
Abstract
Wnt/β-catenin signaling has been broadly implicated in human cancers and experimental cancer models of animals. Aberrant activation of Wnt/β-catenin signaling is tightly linked with the increment of prevalence, advancement of malignant progression, development of poor prognostics, and even ascendence of the cancer-associated mortality. Early experimental investigations have proposed the theoretical potential that efficient repression of this signaling might provide promising therapeutic choices in managing various types of cancers. Up to date, many therapies targeting Wnt/β-catenin signaling in cancers have been developed, which is assumed to endow clinicians with new opportunities of developing more satisfactory and precise remedies for cancer patients with aberrant Wnt/β-catenin signaling. However, current facts indicate that the clinical translations of Wnt/β-catenin signaling-dependent targeted therapies have faced un-neglectable crises and challenges. Therefore, in this study, we systematically reviewed the most updated knowledge of Wnt/β-catenin signaling in cancers and relatively targeted therapies to generate a clearer and more accurate awareness of both the developmental stage and underlying limitations of Wnt/β-catenin-targeted therapies in cancers. Insights of this study will help readers better understand the roles of Wnt/β-catenin signaling in cancers and provide insights to acknowledge the current opportunities and challenges of targeting this signaling in cancers.
Similar content being viewed by others
Introduction
As an evolutionarily conserved signaling that governs numerously vital embryonic and somatic processes, such as cell fate determination, organogenesis, tissue homeostasis, and a variety of pathological conditions, Wnt/β-catenin signaling also plays crucial roles in cancers.1 Aberrant Wnt/β-catenin signaling has been uncovered to be tightly woven with many aspects of cancers, including the onset, progression, malignant transformation, and so on.2,3 Evidence-based medicine has further proved that the abnormal activation of this signaling showed non-negligible effects on cancer-associated mortality.4,5,6 Despite these broad acknowledgments of the significant impact of Wnt/β-catenin signaling on cancers, advances in targeted therapies remain largely incipient. Considering the extremely heavy clinical burden of Wnt/β-catenin-associated cancers globally, it is urgent to comprehensively summarize the up-to-date knowledge of Wnt/β-catenin signaling in cancers and to clarify the current status and challenges of developing Wnt/β-catenin-associated targeted therapies.
Collectively, it is necessary to systematically review the experimental and clinical knowledge of Wnt/β-catenin signaling in cancers and present the status quo of Wnt/β-catenin-targeted therapies in cancers. With the rapid development of modern pharmacology and evidence-based medicine, certain approaches of targeting Wnt/β-catenin signaling in cancers have already achieved the path of clinical trials. These promising advances will endow researchers and clinicians with more choices and fundaments to better manage aberrant Wnt/β-catenin-associated cancers. In this article, we have systematically reviewed the most updated knowledge of Wnt/β-catenin signaling in caners and targeted therapies in accordance. Therefore, we sought to provide readers with the latest progress of Wnt/β-catenin signaling in cancers and demonstrate both opportunities and challenges of Wnt/β-catenin signaling-dependent targeted therapies in cancers.
Brief intro of Wnt/β-catenin
The nomination of Wnt is after wingless in drosophila and int1 in mammalians.7,8 As the prerequisite of understanding the roles of Wnt/β-catenin in cancer, in this part, we will briefly introduce the signaling transduction. Numerous researches have already depicted an integral scene of the core components of β-catenin-dependent Wnt signaling. It consists of extracellular ligands, agonists; trans-membraned receptors/co-receptors; intracellular compounds including disheveled (Dsh in drosophila and Dvl in mammals), degradation complex comprising glycogen synthase kinase 3 β (Gsk3β), casein kinase1α (CK1α), Axin/conductin, and adenomatous polyposis coli (Apc), β-catenin, and transcription factors.9
Extracellular Wnt ligands are inevitable to switch-on downstream signaling cascade. The extracellular secretion and functions of Wnt ligands depend on post-transcriptional modifications.10 The post-transcriptional modifications of Wnt ligands mainly include glycosylation, palmitoylation, and acylation.11,12,13 Especially, the acylation is necessary for extracellular transport and receptor/co-receptors recognition and bounding.14,15
With respect to the receptor/co-receptors of Wnt/β-catenin signaling, including frizzled (Fzd) receptors and Lrp co-receptors. For Fzds family, it owns at least 10 members of G protein-coupled receptor (GPCR).16 The highly conserved cysteine-rich domain (CRD) of Fzds manipulates the ligand recognition and binding.15,17 In addition, as co-receptors to Wnt ligands, Lrps consist of Lrp5 and 6, whose extracellular domains interact with Fzds and then its intracellular domains trigger further signaling transduction.18,19 In addition to receptor/co-receptors, there also exist certain extracellular regulators that can influence the ligand–receptor/co-receptors interaction. For instance, the R-spondins (Rspo), member 1/2/3/4,20 which coordinates with leucine-rich repeat-containing GPCR (Lgr) 4/5/6 to enhance Wnt/β-catenin signaling.21,22,23,24 Specifically, the Rspo-Lgr complex increases Lrp5/6 phosphorylation and inactivates Wnt repressors Rnf43 and Znrf3.23,25,26 Rnf43 and Znrf3 are E3 ubiquitin ligases that mediate the degradation of Fzds.26,27 Furthermore, certain intracellular regulators can also abolish the signaling cascade downstream of ligand–receptor interaction. Dsh, a cytoplasmic protein with three highly conserved sections,28 directly interacts with the C-terminal of Fzds via its PDZ region.29,30
Finally, after receiving the upstream activation signals, β-catenin functions as the ultimate effector.31,32 Without canonical Wnt ligands, β-catenin binds to cadherin of cytoplasmic sides rather than being transported into the nucleus, further phosphorylated and eliminated by degradation complex.32,33,34 Phosphorylated β-catenin is degraded by the ubiquitin-proteasome system to keep the low level of free β-catenin in the cytoplasm.32,33 Conversely, once being activated, intracellular β-catenin is rapidly enriched, and then trans-localized into the nucleus to regulate gene expressions.32,33 Apart from directly manipulating gene transcription as a TF, β-catenin can also form a transcriptional complex with Lef/Tcf via its armadillo repeats regions.32,35 Intriguingly, the degradation complex of β-catenin is concise and complicated. As soon as the degradation complex converts into the active form, it phosphorylates β-catenin for ubiquitination, which sends it to the proteasome. When the degradation complex is deactivated, β-catenin accumulates and influxes into the nucleus to initiate the transcription of downstream genes.
Wnt/β-catenin signaling in cancers
Basing on the subtle summary of Wnt/β-catenin signaling, in this part, we aimed to further demonstrate the roles of Wnt/β-catenin signaling in cancers. Specifically, we will follow the progressively organized structure going behind the signal conduction cascade of this pathway: extracellular, membrane-linked, and intracellular (cytoplasmatic and nuclear) compositions.
Extracellular compositions
Porcupine (Porc)
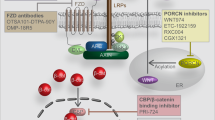
Porc is a membrane-bound O-acyltransferase (MBOAT), by whose palmitoylation function Wnt ligands can be subsequently secreted and recognized.36 After being palmitoylated, Wnt ligands will bind to wntless and be transported into cell membrane from Golgi apparatus37 (Fig. 1). Thereout repressing Porc is a candidate way against tumors with aberrant Wnt/β-catenin activation. Nowadays, inhibitors targeting Porc have been uncovered to be underlyingly beneficial for diverse types of cancers.38

The extracellular components and signaling transduction of Wnt/β-catenin signaling. In this figure, we do not distinguish the autocrine or paracrine patterns of Wnt ligands
Ligands
The Fzds are responsible for recognizing and receiving canonical Wnt ligands, which are defined as β-catenin-dependent ligands. In this review, we focus on the canonical Wnt ligands, including Wnt1, 2, 3, and 3a.
Wnt1
Previous studies have shown that knockout of wg, the homolog gene of mammalian Wnt1, caused the wingless phenotype in Drosophila.7,8 Experimental and clinical analysis further revealed the frequent abnormal upregulations of Wnt1 in massive cancers.39,40,41,42 Abnormal Wnt1 expressing patients comprised the majority of cancer patients of non-small cell lung cancer (NSCLC).43 Nevertheless, a Wnt1-dependent gene, Wnt1-inducible signaling pathway protein 2 (WISP2), was reported to effectively undermine the immunologic evasion of cancer cells.39,44 In addition to the direct involvements of Wnt1 in cancers, certain upstream modulators of Wnt1 can repress the viability of a few sorts of cancer cells, containing RU484, MicroRNA-140-5p, and SJ26.45,46,47
Encouragingly, inhibiting Wnt1 relieved the growth and progression of breast cancer in a transgenic murine model. Conversely, overexpression of Wnt1 promoted the growth of cancers.48,49 Therapeutically, Blocking Wnt1 has been found to strengthen the apoptosis of colorectal cancer (CRC) cells via the utilization of Wif1, Wnt1-specific siRNAs, and neutralizing antibodies.40
Wnt2
Similarly, the overexpression of Wnt2 has also been detected in human fibroadenomas, breast cancer, pancreatic cancer, and CRC.50,51,52,53 Despite the concerns that in CRC (CRC) Wnt2 perhaps accelerated its migration and invasion,52,54 on the contrary, in other kinds of cancers such as gastric, pancreatic, and NSCLC, Wnt2 aggregates the cancer progression.55,56,57 With respect to its therapeutic potentials, direct silence of Wnt2 alleviated the xenograft breast cancer growth and rescued the malignance and the chemo-drugs resistance in breast cancers.58,59
Wnt3
Wnt3 is a homologous gene of Wnt3a, and the similarity is up to 84.2% total amino-acid identity in humans.60 The level of Wnt3 mRNA was obviously upregulated in primary breast and rectal cancer.60 Wnt3 plays a cacoethic role in multiple cancers, such as CRC,61 breast cancer,62,63 NSCLC,64,65 and prostate cancer.66 Specifically, the excitation of Wnt3 speeds up the tumorigenesis of CRC.61 Furthermore, it has been implied to elevate the epithelium–mesenchyme transit (EMT) of breast cancers through Wnt/β-catenin.62,63 In terms of therapeutic choices, the downregulation relieved the progression of CRC by reducing cancer cell proliferation and migration.61 For NSCLC, knockdown of Wnt3 could increase drug sensitivity.64,65 In prostate cancer, inhibiting Wnt3 signaling by the deficiency of trophinin-associated protein attenuated the cancer growth.66 The demethylation of non-coding RNA miR-1247-5p provided an optional remedy for human hepatocellular carcinoma (HCC) via inhibiting Wnt3.67
Wnt3a
As the strongest Wnt/β-catenin stimulator, Wnt3a has been proposed to participate in numerous cancers. For instance, in most solid tumors, Wnt3a promoted the tumorigenesis and progression of CRC, prostate, liver, and lung cancers.68,69,70,71 Mechanistically, Wnt3a enhanced cancer cells proliferation, differentiation, migration, and self-renewal,72,73,74 and conversely inhibits cell apoptosis depending on activating Wnt/β-catenin.68 In leukemia, a study indicated that Wnt3a, activating Wnt/β-catenin, suppressed the proliferation of cancer cells.75
Therapeutically, in prostate cancers, targeting Wnt3a through Traf6 either Tmem64 restrained tumor development.76,77 For liver cancers, targeting Wnt3a by miRNA-195 and miRNA‑214 presented the possibility of cancer management.78,79
Membrane-linked compositions
Receptors and co-receptors
Fzds
Fzds, a subset of seven-transmembrane protein, are the principal receptors of canonical Wnt ligands.80 Fzds, mainly including subfamilies Fzd1/2/7, Fzd3/6, Fzd4, Fzd5/8, and Fzd9/10, are ubiquitously expressed in most animal species but not in plants and single-cell eukaryotes.80 The N-terminal CRD domain of Fzds spontaneously binds to Wnt ligands and Lrp5/6 co-receptors.15,18 The C-terminus of Fzds is localized in the cytosol which recruits and binds to Dsh to intracellularly trigger subsequent signal cascades.81 Fzds and Lrp5/6 are indispensable for Wnt/β-catenin activation, and non-eligibly, Fzds receptors, and Lrp5/6 co-receptors are both oncogenic under certain conditions.82,83,84,85 Except for main canonical Wnt ligands compromising Wnt1, 3, and 3a, there still exists several non-canonical Wnt ligands including Wnt5a, 7a, and 7b known to interact with Fzds under specialized circumstances.86,87,88
Fzd3 could be promotive for the development of Ewing sarcoma and breast cancer.89,90,91 Also, Fzd4 and Fzd5 were reported to be significantly increased in prostate cancer.92,93 Fzd6 highly emerged in CRC, breast cancer, and bladder cancer.94,95,96 Most importantly, Fzd7 is vitally essential in numerous cancers like HCC, breast cancer, gastric cancer, CRC and so on.97 Detailedly, Wnt3a-Fzd7 dependent Wnt/β-catenin signaling exaggerated the tumorigenesis and advancement of HCC.98 Prostate cancer metastasis is related to ERG-induced Fzd8 up-regulation.99 Besides, Fzd10 plays an important positive role in various cancers, at least in CRC development, but the level of Fzd10 is down in metastatic cancers.100 The Fzd10 expression may be a prognostic marker in CRC,100 and the same is true in synovial sarcoma.101
Next, the following part is about to concisely summarize the potential therapeutic values of targeting Fzds in cancers. Fzd1 is related to drug resistance in cancers, subsequently, inhibiting Fzd1 attenuating the resistance.102,103,104 Moreover, repressing Fzd1 via knocking down Fzd1, using rosiglitazone or miR-135b decreased the metastasis of breast cancer.102,103,104,105 For targeting Fzd3, by which some non-coding RNAs, like miRNA-505, miRNA-493, and HOXD cluster antisense RNA 1 relieve cancer progression.106,107,108 And Fzd7 deficiency is enough to combat Apc carcinogenesising.84 Fzd3 has a certain promotion function on Ewing sarcoma, breast cancer.89,90,91 Research shows that Fzd4 expression is extremely high in human prostate cancer cells and some non-coding RNA, like microRNA-505, microRNA-493, and HOXD cluster antisense RNA 1 (HOXD-AS1), relieves cancer progression by Fzd3.92,106,107,108 Fzd5 functions in prostate and gastric cancer,93,94 and Fzd6 in colorectal, breast, and bladder cancer.94,95,96 Fzd7 overexpression is detected in numerous cancers, like hepatocellular, breast, and CRC.97 WNT3A-Fzd7 interaction activates Wnt/β-catenin signaling in human hepatocellular carcinoma cells and promotes tumorigenesis.98 Fzd7 is vitally essential for the development of gastric cancer and it is the major receptor of Wnt ligands.84 Inhibitor targeting Fzd7 is effective for treating gastric cancer, no matter with Apc mutations or not.84 Fzd7 may be an extraordinarily effective therapy for gastric and CRCs.84,109 ERG is overexpression in most prostate cancer and Wnt/β-catenin signaling is activated.99 Prostate cancer metastasis is related to ERG inducing Fzd8 up-regulation.99 Besides, Fzd10 plays an important positive role in various cancers, at least in CRC development, but the level of Fzd10 is down in metastatic cancers.100 The Fzd10 expression may be a prognostic marker in CRC,100 and the same is true in synovial sarcoma.101
Lrps
As co-receptors of Wnt ligands Lrp5/6 are tightly associated with the growth of Wnt-hypersensitive tumors. The single-domain antibody fragments of Lrp5/6 can effectively relieve the development of intestinal cancers.110 Generally, the truncated LRP5 amplified Wnt/β-catenin signaling to severely promote the growth of parathyroid tumors.111 Herein, stabilizing of Lrp5 by Hsp90ab1 enhanced gastric cancer progression.112 Moreover, LRP6 also plays a positive role in various cancers, such as CRC, breast cancer, prostate cancer, and so on.83,113,114,115 Basing on these facts, LRP6 neutralizing antibodies were demonstrated to repress tumorigenesis.116 It is of note that contrarily targeting Lrp5/6 may be not eligible for managing metastasis of breast cancer.
Extracellular repressors
The abolishers of receptor/co-receptors
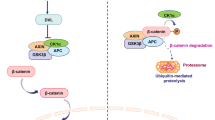
Znrf3 and Rnf43, as transmembrane E3 ubiquitin ligases on cell surface, are negative regulators of Wnt/β-catenin signaling.26,27 Researches indicated that Znrf3/Rnf43 attenuated Wnt signaling by selectively ubiquitinating receptors/co-receptors of Wnts (Fzds and Lrps) to advance proteins degradation.26,27 Rspos, binding with Lgrs, can activate Wnt/β-catenin signaling by dually enhancing Lrp5/6 activity and removing Znrf3/Rnf43 from cell membrane26,27 (Fig. 2). Rspos could as well activate Wnt/β-catenin signaling via interacting with HSPGs independent of Lgrs.117 Interestingly, Rspo2 mutations were unraveled to associate with tetra-amelia syndrome, contributing to the destruction of Rspo2-Lgr binding.118 Current evidence depicted the crucial roles of somatic mutations of Rspos in cancers. In detail, Rspo fusions are regarded as important for tumorigenesis in CRC, by activating Wnt/β-catenin signaling.119 Rspo2/3 chromosome rearrangements can initiate and maintain tumor development absolutely through Wnt signals.120

The membrane-linked components and signaling transduction of Wnt/β-catenin signaling. Ub ubiquitin, ① the switch-on of Fzd/Lrp ubiquitination, ② the switch-ff of Fzd/Lrp ubiquitination via Rspo function
Apart from Rspos, the mutations of Znrf3/Rnf43 were also proposed to participate in CRC. The most common mutations of Znrf3/Rnf43 were missense and truncating mutations, respectively.121 Znrf3 mutations were frequently detected in adrenocortical carcinoma, uterine corpus endometrial carcinoma, and skin cutaneous melanoma. Similarly, Rnf43 mutations were overall found in uterine corpus endometrial carcinoma, stomach adenocarcinoma, colorectal adenocarcinoma, ovarian cancer, and pancreatic ductal adenocarcinoma.121,122 Znrf3 and Rnf43 were not essential in the intestine but dysregulation of Znrf3/Rnf43 was important for the growth of CRC.26,27,123 Mutations of Rnf43, resulting in E3 ubiquitin ligases function loss, promoted CRC development and poor prognosis.38 Inactivating mutations of Rnf43 were related to Wnt dependency. LGK974, targeting on Wnt/β-catenin signaling, alleviated Rnf43 mutation-associated pancreatic cancer cell proliferation, however, which did not affect the non-mutant cancers.124 For CRC, Rspo/Rnf43 dysregulation plays a positive role in development and dominates over Znrf3.125,126 There still needs further researches to explore the relation between Wnt/β-catenin and Znrf3/Rnf43, as well as them in cancers.
The antagonists of receptor/co-receptors
Endogenous repressors of Wnt/β-catenin signaling can be divided into two groups: the reversible and the irreversible ones. The former group including competitively binds to the receptor/co-receptor to block the ligand–receptors interaction, like Dkks, Sost, sfrp, and so on. The latter group functions through different mechanisms. Notum deacetylates Wnts and permanently invalidates the recognition. Tikis hydrolyze Wnts to polymerize ligands and finally abolish ligand function. Considering that there are bare therapeutic analyses of targeting these irreversible repressors in cancer until now, in this part we will not further discuss relative aspects. (for more details, please find in Table 1)
Intracellular compositions
Adenomatous polyposis coli (Apc)
Apc gene locates on chromosome 5q21-q22, containing 8535 amino acids and encoding a cytoplasmic protein around 310 kDa. More than 1500 mutations of Apc were detected over multiple tumors, and majorly in CRC.127 Meanwhile, the most mutant sites were located on exon 15. Over 700 somatic mutations of Apc resulted in various types of cancers by truncating Apc protein that was dependent on nonsense (34%) or frameshift mutation (62%).127,128 Mutations of Apc can cause familial adenomatous polyposis (FAP), which is also the major hereditary carcinogenic factor in CRC progression.129,130 A study showed that around 72% of Apc mutations were detected spreading throughout the Apc gene in early-onset CRC.131
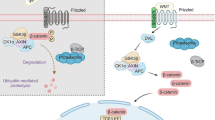
Apc gene has been identified as a vital suppressor in CRC genesis by inactivating Wnt/β-catenin signaling and stabilizing chromosomes. And mutations of Apc directly or indirectly lead to tumorigenesis as well.132 Mechanistically, Apc regulates β-catenin phosphorylation and restrains the nuclear trans-localization of β-catenin95,133 (Fig. 3). The three-hit hypothesis suggests that the abnormal mutation of Apc results in β-catenin abundance and abnormal activation of Wnt/β-catenin signaling in CRC.134 In contrast, the abnormal expression or truncation of Apc weakened the ability of tumor inhibition.135 Studies indicated that R2 and B motifs of Apc were the binding sites of Apc-Gsk3β/Axin, by which complex the diversity and structural stability of Axin were promoted.60 The loss-of-function (LoF) of Apc may be an essential contributor to various cancers, especially CRC.136 At present, experimental rodent models of CRC were initiated by knocking out of Apc.137,138,139

The intracellular components and signaling transduction of Wnt/β-catenin signaling
Apc also suppresses other cancers, such as lung, breast, gastric, and prostate cancers, excepting for CRC.140,141,142,143,144 The DNA methylation of Apc promoter is closely associated with various cancers, like lung cancer and prostatic cancer.145,146 The methylation reduces the normal expression of Apc in cancers, resulting in abnormal activation of the Wnt/β-catenin signaling pathway. Overall, this evidence indicated the repressive role of Apc in cancers, thus making it a promising way to remedy cancers via enhancing Apc function or restore the normal function of Apc.
