
Abstract
Since the clinical approval of imatinib, the discovery of protein kinase downregulators entered a prosperous age. However, challenges still exist in the discovery of kinase downregulator drugs, such as the high failure rate during development, side effects, and drug-resistance problems. With the progress made through multidisciplinary efforts, an increasing number of new approaches have been applied to solve the above problems during the discovery process of kinase downregulators. In terms of in vitro and in vivo drug evaluation, progress was also made in cellular and animal model platforms for better and more clinically relevant drug assessment. Here, we review the advances in drug design strategies, drug property evaluation technologies, and efficacy evaluation models and technologies. Finally, we discuss the challenges and perspectives in the development of kinase downregulator drugs.
Similar content being viewed by others

Introduction
Kinases are crucial mediators of signal transduction processes, and by catalyzing the transfer of phosphates from ATP to other specific molecules, they are key regulators of a variety of cell functions. Protein kinases and lipid kinases constitute the human kinome. A total of 518 protein kinases containing 478 typical and 40 atypical kinases (Fig. 1) have been identified in the human genome.1 In general, kinases contain a eukaryotic protein kinase domain, and atypical kinases lack this protein kinase domain, but still exhibit kinase activity. Based on the sequence similarity of kinase domains, typical kinases are further classified into eight major groups: TK (tyrosine kinase, 90 members), TKL (tyrosine kinase-like kinase, 43 members), STE (STE7-, STE11-, and STE20-related kinase, 47 members), CK1 (casein kinase 1, 12 members), AGC (protein kinase A/G/C-related kinase, 63 members), CAMK (Ca2+/calmodulin-dependent kinase, 74 members), CMGC (CDK/MAPK/GSK/CDK-like-related, 61 members), and RGC (receptor guanylyl cyclase, 5 members). In addition, 40 atypical protein kinases have been identified, including pyruvate dehydrogenase kinases, bromodomain kinases, B cell receptors (BCRs), and PI3 kinase-related kinases.

The KinMap of 518 human kinases. (The picture was obtained from http://www.kinhub.org/kinmap/index.html)
The TK group consists of two families: receptor tyrosine kinases (RTKs) and non-RTKs.2 RTKs are transmembrane proteins that contain an extracellular segment and intracellular kinase domains. RTKs are activated upon dimerization, which is triggered by the binding of ligands to the extracellular segment; examples of RTKs include ErbB/HER family proteins, FLT3, PDGFRs, and FGFRs. Non-RTKs include SRC, ABL, and Janus kinase (JAK) kinases. Most members of the TKL family are serine/threonine kinases, with sequences similar to those of the TK group, and include IRAK, RAF, LIMK, and TGFβ.3 Three families of STE kinases have been identified on the basis of their homology with yeast proteins STE20 (MAP4K), STE11 (MAP3K), and STE7 (MAP2K, MEK), which regulate various signaling pathways.4 CK1 is also a Ser/Thr kinase associated with cytoskeletal function and transcription.5 In the human genome, AGC protein kinases are named after the protein kinase A, G, and C families (the PKA, PKC, and PKG families, respectively), which contain more than 60 protein kinases and are classified into 14 families; they regulate the signaling pathway of infectious diseases.6 Most members of the CAMK group are involved in calcium/calmodulin modulation, and they can be classified into four subfamilies: CaMK I, CaMK II, CaMK III, and CaMK IV.7 However, CHK1 and CHK2 are special classes of CAMK kinases, and both are cell cycle checkpoint kinases. CDKs, MAPK, GSK, and CDK-like kinases constitute the CMGC group, a diverse group of kinases. CDKs are involved in cell cycle progression and transcription. MAPKs play key roles in the proliferation, differentiation, death of cells, which contain three major groups: ERK, JNK, and p38. The functions of GSKs are rather different, which are involved in glycogen metabolism and inflammation.
Kinase can transfer the phosphorylation of specific amino acids using ATP as the source of phosphate, the kinase domain contains special residues that promote the transfer of γ-phosphate from ATP to a protein or lipoprotein and the release of the products; this action requires a conformational change between the active (catalytically competent) and inactive states of the phosphate recipient. Approximately 250 amino acids in the N- and C-lobe constitute the kinase domain. One C-helix and several β-strands constitute the N-lobe; however, C-lobes mainly contain α-helixes that contain critical residues needed for kinase interaction with phosphate. A cleft, called a hinge, separates the N-lobe from the C-lobe, enabling ATP binding and phosphate transfer through residues that form hydrogen bonds with the adenine ring of ATP. The dysregulation of signaling pathways involving these kinases has been found in multiple diseases, e.g., cancer, inflammatory, degenerative, and infectious diseases.8 Tremendous efforts have been devoted to kinase-target drug discovery in the past three decades. As a result, many kinase inhibitors have been approved and are being used in clinical treatment, and others have been added to the rich pipeline of new molecules currently under clinical evaluation or preclinical development. Until August 2021, 3 natural macrocycles targeting mTOR and 68 other kinase inhibitors have been approved by the United States and Korea Food and Drug Administration (FDA),9 and 11 kinase inhibitor drugs have been approved by the National Medical Products Administration of China for pharmacological research, oncological applications, and inflammatory diseases (Fig. 2 and Table 1).

Chemical structures of approved kinase inhibitors
Generally, kinase inhibitors can be categorized into two classes according to whether ATP competition or non-ATP competition or not.3,10 The potencies of kinases that compete for ATP decrease with increasing ATP concentration, and these kinases can be classified as type I and type II inhibitors.3,11 Type I inhibitors bind to the active (DFG-in) conformation, whereas type II inhibitors bind to the inactive (DFG-out) conformation. The potency of non-ATP-competitive kinases is unchanged at different ATP concentrations, and they are also called allosteric inhibitors. Type I inhibitors bind to the ATP-binding pocket through the formation of hydrogen bonds to the kinase “hinge” residues, which are just like the interaction of adenine with the kinase. In addition, hydrophobic interactions in and around the region occupied by the adenine ring of ATP are also important. However, type II inhibitors not only occupy the ATP-binding pocket but also exploit unique hydrogen bonds with the residues of α-helix and DFG motif. In addition, the special hydrophobic interactions formed by the DFG motif of the activation loop being folded away from the conformation required for ATP phosphate transfer can improve the selectivity among the kinases. In most cases, the number of hydrogen bonds between kinases and inhibitors in type II inhibitors is more than that of type I inhibitors. Typically, type I inhibitors have a heterocyclic motif that forms 1–3 hydrogen bonds at the hinge area. Similar to type I inhibitors, the hinge binding part needs heterocycle to form the typical hydrogen bonds with target kinase. Type II inhibitors can be divided into five parts from the chemical structure, e.g., head, hinge binding, linker, hydrogen bond, and tail. The moiety of the head can be hydrophobic or hydrophilic. The linker part needs a hydrogen donor or acceptor, e.g., amido linkage, urea bridge bonds, and so on, which can form special hydrogen bonds with the residues of α-helix and DFG motif. The final “tail” segment contains a hydrophobic moiety that occupies the pocket created by the DFG-out flip. Allosteric inhibitors are described in more detail in the following section. By analyzing the binding modes of small molecules to kinases, we found that most inhibitors adopt type I binding modes, only four MEK inhibitors adopt type III binding modes (Table 1). From the co-crystallization data published on PDB, 29 co-crystal complexes of small molecules and kinases employ type I binding modes, 11 adopt type II binding modes. In addition, nine small molecules are reported as irreversible inhibitors by forming covalent bonds with kinases (Table 1), which will be discussed in more detail in the following sections.
In spite of the booming development of kinase inhibitors, issues with available drugs still exist, such as the high failure rate during development, side effects, and drug-resistance problems. To cope with these, multidisciplinary technologies and new approaches have been applied to solve the above problems during the discovery process of kinase inhibitors. In this review, we summarize the advances from three directions, including advances in drug design strategies, drug property evaluation technologies, and efficacy evaluation models and technologies. Finally, we discuss the challenges and perspectives of the future development of kinase inhibitors.
Advances of kinase inhibitors design strategies
Acquired drug-resistance problems driven by evolution pressure during small-molecule kinase inhibitor treatment become a major unmet clinical need in cancer therapy. Almost all the protein kinases drugs will face drug resistance after a period of time of clinical use. To address the problem of drug resistance, many strategies have been developed, e.g., forming a covalent bond to improve the affinity, degradation mutant kinase by new approaches, and overriding ATP-binding site mutations with allosteric inhibitors.
Covalent binding approach
Traditionally, compared with the traditional reversible binding inhibitors, the induction of covalently binding drugs can reduce inevitable side effects due to possible off-target and improve the specific activity, and covalently binding drugs have been widely used; compound forms a covalent bond with a specific nucleophilic residue on the target protein kinase via reaction with an electrophile. Initially, the interaction of small molecules and kinase is in a dynamic equilibrium state. The equilibrium will be interrupted and tend to complex formation with inhibitor after the conjugation through an irreversible covalent bond (Fig. 3). In addition, the higher (or infinite) binding affinity obtained by covalently binding drugs is known to lead to longer residence time and induce protein degradation, which can completely block signaling pathways. Particularly after the early success of the epidermal growth factor receptor (EGFR) inhibitor afatinib, increasing efforts have been directed to the covalent binding approach in the past decade. The most critical point for designing covalent kinase inhibitors is to find suitable targetable amino acids in the binding pocket. In most cases, covalent binding kinase inhibitors are designed to form chemical bonds with the appropriate cysteine thiols via Michael addition reactions.12,13 To date, eight covalent inhibitors have been approved by the FDA (Fig. 2), all of which are designed to target cysteine thiols. Recently, considerable efforts have been made to explore alternative electrophiles that exhibit irreversible or reversibly covalent binding mechanisms toward other amino acids, such as lysine,14 aspartic acid,15 and others.16

Mechanisms of irreversible covalent inhibitors and the selected examples of electrophilic traps. a Small molecules form a covalent bond with the specific nucleophilic residue of the target protein kinase via reaction with an electrophile; then, the equilibrium will be interrupted and tend to complex formation with the inhibitor after the conjugation through an irreversible covalent bond. b Selected widely used examples of electrophilic traps are shown
Nearly half of eukaryotic kinases contain at least one cysteine residue neighboring the ATP pocket, e.g., EGFR and Bruton tyrosine kinase (BTK). To overcome the first-generation drug resistance caused by gatekeeper T790M mutation, the second-generation EGFR inhibitors (e.g., afatinib,17 dacomitinib,18 neratinib19) and the third-generation EGFR inhibitors (e.g., osimertinib20) were designed, which can irreversibly bind to EGFR C797 residue and each contains an α, β-unsaturated acrylamide electrophilic warhead. However, new point mutation C797S appeared, which disrupted the formation of covalent bonds; thus, new scaffold drugs are needed. In 2013, the first approved BTK inhibitor ibrutinib has a classical covalent binding mode, and binds to BTK in the ATP pocket and forms one covalent bond with Cys481 and four hydrogen bonds with Thr474, Glu475, Tyr476, Met477, and Cys481, respectively (Fig. 4). In 2017, the second BTK inhibitor acalabrutinib was approved. Both BTK inhibitors form a covalent bond with Cys481 residue in the BTK activation site through an acrylamide electrophile warhead.21,22 Acalabrutinib possesses a unique and relatively inert 2-butynamide electrophile, which is thought to be activated by its relative positioning to the electron-withdrawing imidazopyrazine core. However, it should be noted that 11 kinases including EGFR, ITK, BTK, JAK3, and others share an equivalently positioned cysteine residue that can be targeted through the covalent binding approach, which makes it hard to get high selectivity between them.

