
Abstract
Sporadic venous malformations are genetic conditions primarily caused by somatic gain-of-function mutation of PIK3CA or TEK, an endothelial transmembrane receptor signaling through PIK3CA. Venous malformations are associated with pain, bleedings, thrombosis, pulmonary embolism, esthetic deformities and, in severe cases, life-threatening situations. No authorized medical treatment exists for patients with venous malformations. Here, we created a genetic mouse model of PIK3CA-related capillary venous malformations that replicates patient phenotypes. We showed that these malformations only partially signal through AKT proteins. We compared the efficacy of different drugs, including rapamycin, a mTORC1 inhibitor, miransertib, an AKT inhibitor and alpelisib, a PI3Kα inhibitor at improving the lesions seen in the mouse model. We demonstrated the effectiveness of alpelisib in preventing vascular malformations’ occurrence, improving the already established ones, and prolonging survival. Considering these findings, we were authorized to treat 25 patients with alpelisib, including 7 children displaying PIK3CA (n = 16) or TEK (n = 9)-related capillary venous malformations resistant to usual therapies including sirolimus, debulking surgical procedures or percutaneous sclerotherapies. We assessed the volume of vascular malformations using magnetic resonance imaging (MRI) for each patient. Alpelisib demonstrated improvement in all 25 patients. Vascular malformations previously considered intractable were reduced and clinical symptoms were attenuated. MRI showed a decrease of 33.4% and 27.8% in the median volume of PIK3CA and TEK malformations respectively, over 6 months on alpelisib. In conclusion, this study supports PI3Kα inhibition as a promising therapeutic strategy in patients with PIK3CA or TEK-related capillary venous malformations.
Similar content being viewed by others
Introduction
Venous malformations are a disabling condition characterized by low flow and slow growth vascular deformities that can be either localized or diffused. These malformations can be associated with hemorrhage, infections, thrombosis, pulmonary embolism, chronic pain, fatigue, functional impairments, disabilities and disseminated intravascular coagulation.1 Genetically, venous malformations are mainly due to gain-of-function mutations in either, PIK3CA or Tie2R (TEK).1,2 These mutations arise during embryonic development and result in somatic mosaicism.3. The estimated incidence of venous malformations is 1–5 in 10,000 births.4,5 Somatic mutations in TEK account for over 50% of sporadic unifocal venous malformations,2,6 while around 20% of cases are attributed to somatic mutations in PIK3CA.7,8
PI3Kα is a lipid kinase regulating signaling pathways involved in cell metabolism, motility, proliferation and survival.9 PI3Kα is predominantly activated via tyrosine kinase receptors. The PIK3CA gene encodes the 110-kDa catalytic alpha subunit of PI3Kα (p110α), responsible for converting phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) into phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3; or PIP3) at the plasma membrane and subsequently recruits PDK1 through which AKT is phosphorylated on Thr308 residue. As for TEK, also called Tie2, it is a transmembrane receptor for angiopoietin-1 that signals through PI3Kα explaining why a gain-of-function mutation in this gene is associated with a phenotype that resembles the one seen with a gain-of-function mutation in PIK3CA.10
Venous malformations can be isolated or more complex when PIK3CA is the gene involved and are regrouped then under the appellation PIK3CA-related disorders.11,12 Histological examination of venous malformations usually shows thin-walled vessels that are variably dilated, commonly display thrombosis and occasionally contain phleboliths in the lumen and hemosiderin interpositions in the wall. Currently, treatments rely on compression, anticoagulation, sclerotherapy and surgery. Off-label use of rapamycin, an mTORC1 inhibitor, showed encouraging but variable results.13,14
Various mouse models are employed to understand the pathophysiology of capillary venous malformations. They include xenograft models with human endothelial cells bearing TEK or PIK3CA mutations.7,15 as well as mouse models created through a Cre/Lox approach.8,16 that recapitulate more or less faithfully the human disease.
A few years ago, a mouse model of PROS was successfully developed by our group using an inducible ubiquitously expressed Cre recombinase (CAGG CreER).17 The transgene used in this model was more potent than the mutations found in humans, enabling an accurate reproduction of the phenotype observed in PROS. With this mouse model, the effectiveness of alpelisib in the treatment of PROS was demonstrated in preclinical testing and subsequently in patients.17,18,19,20 These findings were further validated in the EPIK P1 clinical trial (NCT04285723), which led to the extension of alpelisib’s approval by the US FDA to patients with PROS aged two years and older.21 During the clinical trial, it was observed that the response to alpelisib varied among different tissues, with potentially significant efficacy seen in vascular tissues, particularly in veins.
Indeed, the recent progress in genetics and the emergence of targeted therapies hold promises for creating new opportunities for patients with venous malformations. Since the mouse models available to study venous malformations only partially reflect the human pathology, our aim here was to develop a novel preclinical model specifically mirroring the most severe manifestation of PIK3CA-related capillary venous malformations and that would enable us to closely examine the effects of alpelisib.
Results
A mouse model of PIK3CA gain of function in endothelial cells
We chose to develop a mouse model with a gain-of-function mutation in PIK3CA that specifically targets endothelial cells. For this purpose, we utilized the transgenic mouse strain R26StopFLP110* that is designed to express a dominant active PIK3CA transgene upon Cre recombination. By crossing these mice with Tie2 Cre mice, we thus generated PIK3CATie2-CreER animals that carry a constitutively overactivated form of PIK3CA in capillaries and vein endothelial cells upon tamoxifen administration. To monitor Cre recombination, we further bred the PIK3CATie2-CreER mice with Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J mice in which an ubiquitously expressed cell membrane-localized tdTomato fluorescent protein is replaced by GFP following Cre recombination.22 For control purposes, littermate mice carrying only the Tie2 Cre recombinase without the PIK3CA transgene were used and will be referred to hereafter as PIK3CAWT mice.
To address developmental concerns, we utilized four-week-old mice and administered a daily dose of 40 mg.kg-1 tamoxifen for five consecutive days to induce Cre recombination. After observing a delay of 2 to 3 weeks following Cre recombination, we noticed progressive and visible generalized subcutaneous capillary and venous malformations in PIK3CATie2-CreER mice (Fig. 1a), accompanied by excessive weight gain (Supplementary Fig. 1a). Both male and female mice exhibited these characteristics (Supplementary Fig. 1a). The malformations showed rapid growth and were associated with a significant reduction in survival compared to controls (Fig. 1b). To further investigate the extent of the malformations, we conducted whole-body T2-weighted magnetic resonance imaging (MRI) at 5 weeks after Cre induction. These studies revealed the presence of disseminated superficial capillaries and deep venous malformations (Fig. 1c and Supplementary Fig. 1b). Necropsy examinations demonstrated subcutaneous hemorrhage all linked to disseminated vascular malformations in the peri-laryngeal space, lower and/or upper limbs, and urogenital area (Supplementary Fig. 1c).