Axin
Axin proteins, including Axin1 and Axin2, maintain β-catenin phosphorylation, thereby inhibiting signaling pathways by assembling the degradation complex with Gsk3β, Apc, and Ck1 (Fig. 3). Intriguingly, even though the Apc has been blocked, therapies targeting Axin could still be effective.147,148 Axin was identified as a suppressor in various cancers through majorly inhibiting Wnt/β-catenin signaling. Axins consist of three functional domains, the RGS domain of amino-terminal, the DIX domain of carboxyl-terminal, and the central region (AxinCR).149 Separately, the RGS domain is responsible for interacting with Apc to phosphorylate β-catenin.150 The DIX domain directly affects Dsh protein,149,151 and AxinCR binds to β-catenin and Gsk3β to regulate Wnt/β-catenin signaling.33,149,152
Some researches indicated that a single point mutation of Axin destroyed the stabilization of the RGS domain, resulting in Axin polymerization. The function of the Apc complex is affected by Axin self-polymerization. Inhibition of Axin and Apc complex together promoted tumor genesis and progression by enhancing Wnt/β-catenin signaling.153,154 Controlling Axin polymerization may be a potential therapeutic choice to suppress cancer development.154,155 Mutations of AxinCR were reported to accelerate the tumorigenesis in the following cancers: HCC, colorectal adenomas, ovarian carcinomas, lung carcinomas, and sporadic medulloblastomas. Therefore, erasing or eliminating the mutations of Axin could a promising method to combat diverse cancers.
Axin1/2 mutation in cancers
Axin1 mutations were detected widely in HCC.156,157 This mutation could phenocopy various tumors in animal models.158,159 Axin2 can partially compensate for the functional impairment caused by Axin1 mutation.155,158,160 Axin2 dysfunction was associated with a variety of tumors, including endometrial cancer,161 CRC,162,163,164,165 lung cancer,166,167 and breast cancer.168,169,170 In these cancers, Axin2 performed suppressive roles by mainly constraining the level of β-catenin.
Tankyrase (Tnks) in cancers
Tankyrase, consisting of two members (Tnks1 and Tnks2 in most species), participates in modulating Wnt/β-catenin signaling.171 Tankyrase1/2 is a poly (ADP-ribose) polymerase (PARP), attaching PAR chains onto substrates by its catalytic action.172,173 The enzymes bind and then PARylate Axins, utterly, the Axins are ubiquitylated and degraded.174,175 Additionally, researches showed that tankyrase enhanced Wnt/β-catenin signaling.176,177 A series of studies further proved that tankyrase could suppress cancer development via inhibiting the Wnt/β-catenin signaling pathway.175,178,179,180
Glycogen synthase kinase 3 (Gsk-3)
In Wnt/β-catenin signaling, generally, Gsk3 controls β-catenin degradation by comprising a degradation complex.95,133,181 Axin, phosphorylated by Gsk3, binds to and phosphorylates β-catenin, resulting in β-catenin degradation by ubiquitin proteasome.95,133,181 It is interesting that Gsk3 and CK1α can phosphorylate Wnt-co-receptor LRP6 to play positive roles in Wnt/β-catenin signaling activation.182,183,184,185 The dissociated Gsk3 from the degradation complex is essential for the nuclear shuttle of β-catenin182,183,184,185 (Fig. 3). So far, targeting Gsk3 may be a potential therapy for cancers. In certain cancers, Gsk3 may crosstalk with several pathways, including PI3K/PTEN/AKT/Gsk-3/mTORC1 and NF-κB pathway, and the details will be discussed in blow.186,187,188 Despite the function of Gsk3β is relatively clear in HCC, targeting Gsk3β in cancers still needs far more researches to elucidate its concise effects.
CK1
First encountered for phosphorylating casein in vitro, the CK1s are serine/threonine kinases that serve as pathway signal transductor in most eukaryotic cells. They are ubiquitously expressed in human tissues all through the developmental and adult period, and the Wnt/β-catenin signaling pathway is just one of the pathways that CK1s impact.189 The mechanisms of CK1 isoforms on regulating Wnt/β-catenin pathway are complicated. Depending on the substrates and subcellular location, CK1s play distinguished roles. To begin with, CK1α is the typical isoform for down-regulates Wnt/β-catenin signaling. As a composition of the degradation complex, CK1α initiates the phosphorylation of β-catenin.190 In fact, CK1δ/ε are also involved in this process by phosphorylating Apc (which strengthens the affinity of Apc to β-catenin), in collaboration with Gsk3.34,191 Furthermore, CK1ε phosphorylates and activities Dvl; however, this action also provokes negative feedback to inhibit Wnt/β-catenin pathway (vide infra).192 In terms of activation, on receiving Wnt ligands, firstly CK1ε phosphorylates Dvl, then CK1γ phosphorylates the cytoplasmatic domain of Lrp6, finally recruiting CK1α and Axin to bind with Lrp6 in the signalosomes,193 thus delivering the signal to downstream. Other mobilization effect includes the phosphorylation of CK1ε and CK1α to Tcf3 and Pygo, respectively.194,195
Dvl
The Dvls family is the transportation hub of Wnt/β-catenin signaling pathway. To adapt to this responsibility, the three highly evolutionally conserved domains of Dvls are the binding sites of various proteins. DIX, a highly conserved domain, is indispensable for the recognition of Axin.196 Moreover, DIX mediated the polymerization of Dvl monomer, which assembles as the anchoring site for Axin and Gsk3β.197 This process may have significant effects on the ligand–receptor/co-receptor internalization. CK1ε phosphorylating mediated DIX-E3 ligase interaction ubiquitinates Dvl, abolishing the polymerization of Dvls, ultimately inhibiting Wnt/β-catenin signaling.192 Next, the PDZ domain lying in the central part of Dvls, essential for the signal conduct from Fzd to downstream molecules, is also the most druggable region in Dvl. CK1 also targets this site.190 Near the C-terminal region is the DEP domain, whose role is still obscure in the canonical Wnt signaling pathway. Despite these three classic domains, Dvls also have a basic region and a proline-rich region, which may have effects in protein–protein interactions (PPI).197 Newish founds also indicated that Dvls shuttled between the membrane and the nucleus, and chances were that two distinct Dvls pools may exist.198,199 However, these discoveries were vague about the druggability of Dvl. So in this review, we suspended this topic aside.
β-catenin and Lef/Tcf in cancers
β-catenin is a multifunctional protein, which is versatile in various cellular events and human diseases. The very core of Wnt/β-catenin is the balance between the phosphorylation/dephosphorylation and degradation of β-catenin. In brief, the destruction complex phosphorylates β-catenin and to degradeβ-catenin by ubiquitin-proteasome system. There are some other kinases are associated with protein phosphorylation, including PP1, PP2A, and PP2C in Wnt/β-catenin signaling pathway. PP1 and PP2C play a positive role in Wnt/β-catenin signaling by dephosphorylating Axin.200,201 PP2A, a principal Ser/Thr phosphatase, involves multiple proteins phosphorylation, and importantly, the kinase malfunction could result in several cancers.202,203,204 PR55α, as a regulatory subunit of PP2A, can enhance the activity of Wnt/β-catenin signaling by regulating PP2A to suppress β-catenin phosphorylation.205 Hsp105, a PP2A regulator, is overexpressed in various tumors to reduce β-catenin degradation.206 Researches showed that PP2A dephosphorylated β-catenin to increase β-catenin accumulation.205,206,207 Therefore, it may be an alternative way to target PP2A in aberrant Wnt/β-catenin signaling cancers.
The stabilization of β-catenin is heavily associated with various cancers. In detail, the mutations of β-catenin are of great significance in tumorigenesis, progression, and prognosis of cancers.208,209 In blow, we will detailedly discuss the mutations of β-catenin in cancers. The constitutive activation form of β-catenin, the exon 3 mutations, is believed to regulate the genesis of hereditary non-polyposis CRC.208 In CRC, there are several mutations of β-catenin, leading to abnormally activated Wnt/β-catenin signaling.209 The mutations of β-catenin are commonly detected in HCC, uterine corpus endometrial carcinoma, adrenocortical carcinoma, and so on.210,211,212,213,214 The mutations of β-catenin majorly are missense that block Gsk3β consensus sites to activate Wnt/β-catenin signaling.215,216 Besides, β-catenin mutations may be a significant carcinogenic factor in endometrial carcinoma.215,216 Apart from the direct regulatory functions on tumorigenesis, the characteristics of β-catenin can also provide an approach to estimate the stage of low-graded, early-staged endometrial cancer recurrence.215,216
In addition to the direct transcription regulation of β-catenin as a transcription factor (TF), it can also form diverse types of transcription complex. β-catenin–Lef/Tcf includes Tcf/Lef, p300/CBP, and other proteins to assist β-catenin in binding to specific DNA sequence217,218,219 (Fig. 3). Tcf binds to Groucho/TLE, CtBP, and histone deacetylase proteins when β-catenin signaling is not activated.217,218,219 Tcf/Lef separates from Groucho/TLE and then composites β-catenin–Lef/Tcf complex, depending on X-linked inhibitor of apoptosis (XIAP) monoubiquitylating Groucho (Gro)/TLE.220 CBP promotes the transactivation of β-catenin/Tcf cooperating with thymine DNA glycosylase.221 In addition, the transcription complex recruits p300/CBP,222,223 Bcl9,224 Pontin522,225 Reptin52,225 Brg-1,226,227 Mllt/Af10-Dot1,228 SOX10,229 p68/p72,230 βTrCp1/Fbw1a,231 FOXM1232,233,234, and yes-associated protein 1 (YAP1).235,236
However, in CRC, HIFα can competitively bound to β-catenin to abolish the Tcf4/β-catenin complex, then enhancing the hypoxia tolerance of cancer cells to increase the survival in an anoxic environment.237 Interestingly, HIF2α binds to the β-catenin–Tcf complex at a different site from HIF1α to recruit p300 and then enhances Wnt/β-catenin signaling.210 The synergistic actions of HIF2α and β-catenin increase the proliferation of renal cell carcinoma cells.210 Accordingly, utilizing Tanshinone IIA can inhibit CRC angiogenesis by means of interrupting HIF-1α/β-catenin/Tcf3/Lef1 signaling.238,239
Besides, HOXB13, SOX4, RUNX3, CDK8, TCTP, and Daxx participate in regulating Wnt/β-catenin signaling to target Tcf in numerous cancers. Specifically, HOXB13 expression was downregulated in colorectal and prostate cancer.240,241 And it inhibited the growth of cancers by reducing Tcf and c-myc protein levels.240,241 RUNX3 inhibited Wnt/β-catenin signaling by comprising of a ternary complex with β-catenin–Tcf and attenuates growth and progression of multiple cancers, especially in gastric cancer.242,243 SOX4 can increase β-catenin–Lef/Tcf transcriptional activity through upregulating Tcf4.244,245 CDK8, an oncogene in CRC, partly functions by co-activating β-catenin–Tcf complex.246 Thus, CDK8 may be a promising target for β-catenin-associated cancers.246 The translationally controlled tumor protein (TCTP) can enhance the transcription complex activity by increasing the ability of β-catenin–Tcf binding and then inducing the growth of glioma tumor.247 In summary, HOXB13 and RUNX3 are identified as suppressors of Wnt/β-catenin signaling by obstructing Tcf4 activity to inhibit tumorigenesis and cancer progression.248,249
Tnik
Traf2- and Nck-interacting kinase (Tnik) is one member of germinal center kinases (GCKs) that can activate the c-Jun N-terminal kinase pathway.139 Tnik is an essential component for Wnt/β-catenin signaling to maintain physiological cell homeostasis.250,251 Tnik can directly interact with β-catenin and Tcf to modulate Wnt/β-catenin signaling250,251 (Fig. 3). Therapeutically, Tnik is a crucial target for the treatment of CRC. In CRC cells, Tnik activates the transcriptional capability of Tcf4 through phosphorylation.250,252 It has been shown that the growth of CRC cells was strictly dependent on Tnik stimulation. Indeed, after knocking down Tnik, the growth of xenograft CRC cells was brought to stall.251 And patients with overexpression of Tnik were manifested with poor postsurgical outcomes.253 Over 80% of CRCs have mutations in Apc,254 which makes the only molecule downstream of Apc a therapeutic target.
The crosstalk of Wnt/β-catenin signaling in cancers
Wnt/β-catenin and Notch signaling
Notch signaling widely interacts with Wnt/β-catenin signaling in cell homeostasis and embryo development.255 Notch signaling also involves in the wingless development with Wnt/β-catenin signaling.256,257 Notch can directly inhibit Armadillo/β-catenin to enhance destruction complex activity.256,257 Besides, LNX2, a regulator of Notch, enhances the cell vitality in Wnt/β-catenin-associated CRC.258 In turn, β-catenin–Lef/Tcf can reciprocally activate Notch signaling by inducing the expression of Jagged1 and Dll1, which are the ligands of Notch signaling.255,259 In addition, β-catenin reduces Notch1 ubiquitination and increases the expression of Hes1 to interfere with Notch signaling transduction.255,259 The synergistic action of Wnt/β-catenin and Notch signalings promotes tumorigenesis and cancer progression. Apparently, the potential remedies of inhibiting this synergistic function can be beneficial in cancer therapies.260,261,262,263
Wnt/β-catenin and Sonic hedgehog (Shh) signaling
The Sonic hedgehog signaling is another important interactor of β-catenin-dependent Wnt signaling.9,264 Smo, a receptor of Shh ligands,265 once being triggered via Shh, can activate Gli activities.264,265,266 Glis, including Gli1-3, are the ultimate effectors of Shh signaling as transcriptional regulators.266 Intriguingly, Gsk3β and CK1α can both phosphorylate and active Glis to reciprocally enhance Wnt/β-catenin signaling activity.267,268 The crosstalk of Shh-Wnt/β-catenin involves the relapse, invasion, and metastasis of certain cancers, so that the repression of this crosstalk may alleviate the progression, migration, and invasion of cancers.269 For instance, cyclopamine, as an inhibitor of Shh signaling, postpones the invasion of CRC by suppressing β-catenin–Tcf transcriptional activity.270
Targeted therapies of Wnt/β-catenin in cancers
Categories of pharmacology
Drugs targeting canonical Wnt signaling can be divided into two major subtypes, which are small-molecule inhibitor (SMI) and monoclonal antibody (mAb). SMIs usually refer to chemically synthesized compounds with a molecular weight of less than 1 kDa. In recent years, taking advantage of the in-depth learning of molecular and biological mechanisms in cancers, targeted drugs with fewer side effects plus better specificity than traditional chemo drugs have been developed. Theirs strengthens include but are not limited to (1) pronounced permeability into tissues and cells owing to the small-molecule weight; (2) larger volume of distribution; (3) diversified methods of drug delivery; (4) generally better oral tolerance and oral bioavailability after chemical modification. In contrast, mAbs cannot be taken orally, and are often administered by injection, which results in poor patient compliance. Even being directly injected, the distribution of mAbs in vivo is relatively limited. They cannot often easily reach therapeutic concentrations in some specialized tissues (such as the brain) as SMIs do. And as the inherent antigenicity of mAbs, they are likely to cause immune responses. mAbs are generally dominantly distributed in the kidneys, followed by the liver and spleen. In addition, they appear with non-linear pharmacokinetic characteristics, longer half-life, and smaller volume of distribution. Though mAbs can only act on extracellular targets, they have still achieved remarkable antitumor performances in comparison to SMIs, which is attributing to their different pharmacological mechanisms.
SMIs can bind to receptors with stronger affinity than the original ligands. On some occasions, they modify the structures of the receptors so that the ligands cannot be recognized. Moreover, certain SMIs may act as ATP analogs and bind to kinase sites in the cytoplasm, making the receptors unrecognizable.
The advantage of mAbs is not only to directly act on the extracellular and membrane-linked targets but also to activate the intrinsic internal immune system to indirectly exert antitumor effects. With respect to the direct functions, mAbs bind to receptors or ligands to block signal recognition or mediate internalization to reduce the density of receptors on the cell membrane surface.271 When acting indirectly, mAbs induce complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), or complement-dependent cell-mediated cytotoxicity (CDCC).272,273 Therefore, in in vivo experiments with a complete immune system, mAbs may show better antitumor effects than in in vitro experiments.274 In addition, mAbs can serve as the vehicle to achieve precision medicine, such as targeting radiogen to tumors.275
To sum up, in practical scenarios, mAbs are more competent for the treatment of hematological tumors than solid tumors as a consequence of their antigenicity and large particles.274 SMIs can target proteins even in the nucleus and penetrate the blood-brain barrier easily, thus preferentially suitable for treating solid tumors. Even if being humanized, mAbs are inevitable to arouse unintended immune reactions. Meanwhile, SMIs have poor antigenicity, but low specificity,276 which also brings a higher risk of side effects. Briefly, SMIs and mAbs exhibit diverse characteristics and various indications of cancers.
In addition to SMIs and mAbs, an under-developed category of Wnt/β-catenin-targeted therapy goes to peptides and peptide-associated modified drugs. Peptides have been put into use as drugs long ago, but obvious limitations delayed their clinical advancements. Peptidomimetic, a kind of peptide-associated modified drug, is much smaller than the parental molecule, yet having low antigenicity, good oral bioavailability, good permeability, and better diffusing to the target. Different from SMIs, their half-life is generally very short and has very rapid excretion. Although this feature reduces the risk of side effects, it contrarily causes difficulties to reach satisfactory concentration.277
Promising preclinical targeted therapies
SMIs targeting Porc
Porc inhibitors are well-acknowledged to block Wnt signaling with a low risk of off-target. Moreover, it was revealed that the exhaustion of canonical Wnt ligands prevented cancer cell proliferation but induced differentiation. Therefore, inhibiting Porc may be a mild therapy that could prevent tumor growth rather than directly causing lethal effects. It has been previously described that Znrf3/Rnf43 are important negative regulators of Wnt signaling. The LoF mutation of Znrf3/Rnf43 was reported to be oncogenic, and Porc inhibitors had shown a remarkable potency in this aspect.27,124,278 Unfortunately, the absence of Apc is likely to induce a Wnt ligand-independent signaling, so tumors with Apc LoF mutations may be resistant to Porc inhibitors.279 And Picco et al. reported that in a CRC cell line (VACO6) with RSPO3 fusion, long-term exposure (3 months) of a Porc inhibitor (LGK974) with incremental doses can induce drug resistance. This kind of drug resistance is accomplished by LoF of Axin1 in the VACO6.160
Contemporarily, there are a growing number of reports showing the side effects of Porc inhibitors, among which bone loss is the most frequent outcome.280,281 Two studies indicated that LGK974, WNT-C5, or ETC159 can lead to bone loss by diminished osteogenesis and increased osteolysis. These bone loss phenotypes were reported to even appear under the condition of effective dosages of Porc inhibitors.282,283,284 Hopefully, this side effect can be relieved by co-administrating diphosphonate (alendronate, zoledronic acid for instance).280 And other adverse effects may be solved by lower doses through the incorporation of other antitumor drugs.
