
Abstract
Background
Globally, one in ten babies is born preterm (<37 weeks), and 1–2% preterm at very low birth weight (VLBW, <1500 g). As adults, they are at increased risk for a plethora of health conditions, e.g., cardiometabolic disease, which may partly be mediated by epigenetic regulation. We compared blood DNA methylation between young adults born at VLBW and controls.
Methods
157 subjects born at VLBW and 161 controls born at term, from the Helsinki Study of Very Low Birth Weight Adults, were assessed for peripheral venous blood DNA methylation levels at mean age of 22 years. Significant CpG-sites (5’—C—phosphate—G—3’) were meta-analyzed against continuous birth weight in four independent cohorts (pooled n = 2235) with cohort mean ages varying from 0 to 31 years.
Results
In the discovery cohort, 66 CpG-sites were differentially methylated between VLBW adults and controls. Top hits were located in HIF3A, EBF4, and an intergenic region nearest to GLI2 (distance 57,533 bp). Five CpG-sites, all in proximity to GLI2, were hypermethylated in VLBW and associated with lower birth weight in the meta-analysis.
Conclusion
We identified differentially methylated CpG-sites suggesting an epigenetic signature of preterm birth at VLBW present in adult life.
Impact
-
Being born preterm at very low birth weight has major implications for later health and chronic disease risk factors.
-
The mechanism linking preterm birth to later outcomes remains unknown.
-
Our cohort study of 157 very low birth weight adults and 161 controls found 66 differentially methylated sites at mean age of 22 years.
-
Our findings suggest an epigenetic mark of preterm birth present in adulthood, which opens up opportunities for mechanistic studies.
Similar content being viewed by others

Background
Globally, ~10% of all children are born preterm (<37 gestational weeks) and 1–2% very preterm (VPT, <32 weeks) or with a very low birth weight (VLBW; <1500 g).1 Cohort studies have shown that adults born preterm at VLBW have poorer blood glucose regulation,2 decreased bone mineral density,3 higher blood pressure,4 poorer lung function,5 poorer cognitive abilities6,7 and a particular “preterm behavioral phenotype” that may predispose to anxiety disorders and depression,8 with many of these also observable in low birth weight (LBW; <2500 g) adults. The differences in these outcomes between individuals born at VLBW range from moderate (~0.5 SD) to large (~0.8 SD) when compared with individuals born at term or with normal birth weight.9
Epigenetic modifications including DNA methylation (DNAm) have been suggested as mediators between early life exposures and later health outcomes.10,11 In utero exposure to maternal and environmental factors have been linked to epigenetic modifications in the offspring, exemplified by maternal smoking associating with epigenetic changes in newborns, children, adolescents, and middle-aged individuals.12,13 Prenatal maternal stress has also been linked to DNAm changes in the offspring.14,15
Individuals born preterm at VLBW serve as a model for studying survivors of extreme adverse early life events. Their comprehensive phenotyping and epigenotyping offer valuable insights into the mechanisms of adverse early life conditions potentially influencing long-term health.16 Differences in DNAm between VLBW or VPT subjects and term controls have been reported in a few studies. Epigenetic changes in VLBW subjects have most clearly been seen in the neonatal period and infancy,17,18 and emerging evidence suggests that some differentially methylated positions (DMPs) are present in adolescence.19 Only few epigenetic studies of VLBW individuals have been published beyond adolescence and they are relatively small-scale,20 with the largest and longest follow-up of 45 extremely low birth weight (ELBW, <1000 g) individuals and 47 controls being reported by Mathewson et al. in 32-year-olds.21 A recent study by Cameron et al. compared DNAm at birth (109 VLBW individuals, 52 controls) and in 28-year-olds (215 VLBW individuals, 96 controls).22 No robust DMPs across these studies have emerged. There is little evidence of overlap of DMPs between studies examining VLBW and LBW individuals. Meta-analyses assessing both gestational age18 and birth weight19 suggest that specific regions in the epigenome may be more likely to be linked to prematurity than others, but results remain inconclusive. As these studies have been conducted in cohorts with varying exposures, it is plausible that the DNAm differences are consequences of adverse pre- and perinatal conditions and not causes for preterm birth.
