung tissue, serum, and bronchoalveolar lavage fluid (BALF) compared with those in the CLP group. In addition, the levels of TNF-α and IL-6 at 24 h in the CLP + ulinastatin group were markedly lower than those in the CLP group. These results suggest that ulinastatin decreases lung injury and increases the survival time of ALI rats by downregulating HMGB1 expression and inhibiting TNF-α and IL-6 levels in serum and bronchoalveolar lavage fluid [104].
CLP increases the wet/dry weight ratio and myeloperoxidase activity in the lung by upregulating HMGB1 and enhancing the number of inflammatory cells, such as neutrophils and macrophages, in the BALF. Moreover, the expression of HMGB1 and RAGE in lung tissues is increased after CLP, whereas ketamine reverses all of the above effects. Ketamine also suppresses the activation of IκB-α, NF-κB, p65, and mitogen-activated protein kinases (MAPK). This finding reveals that ketamine protects rats against HMGB1-RAGE activation in a rat model of sepsis-induced ALI [105]. In a mouse model of ALI, blocking HMGB1 or a myeloid-specific PTEN knockout (PTENM-KO) increases animal survival and body weight, reduces the expression of IL-17 and lung damage, increases TGF-β production, promotes β-catenin signaling, and subsequently induces CD4+CD25+Foxp3+ Tregs to suppress endotoxin-mediated inflammation in LPS- or rHMGB-induced lung injury. Notably, myeloid-specific β-catenin ablation (β-catenin M-KO) results in reduced animal survival and increased lung injury in rHMGB-induced ALI. Furthermore, the disruption of macrophage HMGB1/PTEN or activation of β-catenin significantly increases CD4+CD25+Foxp3+ Tregs, contributing to repressed endotoxin-induced inflammation responses in vitro. These findings suggest that HMGB1/PTEN/β-catenin signaling is a novel pathway that regulates Treg development and provides a potential therapeutic target for sepsis-induced lung injury [106].
Other sepsis-related injuries
HMGB1 also plays a pivotal role in other sepsis-related organ injuries and diseases. In sepsis-induced liver injury models, sepsis-related serum aminotransferase activity, and proinflammatory chemokine levels are reduced by resveratrol pretreatment, which also improves liver histological parameters that are complicated by SIRT1 upregulation and HMGB1 downregulation. Mechanistically, resveratrol suppresses the cytoplasmic translocation of HMGB1 and decreases the inflammatory responses in hepatocytes [107, 108]. In addition, Zheng and colleagues reported that the mRNA transcription levels of IL-1 and IL-6 in renal tubular epithelial cells (RTECs) were significantly increased during sepsis, accompanied by accumulated HMGB1 in renal tissue that entered urine during sepsis. These authors further found that HMGB1 turns RTECs into inflammatory promoters by interacting with TLR4 during sepsis [109]. Bacterial sepsis not only triggers innate immune responses but also extensively stimulates coagulation cascades that leads to disseminated intravascular coagulation (DIC). Type 1 IFNs significantly mediate bacterial infection-induced DIC by enhancing the release of HMGB1 into the blood stream, where it binds and delivers LPS into the cytoplasm of the host cell, consequently activating caspase-11-dependent immune responses and dysfunction of endothelial cells [110].