The binding mode of ibrutinib with BTK (PDB: 5P9J). The N atom and NH2 of the purine ring form three hydrogen bonds with Glu475, Tyr476, and Thr474, respectively, to fix ibrutinib. The carbonyl forms a hydrogen bond with Cys481, at the same time the allyl and Cys481 forms a covalent bond via Michael addition
Some efforts have been made to target active site lysine residues in certain kinases in which cysteine residues are unsuitable for covalent binding. Lysine is an attractive nucleophile because of its prevalence in many active sites and at interfaces mediating protein–protein interactions.23,24,25 Several studies have reported the covalent modification of lysine residues using various warheads. A sulfonyl fluoride-based probe was recently used to covalently combine with Lys514 of FGFR.25 Ester electrophiles were recently reported to form a covalent bond with Lys779 in the P-loop of PI3Kδ.26 Other amino acids, such as tyrosine and aspartic acid, can also serve as potential nucleophilic agents depending on the microenvironment. The first irreversible serine–arginine protein kinase (SRPK1) inhibitor targeted a tyrosine residue with a sulfonyl fluoride warhead.27 An EGFR inhibitor can form a unique covalent interaction with the boronic acid moiety through Asp800 at the EGFR kinase hinge region.28 However, rapid selection pressure inside the cells makes the drug-resistant mutation occur, e.g., patients treated with irreversible kinase inhibitors such as ibrutinib and osimertinib usually develop resistance due to cysteine residue mutation.29,30 Therefore, people have to design new drugs through new strategies to solve the drug-resistance issues.
Protein degradation approach
To show the important role of catalytic kinases in the signal transduction pathway, reports have indicated that kinases play roles in cancers independent of their catalytic function. Notably, traditional small-molecule inhibitors that do not modulate the noncatalytic function of kinases cannot block the proliferation of tumor cells that rely on the noncanonical role of kinases. Promotion of kinase degradation can lead to the total loss of target function, including both catalytic and noncatalytic functions. Thus, using small-molecule kinase inhibitors to downregulate the protein levels of the kinase is an emerging therapeutic modality in cancer therapy. The ubiquitin–proteasome system is one of the main pathways critical for protein degradation. Molecular glue degraders and proteolysis targeting chimeras (PROTACs) (Fig. 5) are the main targeted protein degradation tools used to induce ubiquitin-dependent degradation of proteins through small molecules.

Schematic diagram of targeted protein degradation strategies. PROTACs are heterobifunctional small molecules consisting of a ligand for the target protein and another ligand for the E3 ubiquitin ligase, which are connected via a linker. Molecular glue is a class of small-molecule drugs that promotes the interaction between disease-causing target proteins and ubiquitin ligase complexes and marks the targeted protein for degradation
PROTACs constitute a promising technology for the selective degradation of cellular proteins. PROTACs are heterobifunctional small molecules consisting of a ligand for the target protein and another ligand for the E3 ubiquitin ligase, which are connected via a linker.31 PROTACs recruit the E3 ligase to the protein of interest and trigger ubiquitination and subsequent degradation of the target by the ubiquitin–proteasome system. With the successful development of small-molecule E3 ligands, many ligands for MDM2, VHL, CRBN, and cIAP have been used as the basis for PROTAC development against various cancer targets.32 Since the concept of PROTACs was raised by Deshaies’s team in 2001,33 more than 60 proteins in different protein families and structure classes have been successfully degraded by PROTACs, including transcription-related kinases CDK8 and CDK9,34,35 proto-oncogene kinases BCR-ABL36 and FLT3,37 the inflammation-related kinase PIPK2,38 and the immunity-related kinase TBK1.39 The breakthrough in PROTAC technology led to two orally bioavailable PROTACs (ARV-471 and ARV-110), which have been evaluated in phase 1/2 clinical trials in patients with locally advanced or metastatic ER+/HER2− breast cancer and metastatic castration-resistant prostate cancer (NCT04072952 and NCT03888612); the early data on safety, tolerability, and efficacy are encouraging.
Recently, as a promising new drug design technology, PROTAC attracts more and more attention. PROTACs present several advantages, including induction of isoform-selective degradation, elimination of both enzymatic and scaffolding functions, ability to target members of multicomponent complexes for degradation, and conversion of promiscuous ligands into selective degraders.40 PROTACs have also been proven to overcome drug resistance by degrading targets. BTK plays a crucial role in the BCR signaling pathway and has been validated as a major drug target for B cell-related malignancies.41 Ibrutinib was the first covalent BTK kinase inhibitor approved by the FDA, and it has been approved for the treatment of mantle cell lymphoma (MCL), Waldenström’s macroglobulinemia, chronic lymphocytic leukemia (CLL), etc21 However, BTK C481S drug-resistant mutations have been found in several MCL or CLL patients after long-term treatment of ibrutinib.42 BTK PROTAC P13I consists of ibrutinib with pomalidomide to recruit CRBN, which can effectively degrade wild-type and mutant BTK C481S. Furthermore, P13I efficiently inhibited the proliferation of ibrutinib-resistant BTK C481S and BTK WT DLBCL cell lines. The results of this study suggest that PROTAC may be a promising strategy for treating mutation-induced drug resistance.43
Nevertheless, various challenges have limited PROTAC applications, including difficulties in rational design, metabolic instability, poor cellular permeability, off-target toxicity, and large molecule weights. Researchers have adopted several creative strategies to overcome these barriers. Many efforts have been made to optimize the pharmacokinetic (PK) and pharmacodynamic properties of PROTAC. Since the length, rigidity, and hydrophilicity of the linker play important roles in determining the property of PROTAC. A systematic medicinal chemistry campaign took place to improve the PK properties of BTK PROTAC by optimizing the linker. The results show that the presence of ether moiety in the linker improved the PK characteristics and show good antitumor activity in vivo.44 It is also reported that using triazole as a linker can improve the metabolic stability and antitumor activity in vivo;45 “click chemistry” is used for the rapid synthesis of molecules to induce protein degradation. Through the rapid chemical reaction in cells, two molecules are paired to form a PROTAC. Both of these proteins have lower molecular weights and better cell permeability than the original PROTAC. For a proof-of-concept for this “click chemistry” platform, the JQ-1-IMiD PROTAC was synthesized to successfully degrade the BET bromodomain.46 Besides, many efforts have been made to improve the selectivity of PROTAC. Distinct E3 ligases may have different lysine selectivity profiles, which alter the selectivity of PROTAC with different E3-recruiting moieties as well. Lai et al.47 reported that PROTACs recruiting CRBN can degrade both BCR/ABL and c-Abl, whereas c-Abl can be only degraded by VHL-recruiting PROTAC. Also, a stable ternary complex is another key to achieving the selectivity of PROTACs. c-Abl can be effectively degraded by PROTAC with appropriate linker length between VHL and c-Abl, but longer linkers may reduce degradation efficiency due to the unfavored protein–protein interaction between VHL and c-Abl. To obtain a deeper understanding of the key molecular events in PROTAC discovery and optimization, an experimental map of degradable kinases in all clades of the kinome and chemical leads for more than 200 distinct kinases has been established and presented as a public resource (https://proteomics.fischerlab.org). The large dataset shows that ligase binding moiety, ternary complex formation, linker length, cellular target occupancy, and target expression are important factors that should be considered in PROTAC development. This work provides guidance for predicting proteins that can be degraded and a rational design method for more effective discovery of active degraders.48 In addition, most of the reported PROTACs are based on noncovalent binding to the target protein. Covalent binding can enhance the binding affinity to the targeted protein. Several studies have used covalent ligands against target proteins in the PROTAC design, such as BLK, KRASG12C, and BTK, these targets were successfully degraded by covalent PROTACs.49,50,51 Recently, reversible noncovalent, reversible covalent, and irreversible covalent BTK inhibitors are compared side by side to see how different warheads affect degradation efficiency. The results showed that reversible covalent ligands can significantly improve intracellular accumulation and target binding, and the reversible binding maintains the catalytic properties of PROTACs, making them preferable to irreversible ligands.52
Molecular glue is a class of small-molecule drugs that promotes the interaction between disease-causing target proteins and ubiquitin ligase complexes and marks the targeted protein for degradation. Thalidomide analogs and arylsulfonamides are two classes of drugs that act in molecular glue degradation. Thalidomide is used for the treatment of multiple myeloma (MM). It was discovered that thalidomide works by repurposing the CRL4 CRBN ubiquitin ligase to degrade transcription factors IKZF1/3.53,54 Furthermore, kinase casein kinase Iα can be degraded by thalidomide through the CRL4CRBN ubiquitin ligase.55 Thus, thalidomide and thalidomide analogs can be used to treat myelodysplastic syndrome with a deletion in chromosome 5q in the clinic. The mechanistic findings for thalidomide provided clinical proof-of-concept for targeted protein degradation therapeutics. Indisulam is an arylsulfonamide with anticancer activity. Similar to thalidomide, the mechanism of action is to recruit RBM39 (RNA-binding motif protein 39) to the CRL4DCAF15 E3 ubiquitin ligase, resulting in RBM39 polyubiquitination and proteasome degradation. These degraders work by exploiting the complementary protein–protein interfaces between the receptor and target.
Compared to PRAOTAC, molecular glue has a lower molecular weight, higher cell permeability, and better oral absorption, which makes it more drug-like.56 Although molecular glue degradation agents are very desirable, they are discovered mostly by chance, and the strategies used to identify or design these compounds are limited. Recently, it was found that the CDK inhibitor CR8 can be used as a molecular glue to depredate cyclin K. The compound has a solvent-exposed pyridyl moiety, which enables the formation of a complex between CDK12–cyclin K and the CUL4 adaptor protein DDB1, inducing ubiquitination and degradation of cyclin K.57 This study shows that modifying the surface exposure region where small molecules bind to the target is a reasonable strategy that can be used to develop molecular glue degradation agents for protein targets of interest.58 Also, a rational discovery of molecular glue degraders via scalable chemical profiling has been reported. The authors presented differential chemical profiling in hypomethylated cell lines with a promiscuous deficiency in CRL activity. Using this method, they successfully discovered a new molecular glue degradation agent that can induce cyclin K degradation through the CRL4B ligase complex. This research provides a rational strategy for identifying new cellular active molecular glue with a known mechanism.59
Allosteric binding approach
The majority of approved and preclinical kinase inhibitors are type I or type II compounds that bind to the ATP pockets. However, due to the high sequences and structural similarities among ATP pockets of human kinases, it is difficult for inhibitors to achieve high selectivity, and poor selectivity usually causes side effects. In addition, acquired drug resistance such as mutations of amino acids blocks the drugs from binding to target kinases. Allosteric kinase inhibitors with targets outside the highly conserved ATP pocket have been proposed as promising tools to overcome barriers to using kinase inhibitors, including poor selectivity and drug resistance. Since the first small-molecule allosteric inhibitor trametinib was approved in 2013, a number of clinical trials of more than ten allosteric inhibitors are ongoing, and an increasing number of highly selective and potent preclinical allosteric kinase inhibitors are being developed.
Allosteric inhibitors bind to allosteric pockets that are adjacent to the ATP pocket, but do not overlap with the ATP-binding pocket, also defined as type III inhibitors, or these are distant from the ATP-binding pocket, also defined as type IV inhibitors (Fig. 6). Three type III allosteric MEK1/2 inhibitors, trametinib,60 cobimetinib,61 and binimetinib,62 have been approved for use as single agents or in combinations with Raf inhibitors for the treatment of solid tumors patients, and all bind to the allosteric MT3 site of the kinase domain encompassed by the Lys97, Ser212, and Val127 residues of MEK1.63,64 For example, trametinib fits well with MEK1 in type III binding mode at the allosteric pocket next to the ATP pocket and forms five hydrogen bonds with Asp208, Phe209, Gly210, Val211, Ser212, and Ile216 (Fig. 7). Approximately 20% of patients treated with single-agent trametinib showed specific side effects, such as rash, diarrhea, and lymphedema. To overcome these side effects, the combination of trametinib with the BRAF inhibitor dabrafenib was approved in 2014, but this combination did not decrease the overall toxic effects of trametinib treatment.65 Cobimetinib is a diarylamine compound that led to improved median progression-free survival. Selumetinib has approved in the United States for pediatric patients with neurofibromatosis type 1 plexiform neurofibromas in 2020; however, it failed to show positive therapeutic effects in a phase III trial for the treatment of KRAS mutant lung cancer. In addition, some other allosteric inhibitors with structures similar to that of cobimetinib and binimetinib have been used for the treatment of solid tumors at different stages of clinical evaluation, e.g., selumetinib, pimasertib, and PD0325901. More than ten allosteric small-molecule MEK inhibitors have entered clinical trials.66 Although these type III MEK inhibitors have markedly improved patient survival, allosteric mutations eventually occur, which dramatically reduced binding affinity. A recent study revealed that the allosteric pocket “gatekeeper’ MEK1 V211D mutation situated within the arylamine-binding pocket disrupts the binding of multiple allosteric inhibitors and eventually leads to drug resistance.67 Allosteric EGFR-T790M inhibitors such as EAI001 is a new type of EGFR inhibitor that binds to EGFR kinase adjacent to the ATP-binding site. The crystal structure of the inhibitor–EGFR complex shows that the compound binds an allosteric site created by the displacement of the regulatory C-helix in the inactive conformation of the kinase.68 EAI001 inhibits mutant EGFR L858R/T790M—with low-nanomolar potency in biochemical assays, but as a single agent, it is not effective in blocking EGFR-driven proliferation of cells. In addition, JBJ-04-125-0269 also binds to the allosteric pocket adjacent to the ATP-binding pocket. Some allosteric AKT inhibitors, such as MK2206,70 ARQ902,71 and borussertib,72 binds to the interface of the allosteric pocket of AKT formed by the kinase domain and the PH domain. The complex of borussertib and AKT irreversibly stabilize the inactive PH conformation. Borussertib is also a first-in-class covalent-allosteric AKT inhibitor that is used to treat small cell lung cancer in combination with topotecan in the clinic (NCT04768296). The ABL inhibitors GNF-273 and asciminib (ABL001)74 bind to an allosteric pocket distant from the ATP-binding pocket, both are defined as type IV inhibitors. Asciminib (ABL001) was the first allosteric BCR-ABL inhibitor to enter the clinic. Asciminib binds to the myristate pocket of BCR-ABL and maintains activity against RTK inhibitor (TKI)-resistant ATP-site mutations.