We investigated DNAm differences in adulthood in three steps. First, we compared DNAm differences in 157 young adults born at VLBW and 161 term controls. Second, we validated our findings in relation to birth weight meta-analyzing four independent birth cohorts with an overall sample-size of 2235 individuals spanning an age range of 0–31 years. Third, we extended our exploration of DNAm differences to include secondary exposures (gestational age, birth weight SD, and being born small for gestational age (SGA, <–2 birth weight SD)).
We hypothesized that there are blood DNAm differences between young adults born preterm at VLBW and term controls. We further hypothesized that some of these differences may be seen in unrelated cohorts of varying ages when investigating birth weight as the exposure. We also hypothesized that some of the differences in DNAm may be related to secondary birth exposures.
Material and methods
Discovery study population
Briefly, the original cohort of HeSVA (Helsinki Study of Very Low Weight Adults) consists of 335 children born at VLBW between 1978 and 1985 and treated at the neonatal intensive care unit in Helsinki University Central Hospitals Children’s Hospital, the tertiary center serving the province of Uusimaa, Finland. For the current study, 166 VLBW subjects and 172 term controls, group-matched as described previously by age, sex, and birth hospital attended a follow-up visit in 2004 at mean age of 22.5 years of age.2,3,23 The examination included a fasting blood sample for DNA extraction. In case of multiple pregnancy, we included one subject chosen at random from each family to dilute the possibility of familial bias or inflation, leaving 157 VLBW subjects.
Follow-up cohorts and age groups
Follow-up and validation of the discovery cohort’s findings was performed in four independent cohorts of varying age groups: PREDO (Prediction and Prevention of Preeclampsia and Intrauterine Growth Restriction, n = 817)24 at birth, GLAKU (Glycyrrhizin in Licorice, n = 236)25,26 at mean age of 12 years, NFBC1986 (Northern Finland Birth Cohort 1986, n = 479)27,28 at mean age of 16 years and NFBC1966 (Northern Finland Birth Cohort 1966, n = 703)29 at mean age of 31 years.
PREDO (at birth/newborn subjects)
Briefly, PREDO is a prospective multicenter study of 1079 Finnish women with known risk-factor status for pre-eclampsia and intrauterine growth restriction. The women gave birth to a singleton live child between 2006 and 2010. Fetal cord blood DNAm data were available from 963 children, and 817 samples were available for the current analysis.
GLAKU (12–13-year-old subjects)
GLAKU is an urban-based cohort of 1049 women who gave birth to a singleton live child between March and November 1998. Between 2009-2011, 920 children were invited for a follow-up visit and 451 participated at early adolescence. Of the participating children, peripheral blood DNAm data were available from 236 children.
NFBC1986 and NFBC1966 (16-, 31- and 46-year-old subjects)
NFBC1986 and NFBC1966 are prospective birth cohorts of individuals born in the two northernmost provinces (Oulu and Lapland) of Finland in 1966 and 1986 and followed through life. NFBC1966 comprises of 12,231 children and their parents who were originally recruited based on their expected delivery date being between January 1 and December 31, 1966. NFBC1986 comprises of 9479 children (9432 live births) whose mothers were recruited based on an expected date of birth between July 1, 1985, and June 30, 1986. In the current study, peripheral venous blood DNAm data from 510 (NFBC1986 at 16 years), 722 (NFBC1966 at 31 years) and 705 (NFBC1966 at 46 years) randomly selected individuals were available.
Exposure variables, covariates, DNA extraction, and DNAm data
Fasting peripheral venous EDTA blood samples, anthropometric measurements, and questionnaire data regarding health and lifestyle were collected. Blood samples were stored at -20 C for later analyses. Genomic DNA was extracted from EDTA-anti-coagulated whole peripheral blood by using QIAamp DNA Blood Maxi Kit (Qiagen®).
