
Abstract
The purpose of this study is to investigate the complex connection between apathy and cognitive decline that remains unclear. A total of 1057 non-dementia elderly from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database received up to 13 years of follow-up and were divided into an apathy negative (−) group of 943 participants and an apathy positive (+) group of 114 participants through the Neuropsychiatric Inventory (NPI)-apathy subitem. Cerebrospinal fluid (CSF) AD biomarkers and amyloid β (Aβ) PET were measured, and their longitudinal changes were assessed using linear mixed-effects models. Risk factors for cognitive decline and apathy conversion were explored through the Cox proportional hazards model. Mediation effects of Aβ pathology on cognition were investigated using the causal mediation analysis. Apathy syndrome was associated with faster impairment of cognition and elevation of the Aβ burden. The effects of apathy on cognitive function and life quality were mediated by Aβ pathology, including CSF Aβ42/total tau ratio, and Aβ deposition in the prefrontal regions. Apathy syndrome was the risk factor for cognitive deterioration; meanwhile, frontal Aβ burden was the risk factor for apathy conversion. Apathy syndrome is an early manifestation of cognitive decline and there are bidirectional roles between apathy syndrome and Aβ pathology. Prefrontal Aβ pathology influenced the pathway from apathy to cognitive decline.
Similar content being viewed by others

Introduction
Apathy, defined as a quantitative reduction of goal-directed activity compared to the patient’s previous level of functioning [1], is the most prevalent behavioral and psychological syndrome in up to 88% of patients with Alzheimer’s disease (AD) and the vast majority of frontotemporal dementia [2]. The syndrome persists for at least 4 weeks and affects at least two of the three apathy dimensions, including behavior/cognition, emotion, and social interaction [1]. The occurrence of apathy is associated with a faster cognitive decline for patients and a higher burden for caregivers [3], which causes the isolation and drastic daily routine alterations of families with dementia patients. Although neuropsychiatric symptoms have long been recognized as emerging after dementia, recent reports have found apathy may proceed with cognitive decline, up to 5 years prior to the manifestation of the cognitive convention [3]. Therefore, understanding the effects and pathology of apathy in cognitive decline is necessary to measure dementia progression and explore therapy strategy.
In 2016, a new A/T/N classification scheme based on biomarkers was proposed for making an early AD diagnosis. As cerebrospinal fluid (CSF) biomarkers, Aβ42 stands for amyloid pathology, phosphorylated (p-tau) stands for tau pathology, and total tau (t-tau) stands for neurodegeneration [4]. Few studies had explored the neuropathologic correlations of neuropsychiatric syndromes and apathy in particular [5]. Existing research suggested that AD-type pathology might be a determinant of apathy. Small sample studies revealed that apathy might be correlated with Aβ burden and cognitive decline in Parkinson’s disease [6], AD [3, 7], and non-dementia elderly [8, 9]. Conflicting results reported that apathy scores positively correlated with CSF t-tau and p-tau levels[10], but not Aβ42 [11], or noncorrelation with CSF AD biomarkers [12].
To this day, it is still disputable how apathy syndrome impacts cognitive decline and whether AD-type biomarkers modulate the relationship of apathy with cognitive function. Thus, the present study aimed to examine the effect of apathy on cognitive functions and AD-type biomarkers. In addition, we tested whether the influences of apathy on cognition were mediated by AD pathology.
Materials and methods
Subjects
The data of 2272 adults were downloaded from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu) [13, 14]. A total of 1215 entries were excluded; 949 subjects with missing data, including CSF AD biomarkers, cognitive assessments, and APOE genotype, 239 subjects diagnosed with dementia, and 27 subjects with outliers of AD biomarkers. A total of 1057 non-dementia elderly were eventually involved in this study. At baseline, all the subjects received cognitive assessments and CSF tests. Meanwhile, the AV45 positron emission tomography (PET) data from 663 individuals were downloaded. After 2 years, 774 individuals were willing to be re-assessed, 342 received CSF tests, and 413 were scanned by AV45 PET. In ADNI, cognitively normal subjects had Mini-Mental State Examination (MMSE) scores between 24 and 30, a clinical dementia rating (CDR) of 0, no memory complaints, and Geriatric Depression Scale-15 (GDS-15) score < 7. Mild cognitive impairment (MCI) subjects were diagnosed according to the criteria of Petersen et al. [13], which included MMSE scores between 24 and 30, a CDR of 0.5, a GDS-15 score of <7, a memory complaint verified by an informant, and objective evidence of memory loss. Through follow-ups, dementia patients were diagnosed with an MMSE score between 20 and 26, a CDR of 0.5–1, and a GDS-15 score of <7.