Potential therapeutics
Recently, several novel interventions targeting HMGB1 for sepsis treatment have emerged. Glycyrrhizin (GL), which is also known as glycyrrhizic acid, is a natural triterpene glycoside and the major active component of Gancao. Zhao and colleagues discovered that GL significantly reduced the serum levels of HMGB1 in vivo. GL also ameliorates the release and expression of HMGB1 and HMGB1-related proinflammatory cytokines. Furthermore, GL blocks the interaction between HMGB1 and RAGE or TLR4 and suppresses the downstream MAPKs/NF-κB pathway [111]. Naringin, which is a flavanone glycoside extracted from various plants, has a wide range of pharmacological effects. Naringin increases the expression of HMGB1 and heme oxygenase (HO)-1 in peritoneal macrophages by activating adenosine monophosphate-activated protein kinase, p38, and NF-E2-related factor 2 (Nrf2). Mechanistically, naringin restrains HMGB1 release from LPS-stimulated macrophages and improves survival in CLP-induced septic mice [112]. However, the appropriate dose and intervention time for the use of these inhibitors, such as GL and Naringin, in sepsis are unclear and require further investigation. Moreover, many reports regarding the successful utilization of polyclonal or mouse anti-HMGB1 monoclonal antibodies in experimental inflammatory models have been published. To date, different humanized anti-HMGB1 monoclonal antibodies and HMGB1-neutralizing Affibody molecules have also been identified in many preclinical studies investigating inflammatory conditions. As investigations aiming to identify the conditions under which the concentrations of HMGB1 are suppressed are still ongoing, future clinical investigations focusing on HMGB1 inhibitors may focus on (1) exploring the safety and efficacy of the preventive use of HMGB1 in the clinic; (2) identifying the more accurate mode of inhibitory action and verifying the bioactivity of a given batch of inhibitors; (3) determining how to maintain HMGB1 at appropriate levels to prevent damage without inhibiting tissue regeneration and wound healing as a balanced therapeutic mechanism; and (4) confirming whether the clinical application of therapeutics targeting HMGB1 has other unsatisfactory side effects.
Although abundant preclinical evidence regarding the efficacy of targeting HMGB1 in inflammation is available, a detailed mechanistic understanding of the structure-function interactions between HMGB1 and inflammatory receptors or signaling pathways remains lacking. Studies may provide more preclinical evidence for clinical translation that should include evaluations of (1) whether the upregulation or disparate intracellular location of HMGB1 is a cause or a consequence of the progression of sepsis; (2) which type(s) of post-transcriptional or post-translational regulation contributes to HMGB1 upregulation or activation in sepsis; and (3) the identification of more downstream targets or pathways activated by HMGB1 in mitochondrial dysfunction and inflammation storms.
Surprisingly, HMGB1 is regarded as an essential predictor of organ dysfunction and outcome in patients with severe sepsis. Van and colleagues [113] revealed that the HMGB1 levels were increased in severe sepsis patients but were not correlated with the outcome. Moreover, another investigation showed that there was no difference in the HMGB1 concentrations between patients with septic shock and patients with severe sepsis [114]. Recently, Karlsson et al. [115] enrolled 247 adult patients with severe sepsis. The serum HMGB1 concentrations were elevated in the patients with severe sepsis after 72 h but did not differ between the survivors and nonsurvivors and did not predict hospital mortality. Thus, more work is still needed to elucidate the clinical significance of HMGB1 in sepsis.
Intriguingly, under normal physiological conditions, the serum levels of HMGB1 are attenuated in humans with aging, which contrasts the detrimental effects of increased HMGB1 under pathologic conditions [116]. In many aging studies, the HMGB1 plasma levels were found to be associated with organ failure [117, 118]. Additionally, immunosenescence alters the role of sepsis as a cause of death in the elderly [6]. Here, the inhibition of HMGB1 is pursued as a therapeutic target in sepsis. HMGB1 acts as a late mediator of inflammation and contributes to prolonged and sustained systemic inflammation in a model of aging-related osteoarthritis [119]. Interestingly, studies involving the senescence inhibitor metformin revealed that its direct binding to HMGB1 suppresses the pro-inflammatory activity of HMGB1 [120].