The four types of allosteric kinase inhibitors. Allosteric inhibitors bind to allosteric pockets that are adjacent to the ATP pocket, but do not overlap with the ATP-binding pocket, which is defined as type III inhibitors. The binding pockets are distant from the ATP-binding pocket; these kinds of small molecules are defined as type IV inhibitors. Small molecules that bind the pseudokinase domain (JH2) are defined as type VI inhibitors and these bind the extracellular domain (ECD), which are defined as type VII

The binding mode of trametinib with MEK1 (PDB: 7M0Y). Trametinib binds with MEK1 in type III binding mode at the allosteric pocket next to the ATP pocket and forms five hydrogen bonds with Asp208, Phe209, Gly210, Val211, Ser212, and Ile216
JAK family members contain a distinctive catalytically inactive pseudokinase domain (JH2) that is similar in structure to the JH1 domain and includes an ATP-binding site, but it lacks catalytic activity.75 JAK family proteins play important “auto-inhibitory” roles in regulating the activation of the JH1 kinase domain, which makes this allosteric site an ideal target for the design of selective JAK inhibitors, and this type of binding mode was regarded as type VI. AT9283, a multitarget kinase inhibitor with potent aurora kinase activity, binds the pseudokinase-kinase domain of JAK2.76 Among JAK family members, BMS-986165 has been reported to be a selective allosteric TYK2 inhibitor that exhibits high selectivity, shows excellent PK properties with minimal profiling liabilities, and is efficacious in several murine models of autoimmune disease in clinical trials.77 SSR128129E was the first reported small molecule known to bind the extracellular domain (ECD) of FGFR, and crystallography combined with flexible docking and metadynamics studies demonstrated that SSR128129E binds the nonflexible H2 state of D3 in the ECD of FGFR2,78 which was defined as type VII. In addition, a highly selective DDR2 inhibitor that also targets the ECD of DDR2 inhibits DDR2–collagen binding interactions via allosteric modulation of the kinase.79
Overall, allosteric kinase inhibitors have several significant advantages over traditional orthostatic ATP-binding inhibitors, including improved activity, greater selectivity, lower off-target toxicity, and the ability to overcome the resistance mutations of ATP-binding pocket. Despite the advantages, challenges still exist. For example, as mentioned above, accumulating evidence shows that drug-resistance mutations also occur at the allosteric sites. In addition, the main reason that limits the discovery of allosteric inhibitors may be the finding of allosteric sites and the difficult rational design of allosteric inhibitors. Computational-aided approaches are helpful to address the emerging problems such as the identification of allosteric sites by AlloDriver80 and SBSMMA (structure-based statistical mechanical model of allostery).81 More importantly, a comprehensive understanding of allosteric-resistant mechanisms is crucial so that we can solve the emerging drug-resistance problems with allosteric inhibitors and guide future endeavors to overcome such barriers.
Macrocycles design approaches
In the past few years, macrocycles have attracted increasing attention in the discovery of kinase inhibitors. The macrocycles not only offer an opportunity for chemical novelty than the existing scaffolds but also improve the selectivity and overcome the acquired drug resistance.82 So far, except for the three mTOR inhibitors, only one ALK/ROS1 dual kinase inhibitor lorlatinib has been developed via rational design, which is a third-generation ALK/ROS1 inhibitor and it can overcome gatekeeper and solvent-front mutations.83 To avoid steric hindrance with a gatekeeper and solvent-front mutations, lorlatinib is designed using a rigid amide-linked 12-membered macrocycle based on the structure of crizotinib. The selectivity and activity were also improved at the same time. It is worth noting that lorlatinib has good blood–brain barrier penetration and central nerve system exposure, these properties lead to its applications as an effective agent against metastatic brain cancer.84 Currently, many macrocycles kinase inhibitors entered clinical trials (Fig. 8). Pacritinib, zotiraciclib, and SB1578 are all 2-aminopyrimidine-based macrocycles targeting JAK2/FLT3 kinases. But their clinical applications are different; pacritinib is being evaluated for the treatment of myelofibrosis and lymphoma in phase III85 and zotiraciclib is used for leukemia and MM.86 Different from pacritinib and zotiraciclib, the clinical applications of SB1578 are autoimmune diseases and inflammatory disorders such as psoriasis and rheumatoid arthritis.87 The resorcyclic lactone E6201 is a MEK and FLT3 dual inhibitor88 that entered clinical trial in 2007 for the treatment of plaque psoriasis,89 and then the other application in malignant melanoma was initiated in 2017. To overcome the mutations of TRK, two macrocyclic compounds were designed using macrocycle strategy based on the structure of larotrectinib.90,91 Both selitrectinib and recrotrectinib contain a pyrazolopyrimidine structure and entered clinical trials in 2017 for the treatment of advanced solid tumors. Recrotrectinib, a next-generation ROS1, pan-TRK, and ALK kinase inhibitor, overcomes resistance due to acquired solvent-front mutations involving ROS1, NTRK, and ALK. The macrocycle was introduced to increase conformation rigidity based on the larotrectinib scaffold, and it was used in patients with advanced solid tumors harboring ALK, ROS1, or NTRK1-3 rearrangements.90,91 JNJ-26483327 contains a quinazoline scaffold and entered phase I clinical trial in 2008 as a multitargeted tyrosine kinase inhibitor for the treatment of advanced and/or refractory solid malignancies.92 Although macrocyclic kinase inhibitors offer many advantages such as offering chemical novelty, the ability to overcome drug resistance, and improved selectivity, the challenges in chemical synthesis slow down its structure–activity relationship (SAR) optimization process.

Macrocyclic kinase inhibitors approved and in clinical trials
Advances of property evaluation technologies
Kinase family members share a conserved ATP-binding domain and exhibit high structural similarity, rendering it a great challenge to find selective kinase inhibitors. On account of the function of kinases in normal cells, usually multitarget inhibitors induce side effects due to off-targets. Kinomics selectivity analysis of kinase inhibitors is important for better understanding of inhibitor biological activity, obviating off-target activities, and, in some cases, identifying new targets that may lead to novel therapeutic applications. In the following section, we will review the profiling technologies in three categories including probe-based, probe-free, and catalytic activity-based.
Probe-based selective kinome profiling technology
To evaluate the selectivity and activity of compounds against multiple kinases in a single experiment, a competitive binding assay based on phage displays and chemical probes was developed.93 In this assay, a probe that binds to multiple kinases at the ATP-binding pocket is immobilized on solid beads and incubated with DNA-tagged kinases. Then, a free test compound is introduced to compete with the probe to bind its target kinase, and the selectivity of the test compound is evaluated by quantifying the DNA-tagged kinase associated with the solid bead by real-time quantitative PCR (Fig. 9). Commercial profiling services, such as KINOMEscan, which can test most kinases (with coverage of more than 80% of the “typical” human protein kinome), are available.94 Using this approach, 10 μM 72 kinase inhibitors have been screened against 113 kinases and profiled, with the quantitative binding constant subsequently determining their targets. The compounds were quantified by measuring a selectivity score (S score). The results showed that, compared to type I inhibitors, type II inhibitors exhibited better selectivity.94 This analysis facilitated structural optimization and accelerated the drug development process. However, as it was performed in vitro without ATP or cofactors, the affinity as determined might not be correlated with that in cells.95,96 Loss of activity can be attributed to the protein structure, complex formation in the native environment, low cell permeability, or compound efflux.

Schematic overview of competition binding assay. In this assay, DNA-tagged kinases are incubated with immobilized probes that bind kinases at the ATP-binding pocket. Then, a free test compound is introduced to compete with the probe to bind its target kinase, and the selectivity of a test compound is evaluated by quantifying the DNA-tagged kinase associated with the solid bead by real-time quantitative PCR
The utilization of native proteins in profiling can recapitulate the physiological binding mode most accurately. Several chemical proteomic platforms have been developed for use in direct profiling of endogenous kinases in cellular and tissue proteomes via liquid chromatography-tandem mass spectrometry (LC-MS/MS).
KiNativ is an in situ profiling platform based on a biotinylated ATP acyl phosphate probe, which covalently reacts with conserved lysine residues in the ATP-binding pocket of kinases.97,98 Test compounds are preincubated with a cell lysate, then probes are introduced to enrich the uninhibited kinases. The test inhibitor competes with the probe, and then uninhibited kinase proteins in cell lysates covalently bound with the probe are enriched by streptavidin and quantitated by LC-MS/MS (Fig. 10, left).2,99 Profiling by KiNativ demonstrated that CDKN2A-modulated CDK4 prevents target engagement of palbociclib,100 which correlated with the phenotype in which a high level of CDKN2A is associated with reduced sensitivity to palbociclib.100,101 This result indicated that profiling of CDK kinases in cell lysates is more accurate than in vitro biochemical assays, as CDKs are associated with cofactors that might influence the interaction of compounds and kinases.