A mouse model of PIK3CA-related capillary venous malformations. a Representative photography of PIK3CAWT and PIK3CATie2-CreER mice 4 weeks after Cre recombination. b Kaplan–Meier survival curves of PIK3CAWT and PIK3CATie2-CreER mice (n = 25 per group). c Coronal whole-body T2-weighted fat saturated magnetic resonance images (MRI) of PIK3CAWT and PIK3CATie2-CreER mice (n = 4–6 mice per group) 5 weeks after Cre recombination. Volumetric quantification of the vascular malformations. d Representative hematoxylin and eosin (H&E) staining of the skin of PIK3CAWT and PIK3CATie2-CreER mice and patient with venous malformation (VM). Large irregular and dilated vessels filled with red blood cells were visible in the skin of PIK3CATie2-CreER mice. Scale bar: 40 μm. e Representative coimmunofluorescence staining of Tie2 and GFP in the skin of PIK3CAWT and PIK3CATie2-CreER mice. Scale bar: 10 μm. f GFP is express in CD31+ cells as assessed by flow cytometry experiments of cells isolated from the skin of PIK3CAWT (n = 4 mice). g GFP and podoplanin staining using flow cytometry experiments of cells isolated from the skin of PIK3CAWT (n = 4 mice). h Representative immunofluorescence staining of P-AKTThr308 and P-S6RP in the skin of PIK3CAWT and PIK3CATie2-CreER mice. Scale bar: 10 μm. i Western blot and j quantification of P-AKTSer473 and P-S6RP in the skin of PIK3CAWT and PIK3CATie2-CreER mice (n = 5–8 mice per group). k Flow cytometry experiments showing the percentage of GFP + CD31+ cells isolated from the skin of PIK3CAWT and PIK3CATie2-CreER mice expressing P-AKTSer473 (n = 6–7 mice per group). l Representative immunofluorescence staining of KI67 in the skin of PIK3CAWT and PIK3CATie2-CreER mice. Scale bar: 10 μm and m quantification (n = 4 mice per group). n Flow cytometry experiments showing the percentage of GFP + CD31+ cells isolated from the skin of PIK3CAWT and PIK3CATie2-CreER mice expressing KI67 (n = 3–4 mice per group). o Flow cytometry experiments showing the percentage of Tomato+ cells isolated from the skin of PIK3CAWT and PIK3CATie2-CreER mice expressing KI67 (n = 3–4 mice per group). p Quantification of GFP positive cell surface isolated from PIK3CAWT and PIK3CATie2-CreER mice (n = 5 mice per group). q Complete blood count and D-Dimers measurement in PIK3CAWT and PIK3CATie2-CreER mice (n = 11 per group)
Histological analysis of the capillary and venous malformations revealed dilated and irregular vessels with disorganized tissue structure, endothelial hyperplasia, blood-filled lumens, hemosiderin interposition, and features of hemophagocytosis (Supplementary Fig. 1d). In some mice, local clotting was accompanied by pulmonary embolism (Supplementary Fig. 1e, f). In the malformations, we also noticed varying degrees of adipose tissue overgrowth, lymphatic compounds, and αSMA positive cells (Supplementary Fig. 1f). These abnormalities closely resemble the ones that are seen in patients with capillary venous malformations (Supplementary Fig. 1g).
We then verified Cre recombination through GFP and p110* expression. Coimmunostaining (Fig. 1E and Supplementary Fig. 2a), Western blot (Supplementary Fig. 2b) and flow cytometry experiments confirmed that the Cre recombinase was specifically expressed in CD31 positive cells (97.8 ± 0.31% mean ± SEM, n = 4 mice). Conversely 0.54 ± 0.1% of the cells were GFP + CD31− showing indeed the endothelial cell expression of the Cre recombinase (n = 4 mice) (Fig. 1f). Importantly, no Cre expression was detected in lymphatic endothelial cells using flow cytometry experiments (0.67 ± 0.2%, mean ± SEM, n = 4 mice) (Fig. 1g). Indeed, the presence of adipose tissue overgrowth and lymphatic anomalies suggests potential communication between endothelial cells and surrounding tissues or cell recruitment. In this regard, we noticed infiltration of immune cells within the lesions as indicated by the presence of CD3+ cells (Supplementary Fig. 2c) and F4/80-expressing cells (that is, macrophages). Macrophages were visible either within the lumen or in the close vicinity of dilated vessels (Supplementary Fig. 2d–f), with a significant proportion of CD163, a receptor involved in clearing and endocytosis of hemoglobin/haptoglobin complexes (Supplementary Fig. 2d–f). Flow cytometry experiments showed that the Cre recombinase was not expressed in CD45+ (0.21±0.03% mean ± SEM, n = 4 mice) or CD34+(0.09±0.01%, mean±SEM, n = 4 mice) bone marrow cells (Supplementary Fig. 2g, h).
Coimmunofluorescence experiments using GFP, COUP-TFII, CD31, Podoplanin, Ephrin B2 and Endomucin on 30 μm sections revealed severe disorganization in the 3D structure of the skin vessels (Supplementary Figs. 3 and 4).
Immunofluorescence (Fig. 1h), Western blot (Fig. 1i, j) and flow cytometry studies (Fig. 1k) revealed activation of the AKT and mTOR pathways in the endothelial cells of PIK3CATie2-CreER mice by contrast to PIK3CAWT. Mechanistically, PI3Kα is known to activate cell growth and proliferation. We investigated cell proliferation using KI67 staining (Fig. 1l, m, Supplementary Fig. 5a, b) and flow cytometry experiments. In control mice, we noted that 1.7 ± 0.6% (mean ± SEM, n = 4) of CD31 + GFP+ and 11.5 ± 0.4% (mean ± SEM, n = 4) of the Tomato+ cells co-expressed KI67 (Fig. 1n, o). Remarkably, in PIK3CATie2-CreER mice we observed an elevated proliferation rate in the CD31 + GFP+ population (21.7 ± 6.2% were positive for KI67) as well as in Tomato+ cells (26.6±4.8% were positive for KI67) (Fig. 1n, o). This finding indicates the proliferation of endothelial cells and supports again a potential crosstalk between mutated and surrounding wild-type cells. Using Amnis ImageStream® system, we also found that mutated endothelial cells isolated from PIK3CATie2-CreER mice were hypertrophic by contrast to controls (Fig. 1p and Supplementary Fig. 5c).
Finally, complete blood counts in PIK3CATie2-CreER mice revealed significant regenerative anemia, a low platelet count, and elevated D-Dimers levels, consistent with the possibility of disseminated intravascular coagulation, as well as slight hypereosinophilia. All of these abnormalities are also consistent with what is observed in patients with venous malformations (Fig. 1q).23,24
We thus have successfully established a mouse model that accurately replicates the phenotype of patients who are affected by somatic or inherited forms of capillary venous malformations. This model should offer the advantage of providing mechanistic insight into both the PIK3CA- and TEK-related types of vascular diseases.
We then investigated whether the PIK3CA gain-of-function mutation in capillaries and veins would lead to detectable changes in circulating metabolites that could potentially serve as biomarkers for disease progression. For this purpose, we collected plasma samples from both PIK3CAWT and PIK3CATie2-CreER mice after a 12-hour fast and subjected them to thorough metabolomic analysis. PIK3CATie2-CreER mice were found to exhibit activated anabolic pathways, resulting in the accumulation of various amino acids, including arginine, histidine, phenylalanine, thymidine, threonine, 5-adenosyl-homocysteine, valine, orotic acid, acetyl-lysine, methyl-lysine, lysine, and oxoadipate, along with the metabolic intermediates of these amino acids. Additionally, there was evidence of increased mitochondrial respiration in these mice, as indicated by the plasma accumulation of succinic acid and acetyl-carnitine, and activation of the urea cycle as indicated by higher circulating levels of arginine and urate compared to controls (Supplementary Fig. 6). As such, circulating metabolites could serve as potential biomarkers for monitoring disease progression in PIK3CA-capillary venous malformations.
In humans, a great number of PIK3CA variants have been identified and found to exert varying effects on the recruitment of the AKT pathway.25 To simulate a higher degree of AKT recruitment, we also created PIK3CATie2-HO mice that bear homozygous PIK3CA mutations in capillaries and venous endothelial cells. After Cre recombination, a more severe phenotype emerged, characterized by the rapid occurrence of disseminated and voluminous capillary venous malformations with reduced life expectancy (Supplementary Fig. 7a–c). Whole-body T2-weighted MRI conducted five weeks after induction further confirmed the presence of extensive disseminated venous malformations (Supplementary Fig. 7d). Histological examination revealed severe widespread clotting and pulmonary embolism (Supplementary Figs. 7e and 4f), phosphorylation assays of tissues, robust activation of the AKT pathway (Supplementary Fig. 7g, h) and complete blood count, profound regenerative anemia (Supplementary Fig. 7i).
PIK3CA signals partially through AKT1 and not AKT2 in venous endothelial cells
In mammals, AKT is encoded by three distinct genes: AKT1, AKT2, and AKT3, with AKT1 being the main isoform in endothelial cells. To explore whether PIK3CA gain-of-function mutations primarily signal via AKT or other downstream targets, we conducted a series of experiments using genetically modified mice.