Conclusion
Sepsis remains a common and deadly problem worldwide. The discovery of HMGB1 as a potent cytokine mediator of sepsis has sparked a new field of investigation related to the treatment of sepsis. HMGB1 plays important roles as a DAMP mediator of inflammation involved with TLR and RAGE pathways in the pathogenesis of sepsis. The cell-type specific targeting of inhibitors of HMGB1 will likely be necessary. Here, we propose that the inhibition of HMGB1 might be a potential therapeutic avenue against sepsis and sepsis-related organ injury. Although several lines of evidence have clarified the underlying mechanisms of HMGB1 in sepsis, the development of a successful treatment that includes HMGB1 has not been instituted to date. Further investigations may focus on (1) identifying the relationship between HMGB1 concentrations and the progression of sepsis; (2) determining the unknown mechanisms by which HMGB1 regulates sepsis; and (3) performing a high-quality systematic review or a meta-analysis to provide evidence-based support. Overall, this review presents a detailed view of HMGB1 in sepsis and indicates the potential of HMGB1 as a therapeutic target for sepsis-related organ injury.
References
Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–88.
Hu W, Deng C, Ma Z, Wang D, Fan C, Li T, et al. Utilizing melatonin to combat bacterial infections and septic injury. Br J Pharmacol. 2017;174:754–68.
Hardeland R. Melatonin and inflammation-story of a double-edged blade. J Pineal Res. 2018;65:e12525.
Xu D, Liao S, Li P, Zhang Q, Lv Y, Fu X, et al. Metabolomics coupled with transcriptomics approach deciphering age relevance in sepsis. Aging Dis. 2019;10:854–70.
da Rocha EP, Yokota LG, Sampaio BM, Cardoso Eid KZ, Dias DB, de Freitas FM, et al. Urinary neutrophil gelatinase-associated lipocalin is excellent predictor of acute kidney injury in septic elderly patients. Aging Dis. 2018;9:182–91.
Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. 2016;353:i1585.
Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–4.
Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51.
Wang H, Ward MF, Sama AE. Targeting HMGB1 in the treatment of sepsis. Expert Opin Ther Targets. 2014;18:257–68.
Zeni F, Freeman B, Natanson C. Anti-inflammatory therapies to treat sepsis and septic shock: a reassessment. Crit Care Med. 1997;25:1095–100.
Einck L, Bustin M. The intracellular distribution and function of the high mobility group chromosomal proteins. Exp Cell Res. 1985;156:295–310.
Javaherian K, Liu JF, Wang JC. Nonhistone proteins HMG1 and HMG2 change the DNA helical structure. Science. 1978;199:1345–6.
Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93:865–73.
Andersson U, Tracey KJ. HMGB1 in sepsis. Scand J Infect Dis. 2003;35:577–84.
Gregoire M, Tadie JM, Uhel F, Gacouin A, Piau C, Bone N, et al. Frontline science: HMGB1 induces neutrophil dysfunction in experimental sepsis and in patients who survive septic shock. J Leukoc Biol. 2017;101:1281–7.
Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–50.
Efron PA, Martins A, Minnich D, Tinsley K, Ungaro R, Bahjat FR, et al. Characterization of the systemic loss of dendritic cells in murine lymph nodes during polymicrobial sepsis. J Immunol. 2004;173:3035–43.
Cheng SC, Scicluna BP, Arts RJ, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. 2016;17:406–13.
Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–74.
Buwalda M, Ince C. Opening the microcirculation: can vasodilators be useful in sepsis? Intensive Care Med. 2002;28:1208–17.
Escames G, Lopez LC, Ortiz F, Lopez A, Garcia JA, Ros E, et al. Attenuation of cardiac mitochondrial dysfunction by melatonin in septic mice. FEBS J. 2007;274:2135–47.
Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence. 2014;5:66–72.
Aronis A, Aharoni-Simon M, Madar Z, Tirosh O. Triacylglycerol-induced impairment in mitochondrial biogenesis and function in J774.2 and mouse peritoneal macrophage foam cells. Arch Biochem Biophys. 2009;492:74–81.
Wu Y, Yao YM, Lu ZQ. Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure. J Mol Med (Berl). 2019;97:451–62.
Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet. 2018;392:75–87.
Biemond BJ, Levi M, Ten Cate H, Van der Poll T, Buller HR, Hack CE, et al. Plasminogen activator and plasminogen activator inhibitor I release during experimental endotoxaemia in chimpanzees: effect of interventions in the cytokine and coagulation cascades. Clin Sci (Lond). 1995;88:587–94.