Chemical proteomic strategies for kinase inhibitor’s selectivity analysis in native proteomes. In these technologies, the test compound is preincubated with cell lysate, and then probes are introduced to enrich the uninhibited kinases. For KiNativTM approach (left), biotinylated acyl phosphate probes that bind kinases covalently are utilized; in KinobeadTM (right), uninhibited kinases are precipitated with broad-spectrum kinases inhibitors immobilized on Kinobeads, and then enriched proteins or peptides are analyzed by LC-MS/MS
Kinobeads are used in an approach similar to that of KiNativ. Kinobeads are used to reversibly immobilize broad-spectrum kinase inhibitors as probes. The test compound and kinobeads compete to bind target kinases, inducing a decrease in the target associated with the kinobeads, which is identified through LC-MS/MS analysis as the protein in abundance (Fig. 10, right). Klaeger et al. presented large datasets showing the potency of 243 clinical kinase inhibitors based on this strategy and developed a novel selectivity metric termed CATDS (concentration- and target-dependent selectivity) for evaluating the selectivity of kinase inhibitors. Among the tested compounds, lapatinib, capmatinib, and rabusertib showed the highest selectivity and were applied as selective probes.102 The data revealed new targets for kinase inhibitors in the clinic, particularly non-kinase targets, which may explain the mode of action of these drugs under physiological conditions. For example, the bone osteosarcoma cell line U2OS is sensitive to the SRC/ABL dual inhibitor saracatinib even when the target gene SRC is knocked down. The researchers found that BMPRIA, which participates in bone morphogenesis, was a novel target of saracatinib and might mediate the inhibitory effect of saracatinib in U2OS cells. In addition, these results suggest the prospect of using druggable kinases, such as SIK2, in immune and cancer therapy. Combining the data on the phosphorylated proteome led to further refinement of novel pathways, determination of drug response markers, and enhancements to the theoretical basis of combination therapy.102 Through proper modifications, different kinobeads can be applied to profile potential covalently bound off-target irreversible kinase inhibitors.103 Dittus and co-workers reported a unique kinobead profiling strategy to identify new targets for irreversible kinase inhibitors.29 Using this approach, ibrutinib was found to bind covalently not only to TEC family kinases but also to BLK and ZAK. However, there are several limitations of chemoproteomic methods.104 First, kinases that do not interact with probes cannot be detected. Second, the kinase expression level of the selected cell line or tissue may be too low to be detected. Optimization and combination of the probes or complementary cell lysates can be used to extend the target coverage.97,98,104 The utility of the proteomic analysis method is also limited by low throughput and high cost.
Compared to proteomic analysis, bioluminescence-based analysis is more suitable for higher throughput profiling. NanoBRET is a novel profiling assay that enables investigation into compound selectivity in live cells.105 In this profiling, bioluminescence resonance energy is transferred between target kinases fused to NanoLuc luciferase and a cell-permeable fluorescent tracer that binds to the target kinase.105,106 The binding affinity of the test compound is determined on the basis of its ability to compete with the tracer and thus influence the production of BRET. Recently, NanoBRET profiling of clinical kinase inhibitors was performed with 178 kinases.107 By comparing the results of NanoBRET profiling with biochemical profiling data, crizotinib exhibited improved target selectivity under cellular ATP conditions, and certain off-target proteins were disengaged in live cells at the clinically relevant dose.107 These results indicate that profiling data in live cells might be more accurate than that of in vitro protein level profiling due to the high ATP concentration in cells. In addition, a NanoBRET panel against all 21 human CDKs was established to quantify target occupancy in live cells. The profiling results revealed a comprehensive landscape of the isozyme potency and selectivity of 46 CDKs inhibitors that have entered clinical trials. Among these compounds, BAY-1251152 exhibited the strongest target affinity and selectivity of CDK9, and it was found to be suitable for cellular research as a molecule tool. Most CDK inhibitors display affinities of more than two members of CDKs. For example, BMS-265246, which targets CDK1/2, was found to also be a potent CDK8/19 inhibitor.108 Although probes have been widely used in kinase profiling, the development of chemical probe-free profiling methods might have advantages for expanding the target range, particularly non-kinase targets.
Probe-free thermal-based techniques
Thermal-shift assays (TSAs) determine a shift in protein melting temperatures (Tm). Upon ligand binding, the proteins are thermally stabilized, which induces a shift in the protein melting temperatures.109 This provides the theoretical basis for the development of thermal profiling strategies such as differential scanning fluorimetry (DSF) and thermal proteome profiling (TPP) (Fig. 11).110

Thermal-based techniques applicated in kinase inhibitor profiling. a Differential scanning fluorimetry (DSF) estimates the thermal shift of target protein by fluorescence dye. The dye provides a fluorescence signal when protein unfolds. The dye is quenched when proteins fold natively. b Thermal proteome profiling (TPP) evaluated the change of protein Tm by cellular TSA (CETSA); the soluble native proteins are quantitated by mass spectrometry analysis
DSF detects the Tm shift of proteins by labeling purified proteins with fluorescent dyes (Fig. 11a).110 The dyes are quenched when proteins fold natively. When proteins unfold, the hydrophobic cores are exposed and interact with the dyes which provided a fluorescence signal. The melting curve of proteins was evaluated by the fluorescence intensity as a function of temperature.111 It is easy to perform on RT-PCR instruments, which is suitable for high-throughput screening.111,112 It has been applied in lead compounds discovery and fragment-based ligand design.113,114 By DSF screening against the small-molecule library, p90 RSK inhibitor BI-D1870 was found to induce a significant Tm shift of vaccina-related protein kinase 1 (VRK1).115 Based on this scaffold, a novel VRK1 inhibitor was developed, which showed better potency and selectivity.116 However, using extrinsic dyes in the DSF experiment, the signal-to-noise ratio is often limited by the protein purity and homogeneity, and the high background of the signal often leads to false-positives hits. In addition, this system is not suitable for membrane proteins in buffers containing detergent.117 To solve these limitations, intrinsic fluorescence was utilized in DSF. Using GFP (green fluorescent protein) fused protein in DSF, the stability of protein can be detected through the green fluorescence.118 Recently, nanoDSF technique was developed, which detects the intrinsic tryptophan fluorescence of proteins at 330 and 350nm119 with specially designed monitoring instrument Prometheus NT.48 that is commercially available (NanoTemper Technologies, Munich).
Mikhail and co-workers developed TPP for tracking cancer drugs in living cells (Fig. 11b).120 By combining the cellular TSA with quantitative mass spectrometry analysis, protein denaturation curves and Tm changes, which reflect the thermal stability in cells, were evaluated.120,121,122 Through subsequent comparison of the changes in protein Tm between vehicle- and drug-treated samples, the approach provides an unbiased evaluation of drug target occupancy for various targets, not just for kinases, facilitating the identification of drug efficacy and toxicity markers.120,121Using this strategy, Steffan and co-workers found that brusatol, which has KEAP1/Nrf2 inhibition activity, inhibited global protein synthesis.123 The heme biosynthesis enzyme FECH was reported to be an off-target of vemurafenib and alectinib, which explains why these inhibitors cause phototoxicity as a side effect in the clinic.120 In addition, this thermal profiling strategy can be used to detect potential downstream effectors of target proteins. For example, by profiling dasatinib in intact K562 cells with an active signaling pathway and in cell extracts, Mikhail and co-workers found a thermoshift of CRK/CRKL downstream of BCR-ABL. However, the direct target of dasatinib, BCR-ABL is not stabilized by its binding.120 Therefore, the fact that some proteins are not stabilized by ligand binding might limit the application of this assay. Besides, this assay is not suitable for membrane proteins that are not solubilized in detergent-free cell extraction buffer, low abundant proteins, and proteins with extreme melting temperature.124,125 Recently, the detection limit for membrane proteins has been overcome by new methods such as the application of mild detergent and enrichment of cell surface proteins.125,126 Furthermore, the development of mass spectrometry improved the throughput and sensitivity of this assay.127,128 Although this profiling is expensive and not commercially available, it might have more applications through methodological optimization, allowing the study of the drug–protein interaction in live cells, even in tissue or organs in the future.129,130
In summary, we can see that all of the aforementioned profiles truly detected the binding affinity of compounds with kinases in competitive or uncompetitive ways. However, in some cases, strong binding affinity does not necessarily correlate with potent inhibition of activity.131
Catalytic activity-based profiling approaches
Measurement of kinase catalytic activity can be used to verify the results of a binding assay.131,132 The ADP-Glo™ assay from Promega is a universal assay with accuracy that has been validated with a large kinase panel spanning the kinome, and it detects the generation of ADP catalyzed by kinase.133 It is suitable to use not only for hit compound screening against one target but also for profiling inhibitors against large or small kinase panels using a single platform. In addition, based on time-resolved fluorescence resonance energy transfer technology, different platforms for binding affinity or kinase activity measurement have been developed that can be applied to address high-throughput screening, hit confirmation, and lead optimization programs. However, catalytic activity tested in vitro may reflect interference by many factors. For example, the lack of cofactors and the presence of truncated, fused, or tagged recombinant proteins do not always reflect the physiological kinase form.134 ATP concentration and the peptides used to mimic protein substrates are all factors that will affect the in vitro activity.94
A kinase-dependent Ba/F3 panel provided a strategy to evaluate kinase catalytic activity in vivo. Notably, this phenotype profile characterizes the activity through cell viability. Ba/F3 is a mouse-derived hematopoietic cell line that depends on interleukin-3 (IL-3) for proliferation and survival (Fig. 12).135 Expression of kinase proteins in Ba/F3 cells through a genetic engineering approach activated the kinase-dependent proliferation signaling pathway and rendered the cells IL-3-independent.136 In this way, cell viability was associated with kinase enzymatic activity. Many mutations of oncogenic kinases have been found to be driving forces of cancers and other diseases. By constructing kinase mutation-dependent Ba/F3 cell lines, selectivity and activity profiling between kinases and their mutants is possible, which has been particularly useful for characterizing certain fusion mutations that are not easy to express in vitro.136 Screening randomly mutated kinase-Ba/F3 cells by kinase inhibitors allowed one to identify site mutations that induced drug resistance.135,137 Using this strategy, Cools et al. performed an in vitro screening of FLT3 randomly mutated Ba/F3 cells with FLT3 inhibitors and identified that the FLT3 G697R mutation in the ATP-binding region induced resistance to FLT3 inhibitors such as PKC412.138 This site mutation was later found in patients treated with PKC412, indicating the validity of the in vitro predictions.139 However, for kinases that are not functionally related to the cell growth signaling pathway and cannot maintain the survival of Ba/F3 kinase cells in the absence of IL-3, this approach is unlikely to work.

Engineered Ba/F3 kinase cells for kinase inhibitor selectivity and activity profiling. Ba/F3 cells are rendered IL-3-independent and kinase-dependent by introducing a kinase gene in Ba/F3 cells through a genetic engineering approach. The activity of the kinase inhibitor can be evaluated by cell viability of kinase-Ba/F3 cells after inhibitor treatment
Caliper off-chip mobility-shift assay (MSA) is most frequently used for drug discovery such as small-molecule screening, fragment-based screening, target specificity (profiling), and mechanism of action studies. This assay can be applied to a wide range of proteins, including kinases, phosphatases, lipid kinases, proteases, phosphodiesterases, nucleic acid-binding proteins, and epigenetic targets (HATs, HDACs, and methyltransferases). Caliper off-chip MSA technology uses capillary electrophoresis in a microfluidic environment to analyze enzymatic assays. The Caliper LabChip platform is based on microfluidic chips containing a network of miniaturized, microfabricated channels through which fluids and chemicals are moved to perform experiments. By applying an electric potential difference across the separation channel, fluorescently labeled substrates and products are separated by electrophoresis and detected by light-emitting diode-induced fluorescence. Both the substrate and the formed product were detected and measured for each sample. The amount of product formed is determined by calculating the ratio of product peak heights. For kinetic analysis, the reaction is monitored as it progresses by sequentially “sipping” samples onto the chip at various time intervals.140,141 The kinase activities of many kinase inhibitors have been evaluated by caliper off-chip incubation MSAs. The major advantages of this method are high sensitivity and no use of termination or quenching reagent, but the assay usually takes a long time for detection.
Advances in efficacy assessment models and technology
Drug discovery is an expensive, low efficiency, and highly risky business. An analysis suggests that it costs more than $1 billion to successfully develop a small-molecule kinase inhibitor, and takes 12.5 years on average. Reliable assessment efficacy models and drug screen technologies can accelerate the discovery and development of kinase inhibitors. Target-based drug discovery (TDD), also known as reverse pharmacology, which features anchoring to a defined target kinase or protein, has long been favored by pharmacologists due to its rational basis for drug design. Compared to phenotypic screening, the TDD approach is more target-specific and requires known targets, which are sometimes very difficult to identify. In addition, TDD makes SAR exploration easier and faster, hence offering a better chance to obtain the “best-in-class” inhibitors (Table 2). In recent years, phenotypic-based drug discovery approaches become more and more popular. With this approach, the properties of compounds are measured through a physiologically relevant system such as animal or primary cells, the previous understanding of the molecular mechanism of action is unnecessary, and drug identification follows an unbiased approach (Fig. 13). A well-known example of a drug discovered by phenotypic screening is aspirin, whose mechanism of action was identified after approximately 100 years. The majority of first-in-class drugs identified from 1999 to 2008 were the culmination of phenotypic drug discovery approaches based on either in vitro or in vivo screening models that mimicked the disease state of interest.142,143 Rapamycin, an FDA-approved first-in-class drug, was identified through phenotypic screening in a fungus model and later was developed as an antifungal drug on the basis of its potent immunosuppressant activity, which was identified through phenotypic screening in vivo.144 In 1991, the true mechanistic target of rapamycin immunosuppressant activity was discovered, and the target was mTOR.145 Some unusual kinase activators were also found in this way; e.g., MLR-1023, a potent and selective allosteric LYN kinase activator, was discovered through an in vivo phenotypic screening platform, and MLR-1023 has been used alone or in combination with metformin to treat type 2 diabetes in clinical trials (NCT02317796 and NCT03279263).146 Some kinase inhibitor hits were discovered soon after this approach was implemented; for example, the CDK9 kinase inhibitor MC-180295 was found after the optimization of SNS-032, which had been identified through a phenotypic screening approach.147 Moreover, dual TTK protein kinase/CDC2-like kinase (CLK2) was discovered through in vivo phenotypic screening of a triple-negative breast cancer tumor cell line.148 Novartis reported that the multitarget compound GNF3809-protected rat insulinoma β cells from death induced by proinflammatory factors, which was also discovered by phenotypic screening in vitro.149