First, we interbred AKT1flox/flox with PIK3CATie2R-CreER mice to create a PIK3CATie2R-CreER-AKT1KO model (referred to as PIK3CAAKT1KO). Upon inducing Cre induction, we observed that AKT1 deletion in capillaries and veins led to a moderate extension of lifespan (median survival of 80 days) and only delayed the occurrence of vascular malformations (Fig. 2a and Supplementary Fig. 8a) as MRI performed at 6 weeks still revealed diffused venous malformations (Fig. 2b and Supplementary Fig. 8b). Histologically, the malformations were similar to the ones observed in PIK3CATie2R-CreER mice (Fig. 2c). AKT pathway recruitment exhibited no reduction (Fig. 2d, e), and cell proliferation rates remained unaffected by deletion of AKT1 (Fig. 2f, g). AKT1 deletion did not rescue complete blood count anomalies either (Supplementary Fig. 8c). These findings suggest that the AKT1 isoform only plays a partial role in the PIK3CA-driven phenotype of vascular malformations.

PIK3CA-related capillary venous malformations in PIK3CATie2-CreER mice only partially signal through AKT proteins. a Kaplan–Meier survival curves of PIK3CAWT, PIK3CATie2-CreER, PIK3CAAKT1KO, PIK3CAAKT2KO and PIK3CAAKT1AKT2-KO mice (n = 20 per group). b Coronal whole-body T2 weighted magnetic resonance images (MRI) of PIK3CAWT, PIK3CATie2-CreER, PIK3CAAKT1KO, PIK3CAAKT2KO and PIK3CAAKT1AKT2-KO mice 6 weeks after Cre recombination. Volumetric quantification of the vascular malformations (n = 4–6 mice per group). c Representative hematoxylin and eosin (H&E) staining of the skin of PIK3CAWT, PIK3CATie2-CreER, PIK3CAAKT1KO, PIK3CAAKT2KO and PIK3CAAKT1AKT2-KO mice. Scale bar: 10 μm. d Representative P-AKTSer473 in the skin of PIK3CAWT, PIK3CATie2-CreER, PIK3CAAKT1KO, PIK3CAAKT2KO and PIK3CAAKT1AKT2-KO mice. Scale bar: 10μm.*: Vascular malformation. e Western blot and quantification of P-AKTSer473 and P-S6RP in the skin of PIK3CAWT, PIK3CATie2-CreER, PIK3CAAKT1KO, PIK3CAAKT2KO and PIK3CAAKT1AKT2-KO mice (n = 4–5 mice per group). f Representative immunofluorescence staining of KI67 in the skin of PIK3CAWT, PIK3CATie2-CreER, PIK3CAAKT1KO, PIK3CAAKT2KO and PIK3CAAKT1AKT2-KO mice. Scale bar: 10 μm. g Flow cytometry experiments showing the percentage of GFP + CD31+ cells isolated from the skin of the different mouse models expressing KI67 (n = 3–4 mice per group). (n = 3–7 mice per group). h Western blot and quantification of AKT1 and AKT2 in vessels of PIK3CAWT, PIK3CATie2-CreER and PIK3CAAKT1AKT2-KO mice (n = 3–4 mice per group)
Next, we investigated the role of the AKT2 isoform by interbreeding AKT2−/− with PIK3CATie2R-CreER mice to obtain PIK3CATie2R-CreER-AKT2KO model (referred to as PIK3CAAKT2KO). Following the induction of Cre, we observed that PIK3CAAKT2KO mice exhibited the same clinical, biological, radiological and histological manifestations as PIK3CATie2R-CreER mice (Fig. 2a–c and Supplementary Fig. 8a, c). AKT phosphorylation in tissues and proliferation rates were also modestly affected in PIK3CATie2R-CreER animals (Fig. 2d, e). Thus, AKT2 deletion did not impact the PIK3CA-vascular malformation phenotype.
Finally, we investigated the simultaneous deletion of both AKT1 and AKT2 isoforms by interbreeding PIK3CAAKT1KO with PIK3CAAKT2KO mice. The resulting PIK3CAAKT1AKT2-KO pups were born in the expected Mendelian ratio and did not display any particular phenotype during the first few weeks of life. At 4 weeks of age, we activated Cre recombinase, and over time, PIK3CAAKT1AKT2-KO mice began to develop capillary and venous malformations (Supplementary Fig. 8a). Whole-body MRI performed 6 weeks after Cre recombination revealed diffuse malformations (Fig. 2b and Supplementary Fig. 8b). Similar to PIK3CAAKT1KO mice, AKT1 and AKT2 deletion in capillaries and veins was associated with a moderate extension of lifespan (Fig. 2z). Histology and blood examination confirmed severe venous malformation with intravascular coagulation (Fig. 2c and Supplementary Fig. 8c). Once again, a modest reduction in AKT phosphorylation but not in the proliferation rate were observed (Fig. 2d–g). Western blot analysis showed that while AKT1 and AKT2 were successfully deleted in endothelial cells (Fig. 2h), AKT protein was still expressed, indicating possible compensation by AKT3. Indeed, we concluded that, genetic ablation of both isoforms AKT1 and 2 was associated with delayed occurrence of the malformations.
Targeted therapy for venous malformations in PIK3CA related venous malformations
We proceeded to investigate whether targeted therapies.16 could improve the outcome of PIK3CATie2R-CreER mice. For this purpose, we compared the effects of three pharmacological inhibitors: rapamycin, miransertib, and alpelisib, targeting mTORC1,26 pan AKT, and PI3Kα, respectively. Four weeks after tamoxifen administration, mice were randomly assigned to one of the treatment groups.
Rapamycin, which is used off-label to treat various forms of venous malformations in patients, did not show any impact on the volume and histological aspects of venous malformations, on intravascular coagulation, or on overall survival when administered daily (Fig. 3a–d, Supplementary Fig. 9a, b). Rapamycin led to a reduction in S6RP phosphorylation in tissues, but AKT phosphorylation (Fig. 3e, f) and proliferation rate (Fig. 3g, h) remained very high.

Targeted therapies for capillary venous malformations in PIK3CATie2-CreER mice. a Kaplan–Meier survival curves of PIK3CAWT and PIK3CATie2-CreER mice treated with either vehicle, rapamycin, miransertib or alpelisib (n = 12 per group). b Coronal whole-body T2-weighted magnetic resonance images (MRI) of PIK3CAWT and PIK3CATie2-CreER mice 6 weeks after Cre recombination treated with either vehicle, rapamycin, miransertib or alpelisib. Volumetric quantification of the vascular malformations (n = 3-4 mice per group). c Complete blood count in PIK3CAWT and PIK3CATie2-CreER mice treated with either vehicle, rapamycin, miransertib or alpelisib (n = 3–5 mice per group). d Representative hematoxylin and eosin (H&E) staining of the skin of PIK3CAWT and PIK3CATie2-CreER mice treated with either vehicle, rapamycin, miransertib or alpelisib. Scale bar: 10 μm. e Representative P-AKTThr308 and P-S6RP immunostaining in the skin of PIK3CAWTand PIK3CATie2-CreER mice treated with either vehicle, rapamycin, miransertib or alpelisib. Scale bar: 10 μm. f Western blot and quantification of P-AKTSer473 and P-S6RP of the skin of PIK3CAWTand PIK3CATie2-CreER mice treated with either vehicle, rapamycin, miransertib or alpelisib (n = 3–4 per group). g Representative immunofluorescence staining of KI67 in the skin of PIK3CAWTand PIK3CATie2-CreER mice treated with either vehicle, rapamycin, miransertib or alpelisib. Scale bar: 10 μm. h Proliferation index quantification (n = 3–4 mice per group)
Miransertib, which had shown encouraging preclinical results,16 resulted in a slight reduction in venous malformation volumes (Fig. 3b, Supplementary Figs. 9a, b) and modest lifespan extension (Fig. 3a). However, it did not significantly impact platelet level, anemia or the histological abnormalities (Fig. 3c, d). Western blot and immunofluorescence studies did reveal a reduction in AKT and S6RP phosphorylation (Fig. 3e, f) with a modest impact on proliferation (Fig. 3g, h). These findings suggest again that PIK3CA-capillary venous malformations are not solely dependent on AKT for their development
In contrast to these 2 previous drugs, alpelisib showed significant efficacy. It led to prolonged survival (Fig. 3a), a notable reduction in venous malformations volume as confirmed by MRI (Fig. 3b, Supplementary Fig. 9a, b) and histological analyses (Fig. 3d) and to the reversal of intravascular coagulation biologically (Fig. 3c). In the affected tissues, the AKT pathway and cell proliferation were completely inhibited (Fig. 3e–h). These results demonstrate that alpelisib outperformed rapamycin and miransertib in treating PIK3CATie2R-CreER mice, showing promising potential as a therapeutic option for the management of vascular malformations in particular.