Muscatell KA, Dedovic K, Slavich GM, Jarcho MR, Breen EC, Bower JE, et al. Greater amygdala activity and dorsomedial prefrontal-amygdala coupling are associated with enhanced inflammatory responses to stress. Brain Behav Immun. 2015;43:46–53.
Sonneville R, Verdonk F, Rauturier C, Klein IF, Wolff M, Annane D, et al. Understanding brain dysfunction in sepsis. Ann Intensive Care. 2013;3:15.
Kanczkowski W, Sue M, Zacharowski K, Reincke M, Bornstein SR. The role of adrenal gland microenvironment in the HPA axis function and dysfunction during sepsis. Mol Cell Endocrinol. 2015;408:241–8.
Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Behav Immun. 2012;26:469–79.
Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K, et al. Expression and function of the cholinergic system in immune cells. Front Immunol. 2017;8:1085.
Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–94.
Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai). 2014;46:629–40.
Garcia de la Cadena S, Massieu L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis. 2016;21:763–77.
Jiao G, Hao L, Wang M, Zhong B, Yu M, Zhao S, et al. Upregulation of endoplasmic reticulum stress is associated with diaphragm contractile dysfunction in a rat model of sepsis. Mol Med Rep. 2017;15:366–74.
Ho J, Yu J, Wong SH, Zhang L, Liu X, Wong WT, et al. Autophagy in sepsis: degradation into exhaustion? Autophagy. 2016;12:1073–82.
Schafer ST, Franken L, Adamzik M, Schumak B, Scherag A, Engler A, et al. Mitochondrial DNA: an endogenous trigger for immune paralysis. Anesthesiology. 2016;124:923–33.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20.
Schulte W, Bernhagen J, Bucala R. Cytokines in sepsis: potent immunoregulators and potential therapeutic targets-an updated view. Mediators Inflamm. 2013;2013:165974.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Meziani F, Delabranche X, Asfar P, Toti F. Bench-to-bedside review: circulating microparticles-a new player in sepsis? Crit Care. 2010;14:236.
Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768–76.
Khambu B, Huda N, Chen X, Antoine DJ, Li Y, Dai G, et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J Clin Invest. 2019;129:2163.
Huang W, Tang Y, Li L. HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine. 2010;51:119–26.
Thomas JO, Stott K. H1 and HMGB1: modulators of chromatin structure. Biochem Soc Trans. 2012;40:341–6.
Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799:101–13.
Teo SH, Grasser KD, Thomas JO. Differences in the DNA-binding properties of the HMG-box domains of HMG1 and the sex-determining factor SRY. Eur J Biochem. 1995;230:943–50.
Stros M. DNA bending by the chromosomal protein HMG1 and its high mobility group box domains. Effect of flanking sequences. J Biol Chem. 1998;273:10355–61.
Blair RH, Horn AE, Pazhani Y, Grado L, Goodrich JA, Kugel JF. The HMGB1 C-terminal tail regulates DNA bending. J Mol Biol. 2016;428:4060–72.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5.
Ugrinova I, Pasheva E. HMGB1 protein: a therapeutic target inside and outside the cell. Adv Protein Chem Struct Biol. 2017;107:37–76.
Frank MG, Weber MD, Fonken LK, Hershman SA, Watkins LR, Maier SF. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: A role for the NLRP3 inflammasome. Brain Behav Immun. 2016;55:215–24.
Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–23.
Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–28.
Yang H, Wang H, Ju Z, Ragab AA, Lundback P, Long W, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212:5–14.
Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32.
Gougeon ML, Bras M. Natural killer cells, dendritic cells, and the alarmin high-mobility group box 1 protein: a dangerous trio in HIV-1 infection? Curr Opin HIV AIDS. 2011;6:364–72.
Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180:2531–7.
Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–18.