Peripheral venous blood DNA was available from the discovery cohort’s 157 VLBW unrelated subjects and 161 term controls. Illumina Infinium HumanMethylation450 BeadChip (Illumina, San Diego, California) was used to determine genome wide DNAm levels. Pre-processing was performed using the R-package minfi.30 Raw beta-values were normalized using functional normalization, CpG-sites (5’—C—phosphate—G—3’) failing in more than 50% of the samples (based on a detection p value of 0.10) were removed. Batch-correction was performed using combat31 and non-autosomal CpGs as well as cross-hybridizing CpGs were removed. The final dataset included 428,759 CpG-sites and 157 VLBW cases and 161 controls. CpG-sites were mapped to genomic locations using Illumina’s annotation using the R-package minfi.30 The replication cohort’s methylation analyses are based on cord blood (PREDO, n = 817) and peripheral venous blood (GLAKU, n = 236; NFBC1986 n = 479; and NFBC1966, n = 703) and are described in detail previously.25,32,33 Cell-type proportions were estimated using the Houseman method for peripheral venous blood34 and the Bakulski algorithm for cord blood.35 Further details of cohort-specific methods are detailed in the Supplementary Methods.
Statistical analyses
Analyses were conducted using VLBW/term-dichotomization as the primary exposure for the discovery cohort, and birth weight in grams as a continuous exposure variable for PREDO and GLAKU cohorts. Linear regression was applied adjusting for blood cell type proportions, age at sampling, sex, maternal smoking in pregnancy (classified as smoking/non-smoking during pregnancy) and parental socioeconomic status or highest attained parental education. Beta values are expressed as either positive or negative (VLBW = 0, control = 1, i.e., a positive beta corresponds to more methylation in controls and hypomethylation in the VLBW group). For NFBC cohorts we used the same regression models against birth weight as a continuous variable, as well as a dichotomous prematurity-status (<37 gestational weeks).
A meta-analysis of the results of PREDO, GLAKU, NFBC1986, and NFBC1966 was conducted using the R-package metafor,36 with continuous birth weight as the exposure. NFBC1966 had two time points available, 31 and 46 years of age. The time point of 31 years was used, as more samples were available and it was closer to our discovery cohort’s age. Multiple testing was controlled for using Benjamini-Hochberg false discovery rate (FDR) method. The CpG-sites from the discovery analysis with FDR-adjusted p < 0.05 were included in the meta-analysis. After the meta-analysis, associations in CpG-sites with FDR-adjusted p < 0.05 were considered statistically significant. Exploratory meta-analyses relating to the hypothesis for secondary birth exposures were conducted in the NFBC cohorts for dichotomous SGA-status, as well as birth weight SD and gestational age as continuous variables.
Demographic and clinical data were analyzed with SPSS (version 27) and methylation outcome data analyses and meta-analyses were performed with R statistical environment.
Results
Cohort profiles
Characteristics of our study subjects from the discovery and replication cohorts are described in Table 1. The mean age of our discovery cohort subjects was 22 years. In our validation cohorts, ages ranged from newborn (cord blood, PREDO), early adolescence (mean age of 12 years, GLAKU), adolescence (mean age of 16 years, NFBC1986), and adulthood (mean ages of 31 years and 46 years, NFBC1966).