Alpelisib prevents venous malformations in PIK3CA mouse model
We next aimed to assess whether alpelisib could be effective in both preventing and improving vascular malformations in PIK3CATie2-CreER mice. To conduct a preventive study, we began to administer alpelisib vs. a vehicle 48 h after Cre induction and maintained it for 12 weeks. Alpelisib-treated PIK3CATie2-CreER mice displayed a visibly normal appearance and weight gain profile with extended lifespan compared to vehicle-treated PIK3CATie2-CreER mice (Fig. 4a, Supplementary Fig. 10a, b). MRI conducted after 6 weeks of alpelisib demonstrated the absence of vascular malformations (Fig. 4b, Supplementary Fig. 10c, d) and similar conclusions could be drawn based on necropsy examination (Fig. 4c). Alpelisib also effectively inhibited AKT and mTOR (Fig. 4d, e and Supplementary Fig. 10e), improved vascular anomalies (Supplementary Fig. 11a) and prevented blood cell count abnormalities from occurring (Fig. 4f).

Alpelisib improves and prevent capillary venous malformations in PIK3CATie2-CreER mice. a Kaplan–Meier survival curves of PIK3CAWT and PIK3CATie2-CreER mice treated with either vehicle or preventive alpelisib (n = 12 per group). b Coronal whole-body T2-weighted magnetic resonance images (MRI) of PIK3CAWT and PIK3CATie2-CreER mice 6 weeks after Cre recombination treated with either vehicle, preventive or therapeutic alpelisib (n = 4–6 mice per group). Volumetric quantification of the vascular malformations. c Representative hematoxylin and eosin (H&E) staining of the skin of PIK3CAWT and PIK3CATie2-CreER mice treated with either vehicle, preventive or therapeutic alpelisib. Scale bar: 10 μm. d Representative P-AKTThr308 and P-S6RP immunostaining in the skin of PIK3CAWTand PIK3CATie2-CreER mice treated with either vehicle, preventive or therapeutic alpelisib. Scale bar: 10 μm. e Western blot and quantification of P-AKTSer473 and P-S6RP in skin of PIK3CAWTand PIK3CATie2-CreER mice treated with either vehicle, preventive or therapeutic alpelisib (n = 6–7 mice per group). f Complete blood count of PIK3CAWTand PIK3CATie2-CreER mice treated with either vehicle, preventive or therapeutic alpelisib (n = 5–10 mice). g Representative photography of PIK3CATie2-CreER mice before and two weeks after alpelisib initiation. h Kaplan–Meier survival curves of PIK3CAWT and PIK3CATie2-CreER mice treated with either vehicle or therapeutic alpelisib (n = 12 per group)
For the therapeutic study, alpelisib was administered to PIK3CATie2-CreER mice 4 weeks after Cre induction when vascular malformations were already visible. This treatment led to a rapid decrease in body weight in alpelisib-treated PIK3CATie2-CreER mice (Supplementary Fig. 10a), clinical improvement of the vascular malformations (Fig. 4g and Supplementary Fig. 10b), and extended lifespan (Fig. 4h). MRI performed 6 weeks after the start of alpelisib treatment showed a significant decrease in the volume of vascular malformations compared to vehicle-treated mice (Fig. 4b, Supplementary Fig. 10c, d). At the time of sacrifice, alpelisib-treated PIK3CATie2-CreER mice exhibited small vascular dilation (Fig. 4c), and AKT and S6RP phosphorylation were reduced (Fig. 4d, e and Supplementary Fig. 10e). Similar to the preventive study, blood anomalies and vessel malformations were corrected (Fig. 4f and Supplementary Fig. 11a). Furthermore, we investigated the impact of alpelisib on the plasma metabolites identified as dysregulated in PIK3CATie2-CreER mice. These metabolites completely or partially normalized under alpelisib in both the preventive and therapeutic conditions (Supplementary Figs. 12 and 13), indicating that they hold potential as early biomarkers of disease progression or regression in the mouse model. Alpelisib had no particular impact on the control mice. Finally, to explore whether vascular malformations could recur following the interruption of alpelisib, we induced Cre recombination in 4 male mice, treated them with alpelisib at 8 weeks post tamoxifen induction when they were severely affected for a period of 16 weeks, and then stopped the drug. In the following weeks, the mice restarted to gain body weight with the development of diffuse vascular malformations (Supplementary Fig. 11b). Indeed, alpelisib halted disease development but did not correct the genetic anomaly.
Alpelisib improves capillary venous malformations in patients with PIK3CA or TEK variants
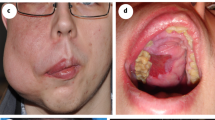
After obtaining encouraging results, we received authorization from the French regulatory agency (ANSM) and The Comité de convenance du CHU de Québec to treat a total of 25 patients, both pediatric and adult, who had venous malformations associated with either PIK3CA mutation (n = 16) or Tie2R (TEK) mutation (n = 9), all demonstrating AKT pathway recruitment within the capillary venous malformations (Fig. 5a–c). However, we observed no difference in KI67 staining between controls and patients (Supplementary Fig. 14). The clinical and demographical characteristics of these patients are summarized in Tables 1 and 2, but briefly there were 10 women, 7 pediatric patients and the mean age was 29.12 years old (SD ± 15.49). All patients were previously heavily treated with multiple surgical, radiological procedures and treatment based on rapamycin for a minimum period of time of 6 months (Tables 1 and 2). Patients were selected because of rapamycin inefficiency (disease progression or uncontrolled symptoms or serious adverse events). All patients underwent a Doppler ultra sound and an MRI before initiating treatment with alpelisib and then a second MRI approximately 6 months after starting the medication. The dosage of alpelisib administered ranged from 50 to 250 mg, adjusted according to age based on the EPIK P1 clinical trial.21 After the introduction of alpelisib, all patients experienced improvements in symptoms such as pain or bleedings. Furthermore, there was a noticeable discoloration and reduction in the volume of vascular malformations observed in the patients (Tables 1 and 2, Fig. 5d, e and Supplementary Fig. 15). Specifically, PIK3CA-related venous malformations showed a mean reduction of 33.4% (SD, ± 22.1) from the baseline at 6 months, while TEK-related venous malformations demonstrated a mean reduction of 27.8% (SD, ±18.9) (Tables 1 and 2, Fig. 5f and Supplementary Fig. 16). Finally, we explored the metabolomic changes in patients following alpelisib introduction. Notably, we identified changes in multiple metabolites that aligned with the ones observed in the mouse model such as aspartate, cystine, glycerol, guanosine, inosine or succinic acid (Supplementary Fig. 17). These metabolic compounds could potentially serve as the foundation for disease improvement biomarkers.