LGK974
Also known as WNT974, LGK974 is a potent SMI targeting Porc. A preclinical study showed the effective performance of LGK974 among variable neoplasm models with good oral tolerance.282 Even this study systematically examined many organs such as the intestine, stomach, and skin, but unfortunately giving an ignorance to bone tissue. Besides, it was discovered that all human head and neck squamous cell carcinoma (HNSCC) cell lines with Notch nonsense mutation were more sensitive to LGK974.282 Inspired by this, a team signed up for a clinical trial attempting to use LGK974 to treat HNSCC patients with Notch LoF mutation285 (Table 2). But this project was abandoned as found retrieval. Another study showed the incredible use of LGK974 in Rnf43 nonsense mutation cell lines in pancreatic ductal adenocarcinoma (PDAC).124 Both of the two clinical studies illustrated a delayed effect of proliferation inhibition by LGK974. Specifically, after a single administration, there still remains some β-catenin in the cytoplasm to initiate the downstream transcriptional activities. LGK974 also preliminarily demonstrated a reliable tumor-suppressive effect in vitro on neuroendocrine tumor and ovarian cancer.286,287
A study using uncovered similar impairment of bone mass in two different doses of LGK974 in mice (3 and 6 mg/kg/d for 7 days).281 Another study expanded the dose range (from 1to 30 mg/kg/d) and showed an all ranges covered bone loss after continuous treatment for 4 weeks.280 What’s worrying is that the dosage of 3 mg/kg/d for mice is necessary to reach a significant tumor repression.282 And the exposure to a high dose of LGK974 (20 mg/kg/d) results in prominent intestinal toxicity.282 Fortunately, as the sustaining treatment of LKG974 is not required for tumor treatment, the practical clinical dosage may not bring so many side effects. Notably, the clinical dosage of LGK974 is still in exploration without explicit results posted.288,289
ETC159
ETC159, originally named ETC-1922159, is a potent orally available SMI for Porc. Madan et al. identified its high efficiency in CRC with Rspo translocations. This study also inspected intestinal tissues, but still neglected bones.283 ETC159 can perform effectively synergistic effects with PI3k/mTor inhibitor GDC-0941 in PDAC with Rnf43 LoF mutation.278 Based on the outstanding behavior of ETC159 in a preclinical experiment, another phase II clinical trial targeting advanced solid tumors has been carried out. Interestingly, part B of this trial evaluates the combination usage of ETC159 and pembrolizumab. But no results have been posted up to date290 (Tabl. 2).
Bone loss has been observed after 4 weeks of administration in a dosage of 3–30 mg/kg/d. The research team adjusts administration frequency from qd to qod to validate the hypothesis of whether the bone loss could be attenuated after a full metabolic cycle of ETC159. Results showed that there were no significant differences in several estimated outcomes of bone between treated daily and treated every other day.280 By the way, ETC131, a similar compound of ETC159, is only used for in vitro assays as a result of its inferior oral bioavailability.283
CGX1321
CGX1321 is a novel Porc inhibitor, yet only a few studies have researched into this small molecule. For experimental analysis, it has been discovered for a good performance in CRC with fused Rspo.291 Further in-depth studies respectively included the combined usage of CGX1321 and immune checkpoint blockade therapy to treat ovarian cancer (but the results were unsatisfactory),292 and liposome encapsulation as a vehicle to deliver CGX1321 to treat cancer stem cells (with expected results).293
As for the clinical trial of CGX1321, it was solely used in solid tumors including gastrointestinal tumors (Table 2). And the combination of CGX1321 with pembrolizumab in CRCs was constructed to evaluate the safety.294 Yet no results have been posted currently.
WNT-C59
This drug is not well-studied when compared with LGK974 or ETC159, but it exclusively targets mammalian Porc. It owns good oral tolerance and efficacy in the MMTV-Wnt1 mouse mammary cancer model,295 the Znrf3/Rnf43−/− mouse CRC model.284 And WNT-C59 satisfactorily prevented tumor growth in mice xenografted with SUNE1 or HNE1 (two nasopharyngeal carcinoma cell lines).296 In addition, WNT-C59 can reverse the resistance of trichostatin A in human pancreatic cells.297 However, when administrated at 10 mg/kg/d for 7 or 21 days, bone loss was observed in mice.281 This should be taken into consideration as the potential side effect of WNT-C59 in cancer therapies.
GNF-1331/GNF-6231
GNF-1331 is a precursor drug, and the optimization of it brings on the discoveries of LGK974 and GNF-6231.298 Currently, GNF-6231 is still a brand-new molecule without clinical studies. But it worked well in MMTV-Wnt1 patient-derived xenograft (PDX) mice model, by reducing Axin2 expression.298
IWP
Relying on the screening approach of the phenotype of inhibition of Wnt production, this kind of molecule was named as “inhibitors of Wnt production (IWP)”. In this study, the research group then verified the exclusive activity of inhibiting Wnt-related Porc function by using IWPs.299 IWP has three family members, IWP-1, −2, and -L6, all of which oppress Wnt/β-catenin signaling by competing for the active sites of Porc with Wntless.299 IWP-2 and IWP-L6 had poor metabolic stability in mice, but at least IWP-L6 showed relatively better stability in human.300 IWPs may synergist with PRI-724 to induce apoptosis in HNSCC through inhibition of both Porc and CBP/β-catenin.301 However, as IWPs have a similar structure to certain CK1 inhibitors, much research have proposed the effects on CK1 isoform suppression of IWPs.302,303 IWP-2 and IWP-4 inhibited the proliferation of various cancer cells via antagonizing Porc and CK1α.303 The growth of gastric cancer cells was restrained via the IWPs-mediated downregulation of Wnt/β-catenin signaling.304 A similar situation occurred in HNSCC cells as well.301
mAbs targeting Wnt receptors and co-receptors
mAbs are remarkably targeted drugs especially for blocking extracellular or membrane-linked proteins. They are characterized by high affinity and low off-target accidents but have longer plasma half-life and lessened clearance rate.305
OMP-18R5/OMP-54F28
Also known as vanticumab, OMP-18R5 is a monoclonal antibody that targets human Fzd1, 2, 5, 7, and 8. And it was reported to inhibit the growth of gastric adenomas in mice models, either with or without Apc LoF mutation.84 OMP-54F28, namely ipafricept, is the chimera of truncated Fzd8 and IgG1 Fc region.306 Ipafricept shares many features with vanticumab. A study has proved the high antitumor effect of ipafricept in MMTV-Wnt1 cancer models. Moreover, the synergistic utilization of ipafricept gemcitabine demonstrated inhibitory function in a pancreatic PDX model, and significantly reduced the cancer stem cells (CSC) frequency. This combined strategy showed superior capabilities of tumor arrest over the sole usage of ipafricept either or gemcitabine.307
Encouragingly, either vanticumab or ipafricept exhibited impressive synergistic therapeutic effects with taxanes (paclitaxel, docetaxel, and cabazitaxel). Fischer et al. discovered that there existed a population of taxane-resistant cancer cells after using paclitaxel. And the successional administration of ipafricept/vanticumab prior to paclitaxel can overcome the drug-resistant effect via a sole administration of paclitaxel. Mechanically, this combined usage of ipafricept/vanticumab and paclitaxel strengthened the mitotic catastrophe.308
The adverse effects of OMPs are similar to Porc inhibitors, but OMPs showed worse impairment of bone mass. As for ipafricept, in a clinical trial in patients with advanced solid tumors, fragility fractures were reported in two participants at 20 mg/kg q3w.309 Another trial recruited patients inflicted with recurrent ovarian cancer, in which a patient experienced a pelvic fracture at 5 mg/kg q3w.310 Studies pointed out that vanticumab had a more severe side effect on bone than ipafricept. In detail, in a clinical trial with metastatic pancreatic cancer, two participants experienced fragility fractures at 7 mg/kg q2w.311 Additionally, in a trial attempted to treat advanced or metastatic HER2- breast cancer, vanticumab led to fragility fractures in 3 patients, including three Grade 2 events and one Grade 3 events of fracture ranking, at the regimen of 7, 14 mg/kg q2w and 8 mg/kg q4w. Surprisingly, this unintended outcome appeared under the critical supervision of bone metabolism outcomes and bone anabolic remedy of diphosphonate treatment. At last, a total of 6 patients experienced fragility fractures, leading to the abolishment of this trial.312 All those fracture events mentioned above were not reported as dose-limited toxicity, because they took place after the 1st 28d-window-phase. Whereas, still no studies could reach the maximum administered dose (MAD) in consideration of bone safety. Overall, though preclinical experiments exhibited a good performance of OMPs, these trials revealed a crisis of applying ipafricept/vanticumab in clinical (Table 2).
OMP-131R10
Also named as rosmantuzumab, OMP-131R10 is a humanized mAb targeting Rspo3. Some malignant hematopoiesis cancers have aberrant Wnt/β-catenin signaling independent to Wnt ligands, due to the redundancy of Rspos in triggering Wnt/β-catenin signaling. Based on this, Salik et al. demonstrated that rosmantuzumab can impair the self-renewal and differentiation of acute myeloid leukemia cells in the PDX model, meanwhile free from influencing normal hematopoietic stem cells.313 For the clinical trial, a phase 1a/b trial about rosmantuzumab in advanced solid tumors and previously treated metastatic CRC is still going on314 (Table 2).
F2.A
F2.A is a newly developed antibody that targets 6 of the 10 human Fzds (Fzd1/2/4/5/7/8). The developer synthesized this compound by firstly identification of anti-Fzd antibodies (F2) with a specific profile matching to OMP-18R5 and secondly using combinatorial antibody engineering to find a variant F2.A with specificity to bounding Fzd4. According to their study, F2.A could selectively bound to Fzd4 without competition with Norrin. Moreover, F2.A had a much better potency when treating Rnf43 mutated PDAC when compared to OMP-18R5 and OMP-54F28.315
DKN-01
DKN-01 is a humanized IgG4 mAb that can bound and block the activity of Dkk1. Physiologically, Wnt/β-catenin signaling activates the transcription of Dkk1, which in turn bounds LRP5/6 and block the recognition of Wnt ligands, forming a negative feedback loop. However, some tumors featured with overwhelmed Wnt/β-catenin signaling are insensitive to Dkk1. In contrast, the superfluous Dkk1 can promote tumor cell proliferation. Though the specific mechanisms are unknown, it was hypothesized that Dkk1 helped tumors escape from immune supervision.316 Indeed, it was reported that an intact immune system was needed for DKN-01 function in a murine model.317 Most clinical trials are researching on DKN-01, most of which focused on the digestive system such as the gastroesophageal, intestine, liver, and biliary tract cancers, and the rest focused on NSCLC, gynecologic malignancies, and multiple myeloma. DKN-01 showed satisfactory tolerance in all clinical trials. Intriguingly, some trials reported a better effect of DKN-01 in Dkk1-overexpressed patients318 (Table 2).
UC-961
UC-961, also called cirmtuzumab, is a first-in-class mAb that targets ROR1. ROR1 is often highly expressed in chronic leukemia lymphoma (CLL) cells. As normal B lymphocytes do not express ROR1, UC-961 has precise and good effects in treating CLL.319
OTSA-101
Fzd10 was found ubiquitously upregulated in synovial sarcoma (SS), but scarcely detectable in normal tissues except the placenta.101 A group of radioimmunoconjugate humanized antibody OTSA-101 was designed to target Fzd10, whose derivations include 211At-OTSA-101, 111In-OTSA-101, and 90Y-OTSA-101. Among them, 111In-OTSA-101 is routinely used as a diagnostic tool. Whereas OTSA-101 only showed a weak antagonistic activity on the growth of SS cells. Afterall, OTSA-101 merely became a putative carrier for targeted SS radiotherapy. In preclinical experiments, a large number of cell death occurred in the PDX murine model on the first day after injection of 211At-OTSA-101. All mice in this interventional group survived, and the tumor volume was greatly diminished. However, 211At-OTSA-101 was easily accumulated in the stomach and its uptake rate of tumor cells was not as good as 111In-OTSA-101. But the inhibitory effect of 211At-OTSA-101 was much better than 90Y-OTSA-101281. Furthermore, 90Y-OTSA-101 showed obvious bone marrow suppression and significant hematotoxicity.320
Peptide mimetics
Foxy-5
Sponsored and developed by WntResearch (https://www.wntresearch.com), foxy-5 is a mimic of WNT5A. Preclinical studies showed that a low level of WNT5A was correlated with a more metastatic or advanced outcome in breast and prostate cancer. In accordance, foxy-5 could prevent metastasis to some extent.321,322 At present, WntResearch has supported five clinical trials registered on NIH and EUCTR, yet with no results posted (Table 2).
CWP232291
CWP232291, abbreviated as CWP291, can decline the transcriptional effect of canonical Wnt signaling. CWP291 was reported to suppress the growth of castration-resistant prostate cancer through the degradation of β-catenin via apoptosis-induced ER stress.323 In fact, CWP232204 is the active form of CWP291 in serum. Currently, CWP291 was put forward in clinical trials of treating acute myeloid leukemia (AML) and refractory myeloma without results updated (Table 2).
SMIs targeting cytoplasmic proteins
Tnks inhibitors
Tankyrase is a poly-ADP polymerase (PARP), which can make Axin1/2 poly-ADP ribosylation, leading to the ubiquitination and degradation of the latter. Therefore, the silence of Tnks will result in the inhibition of the Wnt/β-catenin signaling. According to the binding sites, Tnks inhibitors can be divided into two categories: targeted nicotinamide subsites and adenosine subsites. The nicotinamide domain is ubiquitous in the PARP enzymes, and its specific site inhibitor is XAV939. For the adenosine domain, it is unique to Tnks, and many Tnks inhibitors (G007-LK, NVP-Tnks656, JW55/74, and IWR) target this site.324
Having been developed for a long time, Tnks inhibitors have shown outstanding tumor-suppressive effects in preclinical experiments, but none of them have entered into clinical trials. Of note, they were reported to possibly cause bone loss. Inhibition of Tnks (using either XAV939, IWR-1, or G007-LK) in murine models led to the accumulation of a substrate, SH3 domain-binding protein 2 (SH3BP2), which subsequently enhanced the Rankl-mediated osteoclast formation.325,326 In the human genome, the gain of function (GoF) of SH3BP2 promoted osteoclastogenesis so which led to bone loss.327 Although SH3BP2 can also promote the differentiation and maturation of osteoblasts,328 Tnks inhibitors dominantly showed an osteoclastogenesis effect in vivo, because the concentration needed for stimulating osteoblast is about 10 times higher than that of osteoclasts.326 Not only Tnks, but the family of PARP plays an important role in bone homeostasis. Though theoretically, pan-PARP inhibitors are presumed to have a greater influence on bone, there is currently no convincing evidence to prove their regulations of bone mass.
In addition, Tanaka et al. found that CRC cells (whether established cell lines or patient-derived cells) with Apc-truncated mutations responded well to Tnks inhibitors (XAV939, IWR-1, and G007-LK), especially the mutations with all the 20-amino-acid repeats (the β-catenin binding sites of Apc) obliterated. Conversely, the longer Apc mutation may lead to resistance to Tnks inhibitors. These results suggested an underlying therapeutic value of Tnks for dealing with truncated Apc mutant cancers.329
XAV939 was identified as an inhibitory factor of Wnt/β-catenin signaling, originally in CRC cell lines. This SMI stabilized Axin to increase β-catenin degradation by suppressing Tnks1/2.171 It has been investigated that beside from CRC, XAV939 can constrain certain cancers by inhibiting the Wnt/β-catenin signaling. The combined utilization of low-dose paclitaxel and XAV939 inhibited breast cancer metastasis and the growth of triple-negative breast cancer. This mechanism was contributed to inhibiting Wnt/β-catenin signaling to enhance cancer cell apoptosis and attenuate EMT and angiogenesis.330 In gastric cancer, XAV939 can inhibit the invasion and metastasis of cancer cells.331 XAV939 and RNAi-Tnks1 inhibited the stemness and migration of cancer stem cells and accelerated cell apoptosis in neuroblastoma by attenuating the abnormal status of Wnt/β-catenin signaling.332,333 XAV939 and IWR, the inhibitors of Tnks, repressed the growth of lung cancer and reduced tumorigenesis. Experimental data proved the inhibitory effect of XAV939 and IWR on cancer cell growth in murine lung cancer models.334 XAV939 enhanced the ability of CD4+ lymphocytes in biochemically recurrent prostate cancer cell lines, LNCaP and PC-3.335
JW67 and JW74, two compounds as inhibitory molecules of Wnt/β-catenin signaling, can inhibit the growth of CRC by Axin2 accumulation and β-catenin degradation.336 JW74 and JW55 are Tnks inhibitors that bind to the lower part of donor NAD+ cleft, instead of mimicking nicotinamide. JW55 also worked well in the murine PDX CRC model with Apc mutation.337 G007-LK is an analog of JW74, and G244-LM is analogous to XAV939.171,336 Compound G007-LK and G244-LM can damage the proliferation, colony formation, and growth of CRC cells via activating Axins to suppress Wnt/β-catenin signaling.338 As an adjuvant, G007-LK can also enhance the sensitivity of glioma stem cells to a chemo drug temozolomide and CRC cells to PI3K/EGFR inhibitors.339,340
As mentioned above, IWR compounds were defined based on their anti-Wnt pathway activities in a phenotypic screening assay.299 IWRs, including five molecules, target Wnt-dependent cancers by enhancing Axin capability to suppress Wnt/β-catenin signaling.299,341 The inhibition of IWRs has been identified in various cancers like osteosarcoma, colorectal, breast, lung, and hepatocellular cancers.169,299,342,343,344,345 IWR-1 served as a good adjuvant that reversed the resistance of osteosarcoma to doxorubicin, and in vivo inhibited the growth of subcutaneous PDX osteosarcoma.342 Moreover, IWR-1 could inhibit the EMT of CRC by inhibiting Wnt/β-catenin transduction.343
In the following part, we are going to discuss several agents, NVP-TNKS656, AZ1366, RK-287107, and HLY78, that are Tnks inhibitors with very limited preclinical reports. For instance, NVP-TNKS656 increased the sensitivity of CRC cells to PI3K/AKT inhibitor in vivo, but the increment effect could be reversed by high FOXO3A.346 And NVP-TNKS656 also inhibited the metastatic and invasive EMT hallmarks of hepatoma carcinoma cells.347 AZ1366 is a novel inhibitor of Tnks to constrain NSCLC growth. EGFR-driven NSCLC was suppressed by the synergistic function of AZ1366 and EGFR inhibitors.348 AZ1366 could erase the insensitivity of CRC cells to irinotecan,349 and it coordinated with EGFR inhibitor to control the growth of Wnt-responsive lung cancers in the murine models.348 RK-287107 was claimed to inhibit Tnks1/2 four or eight times more than G007-LK. It could down-regulate the Wnt/β-catenin signaling in Apc-truncated CRC cells, but had little effects in wild-type cancer cells.350 An SMI from the synthetic chemical library of lycorine derivatives, 4-ethyl-5-methyl-5,6-dihydro-[1,3]dioxolo[4,5-j]phenanthridine (HLY78), activated Wnt/β-catenin signaling by targeting the DIX domain of Axin and enhanced the effect of Axin-Lrp5/6.351
CK1 inhibitors
The family of CK1 proteins has several isoforms, of which CK1α/δ/ε are involved in the canonical Wnt signaling, but their roles are distinguished. CK1α serves as a component of the destructive complex and phosphorylates β-catenin. Instead, CK1δ/ε phosphorylates Dvl leading to the stabilization of β-catenin. And CK1δ and CK1ε are also moonlighting proteins that control the circadian clock. IC261 is a selective, ATP-competitive CK1 inhibitor that has shown its high efficiency in handling CRC and glioblastoma cells.352,353 Not only that, IC261 has alternatively biological effects that may lead to the death of cancer cells independent of inhibiting the canonical Wnt pathway.352,354 In an in vitro study, IC261 induced centrosome fragmentation during mitosis independent of CK1δ.355 Other CK1 inhibitors include PF670 and PF480 that do not kill cancer cells.
Also known as umbralisib, TGR-1202 is a dual kinase inhibitor that targets both PI3Kδ and CK1ε. The non-canonical Wnt pathway has been demonstrated to play an important role in CLL with CK1δ/ε overexpression.356 TGR-1202 is exclusively used in hematological malignancies and is currently recruited in clinical trials for treating CLL, Hodgkin’s lymphoma, mantle cell lymphoma, and so on (Table 2).
Pyrvinium, known as an antiparasitic drug, has been approved by FDA as an orphan drug for FAP because of its exclusive CK1α agitation capability. Earlier experiments have shown that pyrvinium inhibited the proliferation of HCT116 and SW480 cell lines by selectively activating CK1α and inhibiting canonical Wnt signaling.194 This activation may be achieved by allosteric regulation, thereby improving the catalytic ability of CK1 without affecting the binding of substrates.357 Although pyrvinium also showed inhibitory ability in other Wnt/β-catenin signaling-driven tumor cell lines (SUM-149/SUM-159,358 and A2278359), its bioavailability was low in tissues other than the intestine.360 In addition, pyrvinium also stimulated CK1γ, leading to Wnt signaling suppression. However, some scholars indicated pyrvinium pamoate lacked the efficacy on CK1, instead, it downregulated AKT by an undiscovered mechanism to activate Gsk3β.361
Gsk3 inhibitors
Ambiguously serving as carcinogenic or cancer suppressor,362 Gsk3 lies downstream of diverse signaling pathways, including the Wnt/β-catenin signaling pathway. Therefore, targeting Gsk3 has very restricted specificity to produce unexpected off-targets. Thus, this review will not thoroughly discuss Gsk3 inhibitors due to their low specificity of Wnt/β-catenin signaling. Therefore, here only lists a typical Gsk3 inhibitor that has shown good performance in inhibiting Wnt/β-catenin signaling.
LY2090314 is a potent Gsk3α/β inhibitor that has demonstrated good cooperation with platinum agents in vitro in the treatment of melanoma,363 AR-V7+ prostate cancer,364 and neuroblastoma.365 But in a phase I clinical trial, the combination of LY2090314 and pemetrexed/carboplatin showed eleven DLTs in ten enrolled patients366 (Table 2). Other Gsk3 inhibitors like ABC1183 and CHIR-99021 are currently under exploring.