Epigenome wide association study (EWAS) of the discovery cohort
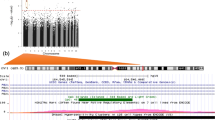
We identified 66 differentially methylated CpGs that were associated with birth at VLBW with a significance of FDR-adjusted p < 0.05 (Table 2). A Manhattan plot of the results is shown in Fig. 1 and a QQ-plot in Fig. 2. The QQ-plot has a lambda value of 1.16, but in EWAS such values are common and may not reflect true inflation.37 Seventeen CpGs were differentially methylated using the epigenome-wide p value thresholds for statistical significance of p < 9 × 10-8 and 23 with p < 2.44 × 10-7, as suggested previously.38,39 The CpG-site with the lowest p value in our study is cg21246516 (p < 5.8 × 10-11), located in an intergenic region between TINAG (dist = 413,877 bp) and FAM83B (dist = 42,693 bp) and is hypermethylated in those with VLBW. Our top DMPs localized to the genomic regions of GLI2, SKIDA1, HIF3A, EBF4, and SOCS1.

The analysis was adjusted for sex, age, cell type proportions, maternal smoking during pregnancy, and parental socioeconomic status. The x-axis is the chromosomal position, and the y-axis is the level of statistical significance on a −log10-scale. The blue line represents the threshold for experiment wide significance at p = 7.6 × 10−6. CpGs highlighted in green represent sites with an epigenome wide significance of p < 9 × 10−8 (red line). The thresholds are based on previous recommendations.38,39

The observed association p values are plotted against the expected null distribution for the discovery analysis.
Meta-analysis in the validation cohorts
We checked whether the top 66 differentially methylated CpG-sites with FDR-adjusted p < 0.05 could be validated in four independent cohorts with an overall age ranging between 0 and 31 years using continuous birth weight as the exposure. We used linear regression analyses and estimated regression coefficients and SEs, describing the linear relationship between birth weight and methylation adjusting for similar factors as in the discovery cohort’s analysis: blood cell type proportions, age, sex, maternal smoking during pregnancy, and parental socioeconomic status (see Supplementary Table 1–5). We then ran meta-analyses across the 66 top-associated CpG-sites to assess whether findings from the HeSVA discovery cohort could be followed up and validated in the cohorts of PREDO, GLAKU, NFBC1986, and NFBC1966. This resulted in five CpGs that were hypermethylated both in the VLBW group and with lower birth weight at FDR-adjusted p < 0.05 (cg20219891, cg13872898, cg00637745, cg14311362 and cg17870997). The forest plots for the meta-analysis of the validating sites are presented in Fig. 3.

Meta-analysis of DNA methylation betas is shown for birth weight (kg) at CpGs across four studies (PREDO, GLAKU, NFBC1986, and NFBC1966) with 95% confidence intervals after FDR-correction by Benjamini-Hochberg method at level 0.05. Adjusted for age, sex, maternal smoking during pregnancy, parental education, technical covariates, and cell type proportions.
Secondary exposure analyses
Exploratory analyses over the 66 top hits from HeSVA were conducted in NFBC1986 and NFBC1966 with being born preterm (<37 gestational weeks) and SGA as dichotomous exposures, and gestational age and birth weight SD as continuous variables. 41 CpGs were differentially methylated with these secondary exposures, with overlapping sites for the primary birth weight exposure. The results of the secondary exposure analyses are described in Supplementary Table 5.
No single CpG of the discovery cohort’s 66 sites was differentially methylated in all cohorts even though they were validated in the meta-analysis. If we only observe samples from beyond infancy, however, seven CpGs show differential methylation with exposures varying among birth weight, birth weight SD, preterm-status and SGA-status. These sites were located in intergenic (GLI2, CTAGE1), intronic, and exonic (EBF4) as well as in UTR3’-regions (SOCS1). The DMPs and their respective cohorts and exposures are described in Supplementary Table 6.
We further conducted exploratory meta-analyses over NFBC1986 and NFBC1966 with being born preterm as the exposure. One CpG (cg14123232, intergenic region with nearest gene being GLI2), was differentially methylated in the meta-analysis at nominal significance (p = 0.044), but this did not survive FDR-correction. Forest plots for prematurity in NFBC1986 and NFBC1966 for the validating sites from the main meta-analysis with birth weight as the exposure, and for the nominally significant site cg14123232 are presented in Supplementary Fig. 1.