ADNI is a multi-site data set designed to test the clinical symptoms, imaging, genetic, and biochemical biomarkers of AD, launched in 2003. Data collection and sharing in ADNI were approved by institutional review boards of all participating institutions and written informed consent was obtained from all participants or their guardians in accordance with the Declaration of Helsinki. The participants are older adults aged 55–90 years. Each participant underwent an in-person interview for health and neuropsychological assessments at baseline and annual follow-up.
Measures
Neuropsychological and neuropsychiatric assessments
Neuropsychological tests included the following measures: global cognition by MMSE, Alzheimer’s Disease Assessment Scale-cognitive sections, CDR, life quality by Functional Activities Questionnaire (FAQ), and depression screening by GDS-15.
Neuropsychiatric symptoms were assessed with the Neuropsychiatric Inventory (NPI), which is an informant-based instrument, measuring the presence (0 = absent, 1 = present), frequency, and severity (1 = mild, 2 = moderate, and 3 = severe) of multiple symptoms including delusions, hallucinations, apathy, agitation, depression, and so on. We used the presence vs. absence of apathy as our dichotomous indicator and divided all subjects into apathy positive (+) and apathy negative (−) groups [15]. The severity ratings had three levels as follows: mild rating defined that the apathy was perceptible but not obvious, moderate rating defined as obvious but not very prominent, and severe rating defined as very prominent change. The questionnaire about apathy absence included eight questions as follows:
-
1.
Does the participant seem less spontaneous and less active than usual?
-
2.
Is the participant less likely to initiate a conversation?
-
3.
Is the participant less affectionate or lacking in emotions compared to his/her usual self?
-
4.
Does the participant contribute less to household chores?
-
5.
Does the participant seem less interested in the activities and plans of others?
-
6.
Has the participant lost interest in friends and family members?
-
7.
Is the participant less enthusiastic about his/her usual interests?
-
8.
Does the participant show any other signs that he/she doesn’t care about doing new things?
CSF AD-type biomarkers
Before analysis, concentrations were all normalized into Z-score and outliers beyond ±3δ were excluded (n = 27). All 1057 subjects had CSF AD biomarkers, including Aβ42, p-tau, and t-tau proteins. The ADNI used the fully automated and highly standardized Roche Elecsys immunoassay to assess AD biomarkers.
All subjects were also binarized into Aβ negative (−) or positive (+) based on whether their CSF Aβ42 was normal or abnormal. Aβ+ individuals had a CSF Aβ42 < 976.6 pg/ml [16]. T-tau and p-tau were expressed in ratio to Aβ42, because they were reported as better predictors of Aβ deposition and cognitive decline [17].
APOE genotype
DNA was extracted with the QIAamp®DNA Blood Mini Kit and amplified by PCR with forward primers 14 5′-ACGGCTGTCCAAGGAGCTG-3′ (rs429358) and 5′-CTCCGCGATGCCGATGAC-3′ 15 (rs7412). APOE genotype was performed through restriction fragment length polymorphism technology.
Regional Aβ PET data
All image acquisition procedures were described in detail on the ADNI website (http://adni.loni.usc.edu/methods/documents/). Briefly, Aβ PET images were acquired in four frames of 5 min each, 50–70 min p.i. for 18F-florbetapir and 90–110 min p.i. for 18F-florbetaben. Regional Aβ PET data were downloaded from the ADNI Laboratory of Neuro Imaging database (adni.loni.usc.edu/methods/pet-analysis). Standardized uptake value ratios (SUVR) were computed using the whole cerebellum as a reference region. Through the Cox proportional hazards model, we selected the following regions of interest: the medial orbitofrontal cortex (mOFC) and the pars orbitalis cortex (POC).
Statistical analyses
Based on the presence of apathy, all the subjects were binarized into apathy positive (+) and apathy negative (−) groups. The data were not normally distributed; therefore, statistical significance was assessed using nonparametric tests. The Mann–Whitney U-test was used for continuous variables and the χ2-test was used for categorical variables to test the differences between two groups. Statistical power was calculated using the PASS software. The alternative hypothesis of two means unequal was adopted with a simulation of 1000.