Dvl inhibitors
Though serving as a critical conductor in the canonical Wnt pathway, only few drugs and studies targeting Dvl are available. Till now, targeting Dvl has already shown a notable potentiality for tumor treatment. For instance, Dvl2 was overexpressed in HCC and reported to link with poor prognosis.367 In comparison to normal adult bronchial/alveolar epithelial and peripheral blood mononuclear cells, Dvl1-3 were found to be exclusively expressed in NSCLC and CLL cells, respectively.368,369 Most drugs targeting Dvls currently are developed to selectively inhibit PDZ-Fzd interaction. Such as FJ9, it was significantly reported to cause apoptosis in melanoma and NSCLC cell lines.370 3289-8625, another Dvl inhibitor, suppressed the growth of PC-3 cells.371 Taken together, the druggability of Dvls remains largely unknown. Urgent studies are demanded to unearth more applicable chances for drugs targeting Dvls in the future.
Agents targeting protein–protein interaction (PPI) in the nucleus
Nuclear localization of β-catenin will initiate downstream gene expressions. This process involves the formation of a key transcription complex, β-catenin–Lef/Tcf. β-catenin-dependent transcriptional regulation is also modulated by the phosphorylation protein Tnik, and various transcription co-factors like CBP, BCL9, CREB, BRG1, etc. These proteins listed above are all potential therapeutic targets of Wnt/β-catenin signaling. Despite the most core position of β-catenin in canonical Wnt signaling, pityingly, among all previously screened SMIs, few can directly bind to β-catenin. This is because, unlike most enzymes or receptors with identifiable binding pockets, the PPI surface of β-catenin is relatively large and flat for small molecules. And the Tcf-binding domain within β-catenin is overlapped with many other proteins, making it difficult to specifically interfere with Tcf/Lef372 (Fig. 4). Moreover, among the limited SMIs (CWP232228 for example) that can directly bind to β-catenin, there lacks evidence of Wnt signaling inhibitory in vivo. In contrast, targeting Tnik showed more stable inhibition of canonical Wnt signaling.

The structural illustration of human β-catenin protein. In this figure we mainly demonstrated the important PPI binding domains and phosphorylation sites of β-catenin. This image was modified from a published research.372
Tnik inhibitors
As mentioned above, Tnik is a critical therapeutic target of CRC. Among the Tnik inhibitors, the most commonly used are aminothiazole-based. Masuda et al. discovered a series of SMIs called NCB, classified as ATP-competitive inhibitors. After a high-throughput screening of the small-molecule compound library, they firstly found NCB-0001 (with a moderate inhibitory effect on canonical Wnt signaling in HEK293 cells), which led to the discovery of N5355 following the structural optimization.252 N5355 significantly reduced the expression of Wnt/β-catenin-dependent genes, such as Axin2 and cMYC, in HCT-116 cell line.373 What’s worth noting, N5355 did not affect the vitality of canonical Wnt signaling independent cell lines, HELA and HEL299.252 This research group subsequently discovered NCB-0005 (or called KY-05009), which markedly inhibited the activation of Wnt/β-catenin signaling mediated by TGF-β1 in A549 cells.373 Also, the NCB-0005 cooperated with the RTK inhibitor dovitinib to impede the growth of multiple myeloma cells.374 Their latest discovery was NCB-0846, which inhibited the growth in a variety of patient-derived CRC xenografted tumors,375 and abrogated the EMT of DLD-1, HCT-116, and A549 cell lines.376,377
As ATP-competitive inhibitors, NCB series drugs may also have inhibitory effects on other kinases. Therefore, it should be concerned that the clinical effects of NCB drugs may not be limited to target canonical Wnt signaling. In addition to CRC, inhibitory effects of NCB drugs have also been reported in other tumors.378,379
Inhibitors targeting β-catenin–-Lef/Tcf complex
Such inhibitors block the PPI between β-catenin and Tcf/Lef. However, the Tcf/Lef recognition domain on β-catenin highly overlaps with that of Axin, Apc, E-cadherin, and other proteins, which may bring potential adverse effects derived from off-targets of canonical Wnt signaling.
In this part, we will mainly discuss the β-catenin–Lef/Tcf complex inhibitors as following: CWP232228, LF3, BC2059, PKF115-584, PKF222-815, and CGP049090. CWP232228 is such an inhibitor, which was reported to induce apoptosis and cell cycle arrest in HCT116 cells,380 also to show effects on liver and breast CSCs.381,382 LF3, a 4-thioureido-benzenesulfonamide derivative, reduced tumor growth and induced differentiation in a CRC xenograft murine model.383 Moreover, BC2059, an anthraquinone oxime-analog repressing Lef1/Tcf4 activity, was elucidated to induce the apoptosis of HL-60, HEL, and K562 significantly.384,385 BC2059 is currently testified in a phase I clinical trial of desmoid tumor (Table 2). Other drugs of β-catenin–Lef/Tcf complex inhibitors, including three fungal derivatives, PKF115-584, PKF222-815, and CGP049090, have shown prominent inhibitory effects towards CLL/AML (PKF115-584 and CGP049090) and HCC (all three agents) cells, through inducing apoptosis.386,387,388 Still, some β-catenin–Tcf/Lef inhibitors need more preclinical experiments to verify their practicabilities, such as PNU74654 and 2,4-diamino-quinazoline derivatives.
Transcription co-factor inhibitors
For less superposition and narrower PPI surface in the recognition domain, inhibitors targeting the terminal domains (unstructured or intrinsically disordered protein regions) are putative to be more specific and druggable. These regions involve the binding of transcriptional co-factors like BCL9/B9C and CBP. Indeed, the SMI 40-fluoro-N-phenyl-[1,10-biphenyl]−3-carboxamide and its derivatives showed a much greater affinity toward β-catenin/BCL9 over β-catenin/E-cadherin.389
For targeting CBP, PRI-724, and ICG-001, a pair of enantiomers, are potent inhibitors of canonical Wnt signaling that antagonize the binding of β-catenin to CBP but not affecting the viability of p300 meanwhile. They have similar effects and sometimes can be used interchangeably. Their preclinical efficiency has been demonstrated in a variable of cancer cells such as HNSCC,301 hepatoma carcinoma,390 and neuroendocrine tumor cells.286 A clinical trial of cancer research is still engaging but without results posted (Table 2, two of the three trials were terminated). However, they seemed to be more applicable in antifibrosis than in antitumor.391,392
BCL9 provides an excellent opportunity for targeting Wnt signaling therapy. In normal cells, BCL9 expression is almost undetectable,393 but it reigns a set of EMT-regulated Wnt target genes in cancer cells.394 And after knocking out Bcl9 and B9l in mice, no obvious harmful phenotypes were found, indicating that these genes are of little importance for mammals to balance Wnt signaling in normal tissues.394 Based on this, Takada et al. developed the stapled peptide stabilized α helix of BCL9 (SAH-BCL9), which can snatch β-catenin from the endogenous Bcl9/Tcf/β-catenin complex with greater affinity. SAH-BCL9 showed a significant antitumor effect in both colo320 in vitro and intraperitoneally injected murine model in vivo.395
Other drugs may target Wnt/β-catenin signaling
Apart from the synthetic compounds, in recent years more and more natural products have been revealed to impact on repressing Wnt/β-catenin signaling.396,397,398,399 The most studied may be resveratrol, a phytoalexin produced when plants are inflicted with injuries or pathogenic attacks. Resveratrol has been enrolled into two phase I clinical trials, investigating its value in the dietary supplement in CRC prophylaxis.400,401 Moreover, silibinin, an extract from the milk thistle seeds, also showed its potentiality in preventing tumorigenesis in a preclinical model.402,403
In addition to natural products, many FDA-approved drugs have expanded their indications to crosstalk with Wnt/β-catenin signaling. Just as mentioned above, as an antiparasitic agent pyrvinium also inhibited Wnt/β-catenin pathway via activating CK1α, or Gsk3β.194,361 Other old drugs like niclosamide to inhibit canonical Wnt signaling by promoting Fzd1 endocytosis,404 celecoxib to induce the degradation of Tcf7,405 and salinomycin to target LRP6.406 Due to the functions of these old drugs for targeting Wnt/β-catenin signaling are still limitedly known, we will not continue to discuss these drugs. For more details, please find in Table 3.
Challenges of Wnt/β-catenin-targeted therapies
Even though drug development in the field of Wnt targeted therapies in cancers has been so prosperous, no exclusive drug has yet been approved by the FDA. This dilemma has aroused an un-neglectable crisis for moving forward the targeted therapies of canonical Wnt signaling in cancers. There are still many challenges before they could be put into large-scale clinical trials. This review crudely divided these drugs into three categories: SMIs, mAbs, and modified peptides. The molecular and structural properties determine the potential diversity of the clinical applications of these three categories. mAbs and peptidomimetics are more suitable for the targets on cell membrane surface, accordingly, SMIs are more suitable for targeting Wnt receptors and intracytoplasmic kinases, and stapled peptides are suitable for disrupting PPI in the nucleus.
For SMI, they are known for their good permeability and oral bioavailability, but off-target effects are common. This is especially inevitable for ATP-competitive drugs. mAbs are known for their unparalleled specificity, but their oral availability is poor. The mAbs have a slow clearance rate and a long half-life, which increases the burden on the liver and kidneys of the patient and increases the side effects. Besides, mAbs also have poor permeability and cannot target intracellular sites. Most importantly, current modifications of mAbs failed to decrease the antigenicity and increase tolerance. For modified peptides, they can achieve good permeability and tolerability, but a larger dose is required for them to maintain the therapeutic concentration.
Considering the ubiquitous and extensive involvements of Wnt/β-catenin signaling in regulating various normal tissue functions, it is ineluctable to accept the unintended byproducts of targeting canonical Wnt signaling. Although it provides an ideal approach to target nuclear β-catenin in aberrant canonical Wnt signaling-associated cancers, it is currently unprocurable to develop such kinds of targeted agents because of the very limited druggability. This crisis of developing the targeted therapies of Wnt/β-catenin signaling in cancers may possibly be overcome via the in-depth analysis of the preexisting molecules. Also, more development strategies such as immunotherapy, the synergistic medications of drugs, the decrement of mAbs weight, and so on could endow us with underlying breakthroughs to realize the canonical Wnt signaling targeted therapies in cancers.
Conclusion and discussion
Identified decades ago, Wnt/β-catenin signaling immediately generated substantial interest in the field of cancer research because of their extensive involvements and intensive roles in regulating numerous aspects of cancers, including the initiation, development, progression, diagnosis. In this study, we revealed that the status quo investigations have depicted an un-neglectable crisis of the Wnt/β-catenin-dependent targeted therapies in cancers. In detail, most of the medications targeting Wnt/β-catenin signaling in cancers have not been enrolled into clinical trials, and even the registered clinical trials remain in the very early phases of which most have not demonstrated satisfactory outcomes (Fig. 5). However, this dilemma clashed with the early evidence in in vitro and in vivo with the optimal performance of targeting Wnt/β-catenin signaling in cancers. The reasons for these poor therapeutic benefits rely on the fact that the current remedies of Wnt/β-catenin signaling-associated targeted therapies in cancers often lack satisfactory efficacy, specificity, and safety. For instance, due to the crucial roles of Wnt/β-catenin signaling in bone, many targeted therapies demonstrated obvious side effects of severe bone loss. Furthermore, certain SMIs and mAbs targeting Wnt/β-catenin signaling showed limited specificity because of the difficulty of identifying the druggable structures and sites of Wnt/β-catenin signaling components. These facts suggest that Wnt/β-catenin signaling targeted therapies in cancers are still lagging behind for a solid clinical translation.

The statistical summary of up-to-date registered clinical trials of targeted therapies via targeting Wnt/β-catenin signaling in cancers. The left panel indicated the stages and the right one showed the phases of clinical trials. P1 phase I, P1b phase IB, P2 phase 2. All clinical trials included were updated until May 2021
Nevertheless, as massive early experimental investigations have already proven the benefits of targeting Wnt/β-catenin signaling in cancers it is still worthy to further analyze the capabilities of developing Wnt/β-catenin signaling targeted therapies in the future (Fig. 6). It should be noticed that future studies are called to solve the current crisis via decreasing the side effects but improving the specificity and safety of Wnt/β-catenin signaling targeted therapies in cancers. To sum up, our review systematically exhibited the strengths and weaknesses of the most updated targeted therapies of Wnt/β-catenin signaling in cancers, aiming to generate a thorough awareness of current challenges and crises. Ultimately, this study sought to provide future studies with the issues and insights that should be taken into account for developing better-targeted therapies of Wnt/β-catenin signaling in cancers.

The overview of status quo registered clinical trials of Wnt/β-catenin signaling-dependent targeted therapies in cancers (updated in May 2021). Within the boxed diagram, the first lane provided the numbers of clinical trials for the specific type of cancers in all clinical trials. From the second to the fifth lane, we provided the types of drugs as SMIs, mAbs, Peptides, and others, and the parts that follow the colon indicted the names of drugs and the targeted components of Wnt/β-catenin signaling were shown in the brackets
References
Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480 (2006).
Clevers, H. & Nusse, R. Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 (2012).
Le, P. N. et al. Wnt signaling dynamics in head and neck squamous cell cancer tumor-stroma interactions. Mol. Carcinog. 58, 398–410 (2019).
Stewart, D. J. Wnt signaling pathway in non-small cell lung cancer. J. Natl Cancer Inst. 106, djt356 (2014).
Wang, W., Smits, R., Hao, H. & He, C. Wnt/β-catenin signaling in liver cancers. Cancers 11, 926 (2019).
Bahrami, A. et al. Therapeutic potential of targeting Wnt/β-catenin pathway in treatment of colorectal cancer: rational and progress. J. Cell. Biochem. 118, 1979–1983 (2017).
Nusse, R. & Varmus, H. E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 31, 99–109 (1982).
Rijsewijk, F. et al. The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 50, 649–657 (1987).
Nusse, R. & Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017).
Brennan, K. R. & Brown, A. M. C. Wnt proteins in mammary development and cancer. J. Mammary Gland Biol. Neoplasia 9, 119–131 (2004).
Kurayoshi, M., Yamamoto, H., Izumi, S. & Kikuchi, A. Post-translational palmitoylation and glycosylation of Wnt-5a are necessary for its signalling. Biochem. J. 402, 515–523 (2007).
Komekado, H., Yamamoto, H., Chiba, T. & Kikuchi, A. Glycosylation and palmitoylation of Wnt-3a are coupled to produce an active form of Wnt-3a. Genes Cells Devoted Mol. Cell. Mech. 12, 521–534 (2007).
Takada, R. et al. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev. Cell 11, 791–801 (2006).
Torres, V. I., Godoy, J. A. & Inestrosa, N. C. Modulating Wnt signaling at the root: porcupine and Wnt acylation. Pharmacol. Ther. 198, 34–45 (2019).
Janda, C. Y., Waghray, D., Levin, A. M., Thomas, C. & Garcia, K. C. Structural basis of Wnt recognition by Frizzled. Science 337, 59–64 (2012).
Zhang, X., Dong, S. & Xu, F. Structural and druggability landscape of Frizzled G protein-coupled receptors. Trends Biochem. Sci. 43, 1033–1046 (2018).
Grainger, S. & Willert, K. Mechanisms of Wnt signaling and control. Wiley Interdiscip. Rev. Syst. Biol. Med. e1422, https://doi.org/10.1002/wsbm.1422 (2018).
MacDonald, B. T. & He, X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 4, a007880 (2012).
Brennan, K., Gonzalez-Sancho, J. M., Castelo-Soccio, L. A., Howe, L. R. & Brown, A. M. C. Truncated mutants of the putative Wnt receptor LRP6/Arrow can stabilize beta-catenin independently of Frizzled proteins. Oncogene 23, 4873–4884 (2004).
de Lau, W. B., Snel, B. & Clevers, H. C. The R-spondin protein family. Genome Biol. 13, 242 (2012).
Carmon, K. S., Gong, X., Lin, Q., Thomas, A. & Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl Acad. Sci. USA 108, 11452–11457 (2011).
Glinka, A. et al. LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep. 12, 1055–1061 (2011).
de Lau, W. et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293–297 (2011).
Ruffner, H. et al. R-Spondin potentiates Wnt/β-catenin signaling through orphan receptors LGR4 and LGR5. PLoS ONE 7, e40976 (2012).
Zebisch, M. et al. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat. Commun. 4, 2787 (2013).
Hao, H.-X. et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 485, 195–200 (2012).
Koo, B.-K. et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488, 665–669 (2012).
Sharma, M., Castro-Piedras, I., Simmons, G. E. & Pruitt, K. Dishevelled: a masterful conductor of complex Wnt signals. Cell. Signal. 47, 52–64 (2018).
Schwarz-Romond, T. et al. The DIX domain of dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol. 14, 484–492 (2007).
Wong, H.-C. et al. Direct binding of the PDZ domain of dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol. Cell 12, 1251–1260 (2003).
Aberle, H., Bauer, A., Stappert, J., Kispert, A. & Kemler, R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16, 3797–3804 (1997).
Valenta, T., Hausmann, G. & Basler, K. The many faces and functions of β-catenin. EMBO J. 31, 2714–2736 (2012).
Xing, Y., Clements, W. K., Kimelman, D. & Xu, W. Crystal structure of a beta-catenin/axin complex suggests a mechanism for the beta-catenin destruction complex. Genes Dev. 17, 2753–2764 (2003).
Nc, H., T, T., Jl, S., Hj, C. & Wi, W. Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol. Cell 15, 511–521 (2004).
Graham, T. A., Weaver, C., Mao, F., Kimelman, D. & Xu, W. Crystal structure of a beta-catenin/Tcf complex. Cell 103, 885–896 (2000).
Gao, X. & Hannoush, R. N. Single-cell imaging of Wnt palmitoylation by the acyltransferase porcupine. Nat. Chem. Biol. 10, 61–68 (2014).
Galli, L. M., Zebarjadi, N., Li, L., Lingappa, V. R. & Burrus, L. W. Divergent effects of porcupine and Wntless on WNT1 trafficking, secretion, and signaling. Exp. Cell Res. 347, 171–183 (2016).
Ho, S. Y. & Keller, T. H. The use of porcupine inhibitors to target Wnt-driven cancers. Bioorg. Med. Chem. Lett. 25, 5472–5476 (2015).
Chiang, K.-C. et al. WNT-1 inducible signaling pathway protein-1 enhances growth and tumorigenesis in human breast cancer. Sci. Rep. 5, 8686 (2015).
He, B. et al. Blockade of Wnt-1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene 24, 3054–3058 (2005).
Lv, J. et al. M2-like tumor-associated macrophages-secreted Wnt1 and Wnt3a promotes dedifferentiation and metastasis via activating β-catenin pathway in thyroid cancer. Mol. Carcinog. 60, 25–37 (2021).
Mizushima, T. et al. Wnt-1 but not epidermal growth factor induces beta-catenin/T-cell factor-dependent transcription in esophageal cancer cells. Cancer Res. 62, 277–282 (2002).
Nakashima, T. et al. Wnt1 overexpression associated with tumor proliferation and a poor prognosis in non-small cell lung cancer patients. Oncol. Rep. 19, 203–209 (2008).
Akalay, I. et al. Targeting WNT1-inducible signaling pathway protein 2 alters human breast cancer cell susceptibility to specific lysis through regulation of KLF-4 and miR-7 expression. Oncogene 34, 2261–2271 (2015).
Benad, P., Rauner, M., Rachner, T. D. & Hofbauer, L. C. The anti-progestin RU-486 inhibits viability of MCF-7 breast cancer cells by suppressing WNT1. Cancer Lett. 312, 101–108 (2011).
Cha, Y. et al. MicroRNA-140-5p suppresses cell proliferation and invasion in gastric cancer by targeting WNT1 in the WNT/β-catenin signaling pathway. Oncol. Lett. 16, 6369–6376 (2018).
Chang, L.-C. et al. Identification of a new class of WNT1 inhibitor: cancer cells migration, G-quadruplex stabilization and target validation. Oncotarget 7, 67986–68001 (2016).
Choi, A.-R. et al. Inhibition of Wnt1 expression reduces the enrichment of cancer stem cells in a mouse model of breast cancer. Biochem. Biophys. Res. Commun. 425, 436–442 (2012).
Debies, M. T. et al. Tumor escape in a Wnt1-dependent mouse breast cancer model is enabled by p19Arf/p53 pathway lesions but not p16 Ink4a loss. J. Clin. Invest. 118, 51–63 (2008).
Huguet, E. L., McMahon, J. A., McMahon, A. P., Bicknell, R. & Harris, A. L. Differential expression of human Wnt genes 2, 3, 4, and 7B in human breast cell lines and normal and disease states of human breast tissue. Cancer Res. 54, 2615–2621 (1994).