Alpelisib improves patients with PIK3CA or TEK-related capillary venous malformations. a Representative Hematoxylin and eosin (H&E) staining and b Immunofluorescence of P-AKTThr308 and P-S6RP in skin biopsies performed in controls and in patients with PIK3CA-related capillary venous malformation. Scale bar: 10 μm. c Immunofluorescence quantification (n = 6 controls and 6 patients). AU Arbitrary units. d Representative photographs of the morphological changes observed in patients with PIK3CA-related capillary venous malformations receiving alpelisib for 6 months. e Representative photographs of the morphological changes observed in patients with TEK-related capillary venous malformations receiving alpelisib for 6 months. f Transversal (upper panel) and coronal (lower panel) T2-weighted fat saturated MRI sequence of patient 1 and 5 before and after alpelisib introduction. In red, segmentation in 2D (left panel) and 3D (right panel). g Percentage change of the volume of the preselected lesion in patients with PIK3CA-related capillary venous malformations (left panel) and TEK-related capillary venous malformations (right panel)
Discussion
In this study, we successfully developed a mouse model of PIK3CA-related capillary venous malformations that accurately mimics the patient phenotype. We discovered that the malformations are partially driven by the AKT pathway. Alpelisib, which was tested in both mice and patients, showed significant efficacy in improving capillary venous malformations. The PIK3CATie2R-CreER mouse model faithfully replicates the clinical, radiological, histological, and biological anomalies observed in individuals affected by venous malformations. Indeed, these malformations can manifest as isolated or diffused occurrences, appearing in various locations such as the head and neck region, trunk, and extremities as observed in the PIK3CATie2R-CreER mouse model. In clinical terms, venous malformations are characterized by a soft, compressible mass presenting a bluish/purple superficial appearance, occasionally featuring palpable thrombi. They can be well-defined or diffuse and infiltrative akin to the PIK3CATie2R-CreER mouse model. At the radiological level, MRI imaging is the mainstay of imaging of venous malformation and lesions identified in the PIK3CATie2R-CreER mouse model are observed in patients. At the histological level, venous malformations are characterized by enlarged venous channels lined by a single flattened layer of endothelial cells surrounded by sparse, irregularly distributed smooth muscle cells. These lesions often demonstrate local hemophagocytosis and thrombosis, features observed in the PIK3CATie2R-CreER mouse model. Biologically, vascular malformations are characterized by the presence of local or disseminated intravascular coagulation. Venous malformations are commonly linked to thrombosis and pulmonary embolism, as seen in our model, alongside instances of chronic bleeding. These bleedings and pulmonary embolisms stand as causes of death within this population. Notably, our mouse model exhibited signs of pulmonary embolism and traits indicative of chronic bleedings. These characteristics highlight the potential of the model to better understand the mechanism of disease progression and to serve as a new platform for drug testing.
The underlying cause of isolated or complex vascular malformations is primarily attributed to genetic activation of the PI3Kα/AKT pathway in endothelial cells. Tie2 is expressed in endothelial cells and has been observed in arteries.27,28 Since we induced the Cre recombinase in adult mice, these findings are in line with a recent report indicating that mutations in PIK3CA acquired postnatally in mice do not result in vascular anomalies.16 Gain-of-function mutations in TEK and PIK3CA are prevalent in the majority of venous malformations.4 Interestingly, TEK directly signals through PI3Kα, explaining the similarity of malformations and their responsiveness to alpelisib.2
Surprisingly, our findings using either genetic manipulations or pharmacological intervention indicate that AKT does not play a critical role in the development of the disease. Although we did not directly compare PIK3CAAKT1AKT2KO mice and PIK3CATie2-CreER mice treated with miransertib, we observed a severe outcome with uncontrolled disease progression in both sets of mice, accompanied by inhibited AKT phosphorylation in vascular malformations, indicating successful target engagement. This implies that PI3Kα likely signals through pathways different from AKT. However, recent reports of AKT3 somatic mutations associated with a vascular phenotype.29,30 suggest a potential partial compensation for the loss of other isoforms. Additional studies will be necessary to provide further clarification.
Our newly created preclinical model is more severe compared to previous models that utilized human variants, thanks to the modified PIK3CA transgene that we are using. This model serves as a powerful tool for drug screening, enabling us to demonstrate the efficacy of alpelisib and its promising results in heavily pretreated patients. The remarkable aspect of this series is the notable clinical and radiological improvement seen in all these patients who were unresponsive to rapamycin treatment and underwent multiple surgical and radiological procedures. These findings validate the high sensitivity of vessels to PI3Kα inhibition.18,31
This work opens new therapeutic avenues for patients with TEK-related vascular malformations, a severe and debilitating condition that currently lacks approved treatment. Presently, patients are managed with laser, surgical, and radiological procedures, along with symptomatic treatments and anticoagulation. If confirmed in randomized controlled trials, such new targeted therapy could prove beneficial for patients with TEK-related vascular malformations, potentially becoming a cornerstone of the new therapeutic strategy for their care.
As a result of real-world evidence from the EPIK P1 clinical trial (NCT04285723), alpelisib has recently received accelerated approval from the US FDA for patients with PROS (age >2 years).21 Alpelisib is linked to adverse events such as hyperglycemia, alopecia, diarrhea, and vomiting. In the EPIK P1 clinical trial, adverse events were less severe and occurred less frequently, likely attributable to the lower dose utilized compared to oncology clinical trial.32 In the EPIK P1 clinical trial, the most common adverse events were grade 1 alopecia (16.7%), diarrhea (15.8%), hyperglycemia (12.3%), and aphthous ulcers (10.5%).32 It is worth noting that the doses of alpelisib that we have used here are based on the real-world data evidence where no pharmacokinetics analyses were available.17,18,20,21,33 The ongoing EPIK P2 clinical trial (NCT04589650) seeks to better define the appropriated dosages, to confirm the initial findings, and MRI follow-ups of these patients will help assess the impact of alpelisib on different tissues, especially venous malformations. Finally, the detection of altered levels of circulating metabolites upon starting alpelisib could lay the groundwork for future identification of biomarkers.
In conclusion, we have presented a novel mouse model of vascular malformations and provided encouraging preliminary evidence of alpelisib’s efficacy in patients with either PIK3CA or TEK-related capillary venous malformations. Further clinical studies are necessary to validate and corroborate our findings.
Methods
Animals
Here, we crossed homozygous R26StopFLP110*.34 (stock no. 012343, Jackson Laboratory) with Tie2 Cre-ER mice.35 (Infrafrontier). Following procedures previously described,20 we obtained R26StopFLP110*+/− × Tie2 Cre-ER + (henceforth PIK3CATie2-CreER) and R26StopFLP110*+/− × Tie2 Cre-ER- (henceforth PIK3CAWT) mice. We also generated Tie2 Cre-ER+ mice with a homozygous R26StopFLP110* mutation (henceforth PIK3CAHO). To generate tissue-specific p110*-transgenic mice, a cloned loxP-flanked neoR-stop cassette was inserted into a modified version of pROSA26-1 followed by the p110* coding sequence, a frt-flanked IRES-EGFP cassette and a bovine polyadenylation sequence (R26StopFLP110*).36 To follow Cre recombination, PIK3CAWT and PIK3CATie2-CreER mice were interbred with Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J mice.
Animals were housed at a constant ambient temperature in a 12-h light cycle and fed ad libitium with regular chow food (2018 Teklad global 18% protein rodent diets, 3.1 Kcal/g, Envigo). Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation approved the animal procedures (APAFIS N°20439-2018121913526398 and 2021110914486827). All appropriate procedures were followed to ensure animal welfare. For induction, all of the mice studied received at the age of 4 weeks a daily dose of 1 mg tamoxifen through oral gavage for 5 consecutive days. Before the sacrifice procedure, mice were harvested over a 12-h period.
PIK3CATie2-CreER mice were interbred with AKT1flox/flox obtained from The Jackson Laboratory (Stock #026474) to generate mice with PIK3CA gain-of-function mutation in venous endothelial cells but lacking AKT1 (PIK3CATie2-CreER-AKT1KO mice; henceforth PIK3CAAKT1KO). PIK3CATie2-CreER mice were also interbred with AKT2-/- mice obtained from The Jackson Laboratory (Stock# 006966) to generate mice with PIK3CA gain-of-function mutation in venous endothelial cells but lacking AKT2 (PIK3CATie2-CreER-AKT2KO mice; henceforth PIK3CAAKT2KO). Lastly, PIK3CAAKT1KO and PIK3CAAKT2KO were interbred to generate mice with PIK3CA gain-of-function mutation in venous cells lacking both AKT genes (PIK3CATie2-CreER-AKT1AKT2-KO mice; henceforth PIK3CAAKT1AKT2-KO).