The drug discovery and development process. Phenotypic-based drug discovery (PDD) approaches are measured through a physiologically relevant system such as animal or primary cells. The molecular mechanism of action is unknown previously; target-based drug discovery (TDD) features anchoring to a defined target kinase or protein for screening a fit compound
Patient-derived tumor cells
In addition to canonical in vivo animal- or in vitro cell line-based phenotypic screening, patient-derived tumor cells (PDCs), derived through a short-term culture of primary cells collected from patients, have recently attracted considerable interest for use in novel phenotypic screening and personalized cancer therapy.150 PDCs are thought to be useful surrogates of patient tumors in the clinic because of their obvious advantages over other preclinical models. First, PDC models faithfully retain the histological and genomic features of primary patient tumors. Second, the labor and time needed to build the PDC models are reduced. Normally, PDC models can be obtained within 2 and 3 weeks, making them more feasible for use in clinical application than other methods. Third, the success rate of model establishment is as high as 73.6% in patients with metastatic cancer. Fourth, PDC models are applicable to high-throughput drug screening. Finally, the concordance between clinical response and drug susceptibility is high.150 We and others have shown that low-passage patient-derived primary cells established from acute myeloid leukemia patients, chronic myeloid leukemia (CML) patients, gastrointestinal stromal tumor (GIST) patients, and sarcoma patients can be used for screening and identifying novel kinase inhibitors in preclinical studies.151,152 Lee et al. tested the therapeutic landscape of 60 molecule-targeted compounds on 462 PDCs across 14 cancer types and identified lineage-specific drug associations, such as gastric cancers and PI3K inhibitors.153 Brodin et al. evaluated the sensitivity of 525 anticancer agents on patient-derived sarcoma cells and found that the drug sensitivity of the patient sarcoma cells in vitro correlated with the response of the patient to the treatment received. They proposed that dasatinib, a c-Src inhibitor that they identified through this research, is an effective drug against sarcomas carrying chromosomal translocation.154
Organoid/3D cultures
The first patient-derived tumor organoid culture was established in 2009, and since then, the explosive growth of organoids as a novel ex vivo model has revolutionized preclinical cancer research over the past decade.155 Organoids are thought to be superior models for identifying and testing novel anticancer drugs due to their unique characteristics; for example, (i) organoids stably retain the genetic and phenotypical features of the parental primary tumors; (ii) organoids from both healthy and tumor tissues can be generated, which allows screening of drugs that specifically target tumor cells while leaving healthy cells unharmed; (iii) organoids can be expanded long term and be cryopreserved, allowing for the generation of living tumor organoid biobanks; (iv) organoids can be generated from 4 to 6 weeks, which is a major advantage for patients with advanced stages of cancer; and (v) organoids are suitable for medium-to high-throughput drug screens.155,156,157
Organoids have been successfully generated from primary tumors of the breast, colon, pancreas, lung, head and neck, ovary, and prostate.155,156,158,159 In fact, there have been some promising results in terms of novel targeted molecular drugs identified with organoid models. Doublecortin-like kinase 1 (DCLK1) is a serine/threonine kinase that is upregulated in a wide range of cancers, including colorectal cancer and gastric cancer. DCLK1 expression is particularly relevant in pancreatic ductal adenocarcinoma (PDAC), where its high expression correlates negatively with PDAC patients’ lifespan.160 Ferguson et al. worked on the development and characterization of DCLK1-IN-1 inhibitors. They used organoid samples isolated from DCLK1-expressing patients as preclinical testing models and found that DCLK1-IN-1 is a potent and highly specific inhibitor of DCLK1 and DCLK2 kinases.161
Patient-derived xenograft
The prevalence of patient-derived xenograft (PDX) models was established in the early 2000s because of significant limitations in cell line-based models that were commonly used in the discovery of novel drugs in clinical trials. PDX models are generated by the removal of primary or metastatic tumor specimens from patients and followed by surgical engraftment into immunocompromised mice; these models have emerged as a useful model for cancer research and have been widely used in novel anticancer agent screening.162 In the PDX model, both the genetic and pathohistological characteristics of tumors are preserved. Tumor heterogeneity was shown to be similar to that of the parental patient tumors. Furthermore, the tumor architecture and tumor microenvironment closely resemble those of the original tumor tissues. All of these characteristics suggest that PDXs are useful preclinical models for mechanistic research on tumor progression and for predicting the therapeutic effects of novel drug candidates.163 To date, multiple PDX models have been established from a wide range of human cancer types, including lung cancer, ovarian cancer, breast cancer, pancreatic cancers, gastric cancer, and CRC, and they have been used in preclinical research.155,164 In addition, PDX models have also been widely used for the development of novel molecular targeted drugs and evaluation of the drug sensitivity patterns of primary tumors. Using PDX models, Wang et al. investigated the antitumor activity of pyrotinib, an irreversible pan-HER TKI, and found that pyrotinib shows potent antiproliferative activity against non-small cell lung cancer with HER2 exon 20 mutations. In a clinical trial, pyrotinib also showed promising efficacy, which validated the PDX findings obtained in preclinical testing.165 BTK is a key mediator of BCR-dependent cell growth signaling and a promising therapeutic target for multiple diseases, particularly in hematopoietic malignancies such as MCL. Li et al. investigated the molecular mechanisms of BTK inhibition with the novel molecular targeted inhibitor BGB-3111 (zanubrutinib) in MCL models.166 They found that BGB-3111 exhibits greater specificity for BTK, and it exhibits excellent efficacy in preclinical MCL cell models and PDX models, which will be helpful in promoting the progress of clinical trials. In addition, PDX models have also been used to evaluate the effect of different molecular targeted drugs against different cancer types, including GISTs, melanoma with BRAF(V600) mutations, and CRC with KRAS G12C mutation.167
DNA-encoded chemical library
A DNA-encoded chemical library (DEL) is a new screen platform seeking bioactive compounds from millions to billions of small molecules with unique DNA sequence tags attached, which is based on the binding affinity screening of pharmaceutically relevant proteins (Fig. 14). This capability allows compound screening and hit selection to be rapidly performed. With more chemical diversity than other methods, the use of a DEL is a highly efficient approach to explore larger chemical spaces for drugs. DELs have been widely adopted and employed in numerous drug discovery programs.168 GSK reported the identification of two compounds, the sEH (soluble epoxide hydrolase) inhibitor GSK2256294 and RIPK1 inhibitor GSK2982772 using the DEL hit-finder approach, and both of these inhibitors are currently being tested in clinical trials.169,170 Although billions of compounds can be screened in a small tube, the limitations of the technology are that compounds should be soluble and target proteins either tagged or immobilized on a solid support should be purified. Moreover, only a few kinds of reactions have been reported for the construction of DEL.171

Schematic diagram of drug screening using a DNA-encoded chemical library (DEL). DEL is a new emerging screen platform for seeking bioactive compounds from millions to billions of small molecules with unique DNA sequence tags attached
Artificial intelligence
It is estimated that there are approximately 1060 compounds in the chemical space, and only a tiny proportion of these molecules have been developed as drugs. Artificial intelligence (AI) is integrated various technologies in a network system that can mimic human intelligence. AI is widely used in drug discovery and development, which is applied in drug discovery and development to improve pharmaceutical products including design, synthesis, repurposing, ADMET prediction, even the clinical trials design, and monitoring. We mainly focus on the application of drug design and repurposing (Fig. 15). Numerous efforts have been made to modify the drug map for a long time, which causes continual increases in the research and development costs of developing a new drug. A computer-assisted drug design (CADD) strategy has been widely used to guide the exploration of the drug space; however, the biological activity of compounds can seldom be predicted precisely. As no formula can be established to precisely describe the interactions between molecules and targets, automating drug research and development through machine learning from a large number of samples represents a new option.172,173 Traditional computational methods rely on manually programmed logical operations to process information and optimize tasks that are difficult or time-consuming for humans to understand or complete, such as performing a linear regression on a set of data. AI, on the contrary, can solve nontrivial tasks on the basis of human-defined rules.