Vider, B. Z. et al. Evidence for the involvement of the Wnt 2 gene in human colorectal cancer. Oncogene 12, 153–158 (1996).
Unterleuthner, D. et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis 23, 159–177 (2020).
Jiang, H. et al. Activation of the Wnt pathway through Wnt2 promotes metastasis in pancreatic cancer. Am. J. Cancer Res. 4, 537–544 (2014).
Kramer, N. et al. Autocrine WNT2 signaling in fibroblasts promotes colorectal cancer progression. Oncogene 36, 5460–5472 (2017).
Zhang, Z., Wang, J. & Dong, X. Wnt2 contributes to the progression of gastric cancer by promoting cell migration and invasion. Oncol. Lett. 16, 2857–2864 (2018).
Xu, Y. et al. Wnt2 protein plays a role in the progression of pancreatic cancer promoted by pancreatic stellate cells. Med. Oncol. Northwood Lond. Engl. 32, 97 (2015).
Lee, S.-B., Park, Y. I., Dong, M.-S. & Gong, Y.-D. Identification of 2,3,6-trisubstituted quinoxaline derivatives as a Wnt2/β-catenin pathway inhibitor in non-small-cell lung cancer cell lines. Bioorg. Med. Chem. Lett. 20, 5900–5904 (2010).
Xiu, D.-H. et al. Long non-coding RNA LINC00968 attenuates drug resistance of breast cancer cells through inhibiting the Wnt2/β-catenin signaling pathway by regulating WNT2. J. Exp. Clin. Cancer Res. 38, 94 (2019).
Dale, T. C. et al. Compartment switching of WNT-2 expression in human breast tumors. Cancer Res. 56, 4320–4323 (1996).
Katoh, M. Molecular cloning and characterization of human WNT3. Int. J. Oncol. 19, 977–982 (2001).
Nie, X. et al. Downregulation of Wnt3 suppresses colorectal cancer development through inhibiting cell proliferation and migration. Front. Pharmacol. 10, 1110 (2019).
Wu, Y. et al. Expression of Wnt3 activates Wnt/β-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol. Cancer Res. 10, 1597–1606 (2012).
Wu, Y. et al. A83-01 inhibits TGF-β-induced upregulation of Wnt3 and epithelial to mesenchymal transition in HER2-overexpressing breast cancer cells. Breast Cancer Res. Treat. 163, 449–460 (2017).
Nakashima, N. et al. Wnt3 gene expression promotes tumor progression in non-small cell lung cancer. Lung Cancer Amst. Neth. 76, 228–234 (2012).
Xing, Z. et al. Wnt3 knockdown sensitizes human non-small cell type lung cancer (NSCLC) cells to cisplatin via regulating the cell proliferation and apoptosis. Eur. Rev. Med. Pharmacol. Sci. 22, 1323–1332 (2018).
Ye, J. et al. TROAP regulates prostate cancer progression via the WNT3/survivin signalling pathways. Oncol. Rep. 41, 1169–1179 (2019).
Chu, Y. et al. miR-1247-5p functions as a tumor suppressor in human hepatocellular carcinoma by targeting Wnt3. Oncol. Rep. 38, 343–351 (2017).
Pashirzad, M. et al. Role of Wnt3a in the pathogenesis of cancer, current status and prospective. Mol. Biol. Rep. 46, 5609–5616 (2019).
Zhang, C. & Wang, Y. Metformin attenuates cells stemness and epithelial‑mesenchymal transition in colorectal cancer cells by inhibiting the Wnt3a/β‑catenin pathway. Mol. Med. Rep. 19, 1203–1209 (2019).
Lee, M. A. et al. Wnt3a expression is associated with MMP-9 expression in primary tumor and metastatic site in recurrent or stage IV colorectal cancer. BMC Cancer 14, 125 (2014).
Kang, D. W. & Min, D. S. Positive feedback regulation between phospholipase D and Wnt signaling promotes Wnt-driven anchorage-independent growth of colorectal cancer cells. PLoS ONE 5, e12109 (2010).
Gui, S. et al. Wnt3a regulates proliferation, apoptosis and function of pancreatic NIT-1 beta cells via activation of IRS2/PI3K signaling. J. Cell. Biochem. 114, 1488–1497 (2013).
Matei, N. et al. Intranasal wnt3a attenuates neuronal apoptosis through Frz1/PIWIL1a/FOXM1 pathway in MCAO rats. J. Neurosci. 38, 6787–6801 (2018).
Doubravská, L. et al. Wnt-expressing rat embryonic fibroblasts suppress Apo2L/TRAIL-induced apoptosis of human leukemia cells. Apoptosis 13, 573–587 (2008).
Nygren, M. K. et al. Wnt3A activates canonical Wnt signalling in acute lymphoblastic leukaemia (ALL) cells and inhibits the proliferation of B-ALL cell lines. Br. J. Haematol. 136, 400–413 (2007).
Aripaka, K. et al. TRAF6 function as a novel co-regulator of Wnt3a target genes in prostate cancer. EBioMedicine 45, 192–207 (2019).
Moon, Y. H., Lim, W. & Jeong, B.-C. Transmembrane protein 64 modulates prostate tumor progression by regulating Wnt3a secretion. Oncol. Lett. 18, 283–290 (2019).
Yang, Y. et al. MicroRNA‑214 targets Wnt3a to suppress liver cancer cell proliferation. Mol. Med. Rep. 16, 6920–6927 (2017).
Yang, Y. et al. MicroRNA-195 acts as a tumor suppressor by directly targeting Wnt3a in HepG2 hepatocellular carcinoma cells. Mol. Med. Rep. 10, 2643–2648 (2014).
Schenkelaars, Q., Fierro-Constain, L., Renard, E., Hill, A. L. & Borchiellini, C. Insights into Frizzled evolution and new perspectives. Evol. Dev. 17, 160–169 (2015).
Tauriello, D. V. F. et al. Wnt/β-catenin signaling requires interaction of the Dishevelled DEP domain and C terminus with a discontinuous motif in Frizzled. Proc. Natl Acad. Sci. USA 109, E812–E820 (2012).
Milovanovic, T. et al. Expression of Wnt genes and Frizzled 1 and 2 receptors in normal breast epithelium and infiltrating breast carcinoma. Int. J. Oncol. 25, 1337–1342 (2004).
Li, Y., Lu, W., He, X., Schwartz, A. L. & Bu, G. LRP6 expression promotes cancer cell proliferation and tumorigenesis by altering beta-catenin subcellular distribution. Oncogene 23, 9129–9135 (2004).
Flanagan, D. J. et al. Frizzled-7 is required for Wnt signaling in gastric tumors with and without Apc mutations. Cancer Res. 79, 970–981 (2019).
Sun, Y., Wang, W. & Zhao, C. Frizzled receptors in tumors, focusing on signaling, roles, modulation mechanisms, and targeted therapies. Oncol. Res. https://doi.org/10.3727/096504020X16014648664459 (2020).
Gazit, A. et al. Human Frizzled 1 interacts with transforming Wnts to transduce a TCF dependent transcriptional response. Oncogene 18, 5959–5966 (1999).
Wang, Z., Shu, W., Lu, M. M. & Morrisey, E. E. Wnt7b activates canonical signaling in epithelial and vascular smooth muscle cells through interactions with Fzd1, Fzd10, and LRP5. Mol. Cell. Biol. 25, 5022–5030 (2005).
Koval, A. & Katanaev, V. L. Wnt3a stimulation elicits G-protein-coupled receptor properties of mammalian Frizzled proteins. Biochem. J. 433, 435–440 (2011).
Endo, Y. et al. Wnt-3a and Dickkopf-1 stimulate neurite outgrowth in Ewing tumor cells via a Frizzled3- and c-Jun N-terminal kinase-dependent mechanism. Mol. Cell. Biol. 28, 2368–2379 (2008).
Hansen, C. et al. Wnt-5a-induced phosphorylation of DARPP-32 inhibits breast cancer cell migration in a CREB-dependent manner. J. Biol. Chem. 284, 27533–27543 (2009).
Mo, D. et al. A tRNA fragment, 5’-tiRNAVal, suppresses the Wnt/β-catenin signaling pathway by targeting FZD3 in breast cancer. Cancer Lett. 457, 60–73 (2019).
Gupta, S. et al. FZD4 as a mediator of ERG oncogene-induced WNT signaling and epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 70, 6735–6745 (2010).
Thiele, S. et al. Role of WNT5A receptors FZD5 and RYK in prostate cancer cells. Oncotarget 9, 27293–27304 (2018).
Dong, D. et al. FZD5 prevents epithelial-mesenchymal transition in gastric cancer. Cell Commun. Signal. 19, 21 (2021).
Rubinfeld, B. et al. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 272, 1023–1026 (1996).
Corda, G. et al. Functional and prognostic significance of the genomic amplification of Frizzled 6 (FZD6) in breast cancer. J. Pathol. 241, 350–361 (2017).
King, T. D., Zhang, W., Suto, M. J. & Li, Y. Frizzled7 as an emerging target for cancer therapy. Cell. Signal. 24, 846–851 (2012).
Kim, M. et al. Functional interaction between Wnt3 and Frizzled-7 leads to activation of the Wnt/beta-catenin signaling pathway in hepatocellular carcinoma cells. J. Hepatol. 48, 780–791 (2008).
Chakravarthi, B. V. S. K. et al. Wnt receptor Frizzled 8 is a target of ERG in prostate cancer. Prostate 78, 1311–1320 (2018).
Scavo, M. P. et al. Frizzled-10 and cancer progression: Is it a new prognostic marker? Oncotarget 9, 824–830 (2018).
Nagayama, S. et al. Therapeutic potential of antibodies against FZD10, a cell-surface protein, for synovial sarcomas. Oncogene 24, 6201–6212 (2005).
Liu, Z. et al. Hypoxia-induced suppression of alternative splicing of MBD2 promotes breast cancer metastasis via activation of FZD1. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-20-2876 (2021).
Zhang, H. et al. Interference of Frizzled 1 (FZD1) reverses multidrug resistance in breast cancer cells through the Wnt/β-catenin pathway. Cancer Lett. 323, 106–113 (2012).
Su, W. et al. miR-135b reverses chemoresistance of non-small cell lung cancer cells by downregulation of FZD1. Biomed. Pharmacother. Biomedecine Pharmacother. 84, 123–129 (2016).
Zhang, H. et al. Suppression of multidrug resistance by rosiglitazone treatment in human ovarian cancer cells through downregulation of FZD1 and MDR1 genes. Anticancer. Drugs 26, 706–715 (2015).
Li, H. et al. WITHDRAWN: MicroRNA-505 modulates cancer proliferation and migration in human non-small cell lung cancer through inverse regulation of FZD4. Lung Cancer Amst. Neth. https://doi.org/10.1016/j.lungcan.2017.03.016 (2017).
Ueno, K. et al. Tumor suppressor microRNA-493 decreases cell motility and migration ability in human bladder cancer cells by downregulating RhoC and FZD4. Mol. Cancer Ther. 11, 244–253 (2012).
Wang, Y., Zhang, W., Wang, Y. & Wang, S. HOXD-AS1 promotes cell proliferation, migration and invasion through miR-608/FZD4 axis in ovarian cancer. Am. J. Cancer Res. 8, 170–182 (2018).
Ueno, K. et al. Frizzled-7 as a potential therapeutic target in colorectal cancer. Neoplasia N. Y. N. 10, 697–705 (2008).
Fenderico, N. et al. Anti-LRP5/6 VHHs promote differentiation of Wnt-hypersensitive intestinal stem cells. Nat. Commun. 10, 365 (2019).
Björklund, P., Akerström, G. & Westin, G. An LRP5 receptor with internal deletion in hyperparathyroid tumors with implications for deregulated WNT/beta-catenin signaling. PLoS Med. 4, e328 (2007).
Wang, H. et al. Hsp90ab1 stabilizes LRP5 to promote epithelial-mesenchymal transition via activating of AKT and Wnt/β-catenin signaling pathways in gastric cancer progression. Oncogene 38, 1489–1507 (2019).
Q, Y. et al. LRP6 promotes invasion and metastasis of colorectal cancer through cytoskeleton dynamics. Oncotarget 8, 109632–109645 (2017).
Liu, C.-C., Prior, J., Piwnica-Worms, D. & Bu, G. LRP6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc. Natl Acad. Sci. USA 107, 5136–5141 (2010).
Tahir, S. A. et al. Caveolin-1-LRP6 signaling module stimulates aerobic glycolysis in prostate cancer. Cancer Res. 73, 1900–1911 (2013).
Ettenberg, S. A. et al. Inhibition of tumorigenesis driven by different Wnt proteins requires blockade of distinct ligand-binding regions by LRP6 antibodies. Proc. Natl Acad. Sci. USA 107, 15473–15478 (2010).
Lebensohn, A. M. & Rohatgi, R. R-spondins can potentiate WNT signaling without LGRs. eLife 7, e33126 (2018).
Szenker-Ravi, E. et al. RSPO2 inhibition of RNF43 and ZNRF3 governs limb development independently of LGR4/5/6. Nature 557, 564–569 (2018).
Seshagiri, S. et al. Recurrent R-spondin fusions in colon cancer. Nature 488, 660–664 (2012).
Han, T. et al. R-Spondin chromosome rearrangements drive Wnt-dependent tumour initiation and maintenance in the intestine. Nat. Commun. 8, 15945 (2017).
Bugter, J. M., Fenderico, N. & Maurice, M. M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 21, 5–21 (2021).
Ryland, G. L. et al. RNF43 is a tumour suppressor gene mutated in mucinous tumours of the ovary. J. Pathol. 229, 469–476 (2013).
Eto, T. et al. Impact of loss-of-function mutations at the RNF43 locus on colorectal cancer development and progression. J. Pathol. 245, 445–455 (2018).
Jiang, X. et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl Acad. Sci. USA 110, 12649–12654 (2013).
Kleeman, S. O. et al. Exploiting differential Wnt target gene expression to generate a molecular biomarker for colorectal cancer stratification. Gut 69, 1092–1103 (2020).
Li, S. et al. Commonly observed RNF43 mutations retain functionality in attenuating Wnt/β-catenin signaling and unlikely confer Wnt-dependency onto colorectal cancers. Oncogene 39, 3458–3472 (2020).
Laurent-Puig, P., Béroud, C. & Soussi, T. APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 26, 269–270 (1998).
Wachsmannova, L., Mego, M., Stevurkova, V., Zajac, V. & Ciernikova, S. Novel strategies for comprehensive mutation screening of the APC gene. Neoplasma 64, 338–343 (2017).
Cao, X., Hong, Y., Eu, K. W., Loi, C. & Cheah, P. Y. Singapore familial adenomatous polyposis (FAP) patients with classical adenomatous polyposis but undetectable APC mutations have accelerated cancer progression. Am. J. Gastroenterol. 101, 2810–2817 (2006).
Aghabozorgi, A. S. et al. Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie 157, 64–71 (2019).
Aitchison, A. et al. APC mutations are not confined to hotspot regions in early-onset colorectal cancer. Cancers 12, 3829 (2020).
Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer Oxf. Engl. 38, 867–871 (2002). 1990.
Pronobis, M. I., Rusan, N. M. & Peifer, M. A novel GSK3-regulated APC:Axin interaction regulates Wnt signaling by driving a catalytic cycle of efficient βcatenin destruction. eLife 4, e08022 (2015).
Singh, I. & Pujol, A. Pathomechanisms underlying X-adrenoleukodystrophy: a three-hit hypothesis. Brain Pathol. Zur. Switz. 20, 838–844 (2010).
Rosin-Arbesfeld, R., Townsley, F. & Bienz, M. The APC tumour suppressor has a nuclear export function. Nature 406, 1009–1012 (2000).
Zhang, L. & Shay, J. W. Multiple roles of APC and its therapeutic implications in colorectal cancer. J. Natl Cancer Inst. 109, djw332 (2017).
De Santis, S. et al. Winnie-APCMin/+ mice: a spontaneous model of colitis-associated colorectal cancer combining genetics and inflammation. Int. J. Mol. Sci. 21, 2972 (2020).
Wechter, W. J. et al. R-flurbiprofen chemoprevention and treatment of intestinal adenomas in the APC(Min)/+ mouse model: implications for prophylaxis and treatment of colon cancer. Cancer Res. 57, 4316–4324 (1997).
Irving, A. A. et al. The utility of Apc-mutant rats in modeling human colon cancer. Dis. Model. Mech. 7, 1215–1225 (2014).
Ohgaki, H. et al. APC mutations are infrequent but present in human lung cancer. Cancer Lett. 207, 197–203 (2004).
Thompson, A. M. et al. Allele loss from 5q21 (APC/MCC) and 18q21 (DCC) and DCC mRNA expression in breast cancer. Br. J. Cancer 68, 64–68 (1993).
Jönsson, M., Borg, A., Nilbert, M. & Andersson, T. Involvement of adenomatous polyposis coli (APC)/beta-catenin signalling in human breast cancer. Eur. J. Cancer Oxf. Engl. 36, 242–248 (2000). 1990.
McKie, A. B., Filipe, M. I. & Lemoine, N. R. Abnormalities affecting the APC and MCC tumour suppressor gene loci on chromosome 5q occur frequently in gastric cancer but not in pancreatic cancer. Int. J. Cancer 55, 598–603 (1993).
Gao, X. et al. High-frequency of loss of expression and allelic deletion of the apc and mcc genes in human prostate-cancer. Int. J. Oncol. 6, 111–117 (1995).
Guo, S. et al. Quantitative assessment of the diagnostic role of APC promoter methylation in non-small cell lung cancer. Clin. Epigenet. 6, 5 (2014).
Chen, Y. et al. APC gene hypermethylation and prostate cancer: a systematic review and meta-analysis. Eur. J. Hum. Genet. 21, 929–935 (2013).
Ikeda, S. et al. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 17, 1371–1384 (1998).
Sakanaka, C., Weiss, J. B. & Williams, L. T. Bridging of beta-catenin and glycogen synthase kinase-3beta by axin and inhibition of beta-catenin-mediated transcription. Proc. Natl Acad. Sci. USA 95, 3020–3023 (1998).
Noutsou, M. et al. Critical scaffolding regions of the tumor suppressor Axin1 are natively unfolded. J. Mol. Biol. 405, 773–786 (2011).
Spink, K. E., Polakis, P. & Weis, W. I. Structural basis of the Axin-adenomatous polyposis coli interaction. EMBO J. 19, 2270–2279 (2000).
Schwarz-Romond, T. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol. 14, 484–492 (2007).
Dajani, R. et al. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. 22, 494–501 (2003).
Anvarian, Z. et al. Axin cancer mutants form nanoaggregates to rewire the Wnt signaling network. Nat. Struct. Mol. Biol. 23, 324–332 (2016).
Cha, P.-H. et al. Small-molecule binding of the axin RGS domain promotes β-catenin and Ras degradation. Nat. Chem. Biol. 12, 593–600 (2016).
Bernkopf, D. B., Brückner, M., Hadjihannas, M. V. & Behrens, J. An aggregon in conductin/axin2 regulates Wnt/β-catenin signaling and holds potential for cancer therapy. Nat. Commun. 10, 4251 (2019).
Satoh, S. et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 24, 245–250 (2000).
Boyault, S. et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 45, 42–52 (2007).
Feng, G. J. et al. Conditional disruption of Axin1 leads to development of liver tumors in mice. Gastroenterology 143, 1650–1659 (2012).
Qiao, Y. et al. Axis inhibition protein 1 (Axin1) deletion-induced hepatocarcinogenesis requires intact β-catenin but not notch cascade in mice. Hepatology 70, 2003–2017 (2019).
Picco, G. et al. Loss of AXIN1 drives acquired resistance to WNT pathway blockade in colorectal cancer cells carrying RSPO3 fusions. EMBO Mol. Med. 9, 293–303 (2017).
Syed, S. M. & Tanwar, P. S. Axin2+ endometrial stem cells: the source of endometrial regeneration and cancer. Mol. Cell. Oncol. 7, 1729681 (2020).
Lammi, L. et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am. J. Hum. Genet. 74, 1043–1050 (2004).
Liu, W. et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat. Genet. 26, 146–147 (2000).
Otero, L. et al. Variations in AXIN2 predict risk and prognosis of colorectal cancer. BDJ Open 5, 13 (2019).
Rosales-Reynoso, M. A. et al. AXIN2 polymorphisms and their association with colorectal cancer in mexican patients. Genet. Test. Mol. Biomark. 20, 438–444 (2016).
Bahl, C., Sharma, S., Singh, N. & Behera, D. Association study between genetic variations in Axin2 gene and lung cancer risk in North Indian population: a multiple interaction analysis. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 39, 1010428317695533 (2017).
Hughes, T. A. & Brady, H. J. M. Regulation of axin2 expression at the levels of transcription, translation and protein stability in lung and colon cancer. Cancer Lett. 233, 338–347 (2006).