Discussion
We hypothesized that there are blood DNAm differences between young adults born preterm at VLBW and term controls. In the HeSVA-cohort, we identified 66 differentially methylated CpG-sites between 157 VLBW individuals and 161 term controls in early adulthood at mean age of 22 years. Our top DMPs localize to the genomic regions: GLI2, SKIDA1, HIF3A, EBF4, and SOCS1. The genes have been previously associated with early life development18,40 and maturation41 as well as with metabolic pathways42,43 suggesting relevant genomic sites possibly connecting adverse health consequences and severe early life exposure.
We further hypothesized that differences seen associating with VLBW could be validated in unrelated cohorts regarding birth weight. Of the 66 sites, five (all annotating to an intergenic region close to GLI2) were validated in a meta-analysis of four independent cohorts using continuous birth weight as main exposure, with subjects aged 0–31 years. Our findings may represent persisting changes in the epigenome that could partly explain differences in health outcomes between individuals born at VLBW and at term. We further explored secondary birth exposures, such as SGA, gestational age, and birth weight SD, and their association with DNAm differences.
The association between preterm birth or low birth weight and later health outcomes has been suggested to be in part mediated by epigenetics. Differences in methylation have been found between VLBW individuals and term controls but their persistence has remained unclear.44 The epigenetic signature of preterm birth at VLBW is present in adult life in our study. This makes preterm birth at VLBW one of the few early life exposures where such a signature can be replicably observed after adolescence. Considering the large number of VLBW or preterm individuals globally, identifying molecular mechanisms between early life exposures and later outcomes is important. Epigenetic differences have been studied in individuals with different degrees of prematurity, usually graded by birth weight or gestational age. Genes annotating to DMPs that have been reported in connection with prematurity associate with the immune system,45 neurologic development,46 cellular differentiation and proliferation,47 metabolism (e.g., IGF2),20,48 fetal development, and birth weight.49 Significant DMPs are also seen in intergenic regions that may act as binding sites for transcription factors. The most significant and compelling CpG-sites of our study are located within or near genes associating with fetal development (GLI2),50 organogenesis (SKIDA1),51 insulin resistance and hypoxia response (HIF3A),52 neural development and B-cell maturation (EBF4),53 and immunological development (SOCS1).40
Our most robust finding is the validation of five DMPs in a meta-analysis of four independent cohorts, suggesting an association between birth weight and DNAm over a wide age range, which may represent long-lasting epigenetic imprint of prenatal factors contributing to birth weight. VLBW or lower birth weight individuals had higher methylation in CpGs cg00637745, cg13872898, cg20219891, cg17870997, and cg14311362. These were validated at FDR < 0.05 in our meta-analysis and are in an intergenic region closest to GLI2. A meta-analysis investigating associations between birth weight and cord blood DNAm (n = 8825) in term subjects found differentially methylated CpGs nearest to GLI2 in childhood (n = 2756), adolescence (n = 2906), and adulthood (n = 1616).19 The five CpGs from our study are present in the meta-analysis which, too, reports a negative association between methylation levels and birth weight at these sites. Cameron et al. report hypermethylation in one CpG site associating with GLI2 (cg20219891), which is one of the sites seen in our discovery cohort and validation meta-analysis.22 Differential methylation of GLI2 has been associated with gestational age in a meta-analysis in cord blood.18 None of the five validated CpGs from our study examining birth weight were reported as differentially methylated.18 GLI2 is a gene in chromosome 2 encoding the protein Gli family zinc finger 2, which is involved in intracell signaling via the Sonic Hedgehog pathway.50 GLI2 is also involved in neural tube development, and embryogenesis, as well as being a potential oncogene.54