Zhao YF, Qiong Z, Zhang JF, Lou ZY, Zu HB, Wang ZG, et al. The synergy of aging and LPS exposure in a mouse model of Parkinson’s disease. Aging Dis. 2018;9:785–97.
Han R, Liu Z, Sun N, Liu S, Li L, Shen Y, et al. BDNF alleviates neuroinflammation in the hippocampus of type 1 diabetic mice via blocking the aberrant HMGB1/RAGE/NF-kappaB pathway. Aging Dis. 2019;10:611–25.
Li FJ, Zhang CL, Luo XJ, Peng J, Yang TL. Involvement of the MiR-181b-5p/HMGB1 pathway in Ang II-induced phenotypic transformation of smooth muscle cells in hypertension. Aging Dis. 2019;10:231–48.
Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001.
Schulman IG, Wang T, Wu M, Bowen J, Cook RG, Gorovsky MA, et al. Macronuclei and micronuclei in Tetrahymena thermophila contain high-mobility-group-like chromosomal proteins containing a highly conserved eleven-amino-acid putative DNA-binding sequence. Mol Cell Biol. 1991;11:166–74.
Zhang X, Wheeler D, Tang Y, Guo L, Shapiro RA, Ribar TJ, et al. Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages. J Immunol. 2008;181:5015–23.
Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–60.
Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, et al. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity. 2018;49:740–53 e7.
Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H, et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci USA. 2014;111:3068–73.
Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. 2003;170:3890–7.
Dominguez Rubio AP, Correa F, Aisemberg J, Dorfman D, Bariani MV, Rosenstein RE, et al. Maternal administration of melatonin exerts short- and long-term neuroprotective effects on the offspring from lipopolysaccharide-treated mice. J Pineal Res. 2017;63:e12439.
Liu Z, Gan L, Xu Y, Luo D, Ren Q, Wu S, et al. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-kappaB/GSDMD signal in mice adipose tissue. J Pineal Res. 2017;63:e12414.
Rahim I, Djerdjouri B, Sayed RK, Fernandez-Ortiz M, Fernandez-Gil B, Hidalgo-Gutierrez A, et al. Melatonin administration to wild-type mice and nontreated NLRP3 mutant mice share similar inhibition of the inflammatory response during sepsis. J Pineal Res. 2017;63:e12410.
Magarinos AM, Schaafsma SM, Pfaff DW. Impacts of stress on reproductive and social behaviors. Front Neuroendocrinol. 2018;49:86–90.
Lee LC, Chen CM, Wang PR, Su MT, Lee-Chen GJ, Chang CY. Role of high mobility group box 1 (HMGB1) in SCA17 pathogenesis. PLoS One. 2014;9:e115809.
Hwang JS, Choi HS, Ham SA, Yoo T, Lee WJ, Paek KS, et al. Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci Rep. 2015;5:15971.
Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128:2435–49.
Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest. 2015;125:4638–54.
Andersson U, Yang H, Harris H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol. 2018;38:40–8.
Tang Y, Zhao X, Antoine D, Xiao X, Wang H, Andersson U, et al. Regulation of posttranslational modifications of HMGB1 during immune responses. Antioxid Redox Signal. 2016;24:620–34.
Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436.
Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:13280.
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, et al. HMGB1 in health and disease. Mol Asp Med. 2014;40:1–116.
Park A, Ra EA, Lee TA, Choi HJ, Lee E, Kang S, et al. HCMV-encoded US7 and US8 act as antagonists of innate immunity by distinctively targeting TLR-signaling pathways. Nat Commun. 2019;10:4670.
Volarevic V, Markovic BS, Jankovic MG, Djokovic B, Jovicic N, Harrell CR, et al. Galectin 3 protects from cisplatin-induced acute kidney injury by promoting TLR-2-dependent activation of IDO1/Kynurenine pathway in renal DCs. Theranostics. 2019;9:5976–6001.
Li T, Jiang S, Lu C, Yang W, Yang Z, Hu W, et al. Melatonin: another avenue for treating osteoporosis? J Pineal Res. 2019;66:e12548.