Aristizabal-Pachon, A. F., Carvalho, T. I., Carrara, H. H., Andrade, J. & Takahashi, C. S. AXIN2 polymorphisms, the β-catenin destruction complex expression profile and breast cancer susceptibility. Asian Pac. J. Cancer Prev. 16, 7277–7284 (2015).
Bao, R. et al. Inhibition of tankyrases induces Axin stabilization and blocks Wnt signalling in breast cancer cells. PLoS ONE 7, e48670 (2012).
Dai, J., Gao, H., Xue, J., Lin, W. & Zheng, L. The association between AXIN2 gene polymorphisms and the risk of breast cancer in chinese women. Genet. Test. Mol. Biomark. 23, 393–400 (2019).
Huang, S.-M. A. et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620 (2009).
Gibson, B. A. & Kraus, W. L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 13, 411–424 (2012).
Haikarainen, T., Krauss, S. & Lehtiö, L. Tankyrases: structure, function and therapeutic implications in cancer. Curr. Pharm. Des. 20, 6472–6488 (2014).
Zhang, Y. et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 13, 623–629 (2011).
Wang, Z. et al. Wnt/Wingless pathway activation is promoted by a critical threshold of axin maintained by the tumor suppressor APC and the ADP-ribose polymerase tankyrase. Genetics 203, 269–281 (2016).
Yang, E. et al. Wnt pathway activation by ADP-ribosylation. Nat. Commun. 7, 11430 (2016).
Wang, W., Tacchelly-Benites, O., Yang, E. & Ahmed, Y. Dual roles for membrane association of Drosophila axin in Wnt signaling. PLoS Genet. 12, e1006494 (2016).
Mariotti, L., Pollock, K. & Guettler, S. Regulation of Wnt/β-catenin signalling by tankyrase-dependent poly(ADP-ribosyl)ation and scaffolding. Br. J. Pharmacol. 174, 4611–4636 (2017).
Riffell, J. L., Lord, C. J. & Ashworth, A. Tankyrase-targeted therapeutics: expanding opportunities in the PARP family. Nat. Rev. Drug Discov. 11, 923–936 (2012).
Kamal, A., Riyaz, S., Srivastava, A. K. & Rahim, A. Tankyrase inhibitors as therapeutic targets for cancer. Curr. Top. Med. Chem. 14, 1967–1976 (2014).
Cormier, K. W. & Woodgett, J. R. Recent advances in understanding the cellular roles of GSK-3. F1000Research 6, 167 (2017).
Yuan, H., Mao, J., Li, L. & Wu, D. Suppression of glycogen synthase kinase activity is not sufficient for leukemia enhancer factor-1 activation. J. Biol. Chem. 274, 30419–30423 (1999).
Zheng, X. et al. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature 438, 873–877 (2005).
Zeng, X. et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development 135, 367–375 (2008).
Davidson, G. et al. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature 438, 867–872 (2005).
McCubrey, J. A. et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget 3, 1068–1111 (2012).
Medunjanin, S. et al. GSK-3β controls NF-kappaB activity via IKKγ/NEMO. Sci. Rep. 6, 38553 (2016).
Liu, C. et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 (2002).
Knippschild, U. et al. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell. Signal. 17, 675–689 (2005).
Cruciat, C. M. Casein kinase 1 and Wnt/β-catenin signaling. Curr. Opin. Cell Biol. 31, 46–55 (2014).
Xing, Y. et al. Crystal structure of a beta-catenin/APC complex reveals a critical role for APC phosphorylation in APC function. Mol. Cell 15, 523–533 (2004).
de Groot, R. E. A. et al. Huwe1-mediated ubiquitylation of dishevelled defines a negative feedback loop in the Wnt signaling pathway. Sci. Signal. 7, ra26 (2014).
Bilic, J. et al. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316, 1619–1622 (2007).
Thorne, C. A. et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 6, 829–836 (2010).
Lee, E., Salic, A. & Kirschner, M. W. Physiological regulation of [beta]-catenin stability by Tcf3 and CK1epsilon. J. Cell Biol. 154, 983–993 (2001).
Kishida, S. et al. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability. Mol. Cell. Biol. 19, 4414–4422 (1999).
Gao, C. & Chen, Y. G. Dishevelled: the hub of Wnt signaling. Cell. Signal. 22, 717–727 (2010).
Habas, R. & Dawid, I. B. Dishevelled and Wnt signaling: is the nucleus the final frontier? J. Biol. 4, 2 (2005).
Itoh, K., Brott, B. K., Bae, G.-U., Ratcliffe, M. J. & Sokol, S. Y. Nuclear localization is required for Dishevelled function in Wnt/beta-catenin signaling. J. Biol. 4, 3 (2005).
Luo, W. et al. Protein phosphatase 1 regulates assembly and function of the beta-catenin degradation complex. EMBO J. 26, 1511–1521 (2007).
Strovel, E. T., Wu, D. & Sussman, D. J. Protein phosphatase 2Calpha dephosphorylates axin and activates LEF-1-dependent transcription. J. Biol. Chem. 275, 2399–2403 (2000).
Cho, U. S. & Xu, W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 445, 53–57 (2007).
Janssens, V., Longin, S. & Goris, J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem. Sci. 33, 113–121 (2008).
Virshup, D. M. Protein phosphatase 2A: a panoply of enzymes. Curr. Opin. Cell Biol. 12, 180–185 (2000).
Zhang, W. et al. PR55 alpha, a regulatory subunit of PP2A, specifically regulates PP2A-mediated beta-catenin dephosphorylation. J. Biol. Chem. 284, 22649–22656 (2009).
Yu, N. et al. HSP105 recruits protein phosphatase 2A to dephosphorylate β-catenin. Mol. Cell. Biol. 35, 1390–1400 (2015).
Su, Y. et al. APC is essential for targeting phosphorylated beta-catenin to the SCFbeta-TrCP ubiquitin ligase. Mol. Cell 32, 652–661 (2008).
Johnson, V. et al. Exon 3 beta-catenin mutations are specifically associated with colorectal carcinomas in hereditary non-polyposis colorectal cancer syndrome. Gut 54, 264–267 (2005).
Morin, P. J. et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275, 1787–1790 (1997).
Abdelmaksoud-Damak, R. et al. Expression and mutation pattern of β-catenin and adenomatous polyposis coli in colorectal cancer patients. Arch. Med. Res. 46, 54–62 (2015).
Voeller, H. J., Truica, C. I. & Gelmann, E. P. Beta-catenin mutations in human prostate cancer. Cancer Res. 58, 2520–2523 (1998).
Ogasawara, N. et al. Mutations and nuclear accumulation of beta-catenin correlate with intestinal phenotypic expression in human gastric cancer. Histopathology 49, 612–621 (2006).
Kizildag, S., Zengel, B., Vardar, E. & Sakizli, M. beta-catenin gene mutation in invasive ductal breast cancer. J. BUON 13, 533–536 (2008).
Roy, D. & Calaf, G. M. Mutation of β-catenin in a radiation and estrogen breast cancer model. Int. J. Oncol. 46, 153–160 (2015).
Wright, K. et al. beta-catenin mutation and expression analysis in ovarian cancer: exon 3 mutations and nuclear translocation in 16% of endometrioid tumours. Int. J. Cancer 82, 625–629 (1999).
Kurnit, K. C. et al. CTNNB1 (beta-catenin) mutation identifies low grade, early stage endometrial cancer patients at increased risk of recurrence. Mod. Pathol. 30, 1032–1041 (2017).
Takemaru, K. I. & Moon, R. T. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 149, 249–254 (2000).
Ono, M. et al. Nuclear receptor/Wnt beta-catenin interactions are regulated via differential CBP/p300 coactivator usage. PLoS ONE 13, e0200714 (2018).
Daniels, D. L. & Weis, W. I. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat. Struct. Mol. Biol. 12, 364–371 (2005).
Hanson, A. J. et al. XIAP monoubiquitylates Groucho/TLE to promote canonical Wnt signaling. Mol. Cell 45, 619–628 (2012).
Jia, Y. et al. Thymine DNA glycosylase promotes transactivation of β-catenin/TCFs by cooperating with CBP. J. Mol. Cell Biol. 6, 231–239 (2014).
Hecht, A., Vleminckx, K., Stemmler, M. P., van Roy, F. & Kemler, R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 19, 1839–1850 (2000).
Lévy, L. et al. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol. Cell. Biol. 24, 3404–3414 (2004).
Kramps, T. et al. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell 109, 47–60 (2002).
Bauer, A. et al. Pontin52 and reptin52 function as antagonistic regulators of beta-catenin signalling activity. EMBO J. 19, 6121–6130 (2000).
Barker, N. et al. The chromatin remodelling factor Brg-1 interacts with beta-catenin to promote target gene activation. EMBO J. 20, 4935–4943 (2001).
Rao, M. et al. Inhibition of cyclin D1 gene transcription by Brg-1. Cell Cycle 7, 647–655 (2008).
Mahmoudi, T. et al. The leukemia-associated Mllt10/Af10-Dot1l are Tcf4/β-catenin coactivators essential for intestinal homeostasis. PLoS Biol. 8, e1000539 (2010).
Zhou, D. et al. SOX10 is a novel oncogene in hepatocellular carcinoma through Wnt/β-catenin/TCF4 cascade. Tumour Biol. 35, 9935–9940 (2014).
Shin, S., Rossow, K. L., Grande, J. P. & Janknecht, R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res. 67, 7572–7578 (2007).
Kimbrel, E. A. & Kung, A. L. The F-box protein beta-TrCp1/Fbw1a interacts with p300 to enhance beta-catenin transcriptional activity. J. Biol. Chem. 284, 13033–13044 (2009).
Gong, A. & Huang, S. FoxM1 and Wnt/β-catenin signaling in glioma stem cells. Cancer Res. 72, 5658–5662 (2012).
Pratheeshkumar, P. et al. FoxM1 and β-catenin predicts aggressiveness in Middle Eastern ovarian cancer and their co-targeting impairs the growth of ovarian cancer cells. Oncotarget 9, 3590–3604 (2018).
Shukla, S. et al. The FOXM1 Inhibitor RCM-1 Decreases Carcinogenesis and Nuclear β-catenin. Mol. Cancer Ther. 18, 1217–1229 (2019).
Tao, J. et al. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 147, 690–701 (2014).
Liu, T. et al. The β-catenin/YAP signaling axis is a key regulator of melanoma-associated fibroblasts. Signal Transduct. Target. Ther. 4, 63 (2019).
Kaidi, A., Williams, A. C. & Paraskeva, C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 9, 210–217 (2007).
Choi, H., Chun, Y.-S., Kim, T.-Y. & Park, J.-W. HIF-2alpha enhances beta-catenin/TCF-driven transcription by interacting with beta-catenin. Cancer Res. 70, 10101–10111 (2010).
Sui, H. et al. Tanshinone IIA inhibits β-catenin/VEGF-mediated angiogenesis by targeting TGF-β1 in normoxic and HIF-1α in hypoxic microenvironments in human colorectal cancer. Cancer Lett. 403, 86–97 (2017).
Jung, C., Kim, R.-S., Lee, S.-J., Wang, C. & Jeng, M.-H. HOXB13 homeodomain protein suppresses the growth of prostate cancer cells by the negative regulation of T-cell factor 4. Cancer Res. 64, 3046–3051 (2004).
Jung, C. et al. HOXB13 is downregulated in colorectal cancer to confer TCF4-mediated transactivation. Br. J. Cancer 92, 2233–2239 (2005).
Ito, K. et al. RUNX3 attenuates beta-catenin/T cell factors in intestinal tumorigenesis. Cancer Cell 14, 226–237 (2008).
Sun, J. et al. RUNX3 inhibits glioma survival and invasion via suppression of the β-catenin/TCF-4 signaling pathway. J. Neurooncol. 140, 15–26 (2018).
Saegusa, M., Hashimura, M., & Kuwata, T. Sox4 functions as a positive regulator of β-catenin signaling through upregulation of TCF4 during morular differentiation of endometrial carcinomas. Lab. Invest. 92, 511–521 (2012).
Sinner, D. et al. Sox17 and Sox4 differentially regulate beta-catenin/T-cell factor activity and proliferation of colon carcinoma cells. Mol. Cell. Biol. 27, 7802–7815 (2007).
Firestein, R. et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 455, 547–551 (2008).
Gu, X. et al. TCTP promotes glioma cell proliferation in vitro and in vivo via enhanced β-catenin/TCF-4 transcription. Neuro Oncol. 16, 217–227 (2014).
Fu, C. A. et al. TNIK, a novel member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and regulates the cytoskeleton. J. Biol. Chem. 274, 30729–30737 (1999).
Taira, K. et al. The Traf2- and Nck-interacting kinase as a putative effector of Rap2 to regulate actin cytoskeleton. J. Biol. Chem. 279, 49488–49496 (2004).
Mahmoudi, T. et al. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 28, 3329–3340 (2009).
Shitashige, M. et al. Traf2- and Nck-interacting kinase is essential for Wnt signaling and colorectal cancer growth. Cancer Res. 70, 5024–5033 (2010).
Sawa, M., Masuda, M. & Yamada, T. Targeting the Wnt signaling pathway in colorectal cancer. Expert Opin. Ther. Targets 20, 419–429 (2016).
Takahashi, H. et al. Prognostic significance of Traf2- and Nck- interacting kinase (TNIK) in colorectal cancer. BMC Cancer 15, 794 (2015).
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Collu, G., Hidalgo-Sastre, A. & Brennan, K. Wnt-Notch signalling crosstalk in development and disease. Cell. Mol. Life Sci. 71, 3553–3567 (2014).
Couso, J. P. & Arias, Martinez A. Notch is required for wingless signaling in the epidermis of Drosophila. Cell 79, 259–272 (1994).
Hayward, P. et al. Notch modulates Wnt signalling by associating with Armadillo/ß-catenin and regulating its transcriptional activity. Dev. Camb. Engl. 132, 1819 (2005).
Camps, J. et al. Genetic amplification of the NOTCH modulator LNX2 upregulates the WNT/β-catenin pathway in colorectal cancer. Cancer Res. 73, 2003–2013 (2013).
Borggrefe, T. et al. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFβ/BMP and hypoxia pathways. Biochim. Biophys. Acta 1863, 303–313 (2016).
Wang, R. et al. Notch and Wnt/β-catenin signaling pathway play important roles in activating liver cancer stem cells. Oncotarget 7, 5754–5768 (2016).
Peignon, G. et al. Complex interplay between β-catenin signalling and Notch effectors in intestinal tumorigenesis. Gut 60, 166–176 (2011).
Saha, S. K., Yin, Y., Chae, H. S. & Cho, S.-G. Opposing regulation of cancer properties via KRT19-mediated differential modulation of Wnt/β-catenin/Notch signaling in breast and colon cancers. Cancers 11, 99 (2019).
Gowrikumar, S. et al. Correction: Upregulated claudin-1 expression promotes colitis-associated cancer by promoting β-catenin phosphorylation and activation in Notch/p-AKT-dependent manner. Oncogene 38, 6566 (2019).
Bangs, F. & Anderson, K. V. Primary cilia and mammalian hedgehog signaling. Cold Spring Harb. Perspect. Biol. 9, a028175 (2017).
Rohatgi, R., Milenkovic, L., Corcoran, R. B. & Scott, M. P. Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc. Natl Acad. Sci. USA 106, 3196–3201 (2009).
Jacob, J. & Briscoe, J. Gli proteins and the control of spinal-cord patterning. EMBO Rep. 4, 761–765 (2003).
Wang, B. & Li, Y. Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc. Natl Acad. Sci. USA 103, 33–38 (2006).
Shin, K. et al. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature 472, 110–114 (2011).
Farahmand, L., Darvishi, B., Majidzadeh-A, K. & Madjid Ansari, A. Naturally occurring compounds acting as potent anti-metastatic agents and their suppressing effects on Hedgehog and WNT/β-catenin signalling pathways. Cell Prolif. 50, (2017).
Qualtrough, D., Rees, P., Speight, B., Williams, A. C. & Paraskeva, C. The Hedgehog inhibitor cyclopamine reduces β-catenin-Tcf transcriptional activity, induces E-cadherin expression, and reduces invasion in colorectal cancer cells. Cancers 7, 1885–1899 (2015).
Harding, J. & Burtness, B. Cetuximab: an epidermal growth factor receptor chemeric human-murine monoclonal antibody. Drugs Today Barc. Spain 1998, 41 (2005).
Iannello, A. & Ahmad, A. Role of antibody-dependent cell-mediated cytotoxicity in the efficacy of therapeutic anti-cancer monoclonal antibodies. Cancer Metastasis Rev. 24, 487–499 (2005).
Gorter, A. & Meri, S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol. Today 20, 576–582 (1999).
Imai, K. & Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 6, 714–727 (2006).
Li, H. K. et al. α-particle therapy for synovial sarcoma in the mouse using an astatine-211-labeled antibody against frizzled homolog 10. Cancer Sci. 109, 2302–2309 (2018).
Huang, S., Armstrong, E. A., Benavente, S., Chinnaiyan, P. & Harari, P. M. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res. 64, 5355–5362 (2004).
Gentilucci, L., Tolomelli, A. & Squassabia, F. Peptides and peptidomimetics in medicine, surgery and biotechnology. Curr. Med. Chem. 13, 2449–2466 (2006).
Zhong, Z. et al. PORCN inhibition synergizes with PI3K/mTOR inhibition in Wnt-addicted cancers. Oncogene 38, 6662–6677 (2019).
Teneggi, V. et al. 152O A phase 1, first-in-human dose escalation study of ETC-159 in advanced or metastatic solid tumours. Ann. Oncol. 27, https://doi.org/10.1093/annonc/mdw579.004 (2016).
Madan, B. et al. Bone loss from Wnt inhibition mitigated by concurrent alendronate therapy. Bone Res. 6, 17 (2018).
Funck-Brentano, T. et al. Porcupine inhibitors impair trabecular and cortical bone mass and strength in mice. J. Endocrinol. 238, 13–23 (2018).
Liu, J. et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl Acad. Sci. USA 110, 20224–20229 (2013).
Madan, B. et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 35, 2197–2207 (2016).
Koo, B.-K., van Es, J. H., van den Born, M. & Clevers, H. Porcupine inhibitor suppresses paracrine Wnt-driven growth of Rnf43;Znrf3-mutant neoplasia. Proc. Natl Acad. Sci. USA 112, 7548–7550 (2015).
University of Michigan Rogel Cancer Center. An open label, non-randomized phase ii trial evaluating WNT974 in patients with metastatic head and neck squamous cell carcinoma. NCT02649530, UMCC 2015.157.
Jin, X.-F., Spoettl, G., Maurer, J., Nölting, S. & Auernhammer, C. J. Inhibition of Wnt/β-catenin signaling in neuroendocrine tumors in vitro: antitumoral effects. Cancers 12, 345 (2020).
Doo, D. W. et al. Inhibition of the Wnt/β-catenin pathway enhances antitumor immunity in ovarian cancer. Ther. Adv. Med. Oncol. 12, 1758835920913798 (2020).
Array BioPharma. A phase Ib/II multi-center, open label, dose escalation study of WNT974, LGX818 and cetuximab in patients with BRAFV600-mutant KRAS wild-type metastatic colorectal cancer harboring wnt pathway mutations. NCT02278133, CWNT974X2102C.
Novartis Pharmaceuticals. A phase I, open-label, dose escalation study of oral LGK974 in patients with malignancies dependent on Wnt ligands. NCT01351103, CLGK974X2101.
EDDC, A. R. E. & PPD. A phase 1A/B study to evaluate the safety and tolerability of ETC-1922159 in advanced solid tumours. NCT02521844, D3-002.
Li, C. et al. Identification of RSPO2 Fusion Mutations and Target Therapy Using a Porcupine Inhibitor. Sci. Rep. 8, 14244 (2018).
Wall, J. A. et al. Manipulating the Wnt/β-catenin signaling pathway to promote anti-tumor immune infiltration into the TME to sensitize ovarian cancer to ICB therapy. Gynecol. Oncol. 160, 285–294 (2021).
Li, C. et al. The delivery of a Wnt pathway inhibitor toward CSCs requires stable liposome encapsulation and delayed drug release in tumor tissues. Mol. Ther. 27, 1558–1567 (2019).
Curegenix Inc. & Merck, S. and D. C. A phase 1 open-label dose escalation and dose expansion study of CGX1321 in subjects with advanced solid tumors and phase 1b study of CGX1321 in combination with pembrolizumab in subjects with advanced gastrointestinal tumors. NCT02675946, CGX1321-101.
Proffitt, K. D. et al. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 73, 502–507 (2013).
Cheng, Y. et al. Wnt-C59 arrests stemness and suppresses growth of nasopharyngeal carcinoma in mice by inhibiting the Wnt pathway in the tumor microenvironment. Oncotarget 6, 14428–14439 (2015).
Wang, B. et al. Reversion of trichostatin A resistance via inhibition of the Wnt signaling pathway in human pancreatic cancer cells. Oncol. Rep. 32, 2015–2022 (2014).