PIK3CAWT and PIK3CATie2-CreER mice were treated with the p110α inhibitor alpelisib [MedChem Express, Germany; 50 mg.kg−1 in 0.5% carboxymethylcellulose (Sigma Aldrich), daily p.o.] or a vehicle [0.5% carboxymethylcellulose (Sigma Aldrich), daily p.o.]. Treatment was started either 1 week (preventive study) or 4 weeks (therapeutic study) following Cre induction. PIK3CAWT and PIK3CATie2-CreER mice were also treated with either the Akt inhibitor miransertib and mTOR inhibitor rapamycin or a vehicle at a concentration of respectively 30 mg/kg and 4 mg/kg (MedChem Express, Germany) both diluted in a solution of 5% DMSO (Sigma Aldrich), 40% PEG-300 (Sigma Aldrich), 5% Tween (Euromedex) and 45% saline (Cooper). Both treatments were administered every day through oral gavage. The last dose of drug or vehicle was administered approximately 3 h before sacrifice. All mice were fasted overnight before blood glucose measurement (Accuchek Performa, Roche Diagnostic), MRI and sacrifice. Every solution was initially prepared over a span of 5–7 days and then systematically remade every 7 days thereafter.
Magnetic resonance imaging (MRI) evaluation
All images were acquired with a 4.7-T small-animal MRI system (BioSpec USR47/40; Bruker BioSpin, Ettlingen, Germany) on the “Plateforme Imageries du Vivant, Université de Paris, PARCC, INSERM, Paris, France.” Mice underwent whole body magnetic resonance imaging (MRI) using 3D T2 weighted sequences with and without fat saturation. Whole body vascular malformations volumetric evaluation on MRI was performed with 3D Slicer software using manual segmentation tools.37
Blood and plasma analyses
Mouse blood samples were collected in EDTA-coated tubes. To measure complete blood count, fresh blood samples were analyzed on a hematology analyzer (ProCyte Dx; IDEXX Laboratories). The remaining blood samples were centrifuged at 500 × g for 10 min to collect plasma.
ELISA & multi-spot assay system (Meso Scale Diagnostics)
Mouse-specific D-dimers were measured in plasma mice samples using enzyme immunoassay kit from Cusabio Biotech (ref# CSB-E13584) following the manufacturer’s instructions and analyzed with a Tecan Infinite® 200 PRO.
Levels of total and phosphorylated (Ser473) AKT protein in mice skin lysates was assessed using Meso Scale Diagnostics (MSD) multi-spot assay system (Phospho(Ser473)/Total Assay Whole Cell Lysate Kit ref K15100D) and read with Meso® QuickPlex SQ 120MM, Model 1300. Data were analyzed with MSD Discovery Workbench and calculated according to manufacturers’ instructions (%Phosphoprotein = ((2*Phospho-signal)/(Phospho-signal + Total signal)) * 100).
Morphological analyses
Mouse tissues (all from abdominal skin) were fixed in 4% paraformaldehyde and paraffin-embedded. Tissue sections (4 µm thick) were stained with hematoxylin and eosin (H&E).
Proliferation index evaluation
Proliferative cells were detected in abdominal skin tissue sections using KI67 immunostaining. Four-µm sections were incubated with a KI67 (Supplementary Table S1) primary antibody, and the appropriate Alexa-Fluor conjugated secondary antibody (Thermo Fisher Scientific). Image acquisition was performed with the Eclipse Ni-E(Nikon) at 400× magnification (numerical aperture of the objective: 0.75). Between 8 to 12 randomly selected fields (with the number depending on the size of the tissue) were taken for each section. Proliferation index was evaluated using QuPath(v0.4.2).38 as well as Cellpose extension.39 and a machine learning object classification method. Briefly, we created a sparse image containing all randomly-acquired images in QuPath software and then used the Cellpose pre-trained model cyto2 to identify all nuclei. Object classification using machine learning was performed and 3 classes were created: DAPI positive, KI67 positive and Other corresponding to potential artefacts.
Machine learning was chosen over the threshold method to overcome tissue staining heterogeneity that would not be as accurate. Consequently, this object classification was performed for each mouse. The ground truth of KI67 classification was determined and validated by the experts according to morphological criteria and the intensity. For each sparse image, we finally have a number of total DAPI-positive only nuclei, and KI67-positive nuclei. To calculate the ratio, we added the number of DAPI-positive and KI67-positive cells to obtain the total number of nuclei, subdivided by KI67-positive only cells. Nuclei classified as Other were excluded and subtracted from calculation.
Immunohistochemistry and immunofluorescence
Paraffin-embedded mouse tissue sections were submitted to antigen retrieval protocols in either citrate or tris-EDTA buffer at high temperature (120 °C) and pressure with a pressure cooker. Four-µm sections were incubated with primary antibodies (Supplementary Table S1) overnight. For immunofluorescence, samples were then incubated with appropriate Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific) and analyzed using the Eclipse Ni-E (Nikon). Serial sectioning has been performed when antibodies were produced from the same species and GFP staining was used to localize the same area to acquire. Thirty-µm sections were submitted to the same protocol and were acquired using a Zeiss LSM 700 confocal microscope and Imaris Viewer 10.1.0 was used to create 3D images and ImageJ for the 2D images.
For immunohistochemistry, samples were then exposed to relevant horseradish peroxidase (HRP)-linked secondary antibodies and analyzed with the E800 microscope (Nikon).
Western blot
Tissues were crushed and then lysed in RIPA lysis buffer supplemented with phosphatase and protease inhibitors. Protein concentrations were determined through the bicinchoninic acid method (Pierce). Protein extracts were analyzed by Western blot where the transfer membrane was incubated with a primary antibody (Supplementary Table S1) followed by the appropriate peroxidase-conjugated secondary antibody (dilution 1:10,000). Chemiluminescence were acquired using a Chemidoc MP and bands were quantitated with the Image Lab Software (Bio-Rad Laboratories).
Tissue digestion
Skin of PIK3CAWT and subcutaneous venous malformations of PIK3CATie2-CreER mice were harvested and rinsed with PBS 1X (Gibco). For immunofluorescence and Western blot experiments, approximately 4 cm2 of abdominal skin was collected from the upper to the lower limbs at the time of sacrifice. For flow cytometry studies, almost all the skin from the front and back, spanning from the neck to the lower limbs, was collected to maximize the available material. After cutting them into small pieces, tissues were incubated in a digestion buffer added with DNAse (0.1 mg/mL), Dispase I (0.8 mg/mL) and Collagenase P (0.2 mg/mL) in 10 mL of RPMI (Gibco) and incubated for 40 min at 37 °C on a GentleMACS system (Miltenyi). Following dissociation, tissues were filtered (70 µm, Clearline), centrifuged 5 min at 250 × g, and resuspended in PBS.
Flow cytometry
After digestion, cells were incubated with conjugated primary antibodies (Supplementary Table S1). Appropriated isotype controls were used.
To isolate bone marrow, we first dissected tibia and femur and then removed skeletal muscles, ligaments, and tendons. Cells were flushed from the marrow using 1-ml syringe containing phosphate-buffered saline (PBS) 1X. Cell suspension was centrifuged at 500 × g within 5 min and filtered (70 µm, Clearline). Cells were incubated with conjugated primary antibodies (Supplementary Table S1). Cells were then incubated with conjugated primary antibody (Supplementary Table S1). Appropriated isotype controls were used.
All samples were analyzed using the Sony ID7000 and Sony ID7000 software. Final analysis and quantification were conducted with the Kaluza software.
Phosphoflow cytometry
Following 10 days of treatment with either the vehicle or alpelisib, subcutaneous skin of PIK3CAWT mice and subcutaneous vascular malformations of PIK3CATie2-CreER mice were harvested and digested. Cells were resuspended in 100 µL PBS in a 96-well round-bottomed plate (ThermoFisher). Cells were incubated with conjugated primary antibodies (Supplementary Table S1) for cell surface markers, rinsed with BD Pharmingen™ Stain Buffer FBS and permeabilized with BD™ Phosphoflow Perm buffer III for 30 min at 4 °C. Staining for intracytoplasmic markers (Supplementary Table S1) was performed for 30 min at 4 °C followed by cell fixation with BD Cytofix™. Appropriated isotype controls were used. Cells were analyzed using the Sony Spectral ID7000 apparatus and all flow data were processed and analyzed with the Sony ID7000 software.