The linear mixed-effect model depicted the longitudinal effects of apathy on the clinical outcome differences in the two groups. Furthermore, we also depicted the longitudinal difference of apathy severity between the Aβ positive (+) and Aβ negative (−) groups. The model included random slope and intercept terms for each participant. Age, education years, sex, and APOE4 genotype were included as covariates. Intracranial volume (ICV) was also adopted as a covariable in the imaging analysis. A Kaplan–Meier plot was constructed to assess the risk of cognitive conversion. A time-dependent Cox proportional hazards model was run to predict the cognitive conversion and apathy conversion. The cognitive conversion was defined as: (1) MCI progressing into dementia and (2) cognitively normal progressing into MCI or dementia. Apathy conversion was defined as apathy negative (−) at baseline progressing into apathy positive (+) during follow-up. Age, education years, sex, APOE4 genotype status, and with or without ICV were included as covariates. Smoothing splines were used to establish fitting curves between conversion stages and multiple biomarkers. The stage of change was calculated as the months away from the onset point of conversion and absolute values of different biomarkers were normalized into Z-score. The change trends of MMSE, Aβ42, and Aβ42/t-tau ratio were the opposites of the other biomarkers, so their negative forms were adopted.
Mediation analysis was used to explore whether Aβ pathology biomarkers mediated the causal pathway from apathy to cognitive decline. We assigned X to be apathy status (apathy severity at baseline), M as the potential mediators (CSF Aβ42/t-tau ratio, frontal lobe Aβ SUVR, mOFC Aβ SUVR, and POC Aβ SUVR), and Y as the outcome (cognitive function and life quality at 2-year follow-up). In this context, we interpreted the total effect as the amplitude of apathy severity in cognitive decline, both directly and through Aβ pathology biomarkers intermediates. To decompose the total effect into the part explained by Aβ pathology biomarkers and the part due to other factors, we analyzed direct and indirect effects by fitting two models: a mediator and an outcome model. The causal association was observed for a possible mediator and we estimated the causal effects of Aβ pathology biomarkers on the cognitive outcome. Age, education years, sex, and APOE4 genotype were included as covariates with or without ICV. This analysis was performed to estimate the total effect, direct effect, indirect effect, and their 95% confidence intervals using the PROCESS macro for SPASS [18] with bootstrapping of 1000 iterations. When the total effect and Sobel test were both significant, the mediation effect was considered to exist.
The statistical significance of all tests was set at a two-sided p-value < 0.05. All analyses were performed using SPSS 17.0. GraphPad Prism and R version 4.0.3 were used for figure preparation.
Results
Participants characteristics
Participant characteristics in ADNI were summarized in Table 1. A total of 1057 individuals without dementia were included in the present study. The participants were in their late midlife (aged 72.72 ± 7.04), with moderate years of education (mean = 16.33 years), and cognitively unimpaired (mean MMSE score = 28.31). Male participants accounted for 53.1%.
Through NPI assessment, individuals were divided into two groups (943 apathy− and 114 apathy+) and 663 participants underwent brain Aβ PET scanning (569 apathy− and 78 apathy+). Participants with apathy tended to be male and APOE ε4 carrier. They also had a more impaired cognitive function and life quality (Fig. 1A1–A3) and higher cerebral Aβ burden (with low statistical power) (Table 1 and Fig. 1B). After 2 years, 774 individuals (698 apathy− and 76 apathy+) received neuropsychological assessments, 342 individuals (306 apathy− and 36 apathy+) received CSF AD biomarkers testing, and 413 individuals (369 apathy− and 44 apathy+) underwent Aβ PET scanning. The analyses of the 2-year follow-up were almost consistent with the result at baseline (Fig. 1C1–C3, D). Due to the large difference in sample size between groups, statistical power was calculated for statically significant analysis. At baseline, the statistical powers of the variables were all above 0.75, except CSF Aβ42 level (0.673); however, at 2-year follow-up, MMSE and Left mOFC Aβ SUVR were only 0.625–0.470, which was due to the small sample size.

We categorized the total sample into apathy− subgroup and apathy+ subgroup. A1, A2, C1, C2 Lower cognitive functions were significant in apathy+ individuals than their normal counterparts. A3, C3 Lower quality of life were significant in apathy+ individuals. B, D Higher CSF Aβ42 (low statistical power) were significant in apathy+ individuals. Dots represented individuals’ clinical outcomes, with violin plots showing their median values and distributions. *p < 0.05, ***p < 0.001.