Artificial intelligence (AI) in kinase inhibitor discovery. AI is integrated in various technologies in a network system that can mimic human intelligence. AI is widely used in drug discovery and development, which is applied in the drug discovery and development to improve pharmaceutical products including design, synthesis, repurposing, ADMET prediction, and even the clinical trials design and monitoring
Many kinase inhibitors with enhanced predicted activities and binding affinities, which can serve as lead compounds for further synthesis and preclinical testing, have been designed through in silico modification. Many new chemical scaffold kinase inhibitors (imidazopyridazine, imidazopyridine, isoquinoline, phenazinamine, etc.) have also been designed by computational approaches and represent good examples of the discovery of new kinase inhibitors.174 ETC-206, an oral MNK 1/2 kinase, was designed from a previous compound through homology modeling, docking, molecular dynamics (MD) simulations, and free energy calculations, and it was investigated for synergism with dasatinib in vivo and is currently in a phase I clinical trial with patients with blast crisis CML.175 CADD techniques (molecular docking, similarity, and pharmacophore searches) were used in the design of the allosteric ABL inhibitor asciminib, which is currently in clinical trials.176
Computational approaches, such as quantitative SAR (QSAR) modeling, ligand- and structure-based virtual screening, molecular docking, MD, quantum mechanics, fragment-based drug design, and machine deep-learning methods, all provide unique insight into the conformational landscape of kinases, structural requirements for inhibitory activity, binding modes, and atomistic mechanisms of allostery, which represent indispensable information for rational de novo design.177 The goal of QSAR modeling in drug discovery is to find a mathematical relationship that connects molecular properties (as encoded in molecular descriptors) with some quantitative activity metric of a compound. QSAR models have become important tools in AI-aided drug design, such as virtual screening and lead optimization. Jiang and co-workers provided new chemical starting points for further structural optimization of FGFR1 inhibitors by combining pharmacophore-based 3D-QSAR analysis and virtual screening approaches.178 Recently, an increasing number of people have developed new QSAR models and applied them to the discovery of kinase inhibitors. Koneru et al. integrated the fragment-based drug discovery technique with QSAR and MD to design the second-generation SRC kinase inhibitor RL-45.179 QSAR has also been used in LBVS to predict the properties of new chemical compounds. Several potential kinase inhibitors, such as a dual ALX/FLT3 inhibitor and a CDK9 inhibitor, were reported on the basis of LBVS methodology.180,181 There are many cases of QSAR models used in the design of kinase inhibitors for VEGFR2,182 EGFR,183 MCL-1,184 IKK-β,185,186 LYN,187 and MER,188 and so on.177,178
Based on QSAR, virtual screening, and deep-learning models, AI-aided de novo drug design became possible. Compared to traditional wet lab-based drug discovery, AI-assisted de novo drug discovery has the advantage of time and cost efficiency as long as the deep-learning techniques used are well developed and basic learning data provided are sufficient. Based on the calculated GScore, the fragment-based drug design method has been used to create new BCR-ABL inhibitors. De novo designed molecules have better inhibitor capacity than the most commonly used tyrosine kinase inhibitors on the market. These molecules have shown strong potential to become drugs capable of inhibiting all mutations, particularly the BCR-ABL-T315I mutant, which is now the leading cause of death due to the difficulty of controlling its inhibitors.189 As an example of a proof-of-concept for developing drug via AI, a preclinical lead compound of DDR1, a kinase target implicated in fibrosis and other diseases, was recently discovered in an AI-assisted de novo drug design approach within 21 days. This approach uses a new deep-learning technique called generative tensorial reinforcement learning, through which synthetic feasibility, novelty, and biological activity can be optimized.190
Silicon-based kinase profiling methods
Recent advances in deep-learning algorithms have made the profiling of small molecules possible. Many in silico modeling approaches have been developed to predict kinase inhibitory activity for large-scale compound libraries. Three online platforms are currently available for use in predicting kinome-wide targets: KinomeX, KinomeFEATURE, and ProfKin. KinomeX is based on an established multitask deep neural network model trained with over 140,000 bioactivity data points for 391 kinases to predict the kinome-wide poly-pharmacology effect of small molecules for designing novel chemical modulators and drug repositioning.191 Extensive computational and experimental validation has been performed using the KinomeFEATURE database to predict the off-targets of small molecules, which is achieved by searching for kinase-binding sites through comparisons of protein structural microenvironments.192 The KinomeFEATURE database and other auxiliary information used for performing kinase pocket similarity analyses can be downloaded from the Stanford Sim TK website (https://simtk.org/projects/kdb). Based on a KinLigDB (kinase-ligand-focused database), ProfKin, which provides the complex structures of 4219 kinase ligands covering 297 human kinases, associated information, binding site type, ligand binding type, interaction fingerprints, downstream signal pathways, and related human diseases, has been exploited as a multifunctional website for structure-based kinase selectivity profiling.193 The website (http://www.lilab-ecust.cn/profkin/) can be used to predict the possible binding modes, which can be helpful for target prediction and mechanistic study.
Challenges and future perspectives
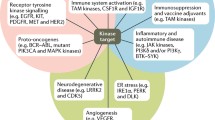
New strategies, technologies, and methods undoubtedly have expanded the kinase inhibitor discovery platform and accelerated the discovery process, increasing the number of diversified preclinical candidates for clinical development. All of these new approaches have been tried in efforts to overcome a specific hurdle or solve an identified problem, such as challenges to time efficiency, cost efficiency, and even knowledge efficacy during the discovery journey, but inevitably, these new methods exhibit their limitations. Because of the evolutionary pressure within cancer cells, drug resistance seems an endless problem. Therefore, to take full advantage of these new approaches, combinations of new approaches and traditional approaches might make these strategies more powerful. Among the approved small-molecule kinase inhibitors, small molecules targeting kinases of DNA-damage response signal pathways such as ATR, ATM, DNA-PK, and CHK1 are still in clinical trial stages; more molecules can be designed of these kinases. Recently, a novel selective ATR kinase inhibitor shows potent antiproliferative activity against a broad spectrum of human tumor cell lines and exhibits strong monotherapy efficacy in cancer xenograft models;194 its antitumor responses were also observed in patients with advanced solid tumors.195 With the quick development of cancer immunotherapy, many kinases involved in tumor immunology such as CSF1R,196 HPK1,197 etc. can be targeted by small molecules. There is still a lot of room for the development of protein degradation technology, e.g., the PKs of PROTACs. In addition, other degradation technology such as lysosome-targeting chimeras for degradation of extracellular proteins,198 merging PROTAC, and molecular glue for degrading may become new directions.199 Research showed that many disease pathology is driven by protein ubiquitination and degradation; thus, DUBTAC (de-ubiquitinase-targeting chimera) platform for targeted protein stabilization of specific proteins was developed, which is contrary to PROTACs.200 The hetero-bivalent system is a new class of molecules that possess three components including an ATP-competitive ligand and pseudo-substrate peptide that are covalently connected through a linker. This unique rational design permits the simultaneous binding to both ATP and peptide binding sites and thus making these compounds possess high selectivity and activity, which can meet the needs of the development of more potent and selective kinase inhibitors.201 In addition, with the development of technology several new promising protein–protein interaction compounds (PPIs) have entered clinical studies. PPIs modulators can be a new strategy to design small molecules and peptide inhibitors targeting specific kinases domains.202,203 Many kinases such as NLRP3, JNK perform their functions by forming complexes with their interacting partners or substrates, and the interactions between them are also involved in the regulation of kinase functions, which provides new opportunities for designing PPI inhibitors that work through blocking these interactions.
In addition to small molecules alone, combinations with large molecules, such as conjugated antibody-drug compounds, may unleash the full potential of small-molecule kinase inhibitors. Despite the variety of innovations applied to kinase inhibitor discovery, from screening, design, and in vitro and in vivo evaluation, the rate of successful development of drugs that can be approved for delivery in the clinic has not significantly improved. One reason for the ultimate failure of most drugs in clinical trials is the result of most innovations being focused on the early discovery stage. Clearing the many hurdles in the preclinical development stage of kinase downregulators also requires extensive empirical knowledge. With more new technologies starting to impact these efforts, such as the use of AI for the prediction of physicochemical and ADMET properties, we believe that more diverse kinase inhibitors will be delivered to the clinic to fulfill the heterogeneous clinical demands in the near future.
References
Manning, G. et al. The protein kinase complement of the human genome. Science 298, 1912–1934 (2002).
Paul, M. K. & Mukhopadhyay, A. K. Tyrosine kinase—role and significance in Cancer. Int. J. Med. Sci. 1, 101–115 (2004).
Duong-Ly, K. C. & Peterson, J. R. The human kinome and kinase inhibition. Curr. Protoc. Pharmacol. Chapter 2, Unit 2.9 (2013).
Morrison, D. K., Murakami, M. S. & Cleghon, V. Protein kinases and phosphatases in the Drosophila genome. J. Cell Biol. 150, F57–F62 (2000).
Gross, S. D. & Anderson, R. A. Casein kinase I: spatial organization and positioning of a multifunctional protein kinase family. Cell Signal. 10, 699–711 (1998).
Arencibia, J. M. et al. AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim. Biophys. Acta 1834, 1302–1321 (2013).
Hudmon, A. & Schulman, H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 364, 593–611 (2002).
Ferguson, F. M. & Gray, N. S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov. 17, 353–377 (2018).
Roskoski, R. Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2021 update. Pharmacol. Res. 165, 105463 (2021).
Knight, Z. A. & Shokat, K. M. Features of selective kinase inhibitors. Chem. Biol. 12, 621–637 (2005).
Liu, Y. & Gray, N. S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2, 358–364 (2006).
Ruddraraju, K. V. & Zhang, Z.-Y. Covalent inhibition of protein tyrosine phosphatases. Mol. BioSyst. 13, 1257–1279 (2017).
Zhao, Z. & Bourne, P. E. Progress with covalent small-molecule kinase inhibitors. Drug Discov. Today 23, 727–735 (2018).
Pettinger, J. et al. An irreversible inhibitor of HSP72 that unexpectedly targets lysine-56. Angew. Chem. Int. Ed. 56, 3536–3540 (2017).
Abdeldayem, A., Raouf, Y. S., Constantinescu, S. N., Moriggl, R. & Gunning, P. T. Advances in covalent kinase inhibitors. Chem. Soc. Rev. 49, 2617–2687 (2020).
Powers, J. C., Asgian, J. L., Ekici, Ö. D. & James, K. E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 102, 4639–4750 (2002).
Li, D. et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27, 4702–4711 (2008).
Engelman, J. A. et al. PF00299804, an irreversible Pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 67, 11924–11932 (2007).
Rabindran, S. K. et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 64, 3958–3965 (2004).
Mullard, A. 2017 FDA drug approvals. Nat. Rev. Drug Discov. 17, 81–85 (2018).
Pan, Z. et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2, 58–61 (2007).
Patel, V. et al. Comparison of acalabrutinib, a selective Bruton tyrosine kinase inhibitor, with ibrutinib in chronic lymphocytic leukemia cells. Clin. Cancer Res. 23, 3734–3743 (2017).
Eliot, A. C. & Kirsch, J. F. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73, 383–415 (2004).
Akçay, G. et al. Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat. Chem. Biol. 12, 931–936 (2016).
Mukherjee, H. et al. A study of the reactivity of S(VI)–F containing warheads with nucleophilic amino-acid side chains under physiological conditions. Org. Biomol. Chem. 15, 9685–9695 (2017).
Dalton, S. E. et al. Selectively targeting the kinome-conserved lysine of PI3Kδ as a general approach to covalent kinase inhibition. J. Am. Chem. Soc. 140, 932–939 (2018).
Hatcher, J. M. et al. SRPKIN-1: a covalent SRPK1/2 inhibitor that potently converts VEGF from pro-angiogenic to anti-angiogenic isoform. Cell Chem. Biol. 25, 460–470 (2018).
Ban, H. S. et al. Discovery of boron-conjugated 4-anilinoquinazoline as a prolonged inhibitor of EGFRtyrosine kinase. Org. Biomol. Chem. 7, 4415–4427 (2009).