Imaging flow cytometry (Amnis ImageStream)
Skin from PIK3CAWT and PIK3CATie2-CreER mice were rinsed in phosphate buffered saline 1X, cut in small pieces and digested as detailed below. After dissociation, cell suspensions were filtered (70 µm, Clearline), centrifuged 10 min at 350 × g, and resuspended in phosphate buffered saline solution 1X supplemented with 2% fetal bovine serum and EDTA at 0.5 m/mol. After these procedures, cells were transferred in microtubes and stained for Tie2 conjugated antibody (Supplementary Table S1). All mice expressed the GFP and Tie2-Cre recombinase, with or without the transgene. Samples were run on the ImageStream ISX mkII (Amnis part of Luminex) apparatus that combines flow cytometry with detailed cell imaging. Magnification (40X) was used for all acquisitions. Data were extracted with the INSPIRE software (Amnis part of Luminex) and analyzed with the IDEAS software (v.6.2, Amnis part of Luminex).
Targeted LC–MS metabolites analyses
Blood samples were obtained in EDTA tubes for plasma analysis and EDTA-free tubes for serum analysis. Plasma and serum were obtained after blood centrifugation at 500 × g for 10 min and immediately snap-frozen in liquid nitrogen. For the LC–MS analyses, metabolites were extracted as previously described.20 Briefly, the extraction solution, which was composed of 50% methanol, 30% ACN, and 20% water, was added to the plasma or serum volume at a ratio of 200 µl per 10 µl. Afterwards, samples were vortexed for 5 min at 4 °C and centrifuged at 16,000 × g for 15 min at 4 °C. The supernatants were collected and stored at −80 °C until the analyses were performed.
LC–MS analyses were conducted using a QExactive Plus Orbitrap mass spectrometer equipped with an Ion Max source and a HESI II probe coupled to a Dionex UltiMate 3000 UPLC system (Thermo). External mass calibration was performed using the standard calibration mixture every 7 d as recommended by the manufacturer. Five microliters of each sample were injected onto guard-column protected (Millipore) Zic‐pHilic columns for liquid chromatography separation. Buffer A was 20 mM ammonium carbonate and 0.1% ammonium hydroxide (pH 9.2) and buffer B acetonitrile. The chromatographic gradient was run at a flow rate of 0.200 μl/min as follows: linear gradient from 80 to 20% B for 0–20 min, linear gradient from 20 to 80% B for 20–20.5 min and held at 80% B for 20.5–28 min. The mass spectrometer was operated in full-scan polarity switching mode with the spray voltage set to 2.5 kV and the heated capillary held at 320 °C. The sheath gas flow was set to 20 units, the auxiliary gas flow to 5 units, and the sweep gas flow to 0 units. The metabolites were detected across a mass range of 75–1000 m/z at a resolution of 35,000 (at 200 m/z) with the AGC target set to 106 and maximum injection time to 250 ms. Lock masses were used to ensure mass accuracy below 5 ppm.
Data were acquired with the Thermo Xcalibur 4.0.27.13 software (Thermo). The peak areas of metabolites were determined using the Thermo TraceFinder 3.3 SP1 software (Thermo) and identified by the exact mass of each singly charged ion and by known retention time in the HPLC column.
Patients
This study was conducted on 25 patients, including 7 children who were followed at Hôpital Necker Enfants Malades (Paris, France).17 and 2 adults who were followed at the L’Hôtel-Dieu de Québec du CHU de Québec – Université Laval (Québec, Canada). The patients followed at Necker were under a protocol that was approved by the Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM). In this study, written informed consent was obtained from all adult patients and from the parents of all pediatric patients. Alpelisib, which was compassionately offered by Novartis, was given every morning during breakfast at an oral dose of 250 mg/day to adults and 50 mg/day to pediatric patients.17
Adverse events were graded according to Common Terminology Criteria for Adverse Events [CTCAE] system (version 4.03) and coded by preferred term using the Medical Dictionary for Regulatory Activities [MedDRA] system (version 24.0). Patients underwent magnetic resonance imaging (MRI) prior to alpelisib introduction and this exam was repeated after 6 months of more of treatment. Patients underwent localized MRI using T2 weighted sequences with fat suppression including fat saturation (FAT-SAT), short TI inversion recovery (STIR) and DIXON sequences. Baselines acquisitions were registered with the follow-up MRI and then cropped to obtain a similar field of view on which segmentation of venous malformation was performed with 3D Slicer as previously described for the MRI in mice. To confirm the venous malformations, all patients underwent Doppler ultrasound prior to drug initiation.
In human, skin biopsies were obtained from patients after informed consent. Control skin was obtained from surgical tissues removed for other reasons. Histology was systematically verified by a pathologist. Paraffin-embedded skin tissue sections were submitted to antigen retrieval protocols in either citrate or tris-EDTA buffer at high temperature (120 °C) and pressure with a pressure cooker. Four-µm sections were incubated with primary antibodies (Supplementary Table S1) overnight. Samples were then incubated with appropriate Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific) and analyzed using the Eclipse Ni-E (Nikon). P-S6RP and P-AKTThr308 staining were quantified using the Ilastik v1.3.3post3 machine learning pixel classification opensource software. Afterwards, mean signal intensity and areas were measured with the Fiji v2.3.0/1.53f51 open-source image processing software (Quantity = Mean intensity × Area) and normalized by tissue area.40
KI67 staining was performed on human paraffin-embedded skin biopsie sections by immunohistochemistry. Proliferation index was evaluated using QuPath(v0.4.2).38 and Stardist extension.41 Then, object classification using machine learning was performed and 3 classes were created: Hematoxylin-positive, DAB-positive corresponding to Ki67-positive nuclei and Artefacts.42,43 To calculate the ratio, we added the number of hematoxylin-positive and DAB-positive cells to obtain the total number of nuclei, subdivided by DAB-positive only cells. Nuclei classified as Artefacts were excluded and subtracted from calculation.
Data analysis and statistics
Data were expressed as means ± SEM. Survival curves were analyzed with the Mantel–Cox (log-rank) test. Differences between experimental groups were evaluated using analysis of variance (ANOVA), followed by the Tukey–Kramer post hoc test for statistically significant differences (P < 0.05). When only two groups were compared, Mann–Whitney tests were used. The statistical analysis was performed using GraphPad Prism software (version 10.0.0).
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper, in the Supplementary Materials or online for the LCM MS raw data.
References
Wassef, M. et al. Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics 136, e203–e214 (2015).
Limaye, N. et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat. Genet. 41, 118–124 (2009).
Kurek, K. C. et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am. J. Hum. Genet. 90, 1108–1115 (2012).
Dompmartin, A., Vikkula, M. & Boon, L. M. Venous malformation: update on aetiopathogenesis, diagnosis and management. Phlebology 25, 224–235 (2010).
Eifert, S., Villavicencio, J. L., Kao, T. C., Taute, B. M. & Rich, N. M. Prevalence of deep venous anomalies in congenital vascular malformations of venous predominance. J. Vasc. Surg. 31, 462–471 (2000).
Soblet, J., Limaye, N., Uebelhoer, M., Boon, L. M. & Vikkula, M. Variable Somatic TIE2 Mutations in Half of Sporadic Venous Malformations. Mol. Syndromol. 4, 179–183 (2013).
Castel, P. et al. Somatic PIK3CA mutations as a driver of sporadic venous malformations. Sci. Transl. Med. 8, 332ra342 (2016).
Limaye, N. et al. Somatic Activating PIK3CA Mutations Cause Venous Malformation. Am. J. Hum. Genet 97, 914–921 (2015).
Bilanges, B., Posor, Y. & Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. https://doi.org/10.1038/s41580-019-0129-z (2019).
Davis, S. et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 87, 1161–1169 (1996).
Canaud, G., Hammill, A. M., Adams, D., Vikkula, M. & Keppler-Noreuil, K. M. A review of mechanisms of disease across PIK3CA-related disorders with vascular manifestations. Orphanet J. Rare Dis. 16, 306 (2021).
Morin, G. M. et al. PIK3CA-Related Disorders: From Disease Mechanism to Evidence-Based Treatments. Annu. Rev. Genomics Hum. Genet. https://doi.org/10.1146/annurev-genom-121222-114518 (2024).
Castillo, S. D. et al. Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans. Sci. Transl. Med. 8, 332ra343 (2016).