Longitudinal effects of apathy on cognitive function, CSF Aβ level, and regional Aβ burden
A total of 1057 individuals underwent an in-person interview at baseline and annual follow-up, and the follow-up time was up to 156 months (n = 5465 person-times in total). The longitudinal effects were used to explore the effects of apathy on cognitive functions, CSF Aβ42 level, and regional brain Aβ deposition. We found that individuals with apathy displayed faster cognition and life quality impairments (Fig. 2A, B1–B4) and faster elevation of CSF Aβ burden (Fig. 2A, C1, C2) after controlling the age, education years, sex, and APOE ε4 status. Moreover, individuals with apathy showed faster Aβ deposition in cortical regions including the frontal lobe, left mOFC, and right POC after controlling the age, education years, sex, APOE ε4 status, and ICV (Fig. 2A, D1–D3). Compared with the 468 Aβ− elderly, the 589 Aβ+ elderly showed faster elevation of apathy severity (β = 0.083, p < 0.0001, Fig. 2E), after controlling the age, education years, sex, and APOE ε4 status (n = 2008 person-times in total).

A Clinical outcomes between apathy- and apathy+ groups based on the linear mixed-effects model after adjusting for age, education years, sex, APOE ε4 status, and ICV (when imaging analysis). B1–B4 The apathy+ group had a lower cognitive function and life quality than the apathy− group. C1, C2 The apathy+ group had decreased CSF Aβ levels than the apathy− group. D1–D3 The apathy+ group had a higher regional Aβ burden. E The Aβ+ group had a faster apathy severity elevation than the Aβ− group.
A total of 68 cortexes, IVC, age, education years, sex, and APOE ε4 status were included as covariables to identify risk regions of Aβ deposition for apathy conversion through the Cox proportional hazards model. We found that the left mOFC (hazard ratio (HR) = 840.409, 95% confidence interval (95% CI) = 1.867–378243.514, p = 0.031) and the right POC (HR = 85.113, 95% CI = 1.511–4782.305, p = 0.031) were the risk regions.
Causal mediation analyses
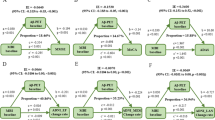
We investigated whether apathy severity contributed to cognitive impairments via modulating Aβ pathology. We did not find the mediation pathway of Aβ42, t-tau, or p-tau alone from apathy to cognitive decline. However, we found the mediation effect of other Aβ pathology biomarkers, including CSF Aβ42/t-tau ratio (Fig. 3A1, A2), frontal lobe Aβ burden (Fig. 3B1, B2), left mOFC Aβ burden (Fig. 3C1, C2), and right POC Aβ burden (Fig. 3D1, D2), which mediated the association between apathy severity and cognitive impairment after 2 years (ADAS-13 and FAQ). The effect was considered partial mediation, with the proportion of mediation varying from 7.92% to 22.80%.

Aβ pathology included the CSF Aβ/t-tau ratio (A1, A2), frontal lobe Aβ SUVR (B1, B2), left mOFC Aβ SUVR (C1, C2), and right POC Aβ SUVR (D1, D2). Blue lines showed the total effect (c) of apathy on 2-year follow-up cognitive functions, blue dotted lines showed the direct effect (c’), and green lines depicted the mediation effect (a*b) of Aβ pathology. Path weights were only shown for significant paths and were expressed as effect and p-value.
Apathy and cognitive conversion risk
In the cohort, 953 follow-up participants (aged 72.94 ± 7.05, 54.1% males with follow-up duration: 43.52 ± 31.84 months, maximum = 156 months) were included for exploring the incident cognitive decline. Among them, 69 cognitively normal subjects developed MCI or dementia, 204 MCI subjects developed dementia, and 680 participants had censored data. Furthermore, 858 subjects with apathy, aged 72.92 ± 7.01 and 51.5% males, were included to explore the apathy conversion risk during the follow-up. Among them, 227 subjects without apathy at baseline developed apathy syndrome and 631 subjects had censored data.