Chu KA, Wang SY, Yeh CC, Fu TW, Fu YY, Ko TL, et al. Reversal of bleomycin-induced rat pulmonary fibrosis by a xenograft of human umbilical mesenchymal stem cells from Wharton’s jelly. Theranostics. 2019;9:6646–64.
Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107:11942–7.
Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–24.
Ramasamy R, Yan SF, Schmidt AM. Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci. 2011;1243:88–102.
Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2019;130:1802.
Li T, Jiang S, Han M, Yang Z, Lv J, Deng C, et al. Exogenous melatonin as a treatment for secondary sleep disorders: a systematic review and meta-analysis. Front Neuroendocrinol. 2019;52:22–8.
Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9.
Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–61.
LeBlanc PM, Doggett TA, Choi J, Hancock MA, Durocher Y, Frank F, et al. An immunogenic peptide in the A-box of HMGB1 protein reverses apoptosis-induced tolerance through RAGE receptor. J Biol Chem. 2014;289:7777–86.
Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21:1229–39.
Yu H, Schwarzer K, Forster M, Kniemeyer O, Forsbach-Birk V, Straube E, et al. Role of high-mobility group box 1 protein and poly(ADP-ribose) polymerase 1 degradation in Chlamydia trachomatis-induced cytopathicity. Infect Immun. 2010;78:3288–97.
Park I, Kim M, Choe K, Song E, Seo H, Hwang Y, et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur Respir J. 2019;53:1800786.
Neudecker V, Brodsky KS, Clambey ET, Schmidt EP, Packard TA, Davenport B, et al. Neutrophil transfer of miR-223 to lung epithelial cells dampens acute lung injury in mice. Sci Transl Med. 2017;9:eaah5360.
Zhang Y, Li X, Grailer JJ, Wang N, Wang M, Yao J, et al. Melatonin alleviates acute lung injury through inhibiting the NLRP3 inflammasome. J Pineal Res. 2016;60:405–14.
Brandenberger C, Kling KM, Vital M, Christian M. The role of pulmonary and systemic immunosenescence in acute lung injury. Aging Dis. 2018;9:553–65.
Sevransky JE, Martin GS, Shanholtz C, Mendez-Tellez PA, Pronovost P, Brower R, et al. Mortality in sepsis versus non-sepsis induced acute lung injury. Crit Care. 2009;13:R150.
Lan KC, Chao SC, Wu HY, Chiang CL, Wang CC, Liu SH, et al. Salidroside ameliorates sepsis-induced acute lung injury and mortality via downregulating NF-kappaB and HMGB1 pathways through the upregulation of SIRT1. Sci Rep. 2017;7:12026.
Wang Q, Wu X, Tong X, Zhang Z, Xu B, Zhou W. Xuebijing ameliorates sepsis-induced lung injury by downregulating HMGB1 and RAGE expressions in mice. Evid Based Complement Altern Med. 2015;2015:860259.
Wang SY, Li ZJ, Wang X, Li WF, Lin ZF. Effect of ulinastatin on HMGB1 expression in rats with acute lung injury induced by sepsis. Genet Mol Res. 2015;14:4344–53.
Li K, Yang J, Han X. Ketamine attenuates sepsis-induced acute lung injury via regulation of HMGB1-RAGE pathways. Int Immunopharmacol. 2016;34:114–28.
Zhou M, Fang H, Du M, Li C, Tang R, Liu H, et al. The modulation of regulatory T cells via HMGB1/PTEN/beta-catenin axis in LPS induced acute lung injury. Front Immunol. 2019;10:1612.
Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. 2003;9:37–45.
Xu W, Lu Y, Yao J, Li Z, Chen Z, Wang G, et al. Novel role of resveratrol: suppression of high-mobility group protein box 1 nucleocytoplasmic translocation by the upregulation of sirtuin 1 in sepsis-induced liver injury. Shock. 2014;42:440–7.