Adams, D. M. et al. Efficacy and Safety of Sirolimus in the Treatment of Complicated Vascular Anomalies. Pediatrics 137, e20153257 (2016).
Goines, J. et al. A xenograft model for venous malformation. Angiogenesis 21, 725–735 (2018).
Kobialka, P. et al. The onset of PI3K-related vascular malformations occurs during angiogenesis and is prevented by the AKT inhibitor miransertib. EMBO Mol. Med. 14, e15619 (2022).
Venot, Q. et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 558, 540–546 (2018).
Delestre, F. et al. Alpelisib administration reduced lymphatic malformations in a mouse model and in patients. Sci. Transl. Med. 13, eabg0809 (2021).
Morin, G. et al. Treatment of two infants with PIK3CA-related overgrowth spectrum by alpelisib. J. Exp. Med. 219 https://doi.org/10.1084/jem.20212148 (2022).
Ladraa, S. et al. PIK3CA gain-of-function mutation in adipose tissue induces metabolic reprogramming with Warburg-like effect and severe endocrine disruption. Sci. Adv. 8, eade7823 (2022).
Canaud, G. et al. Alpelisib for Treatment of Patients With PIK3CA-Related Overgrowth Spectrum (PROS). Genet. Med. 25, 100969 (2023).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Behravesh, S. et al. Venous malformations: clinical diagnosis and treatment. Cardiovasc. Diagn. Ther. 6, 557–569 (2016).
Gupta, A. & Kozakewich, H. Histopathology of vascular anomalies. Clin. Plast. Surg. 38, 31–44 (2011).
Mussa, A. et al. Genotypes and phenotypes heterogeneity in PIK3CA-related overgrowth spectrum and overlapping conditions: 150 novel patients and systematic review of 1007 patients with PIK3CA pathogenetic variants. J. Med. Genet. 60, 163–173 (2023).
Canaud, G. et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N. Engl. J. Med. 371, 303–312 (2014).
Wong, A. L. et al. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ. Res. 81, 567–574 (1997).
Petkova, M. et al. Immune-interacting lymphatic endothelial subtype at capillary terminals drives lymphatic malformation. J. Exp. Med. 220 https://doi.org/10.1084/jem.20220741 (2023).
Bolli, A. et al. Mosaic pathogenic variants in AKT3 cause capillary malformation and undergrowth. Am. J. Med. Genet. A 191, 1442–1446 (2023).
Davies, O. M. T. et al. Cutaneous vascular anomalies associated with a mosaic variant of AKT3: Genetic analysis continues to refine the diagnosis, nomenclature, and classification of vascular anomalies. J. Am. Acad. Dermatol. 87, 162–164 (2022).
Garneau, A. P., Haydock, L., Tremblay, L. E. & Isenring, P. Somatic non-cancerous PIK3CA-related overgrowth syndrome treated with alpelisib in North America. J. Mol. Med. 99, 311–313 (2021).
Andre, F. et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 380, 1929–1940 (2019).
Bayard, C. et al. Hemifacial myohyperplasia is due to somatic muscular PIK3CA gain-of-function mutations and responds to pharmacological inhibition. J. Exp. Med. 220 https://doi.org/10.1084/jem.20230926 (2023).
Klippel, A. et al. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol. Cell. Biol. 16, 4117–4127 (1996).
Kisanuki, Y. Y. et al. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230–242 (2001).
Srinivasan, L. et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell 139, 573–586 (2009).
Fedorov, A. et al. 3D Slicer as an image computing platform for the Quantitative Imaging Network. Magn. Reson. Imaging 30, 1323–1341 (2012).
Bankhead, P. et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Stringer, C., Wang, T., Michaelos, M. & Pachitariu, M. Cellpose: a generalist algorithm for cellular segmentation. Nat. methods 18, 100–106 (2021).
Berg, S. et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232 (2019).
Schmidt, U., Weigert, M., Broaddus, C. & Myers, G. Cell Detection with Star-Convex Polygons. Lect. Notes Comput Sc. 11071, 265–273 (2018).
Greenwald, N. F. et al. Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat. Biotechnol. 40, 555–565 (2022).
Pecot, T., Cuitino, M. C., Johnson, R. H., Timmers, C. & Leone, G. Deep learning tools and modeling to estimate the temporal expression of cell cycle proteins from 2D still images. PLoS Comput. Biol. 18, e1009949 (2022).
Acknowledgements
This study was supported by the European Research Council (CoG 2020 grant number 101000948 awarded to GC), the Agence Nationale de la Recherche – Programme d’Investissements d’Avenir (ANR-18-RHUS-005 to GC) and the Agence Nationale de la Recherche – Programme de Recherche Collaborative (19-CE14-0030-01 to GC). This work was also supported by the CLOVES SYNDROME COMMUNITY (West Kennebunk, USA), Association Syndrome de CLOVES (Nantes, France), Fondation d’entreprise IRCEM (Roubaix, France), Fonds de dotation Emmanuel BOUSSARD (Paris, France), the Fondation DAY SOLVAY (Paris, France), the Fondation TOURRE (Paris, France) to GC, the Fondation BETTENCOURT SCHUELLER (Paris, France) to GC, the Fondation Simone et Cino DEL DUCA (Paris, France), the Fondation Line RENAUD-Loulou GASTE (Paris, France, the Fondation Schlumberger pour l’Education et la Recherche (Paris, France), the Association Robert Debré pour la Recherche Médicale awarded to GC, WonderFIL smiles - a Facial Infiltrating Lipomatosis community (Norway), INSERM, Assistance Publique Hôpitaux de Paris and Université Paris Cité. We are also very grateful to our generous donors. In vivo preclinical imaging was performed at the Life Imaging Facility of the University of Paris (Plateforme Imageries du Vivant), supported by France Life Imaging (grant ANR-11-INBS-0006) and Infrastructures BiologieSanté (IBISA). This study was supported by a grant from MSD Avenir (Signalopathies) awarder to GC.
Author information
Authors and Affiliations
Contributions
Lola Zerbib performed the experiments, analyzed the data and elaborated figures. Sophia Ladraa, Charles Bayard, Marina Firpion, Amandine Thomas, Michael Dussiot and Quitterie Venot participated in some of the experiments. Antoine Fraissenon, Laurent Guibaud, Gwennhael Autret and Bertrand Tavitian designed and analyzed MRI experiments. Sanela Protic and Clément Hoguin were in charge of mice experiments including genotyping, breeding, tamoxifen administration and sacrifice. Sylvie Fraitag and Jean-Paul Duong analyzed mice and human tissues section. Vahid Asnafi, Sophie Kaltenbach, Estelle Balducci, Coline Lefevre and Patrick Villarese performed molecular diagnosis in patients. Nicolas Goudin provided help to quantify immunofluorescence expression in tissues. Ivan Nemazanyy performed the metabolomic analysis. Christine Broissand, Christophe Legendre, Nadia Arzouk, Véronique Minard, Caroline Chopinet, Tristan Mirault and Paul Isenring followed patients. Denis Adams gave input in the manuscript and figures. Guillaume Canaud followed the patients, provided the conceptual framework, designed the study, supervised the project, and wrote the paper. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
A patent application (“BYL719 (alpelisib) for use in the treatment of PIK3CA-related overgrowth spectrum” #WO2017140828A1) has been filed by INSERM (Institut National de la Santé et de la Recherche Médicale), Centre National De La Recherche Scientifique (CNRS), Université Paris Cité, and Assistance Publique-Hôpitaux De Paris (AP-HP) for the use of BYL719 (alpelisib) in the treatment of PIK3CA-related overgrowth spectrum (PROS/CLOVES syndrome). Dr. Canaud is the inventor. This patent is licensed to Novartis. Dr. Canaud receives or has received consulting fees from Novartis, Vaderis, Alkermes, IPSEN and BridgeBio. The other authors declare no other competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zerbib, L., Ladraa, S., Fraissenon, A. et al. Targeted therapy for capillary-venous malformations. Sig Transduct Target Ther 9, 146 (2024). https://doi.org/10.1038/s41392-024-01862-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01862-9
- Springer Nature Limited