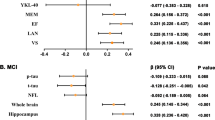
The results of the Kaplan–Meier analysis and the log-rank test showed a significant difference in the cumulative proportion of individuals free of cognitive deterioration between apathy− and apathy+ individuals (χ2 = 27.548, p < 0.001) (Fig. 4B). Individuals with apathy, higher CSF Aβ42 level, or frontal lobe Aβ deposition had an increased risk of cognitive conversion compared to those without apathy through the Cox proportional hazards model with age, education years, sex, APOE, and with or without ICV as covariables (Fig. 4A1, A2). Moreover, subjects with higher frontal Aβ deposition had a higher risk for apathy conversion with age, education years, sex, APOE, and ICV as covariables (Fig. 4A3).

A1, A2 Cox proportional hazards model estimated apathy and Aβ pathology risks for cognitive conversion after adjusting for age, education years, sex, APOE ε4 status, and ICV (when imaging analysis). A3 The frontal Aβ burden was assessed for apathy conversion after adjusting for age, education years, sex, APOE ε4 status, and ICV. B Compared to subjects without apathy, those with apathy were associated with a higher risk of cognitive conversion through the Kaplan–Meier curve. C2 The temporal sequence of biomarkers showed that apathy syndrome began as early as 5–6 years before the onset of cognitive convention. C1 During the process of apathy conversion, Aβ pathology biomarkers elevated slowly at 10 years before apathy onset and changed markedly at 3 years after apathy.
The temporal course of biomarkers
A smoothing spline was used to establish fitting curves to indicate the temporal course of different biomarkers during cognitive decline and apathy conversion. A total of 69 NC that developed MCI or dementia and 204 MCI that developed dementia were included for analysis of cognitive deterioration, whereas 114 apathy+ at baseline and 227 apathy− developing into apathy+ during follow-up were included for analysis of apathy conversion.
“0” on the X-axis indicated the onset point of conversion and values on both sides of “0” indicated the months away from the conversion point. During the process of cognitive decline, AD pathology biomarkers preceded the other biomarkers. Apathy syndrome began almost simultaneously with the turning point of cognitive tests, as early as about 5–6 years before cognitive conversion (Fig. 4C2). During the process of apathy conversion, Aβ pathology biomarkers elevated slowly at 10 years before apathy onset and changed markedly 3 years after apathy (Fig. 4C1).
Discussion
The present study had three main findings. First, apathy syndrome could significantly elevate the risk of cognitive decline and brain Aβ burden. Second, the influences of apathy severity on cognition and life quality were associated with Aβ pathology in the prefrontal regions. Third, apathy syndrome began 5–6 years before cognitive conversion and the brain Aβ burden elevated slowly up to 10 years before the onset of apathy.
Our results were consistent with previous longitudinal findings that demonstrated apathy syndrome predicting cognitive conversion. A study of 332 MCI elderly over 3 years showed a higher incidence of dementia with HR of 1.62 in subjects with apathy [19] and a study of 873 elderly over 2 years found that apathy was associated with incident cognitive decline [20]. Similarly, in the present study, a larger and longer follow-up cohort, we found non-dementia subjects with apathy had a higher risk of cognitive conversion with HR of 1.93–2.08. In addition, our analysis revealed that apathy predicted a higher cerebral Aβ burden, which aligned with some previous studies [8, 9], despite other negative findings [11, 12]. The first postmortem study of AD that explored apathy pathology nearly 30 years ago showed a combination of apathy with increased neurofibrillary tangle burden [21]. A study of 157 non-dementia subjects followed for up to 4 years found that apathy correlated with Aβ burden through 18F-flutemetamol PET [8]. Another study of 413 participants on CSF examination followed up for 3 years found a significant correlation between lower Aβ42 and greater rate of apathy [18]. Lanctôt et al. [22] thought the association with p-tau burden was more consistent across all stages of AD and Aβ burden was associated with apathy earlier in the disease process. Our study found that apathy subjects had a higher brain Aβ burden during the longitudinal observation. Apathy syndrome and brain Aβ burden, including CSF Aβ42/t-tau ratio and frontal lobe Aβ deposition, were risk factors for cognitive decline. Moreover, frontal lobe Aβ deposition manifested to be the risk factor for apathy conversion and Aβ-positive individuals showed greater apathy severity through the longitudinal analysis results. All the above findings suggested the bidirectional roles of apathy and Aβ pathology.