Zheng S, Pan Y, Wang C, Liu Y, Shi M, Ding G. HMGB1 turns renal tubular epithelial cells into inflammatory promoters by interacting with TLR4 during sepsis. J Interferon Cytokine Res. 2016;36:9–19.
Yang X, Cheng X, Tang Y, Qiu X, Wang Z, Fu G, et al. The role of type 1 interferons in Gram-negative bacteria-induced coagulation. Blood. 2020;135:1087–100.
Zhao F, Fang Y, Deng S, Li X, Zhou Y, Gong Y, et al. Glycyrrhizin protects rats from sepsis by blocking HMGB1 signaling. Biomed Res Int. 2017;2017:9719647.
Gil M, Kim YK, Hong SB, Lee KJ. Naringin decreases TNF-alpha and HMGB1 release from LPS-stimulated macrophages and improves survival in a CLP-induced sepsis mice. PLoS One. 2016;11:e0164186.
van Zoelen MA, Laterre PF, van Veen SQ, van Till JW, Wittebole X, Bresser P, et al. Systemic and local high mobility group box 1 concentrations during severe infection. Crit Care Med. 2007;35:2799–804.
Sunden-Cullberg J, Norrby-Teglund A, Rouhiainen A, Rauvala H, Herman G, Tracey KJ, et al. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med. 2005;33:564–73.
Karlsson S, Pettila V, Tenhunen J, Laru-Sompa R, Hynninen M, Ruokonen E. HMGB1 as a predictor of organ dysfunction and outcome in patients with severe sepsis. Intensive Care Med. 2008;34:1046–53.
Fu GX, Chen AF, Zhong Y, Zhao J, Gu YJ. Decreased serum level of HMGB1 and MyD88 during human aging progress in healthy individuals. Aging Clin Exp Res. 2016;28:175–80.
Hatada T, Wada H, Nobori T, Okabayashi K, Maruyama K, Abe Y, et al. Plasma concentrations and importance of High Mobility Group Box protein in the prognosis of organ failure in patients with disseminated intravascular coagulation. Thromb Haemost. 2005;94:975–9.
Bianchi ME, Crippa MP, Manfredi AA, Mezzapelle R, Rovere Querini P, Venereau E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol Rev. 2017;280:74–82.
Schierbeck H, Pullerits R, Pruunsild C, Fischer M, Holzinger D, Laestadius A, et al. HMGB1 levels are increased in patients with juvenile idiopathic arthritis, correlate with early onset of disease, and are independent of disease duration. J Rheumatol. 2013;40:1604–13.
Horiuchi T, Sakata N, Narumi Y, Kimura T, Hayashi T, Nagano K, et al. Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J Biol Chem. 2017;292:8436–46.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81871607, 82070422, 81500202, and 81600306), Key R & D Projects of Hainan Province (ZDYF2020123), and Natural Science Foundation of Shaanxi Province (2020JM-386 and 2018JM3042).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Deng, C., Zhao, L., Yang, Z. et al. Targeting HMGB1 for the treatment of sepsis and sepsis-induced organ injury. Acta Pharmacol Sin 43, 520–528 (2022). https://doi.org/10.1038/s41401-021-00676-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00676-7
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Immune Response Gene-1 [IRG1]/itaconate protect against multi-organ injury via inhibiting gasdermin D-mediated pyroptosis and inflammatory response
Inflammopharmacology (2024)
-
Association between high-mobility group box 1 levels and preeclampsia: a systematic review and meta-analysis
Journal of Assisted Reproduction and Genetics (2024)
-
Protective effect of zerumbone on sepsis-induced acute lung injury through anti-inflammatory and antioxidative activity via NF-κB pathway inhibition and HO-1 activation
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Association between ICU admission (neutrophil + monocyte)/lymphocyte ratio and 30-day mortality in patients with sepsis: a retrospective cohort study
BMC Infectious Diseases (2023)
-
HMGB1 is involved in viral replication and the inflammatory response in coxsackievirus A16-infected 16HBE cells via proteomic analysis and identification
Virology Journal (2023)