Cheng, D. et al. Discovery of pyridinyl acetamide derivatives as potent, selective, and orally bioavailable porcupine inhibitors. ACS Med. Chem. Lett. 7, 676–680 (2016).
Chen, B. et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 5, 100–107 (2009).
Wang, X. et al. The development of highly potent inhibitors for porcupine. J. Med. Chem. 56, 2700–2704 (2013).
Kleszcz, R., Szymańska, A., Krajka-Kuźniak, V., Baer-Dubowska, W. & Paluszczak, J. Inhibition of CBP/β-catenin and porcupine attenuates Wnt signaling and induces apoptosis in head and neck carcinoma cells. Cell. Oncol. Dordr. 42, 505–520 (2019).
Liu, C. et al. Newly developed CK1-specific inhibitors show specifically stronger effects on CK1 mutants and colon cancer cell lines. Int. J. Mol. Sci. 20, 6184 (2019).
García-Reyes, B. et al. Discovery of inhibitor of Wnt production 2 (IWP-2) and related compounds as selective ATP-competitive inhibitors of casein kinase 1 (CK1) δ/ε. J. Med. Chem. 61, 4087–4102 (2018).
Mo, M.-L. et al. Inhibition of the Wnt palmitoyltransferase porcupine suppresses cell growth and downregulates the Wnt/β-catenin pathway in gastric cancer. Oncol. Lett. 5, 1719–1723 (2013).
Gurney, A. et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl Acad. Sci. USA 109, 11717–11722 (2012).
Le, P. N., McDermott, J. D. & Jimeno, A. Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP-54F28. Pharmacol. Ther. 146, 1–11 (2015).
Hoey, T. (ed.) Development of FZD8-Fc (OMP-54 F28), aWnt signaling antagonist that inhibits tumor growth and reduces tumor initiating cell frequency. Presented at AACR Annual Meeting; 2013 April; Washington DC (2013).
Fischer, M. M. et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci. Adv. 3, e1700090 (2017).
Jimeno, A. et al. A first-in-human phase I study of the anticancer stem cell agent ipafricept (OMP-54F28), a decoy receptor for Wnt ligands, in patients with advanced solid tumors. Clin. Cancer Res. 23, 7490–7497 (2017).
Moore, K. N. et al. A phase 1b dose escalation study of ipafricept (OMP54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 154, 294–301 (2019).
Davis, S. L. et al. A phase 1b dose escalation study of Wnt pathway inhibitor vantictumab in combination with nab-paclitaxel and gemcitabine in patients with previously untreated metastatic pancreatic cancer. Invest. N. Drugs 38, 821–830 (2020).
Diamond, J. R. et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 184, 53–62 (2020).
Salik, B. et al. Targeting RSPO3-LGR4 signaling for leukemia stem cell eradication in acute myeloid leukemia. Cancer Cell 38, 263–278.e6 (2020).
Bendell, J. et al. Initial results from a phase 1a/b study of OMP-131R10, a first-in-class anti-RSPO3 antibody, in advanced solid tumors and previously treated metastatic colorectal cancer (CRC). Eur. J. Cancer 69, S29–S30 (2016).
Pavlovic, Z. et al. A synthetic anti-Frizzled antibody engineered for broadened specificity exhibits enhanced anti-tumor properties. mAbs 10, 1157–1167 (2018).
Wall, J. A., Klempner, S. J. & Arend, R. C. The anti-DKK1 antibody DKN-01 as an immunomodulatory combination partner for the treatment of cancer. Expert Opin. Investig. Drugs 29, 639–644 (2020).
Kagey, M. et al. High tumor expression of DKK1 is associated with improved clinical benefit and longer progression free survival across multiple solid tumors when treated with a target anti-DKK1 antibody (DKN-01). J. Immunother. Cancer 7, 282 (2019).
Leap Therapeutics, Inc. A phase 2 study evaluating the efficacy and safety of DKN-01 as a monotherapy or in combination with paclitaxel in patients with recurrent epithelial endometrial, epithelial ovarian cancer, or carcinosarcoma. NCT03395080, DEK-DKK1-P204.
Choi, M. Y. et al. Pre-clinical specificity and safety of UC-961, a first-in-class monoclonal antibody targeting ROR1. Clin. Lymphoma Myeloma Leuk. 15(Suppl), S167–S169 (2015).
Giraudet, A.-L. et al. A first-in-human study investigating biodistribution, safety and recommended dose of a new radiolabeled MAb targeting FZD10 in metastatic synovial sarcoma patients. BMC Cancer 18, 646 (2018).
Säfholm, A. et al. The Wnt-5a-derived hexapeptide Foxy-5 inhibits breast cancer metastasis in vivo by targeting cell motility. Clin. Cancer Res. 14, 6556–6563 (2008).
Canesin, G. et al. Treatment with the WNT5A-mimicking peptide Foxy-5 effectively reduces the metastatic spread of WNT5A-low prostate cancer cells in an orthotopic mouse model. PLoS ONE 12, e0184418 (2017).
Pak, S. et al. The small molecule WNT/β-catenin inhibitor CWP232291 blocks the growth of castration-resistant prostate cancer by activating the endoplasmic reticulum stress pathway. J. Exp. Clin. Cancer Res. 38, 342 (2019).
Gunaydin, H., Gu, Y. & Huang, X. Novel binding mode of a potent and selective tankyrase inhibitor. PLoS ONE 7, e33740 (2012).
Mukai, T., Fujita, S. & Morita, Y. Tankyrase (PARP5) inhibition induces bone loss through accumulation of its substrate SH3BP2. Cells 8, 195 (2019).
Fujita, S. et al. Pharmacological inhibition of tankyrase induces bone loss in mice by increasing osteoclastogenesis. Bone 106, 156–166 (2018).
Ueki, Y. et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat. Genet. 28, 125–126 (2001).
Levaot, N. et al. 3BP2-deficient mice are osteoporotic with impaired osteoblast and osteoclast functions. J. Clin. Invest. 121, 3244–3257 (2011).
Tanaka, N. et al. APC mutations as a potential biomarker for sensitivity to tankyrase inhibitors in colorectal cancer. Mol. Cancer Ther. 16, 752–762 (2017).
Shetti, D. et al. Low dose of paclitaxel combined with XAV939 attenuates metastasis, angiogenesis and growth in breast cancer by suppressing Wnt signaling. Cells 8, 892 (2019).
Wang, J. et al. Bufalin inhibits gastric cancer invasion and metastasis by down-regulating Wnt/ASCL2 expression. Oncotarget 9, 23320–23333 (2018).
Tian, X. et al. XAV939 inhibits the stemness and migration of neuroblastoma cancer stem cells via repression of tankyrase 1. Int. J. Oncol. 45, 121–128 (2014).
Tian, X.-H. et al. XAV939, a tankyrase 1 inhibitior, promotes cell apoptosis in neuroblastoma cell lines by inhibiting Wnt/β-catenin signaling pathway. J. Exp. Clin. Cancer Res. 32, 100 (2013).
Busch, A. M. et al. In Experimental and Molecular Therapeutics (American Association for Cancer Research, 2012).
Stakheev, D. et al. The WNT/β-catenin signaling inhibitor XAV939 enhances the elimination of LNCaP and PC-3 prostate cancer cells by prostate cancer patient lymphocytes in vitro. Sci. Rep. 9, 4761 (2019).
Waaler, J. et al. Novel synthetic antagonists of canonical Wnt signaling inhibit colorectal cancer cell growth. Cancer Res. 71, 197–205 (2011).
Waaler, J. et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 72, 2822–2832 (2012).
Lau, T. et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 73, 3132–3144 (2013).
Solberg, N. T. et al. TANKYRASE inhibition enhances the antiproliferative effect of PI3K and EGFR inhibition, mutually affecting β-CATENIN and AKT signaling in colorectal cancer. Mol. Cancer Res. MCR 16, 543–553 (2018).
Kierulf-Vieira, K. S. et al. A small-molecule tankyrase inhibitor reduces glioma stem cell proliferation and sphere formation. Cancers 12, 1630 (2020).
Kulak, O. et al. Disruption of Wnt/β-catenin signaling and telomeric shortening are inextricable consequences of tankyrase inhibition in human cells. Mol. Cell. Biol. 35, 2425–2435 (2015).
Martins-Neves, S. R. et al. IWR-1, a tankyrase inhibitor, attenuates Wnt/β-catenin signaling in cancer stem-like cells and inhibits in vivo the growth of a subcutaneous human osteosarcoma xenograft. Cancer Lett. 414, 1–15 (2018).
Lee, S. C., Kim, O.-H., Lee, S. K. & Kim, S.-J. IWR-1 inhibits epithelial-mesenchymal transition of colorectal cancer cells through suppressing Wnt/β-catenin signaling as well as survivin expression. Oncotarget 6, 27146–27159 (2015).
Busch, A. M. et al. Evidence for tankyrases as antineoplastic targets in lung cancer. BMC Cancer 13, 211 (2013).
Ma, L. et al. Tankyrase inhibitors attenuate WNT/β-catenin signaling and inhibit growth of hepatocellular carcinoma cells. Oncotarget 6, 25390–25401 (2015).
Huang, J., Qu, Q., Guo, Y., Xiang, Y. & Feng, D. Tankyrases/β-catenin signaling pathway as an anti-proliferation and anti-metastatic target in hepatocarcinoma cell lines. J. Cancer 11, 432–440 (2020).
Arqués, O. et al. Tankyrase inhibition blocks Wnt/β-catenin pathway and reverts resistance to PI3K and AKT inhibitors in the treatment of colorectal cancer. Clin. Cancer Res. 22, 644–656 (2016).
Scarborough, H. A. et al. AZ1366: an inhibitor of tankyrase and the canonical Wnt pathway that limits the persistence of non–small cell lung cancer cells following EGFR inhibition. Clin. Cancer Res. 23, 1531–1541 (2017).
Quackenbush, K. S. et al. The novel tankyrase inhibitor (AZ1366) enhances irinotecan activity in tumors that exhibit elevated tankyrase and irinotecan resistance. Oncotarget 7, 28273–28285 (2016).
Mizutani, A. et al. RK-287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Sci. 109, 4003–4014 (2018).
Wang, S. et al. Small-molecule modulation of Wnt signaling via modulating the Axin-LRP5/6 interaction. Nat. Chem. Biol. 9, 579–585 (2013).
Liu, M. et al. IC261, a specific inhibitor of CK1δ/ε, promotes aerobic glycolysis through p53-dependent mechanisms in colon cancer. Int. J. Biol. Sci. 16, 882–892 (2020).
Varghese, R. T. et al. Casein kinase 1 epsilon regulates glioblastoma cell survival. Sci. Rep. 8, 13621 (2018).
Cheong, J. K. et al. IC261 induces cell cycle arrest and apoptosis of human cancer cells via CK1δ/ɛ and Wnt/β-catenin independent inhibition of mitotic spindle formation. Oncogene 30, 2558–2569 (2011).
Yuan, F. et al. IC261 suppresses progression of hepatocellular carcinoma in a casein kinase 1 δ/ε independent manner. Biochem. Biophys. Res. Commun. 523, 809–815 (2020).
Janovska, P. et al. Autocrine signaling by Wnt-5a deregulates chemotaxis of leukemic cells and predicts clinical outcome in chronic lymphocytic leukemia. Clin. Cancer Res. Clin. Cancer Res. 22, 459–469 (2015).
Shen, C. et al. The CK1α activator pyrvinium enhances the catalytic efficiency (kcat/ Km) of CK1α. Biochemistry 58, 5102–5106 (2019).
Xu, W. et al. The antihelmintic drug pyrvinium pamoate targets aggressive breast cancer. PLoS ONE 8, e71508 (2013).
Zhang, C., Zhang, Z., Zhang, S., Wang, W. & Hu, P. Targeting of Wnt/β-catenin by anthelmintic drug pyrvinium enhances sensitivity of ovarian cancer cells to chemotherapy. Med. Sci. Monit. 23, 266–275 (2017).
Smith, T. C., Kinkel, A. W., Gryczko, C. M., & Goulet, J. R. Absorption of pyrvinium pamoate. Clin. Pharmacol. Ther. 19, 802–806 (1976).
Venerando, A., Girardi, C., Ruzzene, M. & Pinna, L. A. Pyrvinium pamoate does not activate protein kinase CK1, but promotes Akt/PKB down-regulation and GSK3 activation. Biochem. J. 452, 131–137 (2013).
Takahashi-Yanaga, F. Activator or inhibitor? GSK-3 as a new drug target. Biochem. Pharmacol. 86, 191–199 (2013).
Atkinson, J. M. et al. Activating the Wnt/β-catenin pathway for the treatment of melanoma-application of LY2090314, a novel selective inhibitor of glycogen synthase kinase-3. PLoS ONE 10, e0125028 (2015).
Nakata, D. et al. Glycogen synthase kinase-3 inhibitors suppress the AR-V7-mediated transcription and selectively inhibit cell growth in AR-V7-positive prostate cancer cells. Prostate 77, 955–961 (2017).
Kunnimalaiyaan, S., Schwartz, V. K., Jackson, I. A., Clark Gamblin, T. & Kunnimalaiyaan, M. Antiproliferative and apoptotic effect of LY2090314, a GSK-3 inhibitor, in neuroblastoma in vitro. BMC Cancer 18, 560 (2018).
Gray, J. E. et al. A first-in-human phase I dose-escalation, pharmacokinetic, and pharmacodynamic evaluation of intravenous LY2090314, a glycogen synthase kinase 3 inhibitor, administered in combination with pemetrexed and carboplatin. Invest. N. Drugs 33, 1187–1196 (2015).
Zhang, C. et al. Overexpression of dishevelled 2 is involved in tumor metastasis and is associated with poor prognosis in hepatocellular carcinoma. Clin. Transl. Oncol. 19, 1507–1517 (2017).
Wei, Q. et al. Dishevelled family proteins are expressed in non-small cell lung cancer and function differentially on tumor progression. Lung Cancer Amst. Neth. 62, 181–192 (2008).
Khan, A. S. et al. Dishevelled proteins are significantly upregulated in chronic lymphocytic leukaemia. Tumour Biol. 37, 11947–11957 (2016).
Fujii, N. et al. An antagonist of dishevelled protein-protein interaction suppresses beta-catenin-dependent tumor cell growth. Cancer Res. 67, 573–579 (2007).
Grandy, D. et al. Discovery and characterization of a small molecule inhibitor of the PDZ domain of dishevelled. J. Biol. Chem. 284, 16256–16263 (2009).
Jamieson, C., Sharma, M. & Henderson, B. R. Targeting the β-catenin nuclear transport pathway in cancer. Semin. Cancer Biol. 27, 20–29 (2014).
Masuda, M., Sawa, M. & Yamada, T. Therapeutic targets in the Wnt signaling pathway: Feasibility of targeting TNIK in colorectal cancer. Pharmacol. Ther. 156, 1–9 (2015).
Lee, Y., Jung, J. I., Park, K. Y., Kim, S. A. & Kim, J. Synergistic inhibition effect of TNIK inhibitor KY-05009 and receptor tyrosine kinase inhibitor dovitinib on IL-6-induced proliferation and Wnt signaling pathway in human multiple myeloma cells. Oncotarget 8, 41091–41101 (2017).
Yamada, T. & Masuda, M. Emergence of TNIK inhibitors in cancer therapeutics. Cancer Sci. 108, 818–823 (2017).
Masuda, M. et al. TNIK inhibition abrogates colorectal cancer stemness. Nat. Commun. 7, 12586 (2016).
Sugano, T. et al. Pharmacological blockage of transforming growth factor-β signalling by a Traf2- and Nck-interacting kinase inhibitor, NCB-0846. Br. J. Cancer 124, 228–236 (2021).
Sekita, T. et al. Feasibility of targeting Traf2-and-Nck-interacting kinase in synovial sarcoma. Cancers 12, 1258 (2020).
Lee, R. S. et al. Characterization of the ERG-regulated kinome in prostate cancer identifies TNIK as a potential therapeutic target. Neoplasia 21, 389–400 (2019).
Kim, J. Y., Park, G., Krishnan, M., Ha, E. & Chun, K.-S. Selective Wnt/β-catenin small-molecule inhibitor CWP232228 impairs tumor growth of colon cancer. Anticancer Res. 39, 3661–3667 (2019).
Kim, J.-Y. et al. CWP232228 targets liver cancer stem cells through Wnt/β-catenin signaling: a novel therapeutic approach for liver cancer treatment. Oncotarget 7, 20395–20409 (2016).
Jang, G.-B. et al. Wnt/β-catenin small-molecule inhibitor CWP232228 preferentially inhibits the growth of breast cancer stem-like cells. Cancer Res. 75, 1691–1702 (2015).
Fang, L. et al. A small-molecule antagonist of the β-catenin/TCF4 interaction blocks the self-renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res. 76, 891–901 (2016).
Fiskus, W. et al. Abstract C144: Treatment with β-catenin antagonist BC2059 exhibits single agent efficacy and exerts superior activity with tyrosine kinase inhibitor (TKI) or histone deacetylase (HDAC) inhibitor against human AML, CML, and myeloproliferative neoplasm (MPN) progenitor cells. Mol. Cancer Ther. 10, C144–C144 (2011).
Fiskus, W. et al. Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia 29, 1267–1278 (2015).
Gandhirajan, R. K. et al. Small molecule inhibitors of Wnt/beta-catenin/lef-1 signaling induces apoptosis in chronic lymphocytic leukemia cells in vitro and in vivo. Neoplasia 12, 326–335 (2010).
Minke, K. S. et al. Small molecule inhibitors of WNT signaling effectively induce apoptosis in acute myeloid leukemia cells. Eur. J. Haematol. 82, 165–175 (2009).
Wei, W., Chua, M.-S., Grepper, S. & So, S. Small molecule antagonists of Tcf4/beta-catenin complex inhibit the growth of HCC cells in vitro and in vivo. Int. J. Cancer 126, 2426–2436 (2010).
Zhang, M., Wang, Z., Zhang, Y., Guo, W. & Ji, H. Structure-based optimization of small-molecule inhibitors for the β-catenin/b-cell lymphoma 9 protein–protein interaction. J. Med. Chem. 61, 2989–3007 (2018).
Gabata, R. et al. Anti-tumor activity of the small molecule inhibitor PRI-724 against β-catenin-activated hepatocellular carcinoma. Anticancer Res. 40, 5211–5219 (2020).
Hirakawa, T. et al. β-catenin signaling inhibitors ICG-001 and C-82 improve fibrosis in preclinical models of endometriosis. Sci. Rep. 9, 20056 (2019).
Kimura, K. et al. Safety, tolerability, and preliminary efficacy of the anti-fibrotic small molecule PRI-724, a CBP/β-catenin inhibitor, in patients with hepatitis C virus-related cirrhosis: a single-center, open-label, dose escalation phase 1 trial. EBioMedicine 23, 79–87 (2017).
Mani, M. et al. BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res. 69, 7577–7586 (2009).
Deka, J. et al. Bcl9/Bcl9l are critical for Wnt-mediated regulation of stem cell traits in colon epithelium and adenocarcinomas. Cancer Res. 70, 6619–6628 (2010).
Takada, K. et al. Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med. 4, 148ra117 (2012).
Ashrafizadeh, M., Ahmadi, Z., Farkhondeh, T. & Samarghandian, S. Resveratrol targeting the Wnt signaling pathway: A focus on therapeutic activities. J. Cell. Physiol. 235, 4135–4145 (2020).
Flaig, T. W. et al. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest. New Drugs 25, 139–146 (2007).
Lu, W., Lin, C. & Li, Y. Rottlerin induces Wnt co-receptor LRP6 degradation and suppresses both Wnt/β-catenin and mTORC1 signaling in prostate and breast cancer cells. Cell. Signal. 26, 1303–1309 (2014).
Lai, C.-S. et al. Tetrahydrocurcumin is more effective than curcumin in preventing azoxymethane-induced colon carcinogenesis. Mol. Nutr. Food Res. 55, 1819–1828 (2011).
Nguyen, A. V. et al. Results of a phase I pilot clinical trial examining the effect of plant-derived resveratrol and grape powder on Wnt pathway target gene expression in colonic mucosa and colon cancer. Cancer Manag. Res. 1, 25–37 (2009).
University of California, I. & Gateway for Cancer Research. Phase I Biomarker Study of Dietary Grape-derived Low Dose Resveratrol for Colon Cancer Prevention. NCT00578396, OCRT07046.
Villota, H., Röthlisberger, S. & Pedroza-Díaz, J. Modulation of the canonical Wnt signaling pathway by dietary polyphenols, an opportunity for colorectal cancer chemoprevention and treatment. Nutr. Cancer 1–20, https://doi.org/10.1080/01635581.2021.1884730 (2021).
Sangeetha, N. et al. Oral supplementation of silibinin prevents colon carcinogenesis in a long term preclinical model. Eur. J. Pharmacol. 643, 93–100 (2010).
Chen, M. et al. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 48, 10267–10274 (2009).