Our mediation analysis showed that apathy severity led to cognitive impairment at a 2-year follow-up through the mediation effects of brain Aβ burden. First, CSF Aβ42/t-tau ratio and Aβ deposition in the prefrontal regions, including the left mOFC and right POC, could play the mediation role. Gad A found that Aβ burden in the cortical regions modified the association between cognitive impairment and p-tau, and the association was stronger in individuals with greater Aβ burden [18]. Although we did not find the effects of CSF p-tau in the present study, further tau PET analysis might provide more information. Second, we found the effect of apathy at the 2-year follow-up cognitive function but not at baseline cognitive function, which suggested that the influence of apathy syndrome was long term. During the progress of cognitive deterioration, CSF Aβ decreased first, followed by cortical Aβ deposition, CSF p-tau elevation, Fluro-deoxyglucose PET decrease, hippocampus atrophy, and cognitive impairment in order [23], which could explain the delayed and long-term effects induced by Aβ pathology. The effects of medication induced by Aβ pathology varied from 7.92% to 22.8%. In addition, by analyzing the temporal course of biomarkers, apathy syndrome began 5–6 years before cognitive conversion. This finding was similar to a small sample study that included 76 healthy elderly with a mean age of 69.9 years. It found that apathy scores and rates increased over 5 years, and apathy changes were associated with informant ratings of cognitive decline in the years prior to baseline assessment [24].
The mechanism by which Aβ pathology is involved in apathy contributing to cognitive impairment remains unclear. Some researchers thought that cored amyloid plaques damaged the dopamine transporter and caused impaired motivation [25], and others found apathy occurred due to lesions affecting the medial and orbital parts of the prefrontal cortex [26]. Previous findings showed that atrophy of mOFC and white matter abnormalities within mOFC were associated with apathy [27, 28]. In the present study, we found that Aβ deposition in the prefrontal regions, including the left mOFC and right POC, began up to 10 years before apathy onset through a time-biomarker fitting curve and was also a risk factor for apathy conversion through Cox proportional hazards analysis. These findings suggested that these two prefrontal regions with high Aβ burdens were involved in the mechanism of apathy. Although there was no report about POC in apathy, atrophy [28] and dysconnectivity [29] in pars orbitalis gyrus were found in cognitively deteriorated patients with schizophrenia and attention-deficit/hyperactivity disorder [30].
Moreover, amyloid pathology was found to play a mediation role in associating minimal depressive symptoms with cognitive impairments in the non-dementia population [31]. Lower Aβ42 and higher p-tau were also confirmed to be related to an increased probability of depression and apathy over time [32]. There was an overlap between apathy and depression, which shared common clinical features in AD. Diminished interest, psychomotor retardation, fatigue/hypersomnia, and lack of insight were similar in both syndromes. However, symptoms such as dysphoria, suicidal ideation, self-criticism, hopelessness, and pessimism were unique to depression [22]. The similar symptoms of apathy and depression could be due to the same neuropathology pathway, and accurate diagnostic strategy and longitudinal observation could give more distinguishment and deeper insight.
There are limitations to this study. The studied sample was restricted to those with apathy syndrome. The statistical power of some variables was <0.75 at baseline and 2-year follow-up, which was due to the small sample size. The effect size of apathy might be underestimated given the low incident rate of apathy in non-dementia population. Aβ PET analysis was adopted, but not tau PET imaging, which weakened the exploration of tau pathology in the mechanism of apathy. Cognitively normal and MCI subjects were included in this study, which might have affected the population heterogeneity bias. As a theoretical statistical analysis, the final fact about the mediation effect of Aβ pathology on the association between apathy and cognitive decline still needs to be verified by multi-dimensional analysis in the future study. NPI-apathy scale has certain weakness such as data being acquired from the informant, which might bring the recall bias.
In summary, this study indicated that apathy syndrome was an early manifestation of cognitive decline, which could help define high-risk populations suitable for early prevention of dementia. There were bidirectional roles between apathy syndrome and Aβ pathology, and prefrontal Aβ burden influenced the pathway from apathy to cognitive decline.
Data collection and sharing in ADNI were approved by institutional review boards of all the participating institutions in accordance with the Declaration of Helsinki.
Data availability
A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wpcontent/uploads/how_to_apply/ADNI_Acknow-ledgement_List.pdf.
References
Robert P, Lanctôt KL, Agüera-Ortiz L, Aalten P, Bremond F, Defrancesco M, et al. Is it time to revise the diagnostic criteria for apathy in brain disorders? The 2018 international consensus group. Eur Psychiatry. 2018;54:71–6.
Huey E, Lee S, Cheran G, Grafman J, Devanand D. Brain regions involved in arousal and reward processing are associated with apathy in Alzheimer’s disease and frontotemporal dementia. J Alzheimers Dis. 2017;55:551–8.