Jain, P. et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood 125, 2062–2067 (2015).
Thress, K. S. et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 21, 560–562 (2015).
Lai, A. C. & Crews, C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114 (2017).
An, S. & Fu, L. Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 36, 553–562 (2018).
Sakamoto, K. M. et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001).
Robb, C. M. et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 53, 7577–7580 (2017).
Olson, C. M. et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 14, 163–170 (2018).
Shimokawa, K. et al. Targeting the allosteric site of oncoprotein BCR-ABL as an alternative strategy for effective target protein degradation. ACS Med. Chem. Lett. 8, 1042–1047 (2017).
Huang, H. T. et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 25, 88–99 e86 (2018).
Bondeson, D. P. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015).
Crew, A. P. et al. Identification and characterization of Von Hippel-Lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-binding kinase 1. J. Med. Chem. 61, 583–598 (2018).
Nalawansha, D. A. & Crews, C. M. PROTACs: an emerging therapeutic modality in precision medicine. Cell Chem. Biol. 27, 998–1014 (2020).
Davis, R. E. et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92 (2010).
Woyach, J. A. et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 370, 2286–2294 (2014).
Sun, Y. et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 28, 779–781 (2018).
Jaime-Figueroa, S., Buhimschi, A. D., Toure, M., Hines, J. & Crews, C. M. Design, synthesis and biological evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett. 30, 126877 (2020).
Xia, L. W. et al. Triazol: a privileged scaffold for proteolysis targeting chimeras. Fut. Med. Chem. 11, 2919–2973 (2019).
Wurz, R. P. et al. A “Click Chemistry Platform” for the rapid synthesis of bispecific molecules for inducing protein degradation. J. Med. Chem. 61, 453–461 (2018).
Lai, A. C. et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 55, 807–810, https://doi.org/10.1002/anie.201507634 (2016).
Donovan, K. A. et al. Mapping the degradable kinome provides a resource for expedited degrader development. Cell 183, 1714–1731.e1710 (2020).
Bond, M. J., Chu, L., Nalawansha, D. A., Li, K. & Crews, C. M. Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent. Sci. 6, 1367–1375 (2020).
Gabizon, R. et al. Efficient targeted degradation via reversible and irreversible covalent PROTACs. J. Am. Chem. Soc. 142, 11734–11742 (2020).
Xue, G. et al. Protein degradation through covalent inhibitor-based PROTACs. Chem. Commun. 56, 1521–1524 (2020).
Guo, W. H. et al. Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry. Nat. Commun. 11, 4268 (2020).
Lu, G. et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014).
Kronke, J. et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014).
Petzold, G., Fischer, E. S. & Thoma, N. H. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature 532, 127–130 (2016).
Pike, A., Williamson, B., Harlfinger, S., Martin, S. & McGinnity, D. F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov. Today 25, 1793–1800 (2020).
Han, T. et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, eaal3755 (2017).
Slabicki, M. et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 585, 293–297 (2020).
Mayor-Ruiz, C. et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 16, 1199–1207 (2020).
Haling, JacobR. et al. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 26, 402–413 (2014).
Garnock-Jones, K. P. Cobimetinib: first global approval. Drugs 75, 1823–1830 (2015).
Rose, A. A. N. Encorafenib and binimetinib for the treatment of BRAF V600E/K-mutated melanoma. Drugs Today 55, 247 (2019).
Lu, X., Smaill, J. B. & Ding, K. New promise and opportunities for allosteric kinase inhibitors. Angew. Chem. Int. Ed. 59, 13764–13776 (2020).
Khan, Z. M. et al. Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 588, 509–514 (2020).
Robert, C. et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 372, 30–39 (2014).
Roskoski, R. Allosteric MEK1/2 inhibitors including cobimetanib and trametinib in the treatment of cutaneous melanomas. Pharmacol. Res. 117, 20–31 (2017).
Gao, Y. et al. V211D mutation in MEK1 causes resistance to MEK inhibitors in colon cancer. Cancer Discov. 9, 1182–1191 (2019).
Jia, Y. et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 534, 129–132 (2016).
To, C. et al. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov. 9, 926–943 (2019).
Yuan-Yuan et al. Effects of an oral allosteric AKT inhibitor (MK-2206) on human nasopharyngeal cancer in vitro and in vivo. Drug Des. Devel. Ther. 8, 1827–1837 (2014).
Ashwell, M. A. et al. Discovery and optimization of a series of 3-(3-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amines: orally bioavailable, selective, and potent ATP-independent Akt inhibitors. J. Med. Chem. 55, 5291–5310 (2012).
Weisner, J. et al. Preclinical efficacy of covalent-allosteric AKT inhibitor borussertib in combination with trametinib in KRAS-mutant pancreatic and colorectal cancer. Cancer Res. 79, 2367–2378 (2019).
Zhang, J. et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463, 501–506 (2010).
Joseph, S. et al. Discovery of Asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J. Med. Chem. 61, 8120–8135 (2018).
Lupardus, P. J. et al. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl Acad. Sci. USA 111, 8025–8030 (2014).
McNally, R. et al. Discovery and structural characterization of ATP-site ligands for the wild-type and V617F mutant JAK2 pseudokinase domain. ACS Chem. Biol. 14, 587–593 (2019).
Wrobleski, S. T., Moslin, R., Lin, S., Zhang, Y. & Weinstein, D. S. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J. Med. Chem. 62, 8973–8995 (2019).
Herbert, C. et al. Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell 23, 489–501 (2013).
Grither, W. R. & Longmore, G. D. Inhibition of tumor–microenvironment interaction and tumor invasion by small-molecule allosteric inhibitor of DDR2 extracellular domain. Proc. Natl Acad. Sci. USA 115, E7786–E7794 (2018).
Lu, S., Shen, Q. & Zhang, J. Allosteric methods and their applications: facilitating the discovery of allosteric drugs and the investigation of allosteric mechanisms. Acc. Chem. Res. 52, 492–500 (2019).
Guarnera, E., Tan, Z. W., Zheng, Z. & Berezovsky, I. N. AlloSigMA: allosteric signaling and mutation analysis server. Bioinformatics 33, 3996–3998 (2017).
Amrhein, J. A., Knapp, S. & Hanke, T. Synthetic opportunities and challenges for macrocyclic kinase inhibitors. J. Med. Chem. 64, 7991–8009 (2021).
Zou, H. et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc. Natl Acad. Sci. USA 112, 3493–3498 (2015).
Johnson, T. W. et al. Discovery of (10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 57, 4720–4744 (2014).
Mesa, R. A. et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol. 4, e225–e236 (2017).
Pallis, M. et al. The multi-kinase inhibitor TG02 overcomes signalling activation by survival factors to deplete MCL1 and XIAP and induce cell death in primary acute myeloid leukaemia cells. Br. J. Haematol. 159, 191–203 (2012).
Madan, B. et al. SB1578, a novel inhibitor of JAK2, FLT3, and c-Fms for the treatment of rheumatoid arthritis. J. Immunol. 189, 4123–4134 (2012).
Zhang, W. et al. The dual MEK/FLT3 inhibitor E6201 exerts cytotoxic activity against acute myeloid leukemia cells harboring resistance-conferring FLT3 mutations. Cancer Res. 76, 1528–1537 (2016).
Hanke, T., Merk, D., Steinhilber, D., Geisslinger, G. & Schubert-Zsilavecz, M. Small molecules with anti-inflammatory properties in clinical development. Pharmacol. Ther. 157, 163–187 (2016).
Drilon, A. et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 7, 963–972 (2017).
Drilon, A. et al. Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that potently inhibits ROS1/TRK/ALK solvent-front mutations. Cancer Discov. 8, 1227–1236 (2018).
Konings, I. R. et al. Phase I and pharmacological study of the broad-spectrum tyrosine kinase inhibitor JNJ-26483327 in patients with advanced solid tumours. Br. J. Cancer 103, 987–992 (2010).
Sche, P. P., McKenzie, K. M., White, J. D. & Austin, D. J. Display cloning: functional identification of natural product receptors using cDNA-phage display. Chem. Biol. 6, 707–716 (1999).
Davis, M. I. et al. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 (2011).
Fabian, M. A. et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336 (2005).
Gugliotta, L. et al. Combination therapy of hydroxycarbamide with anagrelide in patients with essential thrombocythemia in the evaluation of Xagrid(R) efficacy and long-term safety study. Haematologica 99, 679–687 (2014).
Patricelli, M. P. et al. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 46, 350–358 (2007).
Zhao, Q. et al. Broad-spectrum kinase profiling in live cells with lysine-targeted sulfonyl fluoride probes. J. Am. Chem. Soc. 139, 680–685 (2017).
Patricelli, M. P. et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 18, 699–710 (2011).
Nomanbhoy, T. K. et al. Chemoproteomic evaluation of target engagement by the cyclin-dependent kinase 4 and 6 inhibitor palbociclib correlates with cancer cell response. Biochemistry 55, 5434–5441 (2016).
Green, J. L. et al. Direct CDKN2 modulation of CDK4 alters target engagement of CDK4 inhibitor drugs. Mol. Cancer Ther. 18, 771–779 (2019).
Klaeger, S. et al. The target landscape of clinical kinase drugs. Science 358, eaan4368 (2017).
Dittus, L., Werner, T., Muelbaier, M. & Bantscheff, M. Differential Kinobeads profiling for target identification of irreversible kinase inhibitors. ACS Chem. Biol. 12, 2515–2521 (2017).
Medard, G. et al. Optimized chemical proteomics assay for kinase inhibitor profiling. J. Proteome Res. 14, 1574–1586 (2015).
Machleidt, T. et al. NanoBRET—a novel BRET platform for the analysis of protein–protein interactions. ACS Chem. Biol. 10, 1797–1804 (2015).
Robers, M. B. et al. Target engagement and drug residence time can be observed in living cells with BRET. Nat. Commun. 6, 10091 (2015).
Vasta, J. D. et al. Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell Chem. Biol. 25, 206–214 (2018).
Wells, C. I. et al. Quantifying CDK inhibitor selectivity in live cells. Nat. Commun. 11, 2743 (2020).
Huynh, K. & Partch, C. L. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 79, 28.9.1–28.9.14 (2015).
Vivoli, M., Novak, H. R., Littlechild, J. A. & Harmer, N. J. Determination of protein-ligand interactions using differential scanning fluorimetry. J. Vis. Exp. 13, 51809 (2014).
Niesen, F. H., Berglund, H. & Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 (2007).
Pantoliano, M. W. et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 6, 429–440 (2001).
Elkins, J. M. et al. Comprehensive characterization of the published kinase inhibitor set. Nat. Biotechnol. 34, 95–103 (2016).
Valenti, D. et al. Set-up and screening of a fragment library targeting the 14-3-3 protein interface. MedChemComm 10, 1796–1802 (2019).
Counago, R. M. et al. Structural characterization of human vaccinia-related kinases (VRK) bound to small-molecule inhibitors identifies different P-loop conformations. Sci. Rep. 7, 7501 (2017).
Serafim, R. A. M. et al. Development of pyridine-based inhibitors for the human vaccinia-related kinases 1 and 2. ACS Med. Chem. Lett. 10, 1266–1271 (2019).
Gao, K., Oerlemans, R. & Groves, M. R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 12, 85–104 (2020).
Moreau, M. J., Morin, I. & Schaeffer, P. M. Quantitative determination of protein stability and ligand binding using a green fluorescent protein reporter system. Mol. Biosyst. 6, 1285–1292 (2010).
Alexander, C. G. et al. Novel microscale approaches for easy, rapid determination of protein stability in academic and commercial settings. Biochim. Biophys. Acta 1844, 2241–2250 (2014).
Savitski, M. M. et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784 (2014).
Franken, H. et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 10, 1567–1593 (2015).
Martinez Molina, D. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 (2013).
Vartanian, S. et al. Application of mass spectrometry profiling to establish brusatol as an inhibitor of global protein synthesis. Mol. Cell Proteom. 15, 1220–1231 (2016).
Mateus, A. et al. Thermal proteome profiling in bacteria: probing protein state in vivo. Mol. Syst. Biol. 14, e8242 (2018).
Reinhard, F. B. et al. Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat. Methods 12, 1129–1131 (2015).
Kalxdorf, M. et al. Cell surface thermal proteome profiling tracks perturbations and drug targets on the plasma membrane. Nat. Methods 18, 84–91 (2021).
Meier, F. et al. Parallel accumulation-serial fragmentation (PASEF): multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. J. Proteome Res. 14, 5378–5387 (2015).
Meier, F., Geyer, P. E., Virreira Winter, S., Cox, J. & Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 min. Nat. Methods 15, 440–448 (2018).
Mateus, A., Maatta, T. A. & Savitski, M. M. Thermal proteome profiling: unbiased assessment of protein state through heat-induced stability changes. Proteome Sci. 15, 13 (2016).
Becher, I. et al. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 12, 908–910 (2016).
Zhang, C., Habets, G. & Bollag, G. Interrogating the kinome. Nat. Biotechnol. 29, 981–983 (2011).
Rudolf, A. F., Skovgaard, T., Knapp, S., Jensen, L. J. & Berthelsen, J. A comparison of protein kinases inhibitor screening methods using both enzymatic activity and binding affinity determination. PLoS ONE 9, e98800 (2014).