Egashira, I. et al. Celecoxib and 2,5‐dimethylcelecoxib inhibit intestinal cancer growth by suppressing the Wnt/β‐catenin signaling pathway. Cancer Sci. 108, 108–115 (2016).
Lu, W. & Li, Y. Salinomycin suppresses LRP6 expression and inhibits both Wnt/β-catenin and mTORC1 signaling in breast and prostate cancer cells. J. Cell. Biochem. 115, 1799–1807 (2014).
Li, L., Mao, J., Sun, L., Liu, W. & Wu, D. Second cysteine-rich domain of Dickkopf-2 activates canonical Wnt signaling pathway via LRP-6 independently of dishevelled. J. Biol. Chem. 277, 5977–5981 (2002).
Ahn, V. E. et al. Structural basis of Wnt signaling inhibition by Dickkopf binding to LRP5/6. Dev. Cell 21, 862–873 (2011).
Bu, Q. et al. The crystal structure of full-length Sizzled from Xenopus laevis yields insights into Wnt-antagonistic function of secreted Frizzled-related proteins. J. Biol. Chem. 292, 16055–16069 (2017).
Bhat, R. A., Stauffer, B., Komm, B. S. & Bodine, P. V. N. Structure-function analysis of secreted frizzled-related protein-1 for its Wnt antagonist function. J. Cell. Biochem. 102, 1519–1528 (2007).
Kerekes, K., Bányai, L. & Patthy, L. Wnts grasp the WIF domain of Wnt Inhibitory Factor 1 at two distinct binding sites. FEBS Lett. 589, 3044–3051 (2015).
Semënov, M., Tamai, K. & He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 280, 26770–26775 (2005).
Piccolo, S. et al. The head inducer Cerberus is a multifunctional antagonist of Nodal, BMP and Wnt signals. Nature 397, 707–710 (1999).
Ahn, Y., Sims, C., Logue, J. M., Weatherbee, S. D. & Krumlauf, R. Lrp4 and Wise interplay controls the formation and patterning of mammary and other skin appendage placodes by modulating Wnt signaling. Development 140, 583–593 (2013).
Zhu, W. et al. IGFBP-4 is an inhibitor of canonical Wnt signalling required for cardiogenesis. Nature 454, 345–349 (2008).
Kakugawa, S. et al. Notum deacylates Wnt proteins to suppress signalling activity. Nature 519, 187–192 (2015).
Zhang, X. et al. Tiki1 is required for head formation via Wnt cleavage-oxidation and inactivation. Cell 149, 1565–1577 (2012).
OncoMed Pharmaceuticals, Inc. A phase 1 dose escalation study of OMP-18R5 in subjects with solid tumors. NCT01345201, 18R5-001.
OncoMed Pharmaceuticals, Inc. A phase 1b dose escalation study of vantictumab (OMP-18R5) in combination with paclitaxel in patients with locally recurrent or metastatic breast cancer. NCT01973309, 18R5-002.
OncoMed Pharmaceuticals, Inc. A phase 1b dose escalation study of vantictumab (OMP-18R5) in combination with nab-paclitaxel and gemcitabine in patients with previously untreated stage IV pancreatic cancer. NCT02005315, 18R5-003R5.
OncoMed Pharmaceuticals, Inc. & Bayer. A phase 1 dose escalation study of OMP-54F28 in subjects with solid tumors. NCT01608867, 54F28-001F28.
OncoMed Pharmaceuticals, Inc. A phase 1b dose escalation study of OMP-54F28 in combination with sorafenib in patients with hepatocellular cancer. NCT02069145, 54F28-004.
OncoMed Pharmaceuticals, Inc. A phase 1b dose escalation study of OMP-54F28 in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. NCT02092363, 54F28-003.
OncoMed Pharmaceuticals, Inc. A phase 1b dose escalation study of omp-54f28 in combination with nab-paclitaxel and gemcitabine in patients with previously untreated stage IV pancreatic cancer. NCT02050178, 54F28-002.
WntResearch AB. Phase I dose escalating study to evaluate the safety, tolerability, anti-tumour activity and pharmacokinetic and pharmacodynamic profiles of foxy-5 in patients with metastatic breast, colon or prostate cancer. NCT02020291, SMR-2562.
WntResearch AB. Phase Ib dose escalating study to evaluate the safety, tolerability and pharmacodynamic response of foxy-5 in patients with metastatic breast-, colon- or prostate cancer. NCT02655952, SMR-3164.
WntResearch A. B. et al. A randomized, multicentre, open-label controlled phase II trial of foxy-5 as neo-adjuvant therapy in subjects with Wnt-5a low colon cancer. NCT03883802, SMS-0472B.
Leap Therapeutics, Inc. A Pilot Study of DKN-01 and Lenalidomide (Revlimid®)/Dexamethasone Versus Lenalidomide/Dexamethasone in Patients with Relapsed or Refractory Multiple Myeloma. https://clinicaltrials.gov/ct2/show/NCT01711671 (2017).
Leap Therapeutics, Inc. A Two Part Phase 1 Multicenter Open-label Study of DKN-01 Given Intravenously. Part A: Dose-Escalation in Patients with Multiple Myeloma or Advanced Solid Tumors. Part B: Expansion Cohort in Patients With Relapsed/Refractory Non-Small Cell Lung Cancer (NSCLC). https://clinicaltrials.gov/ct2/show/NCT01457417 (2016).
Goyal, L. et al. Phase I and biomarker study of the Wnt pathway modulator DKN-01 in combination with gemcitabine/cisplatin in advanced biliary tract cancer. Clin. Cancer Am. 26, 6158–6167 (2020).
Lipika, G. A Single Arm Phase II Study of the Combination of DKN-01 and Nivolumab in Previously Treated Patients With Advanced Biliary Tract Cancer (BTC). https://clinicaltrials.gov/ct2/show/NCT04057365 (2021).
Marquardt, D. J. U. A Phase I/II Multicenter, Open-label Study of DKN-01 to Investigate the Anti-tumor Activity and Safety of DKN-01 in Patients With Hepatocellular Carcinoma and WNT Signaling Alterations. https://clinicaltrials.gov/ct2/show/NCT03645980 (2020).
Thomas Kipps. A Phase I clinical trial to determine the safety and tolerability of UC-961 (Cirmtuzumab), an anti-ROR1 monoclonal antibody, for the treatment of patients with relapsed or refractory chronic lymphocytic leukemia who are ineligible for chemotherapy. NCT02222688, #140141.
University of California, S. D., California Institute for Regenerative Medicine & Oncternal Therapeutics, I. A phase 1b/2 study of the ROR1-targeting monoclonal antibody, Cirmtuzumab (UC-961), and the bruton tyrosine kinase inhibitor, ibrutinib, in patients with B-cell lymphoid malignancies. NCT03088878, 170127.
MD, B. P. A Phase 1b Pilot Clinical Trial of Cirmtuzumab, an Anti-ROR1 Monoclonal Antibody, in Combination With Paclitaxel for the Treatment of Patients With Metastatic, or Locally Advanced, Unresectable Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT02776917 (2021).
Choi, M. Y. et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell 22, 951–959.e3 (2018).
H. Lee Moffitt Cancer Center and Research Institute. A Phase 2 Study to Assess the Efficacy, Safety, Pharmacokinetic and Pharmacodynamic Parameters of Umbralisib in Treatment Naïve Patients With Chronic Lymphocytic Leukemia (CLL). https://clinicaltrials.gov/ct2/show/NCT04163718 (2021).
University of Washington. A Phase II Trial of Umbralisib and Pembrolizumab in Patients With Relapsed or Refractory Classical Hodgkin Lymphoma. https://clinicaltrials.gov/ct2/show/NCT03776864 (2021).
Mehta, A. Phase Ib/II Trial of Ublituximab and Umbralisib With CHOP (U2-CHOP) Followed by U2 Maintenance (U2-CHOP-U2) in Previously Untreated Mantle Cell Lymphoma (MCL). https://clinicaltrials.gov/ct2/show/NCT04692155 (2021).
Eli Lilly and Company. A Phase 2 Study of LY2090314 in Participants With Acute Leukemia. https://clinicaltrials.gov/ct2/show/NCT01214603 (2018).
Eli Lilly and Company. Phase 1 Dose Escalation Study of LY2090314 in Patients With Advanced or Metastatic Cancer in Combination With Pemetrexed and Carboplatin. https://clinicaltrials.gov/ct2/show/NCT01287520 (2018).
Rizzieri, D. A. et al. An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia. Leuk. Lymphoma 57, 1800–1806 (2016).
Prism Pharma Co., Ltd. A Phase Ia/Ib Clinical Trial of PRI-724 in Patients With Advanced Solid Tumors. https://clinicaltrials.gov/ct2/show/NCT01302405 (2017).
Prism Pharma Co., Ltd. Phase Ib Multicenter, Cohort Dose Escalation Trial to Determine the Safety, Tolerance and Preliminary Antineoplastic Activity of Gemcitabine Administered in Combination With Continuous Intravenous Doses of PRI-724, a CBP/ β- Catenin Inhibitor, to Patients With Advanced or Metastatic Pancreatic Adenocarcinoma Eligible for Second-Line Therapy After Failing First-Line Therapy With FOLFIRINOX (or FOLFOX). https://clinicaltrials.gov/ct2/show/NCT01764477 (2017).
University of Southern California. PRIMIER*: Randomized Phase II Trial of mFOLFOX6/Bevacizumab With or Without PRI-724 as First Line Treatment for Metastatic Colorectal Cancer. https://clinicaltrials.gov/ct2/show/NCT02413853 (2017).
Iterion Therapeutics. Phase 1 Trial of BC2059 (Tegavivint) in Patients With Unresectable Desmoid Tumor. https://clinicaltrials.gov/ct2/show/NCT03459469 (2021).
Jeong, J. B., Lee, J. & Lee, S.-H. TCF4 is a molecular target of resveratrol in the prevention of colorectal cancer. Int. J. Mol. Sci. 16, 10411–10425 (2015).
Cilibrasi, C. et al. Resveratrol impairs glioma stem cells proliferation and motility by modulating the Wnt signaling pathway. PLoS ONE 12, e0169854 (2017).
Liu, Z.-L. et al. Inactivated Wnt signaling in resveratrol-treated epidermal squamous cancer cells and its biological implication. Oncol. Lett. 14, 2239–2243 (2017).
Mojsin, M., Vicentic, J. M., Schwirtlich, M., Topalovic, V. & Stevanovic, M. Quercetin reduces pluripotency, migration and adhesion of human teratocarcinoma cell line NT2/D1 by inhibiting Wnt/β-catenin signaling. Food Funct. 5, 2564–2573 (2014).
Baruah, M. M., Khandwekar, A. P. & Sharma, N. Quercetin modulates Wnt signaling components in prostate cancer cell line by inhibiting cell viability, migration, and metastases. Tumour Biol. 37, 14025–14034 (2016).
Shan, B.-E., Wang, M.-X. & Li, R. Quercetin inhibit human SW480 colon cancer growth in association with inhibition of cyclin D1 and survivin expression through Wnt/beta-catenin signaling pathway. Cancer Invest. 27, 604–612 (2009).
Amado, N. G. et al. Isoquercitrin suppresses colon cancer cell growth in vitro by targeting the Wnt/β-catenin signaling pathway. J. Biol. Chem. 289, 35456–35467 (2014).
Jaiswal, A. S., Marlow, B. P., Gupta, N. & Narayan, S. Beta-catenin-mediated transactivation and cell-cell adhesion pathways are important in curcumin (diferuylmethane)-induced growth arrest and apoptosis in colon cancer cells. Oncogene 21, 8414–8427 (2002).
Prasad, C. P., Rath, G., Mathur, S., Bhatnagar, D. & Ralhan, R. Potent growth suppressive activity of curcumin in human breast cancer cells: modulation of Wnt/beta-catenin signaling. Chem. Biol. Interact. 181, 263–271 (2009).
Leow, P.-C., Tian, Q., Ong, Z.-Y., Yang, Z. & Ee, P.-L. R. Antitumor activity of natural compounds, curcumin and PKF118-310, as Wnt/β-catenin antagonists against human osteosarcoma cells. Invest. N. Drugs 28, 766–782 (2010).
Teiten, M.-H. et al. Anti-proliferative potential of curcumin in androgen-dependent prostate cancer cells occurs through modulation of the Wingless signaling pathway. Int. J. Oncol. 38, 603–611 (2011).
Hu, P. et al. Both glypican-3/Wnt/β-catenin signaling pathway and autophagy contributed to the inhibitory effect of curcumin on hepatocellular carcinoma. Dig. Liver Dis. 51, 120–126 (2019).
Kim, H. J., Park, S. Y., Park, O. J. & Kim, Y.-M. Curcumin suppresses migration and proliferation of Hep3B hepatocarcinoma cells through inhibition of the Wnt signaling pathway. Mol. Med. Rep. 8, 282–286 (2013).
Wang, J.-Y. et al. Curcumin inhibits the growth via Wnt/β-catenin pathway in non-small-cell lung cancer cells. Eur. Rev. Med. Pharmacol. Sci. 22, 7492–7499 (2018).
Lu, W. et al. Silibinin inhibits Wnt/β-catenin signaling by suppressing Wnt co-receptor LRP6 expression in human prostate and breast cancer cells. Cell. Signal. 24, 2291–2296 (2012).
Kaur, M. et al. Silibinin suppresses growth of human colorectal carcinoma SW480 cells in culture and xenograft through down-regulation of beta-catenin-dependent signaling. Neoplasia 12, 415–424 (2010).
Zhang, X. et al. Wnt blockers inhibit the proliferation of lung cancer stem cells. Drug Des. Devel. Ther. 9, 2399–2407 (2015).
Zhu, Y. et al. Rottlerin as a novel chemotherapy agent for adrenocortical carcinoma. Oncotarget 8, 22825–22834 (2017).
Zhu, J. et al. miR-19 targeting of GSK3β mediates sulforaphane suppression of lung cancer stem cells. J. Nutr. Biochem. 44, 80–91 (2017).
Du, Y.-Y., Liu, X. & Shan, B.-E. Periplocin extracted from cortex periplocae induces apoptosis of SW480 cells through inhibiting the Wnt/beta-catenin signaling pathway. Ai Zheng Aizheng Chin. J. Cancer 28, 456–460 (2009).
Li, X. et al. Henryin, an ent-kaurane diterpenoid, inhibits Wnt signaling through interference with β-catenin/TCF4 interaction in colorectal cancer cells. PLoS ONE 8, e68525 (2013).
Shrivastava, S. et al. Cardamonin, a chalcone, inhibits human triple negative breast cancer cell invasiveness by downregulation of Wnt/β-catenin signaling cascades and reversal of epithelial-mesenchymal transition. BioFactors 43, 152–169 (2017).
Williams, S. P. et al. Indirubins decrease glioma invasion by blocking migratory phenotypes in both the tumor and stromal endothelial cell compartments. Cancer Res. 71, 5374–5380 (2011).
Hui, H. et al. Dihydroartemisinin suppresses growth of squamous cell carcinoma A431 cells by targeting the Wnt/β-catenin pathway. Anticancer. Drugs 27, 99–105 (2016).
Tang, L. et al. Shizukaol D, a dimeric sesquiterpene isolated from Chloranthus serratus, represses the growth of human liver cancer cells by modulating Wnt signalling pathway. PLoS ONE 11, e0152012 (2016).
Lee, S.-H., Richardson, R. L., Dashwood, R. H. & Baek, S. J. Capsaicin represses transcriptional activity of β-catenin in human colorectal cancer cells. J. Nutr. Biochem. 23, 646–655 (2012).
La de, R. M. et al. An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun. 3, 680 (2012).
Duda, P. Targeting GSK3 and associated signaling pathways involved in cancer. Cells 9, 1110 (2020).
Osada, T. et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 71, 4172–4182 (2011).
Monin, M. B. et al. The anthelmintic niclosamide inhibits colorectal cancer cell lines via modulation of the canonical and noncanonical Wnt signaling pathway. J. Surg. Res. 203, 193–205 (2016).
Arend, R. C. et al. Inhibition of Wnt/β-catenin pathway by niclosamide: a therapeutic target for ovarian cancer. Gynecol. Oncol. 134, 112–120 (2014).
Lu, W. et al. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. PLoS ONE 6, e29290 (2011).
Fako, V. et al. Inhibition of wnt/β-catenin signaling in hepatocellular carcinoma by an antipsychotic drug pimozide. Int. J. Biol. Sci. 12, 768–775 (2016).
Ren, Y., Tao, J., Jiang, Z., Guo, D. & Tang, J. Pimozide suppresses colorectal cancer via inhibition of Wnt/β-catenin signaling pathway. Life Sci. 209, 267–273 (2018).
Lu, D. et al. Ethacrynic acid exhibits selective toxicity to chronic lymphocytic leukemia cells by inhibition of the Wnt/beta-catenin pathway. PLoS ONE 4, e8294 (2009).
Wu, W. et al. High LEF1 expression predicts adverse prognosis in chronic lymphocytic leukemia and may be targeted by ethacrynic acid. Oncotarget 7, 21631–21643 (2016).
Al-Dali, A. M., Weiher, H. & Schmidt-Wolf, I. G. H. Utilizing ethacrynic acid and ciclopirox olamine in liver cancer. Oncol. Lett. 16, 6854–6860 (2018).
Wu, D. et al. Salinomycin inhibits proliferation and induces apoptosis of human nasopharyngeal carcinoma cell in vitro and suppresses tumor growth in vivo. Biochem. Biophys. Res. Commun. 443, 712–717 (2014).
Wang, F. et al. Salinomycin inhibits proliferation and induces apoptosis of human hepatocellular carcinoma cells in vitro and in vivo. PLoS ONE 7, e50638 (2012).
Tai, W.-P., Hu, P.-J., Wu, J. & Lin, X.-C. The inhibition of Wnt/β-catenin signaling pathway in human colon cancer cells by sulindac. Tumori 100, 97–101 (2014).
Han, A. et al. Sulindac suppresses beta-catenin expression in human cancer cells. Eur. J. Pharmacol. 583, 26–31 (2008).
Huang, C. et al. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E2 and down-regulating the Wnt pathway activity. Oncotarget 8, 115254–115269 (2017).
Maier, T. J., Janssen, A., Schmidt, R., Geisslinger, G. & Grösch, S. Targeting the beta-catenin/APC pathway: a novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB. FASEB J. 19, 1353–1355 (2005).
Deng, Y. et al. Celecoxib downregulates CD133 expression through inhibition of the Wnt signaling pathway in colon cancer cells. Cancer Invest. 31, 97–102 (2013).
Verkaar, F., van der Doelen, A. A., Smits, J. F. M., Blankesteijn, W. M. & Zaman, G. J. R. Inhibition of Wnt/β-catenin signaling by p38 MAP kinase inhibitors is explained by cross-reactivity with casein kinase Iδ/ɛ. Chem. Biol. 18, 485–494 (2011).
Wang, J.-S. et al. Lithium inhibits proliferation of human esophageal cancer cell line Eca-109 by inducing a G2/M cell cycle arrest. World J. Gastroenterol. 14, 3982–3989 (2008).
Yeung, J. et al. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 18, 606–618 (2010).
Chon, H. J. et al. Traf2- and Nck-interacting kinase (TNIK) is involved in the anti-cancer mechanism of dovitinib in human multiple myeloma IM-9 cells. Amino Acids 48, 1591–1599 (2016).
Park, S. & Chun, S. Streptonigrin inhibits β-catenin/Tcf signaling and shows cytotoxicity in β-catenin-activated cells. Biochim. Biophys. Acta 1810, 1340–1345 (2011).
Acknowledgements
The work is supported by the National Natural Science Foundation of China 81825005 and State Key Laboratory of Oral Diseases SKLOD202007 (L.Y.).
Author information
Authors and Affiliations
Contributions
F.Y. conceived the study, F.Y., C.Y., F.L., Y.Z., L.Yao., C.W., and Y.W. performed literature searching and summary; C.Y., F.Y., and F.L. wrote the manuscript; and C.W. and L.Ye. edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, F., Yu, C., Li, F. et al. Wnt/β-catenin signaling in cancers and targeted therapies. Sig Transduct Target Ther 6, 307 (2021). https://doi.org/10.1038/s41392-021-00701-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00701-5
- Springer Nature Limited
This article is cited by
-
SALL4 in gastrointestinal tract cancers: upstream and downstream regulatory mechanisms
Molecular Medicine (2024)
-
Signaling pathways in colorectal cancer implications for the target therapies
Molecular Biomedicine (2024)
-
Ubiquitin ligase subunit FBXO9 inhibits V-ATPase assembly and impedes lung cancer metastasis
Experimental Hematology & Oncology (2024)
-
Aerobic exercise training engages the canonical wnt pathway to improve pulmonary function and inflammation in COPD
BMC Pulmonary Medicine (2024)
-
F. Nucleatum enhances oral squamous cell carcinoma proliferation via E-cadherin/β-Catenin pathway
BMC Oral Health (2024)