Vergallo A, Giampietri L, Pagni C, Giorgi FS, Nicoletti V, Miccoli M, et al. Association between CSF beta-amyloid and apathy in early-stage Alzheimer disease. J Geriatr Psychiatry Neurol. 2019;32:164–9.
Delmotte K, Schaeverbeke J, Poesen K, Vandenberghe R. Prognostic value of amyloid/tau/neurodegeneration (ATN) classification based on diagnostic cerebrospinal fluid samples for Alzheimer’s disease. J Alzheimers Res Ther. 2021;13:84
Lanctôt KL, Agüera-Ortiz L, Brodaty H, Francis PT, Geda YE, Ismail Z, et al. Apathy associated with neurocognitive disorders: recent progress and future directions. Alzheimers Dement. 2017;13:84–100.
Zhou Z, Müller M, Kanel P, Chua J, Kotagal V, Kaufer DI, et al. Apathy rating scores and β-amyloidopathy in patients with Parkinson disease at risk for cognitive decline. Neurology. 2020;94:e376–83.
Mori T, Shimada H, Shinotoh H, Hirano S, Eguchi Y, Yamada M, et al. Apathy correlates with prefrontal amyloid β deposition in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2014;85:449–55.
Johansson M, Stomrud E, Lindberg O, Westman E, Johansson PM, van Westen D, et al. Apathy and anxiety are early markers of Alzheimer’s disease. Neurobiol Aging. 2020;85:74–82.
Marshall GA, Donovan NJ, Lorius N, Gidicsin CM, Maye J, Pepin LC, et al. Apathy is associated with increased amyloid burden in mild cognitive impairment. J Neuropsychiatry Clin Neurosci. 2013;25:302–7.
Cotta Ramusino M, Perini G, Vaghi G, Dal Fabbro B, Capelli M, Picascia M, et al. Correlation of frontal atrophy and CSF tau levels with neuropsychiatric symptoms in patients with cognitive impairment: a memory clinic experience. Front Aging Neurosci. 2021;13:595758.
Skogseth R, Mulugeta E, Jones E, Ballard C, Rongve A, Nore S, et al. Neuropsychiatric correlates of cerebrospinal fluid biomarkers in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2008;25:559–63.
Donovan NJ, Wadsworth LP, Lorius N, Locascio JJ, Rentz DM, Johnson KA, et al. Regional cortical thinning predicts worsening apathy and hallucinations across the Alzheimer disease spectrum. Am J Geriatr Psychiatry. 2014;22:1168–79.
Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–9.
Trojanowski JQ, Vandeerstichele H, Korecka M, Clark CM, Aisen PS, Petersen RC, et al. Update on the biomarker core of the Alzheimer’s Disease Neuroimaging Initiative subjects. Alzheimers Dement. 2010;6:230–8.
Jang J, Ho J, Blanken A, Dutt S, Nation D. Affective neuropsychiatric symptoms as early signs of dementia risk in older adults. J Alzheimers Dis. 2020;77:1195–207.
Suárez-Calvet M, Morenas-Rodríguez E, Kleinberger G, Schlepckow K, Araque Caballero MÁ, Franzmeier N, et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol Neurodegener. 2019;14:1.
Xu W, Feng W, Shen XN, Bi YL, Ma YH, Li JQ, et al. Amyloid pathologies modulate the associations of minimal depressive symptoms with cognitive impairments in older adults without. Dementia. 2021;89:766–75.
Hayes A, Rockwood N. Regression-based statistical mediation and moderation analysis in clinical research: observations, recommendations, and implementation. Behav Res Ther. 2017;98:39–57.
Pink A, Stokin GB, Bartley MM, Roberts RO, Sochor O, Machulda MM, et al. Neuropsychiatric symptoms, APOE ε4, and the risk of incident dementia: a population-based study. Neurology. 2015;84:935–43.
Nunes PV, Schwarzer MC, Leite R, Ferretti-Rebustini R, Pasqualucci CA, Nitrini R, et al. Neuropsychiatric inventory in community-dwelling older adults with mild cognitive impairment and dementia. J Alzheimers Dis. 2019;68:669–78.
Förstl H, Burns A, Levy R, Cairns N, Luthert P, Lantos P. Neuropathological correlates of behavioural disturbance in confirmed Alzheimer’s disease. Br J Psychiatry. 1993;163:364–8.