Tanega, C. et al. Comparison of bioluminescent kinase assays using substrate depletion and product formation. Assay. Drug Dev. Technol. 7, 606–614 (2009).
Anastassiadis, T., Deacon, S. W., Devarajan, K., Ma, H. & Peterson, J. R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045 (2011).
Daley, G. Q. & Baltimore, D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc. Natl Acad. Sci. USA 85, 9312–9316 (1988).
Melnick, J. S. et al. An efficient rapid system for profiling the cellular activities of molecular libraries. Proc. Natl Acad. Sci. USA 103, 3153–3158 (2006).
Kesarwani, M., Huber, E., Kincaid, Z. & Azam, M. A method for screening and validation of resistant mutations against kinase inhibitors. J. Vis. Exp. 94, 51984 (2014).
Cools, J. et al. Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. 64, 6385–6389 (2004).
Pauwels, D., Sweron, B. & Cools, J. The N676D and G697R mutations in the kinase domain of FLT3 confer resistance to the inhibitor AC220. Haematologica 97, 1773–1774 (2012).
Perrin, D., Frémaux, C., Besson, D., Sauer, W. H. & Scheer, A. A microfluidics-based mobility shift assay to discover new tyrosine phosphatase inhibitors. J. Biomol. Screen. 11, 996–1004 (2006).
Perrin, D., Frémaux, C. & Shutes, A. Capillary microfluidic electrophoretic mobility shift assays: application to enzymatic assays in drug discovery. Expert Opin. Drug Discov. 5, 51–63 (2010).
Swinney, D. C. & Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 10, 507–519 (2011).
Swinney, D. C. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther. 93, 299–301 (2013).
Martel, R. R., Klicius, J. & Galet, S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can. J. Physiol. Pharm. 55, 48–51 (1977).
Choi, J., Chen, J., Schreiber, S. L. & Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273, 239–242 (1996).
Saporito, M. S., Ochman, A. R., Lipinski, C. A., Handler, J. A. & Reaume, A. G. MLR-1023 is a potent and selective allosteric activator of lyn kinase in vitro that improves glucose tolerance in vivo. J. Pharmacol. Exp. Ther. 342, 15–22 (2012).
Zhang, H. et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 175, 1244–1258.e1226 (2018).
Riggs, J. R. et al. The discovery of a dual TTK protein kinase/CDC2-like kinase (CLK2) inhibitor for the treatment of triple negative breast cancer initiated from a phenotypic screen. J. Med. Chem. 60, 8989–9002 (2017).
Huang, Z. et al. Discovery of 5-(3,4-difluorophenyl)-3-(pyrazol-4-yl)-7-azaindole (GNF3809) for β-cell survival in type 1 diabetes. ACS Omega 4, 3571–3581 (2019).
Mitra, A., Mishra, L. & Li, S. Technologies for deriving primary tumor cells for use in personalized cancer therapy. Trends Biotechnol. 31, 347–354 (2013).
Liang, X. et al. Discovery of N-(4-(6-acetamidopyrimidin-4-yloxy)phenyl)-2-(2-(trifluoromethyl)phenyl)acetamide (CHMFL-FLT3-335) as a potent FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD) mutant selective inhibitor for acute myeloid leukemia. J. Med. Chem. 62, 875–892 (2019).
Wu, Y. et al. Discovery of 2-(4-chloro-3-(trifluoromethyl)phenyl)-N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)phenyl)acetamide (CHMFL-KIT-64) as a novel orally available potent inhibitor against broad-spectrum mutants of c-KIT kinase for gastrointestinal stromal tumors. J. Med. Chem. 62, 6083–6101 (2019).
Lee, J.-K. et al. Pharmacogenomic landscape of patient-derived tumor cells informs precision oncology therapy. Nat. Genet. 50, 1399–1411 (2018).
Brodin, B. A. et al. Drug sensitivity testing on patient-derived sarcoma cells predicts patient response to treatment and identifies c-Sarc inhibitors as active drugs for translocation sarcomas. Br. J. Cancer 120, 435–443 (2019).
Bleijs, M., van de Wetering, M., Clevers, H. & Drost, J. Xenograft and organoid model systems in cancer research. EMBO J. 38, e101654 (2019).
Drost, J. & Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 18, 407–418 (2018).
Tuveson, D. & Clevers, H. Cancer modeling meets human organoid technology. Science 364, 952–955 (2019).
Palechor-Ceron, N., Krawczyk, E., Dakic, A. & Liu, X. Conditional reprogramming for patient-derived cancer models and next-generation living biobanks. Cells 8, 1327 (2019).
Kretzschmar, K. Cancer research using organoid technology. J. Mol. Med. 99, 501–515 (2021).
Ferguson, F. M. et al. Discovery of a selective inhibitor of doublecortin like kinase 1. Nat. Chem. Biol. 16, 635–643 (2020).
Ferguson, F. M., Nabet, B., Raghavan, S., Liu, Y. & Gray, N. S. Discovery of a selective inhibitor of doublecortin like kinase 1. Nat. Chem. Biol. 16, 1–9 (2020).
Abdirahman, S. M., Christie, M., Preaudet, A., Burstroem, M. C. U. & Putoczki, T. L. A Biobank of colorectal cancer patient-derived xenografts. Cancers 12, 2340 (2020).
Goto, T. Patient-derived tumor xenograft models: toward the establishment of precision cancer medicine. J. Pers. Med. 10, 64 (2020).
Yoshida, G. J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 13, 4 (2020).
Wang, Y. et al. HER2 exon 20 insertions in non-small cell lung cancer are sensitive to the irreversible pan-HER receptor tyrosine kinase inhibitor pyrotinib. Ann. Oncol. 30, 447–455 (2018).
Li, C. J., Liu, Y., Bell, T., Wang, J. & Wang, M. Novel Bruton’s tyrosine kinase inhibitor BGB-3111 demonstrates potent activity in mantle cell lymphoma. Blood 128, 5374 (2016).
Pauli, C., Hopkins, B. D., Prandi, D., Shaw, R. & Rubin, M. A. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 7, 462 (2017).
Lerner, R. A. & Brenner, S. DNA-encoded compound libraries as open source: a powerful pathway to new drugs. Angew. Chem. Int. Ed. 56, 1164–1165 (2017).
Belyanskaya, S. L., Ding, Y., Callahan, J. F., Lazaar, A. L. & Israel, D. I. Discovering drugs with DNA-encoded library technology: from concept to clinic with an inhibitor of soluble epoxide hydrolase. ChemBioChem 18, 837–842 (2017).
Harris, P. A. et al. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 60, 1247–1261 (2017).
Madsen, D. et al. An overview of DNA-encoded libraries: a versatile tool for drug discovery. Prog. Med. Chem. 59, 181–249 (2020).
Yang, X., Wang, Y., Byrne, R., Schneider, G. & Yang, S. Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev. 119, 10520–10594 (2019).
Cavasotto, C. N. & Di Filippo, J. I. Artificial intelligence in the early stages of drug discovery. Arch. Biochem. Biophys. 698, 108730 (2021).
Gagic, Z., Ruzic, D., Djokovic, N., Djikic, T. & Nikolic, K. In silico methods for design of kinase inhibitors as anticancer drugs. Front. Chem. 7, 873 (2020).
Kannan, S. et al. Correction to probing the binding mechanism of MNK inhibitors by docking and molecular dynamics simulations. Biochemistry 56, 5964–5965 (2017).
El Rashedy, A. A., Olotu, F. A. & Soliman, M. E. S. Dual drug targeting of mutant Bcr-Abl induces inactive conformation: new strategy for the treatment of chronic myeloid leukemia and overcoming monotherapy resistance. Chem. Biodivers. 15, e1700533 (2018).
Fang, C. & Xiao, Z. Receptor-based 3D-QSAR in drug design: methods and applications in kinase studies. Curr. Top. Med. Chem. 16, 1463–1477 (2016).
Gagic, Z., Ruzic, D., Djokovic, N., Djikic, T. & Nikolic, K. In silico methods for design of kinase inhibitors as anticancer drugs. Front. Chem. 7, 873 (2019).
Koneru, J. K., Sinha, S. & Mondal, J. In silico reoptimization of binding affinity and drug-resistance circumvention ability in kinase inhibitors: a case study with rl-45 and src kinase. J. Phys. Chem. 123, 6664–6672 (2019).
Myers, S. H., Temps, C., Houston, D. R., Brunton, V. G. & Unciti-Broceta, A. Development of potent inhibitors of receptor tyrosine kinases by ligand-based drug design and target-biased phenotypic screening. J. Med. Chem. 61, 2104–2110 (2018).
Hussain, A. & Verma, C. K. Ligand- and structure-based pharmacophore modeling, docking study reveals 2-[[4-[6-(isopropylamino)pyrimidin-4-yl]-1H-pyrrolo[2,3-b] pyridin-6-yl] amino] ethanol as a potential anticancer agent of CDK9/cyclin T1 kinase. J. Cancer Res. Ther. 15, 1131–1140 (2019).
Rathi, E., Kumar, A., Suvarna & Kini, G. Design of potential inhibitors and prediction of their activity by the structural insight of VEGFR2 inhibitors: atom-based 3D-QSAR, fingerprint-based 2D QSAR and off-target analysis. ChemistrySelect 5, 689–703 (2020).
Zhao, M. et al. 2D-QSAR and 3D-QSAR analyses for EGFR inhibitors. BioMed. Res. Int. 2017, 4649191 (2017).
Balazs, A. Y. S. et al. Free ligand 1D NMR conformational signatures to enhance structure based drug design of a Mcl-1 inhibitor (AZD5991) and other synthetic macrocycles. J. Med. Chem. 62, 9418–9437 (2019).
Long, W., Liu, P., Li, Q., Xu, Y. & Gao, J. 3D-QSAR studies on a class of IKK-2 inhibitors with GALAHAD used to develop molecular alignment models. QSAR Comb. Sci. 27, 1113–1119 (2008).
Lauria, A. et al. IKK-β inhibitors: an analysis of drug–receptor interaction by using molecular docking and pharmacophore 3D-QSAR approaches. J. Mol. Graph. Model. 29, 72–81 (2010).
Naboulsi, I., Aboulmouhajir, A., Kouisni, L., Bekkaoui, F. & Yasri, A. Combining a QSAR approach and structural analysis to derive an SAR map of Lyn kinase inhibition. Molecules 23, 3271 (2018).
Yu, Z. et al. 3D-QSAR modeling and molecular docking study on Mer kinase inhibitors of pyridine-substituted pyrimidines. Mol. Divers. 19, 135–147 (2015).
Pereira, W. & Camps, I. De novo design of new inhibitor of mutated tyrosine-kinase for the myeloid leukemia treatment. Curr. Pharm. Des. 22, 5057–5064 (2016).
Zhavoronkov, A. et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37, 1038–1040 (2019).
Li, Z. et al. KinomeX: a web application for predicting kinome-wide polypharmacology effect of small molecules. Bioinformatics 35, 5354–5356 (2019).
Lo, Y.-C. et al. Computational analysis of kinase inhibitor selectivity using structural knowledge. Bioinformatics 35, 235–242 (2018).
Shen, Z., Yan, Y. H., Yang, S., Zhu, S. & Li, H. ProfKin: a comprehensive web server for structure-based kinase selectivity profiling. Eur. J. Med. Chem. 225, 113772 (2020).
Wengner, A. M. et al. The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage-inducing or repair-compromising therapies in preclinical cancer models. Mol. Cancer Ther. 19, 26–38 (2020).
Yap, T. A. et al. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J. Clin. Oncol. 38, 3195–3204 (2020).
Xun, Q., Wang, Z., Hu, X., Ding, K. & Lu, X. Small-molecule CSF1R inhibitors as anticancer agents. Curr. Med. Chem. 27, 3944–3966 (2020).
Sawasdikosol, S. & Burakoff, S. A perspective on HPK1 as a novel immuno-oncology drug target. eLife 9, e55122 (2020).
Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
Yang, Z. et al. Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently. Cell Res. https://doi.org/10.1038/s41422-021-00533-6 (2021).
Henning, N. J. et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Preprint at bioRxiv 2021.04.30.441959. https://doi.org/10.1101/2021.04.30.441959 (2021).
Lee, S. et al. Recent advances in development of hetero-bivalent kinase inhibitors. Eur. J. Med. Chem. 216, 113318 (2021).
Jenardhanan, P., Panneerselvam, M. & Mathur, P. Targeting kinase interaction networks: a new paradigm in PPI based design of kinase inhibitors. Curr. Med. Chem. 19, 467–485 (2019).
Lu, H. et al. Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Sig. Transduct. Target Ther. 5, 213 (2020).
Harris, P. A. et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methylbenzenesulfonamide(Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J. Med. Chem. 51, 4632–4640 (2008).
Kesarwani, M. et al. Targeting substrate-site in Jak2 kinase prevents emergence of genetic resistance. Sci. Rep. 5, 14538 (2015).
Gilmartin, A. G. et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res. 17, 989–1000 (2011).
Barf, T. et al. Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J. Pharmacol. Exp. Ther. 363, 240 (2017).
Liu, X., Feng, D., Zheng, M., Cui, Y. & Zhong, D. Characterization of covalent binding of tyrosine kinase inhibitors to plasma proteins. Drug Metab. Pharmacokinet. 35, 456–465 (2020).
Yan, W. et al. Insights into current tropomyosin receptor kinase (TRK) inhibitors: development and clinical application. J. Med. Chem. 62, 1731–1760 (2019).
Xu, W. et al. Safety, tolerability and efficacy of orelabrutinib, once a day, to treat Chinese patients with relapsed or refractory chronic lymphocytic leukemia/small cell leukemia. Blood 134, 4319–4319 (2019).
Wu, L. et al. Discovery of pemigatinib: a potent and selective fibroblast growth factor receptor (FGFR) inhibitor. J. Med. Chem. 64, 10666–10679 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81903650), the Natural Science Foundation of Anhui Province (Grant Nos. 1808085MH274 and 2008085MH274), the China Postdoctoral Science Foundation (Grant Nos. 2020M671916 and 2019M652057), the Postdoctoral Science Foundation of Anhui Province (Grant Nos. 2019B326 and 2019B300), the Frontier Science Key Research Program of CAS (Grant No. QYZDB-SSW-SLH037), and the CASHIPS Director’s Found (Grant Nos. BJPY2019A03 and YZJJZX202011).
Author information
Authors and Affiliations
Contributions
B.W., H.W., C.H and H.W. drafted the manuscript. W.W. and J.L. discussed and revised the manuscript. Q.L. organized and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, B., Wu, H., Hu, C. et al. An overview of kinase downregulators and recent advances in discovery approaches. Sig Transduct Target Ther 6, 423 (2021). https://doi.org/10.1038/s41392-021-00826-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00826-7
- Springer Nature Limited
This article is cited by
-
A novel in silico scaffold-hopping method for drug repositioning in rare and intractable diseases
Scientific Reports (2023)