Lanctôt KL, Agüera-Ortiz L, Brodaty H, Francis PT, Geda YE, Ismail Z, et al. Apathy associated with neurocognitive disorders: Recent progress and future directions. Alzheimers Dement. 2017;13:84–100.
Wang HF, Shen XN, Li JQ, Suckling J, Tan CC, Wang YJ, et al. Clinical and biomarker trajectories in sporadic Alzheimer’s disease: a longitudinal study. Alzheimers Dement. 2020;12:e12095.
Brodaty H, Altendorf A, Withall A, Sachdev P. Do people become more apathetic as they grow older? A longitudinal study in healthy individuals. Int Psychogeriatr. 2010;22:426–36.
Hamaguchi T, Tsutsui-Kimura I, Mimura M, Saito T, Saido TC, Tanaka KF. App mice overall do not show impaired motivation, but cored amyloid plaques in the striatum are inversely correlated with motivation. Neurochem Int. 2019;129:104470.
Baggio HC, Seguro B, Garrido-Millan JL, Marti MJ, Compta Y, Valldeoriola F. et al. Resting-state frontostriatal functional connectivity in Parkinson’s disease-related apathy. Mov Disord. 2015;30:671–9.
Chan NK, Gerretsen P, Chakravarty MM, Blumberger DM, Caravaggio F, Brown E, et al. Structural brain differences between cognitively impaired patients with and without apathy. Am J Geriatr Psychiatry. 2021;29:319–32.
Ohtani T, Bouix S, Hosokawa T, Saito Y, Eckbo R, Ballinger T. et al. Abnormalities in white matter connections between orbitofrontal cortex and anterior cingulate cortex and their associations with negative symptoms in schizophrenia: a DTI study. Schizophr Res. 2014;157:190–7.
Wei Y, Collin G, Mandl R, Cahn W, Keunen K, Schmidt R, et al. Cortical magnetization transfer abnormalities and connectome dysconnectivity in schizophrenia. Schizophr Res. 2018;192:172–8.
Nickel K, van Elst LT, Manko J, Unterrainer J, Rauh R, Klein C. et al. Inferior frontal gyrus volume loss distinguishes between autism and (comorbid) attention-deficit/hyperactivity disorder-a FreeSurfer analysis in children. Front Psychiatry.2018;9:521
Xu W, Feng W, Shen XN, Bi YL, Ma YH, Li JQ, et al. Amyloid pathologies modulate the associations of minimal depressive symptoms with cognitive impairments in older adults without dementia. Biol Psychiatry. 2021;89:766–75.
Banning L, Ramakers I, Rosenberg P, Lyketsos C, Leoutsakos J. Alzheimer’s disease biomarkers as predictors of trajectories of depression and apathy in cognitively normal individuals, mild cognitive impairment, and Alzheimer’s disease dementia. Int J Geriatr Psychiatry. 2021;36:224–34.
Acknowledgements
Data collection and sharing for the ADNI data section was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai, Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer, Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro 6 Imaging at the University of Southern California. The data used to prepare this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report.
Funding
This study was supported by grants of Special Sub Project of Strategic Leading Science and Technology of Chinese Academy of Sciences (XDA12040101), National Natural Science Foundation of China (81301139), Clinical Research Center Project of Shanghai Mental Health Center (CRC2017ZD02), National Science and Technology Support Program of China (2009BAI77B03), Shanghai Clinical Research Center for Mental Health (19MC1911100), Scientific Project of Shanghai Municipal Commission of Science and Technology (18401970602), Cultivation of Multidisciplinary Interdisciplinary Project in Shanghai Jiaotong University (YG2019QNA10), Curriculum Reform of Medical College of Shanghai Jiaotong University, and Feixiang Program of Shanghai Mental Health Center (2020-FX-03).
Author information
Authors and Affiliations
Consortia
Contributions
L.S. wrote the manuscript. L.S. and W.L. analyzed the data. G.L. helped revise the manuscript. S.X. designed and supervised the experiment. The ADNI database provided data support for this study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All subjects provided written consent to publish the findings.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, L., Li, W., Li, G. et al. Prefrontal Aβ pathology influencing the pathway from apathy to cognitive decline in non-dementia elderly. Transl Psychiatry 11, 534 (2021). https://doi.org/10.1038/s41398-021-01653-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01653-8
- Springer Nature Limited