Three CpGs (cg03610228, cg04707519, and cg04714110) associating with SKIDA1 were among our discovery cohort’s top hits with a positive association between DNAm levels and VLBW status. These sites, however, were not validated in the meta-analysis examining continuous birth weight. A positive association between DNAm levels and gestational age has previously been reported for cg03610228, cg04707519, and cg04714110 in infants.55 A positive association between DNAm levels and birth weight for cg04714110 has been observed in cord blood.19 SKIDA1 is a gene in chromosome 10 encoding the protein SKI/DACH domain containing 1 (also known as DLN-1; C10orf140), which has a predicted localization in the nucleus and cytosol, which may reflect molecular trafficking between the two. It has been reported to have a role in autoimmune disorders.56 Animal models suggest involvement in organogenesis and brain maturation,51 and gastrulation and neurulation.41 The importance of SKIDA1 is highlighted by SKIDA1 knockout mice being subviable.57
We observed hypomethylation with VLBW of three epigenome-wide CpG-sites (cg27146050, cg16672562, and cg22891070) annotated to the gene HIF3A. These sites are also differentially methylated in NFBC1966 at 31 years of age, although they were not validated in the subsequent meta-analysis. Cameron et al. also report hypomethylation in three sites associating with HIF3A in 28-year-old VLBW individuals when compared to term controls.22 Our sites are among these. Differential methylation of HIF3A has been associated with gestational age in cord blood.18,58 HIF3A is a gene in chromosome 19 encoding the protein hypoxia inducible factor 3 subunit alpha, which is involved in regulating adaptive processes linked to low oxygen and metabolism.59 Such processes play roles in the regulation of glucose metabolism and cardiometabolic risk factors.60 HIF3A is largely expressed in placental, fat and spleen tissues.61 Hypermethylation of HIF3A has been linked to weight and BMI gain42,62 and to insulin resistance in gestational and type 2 diabetes52 and adipose tissue differentiation and dysfunction.63 Cord blood HIF3A methylation levels have also been associated with pre-eclampsia,64 and elevated blood pressure in childhood,65 suggesting a possibility of being a mediating element of cardiovascular risk factors. Our findings are in concordance with previously reported increased cardiometabolic risk factors in VLBW and ELBW adults.2,66
A fourth gene of interest is EBF4, as four intronic CpGs (cg24263062, cg13518079, cg14959908, and cg05857996) and one exonic CpG (cg05825244) are among our top hits with hypermethylation in the VLBW group and lower birth weight. The EBF4-associated CpGs are present in all our replication cohorts excluding infancy with varying early life exposures (Supplementary Table 7). EBF4 is a gene in chromosome 20 encoding the protein early B-cell factor transcription factor 4, which has been associated with neural development and B-cell maturation.53 Few publications regarding methylation of EBF4 and biological development or growth have been published, but EBF4 methylation has been associated with BMI in adults.43 Differential methylation of EBF4 has been positively associated with gestational age, also in adolescence,18 but in opposite direction to our study. Cameron et al. report four CpG-sites associating with EBF4 being differentially methylated between VLBW participants and term controls in 28-year-olds22; four of these sites (cg24263062, cg05857996, cg13518079, and cg05825244) overlap with our results.
The CpG (cg10784813) associating with SOCS1 is hypermethylated in VLBW, which is in line with Küpers et al. reporting a negative association between DNAm levels and birth weight in cg10784813.19 Alongside GLI2 it is one of the few DMPs present in all our time points from adolescence onwards, albeit with varying exposures making its validity uncertain (Supplementary Table 7). SOCS1 is a gene in chromosome 16 encoding the protein suppressor of cytokine signaling 1, which deals with inflammatory reactions and is particularly present in lymphatic tissue.67 SOCS1 deficiency is associated with perinatal lethality, presumably due to its role in immunology.40 Differential methylation of SOCS1 in cord blood has a positive association with gestational age reported by Merid et al.18 who also report replicating their finding in an independent sample by Hannon et al.13
Previous meta-analyses concerning birth weight and gestational age have found DMPs in the same regions as in our study,18,19,68 but most studies have been conducted in neonates and from cord blood. In the meta-analysis by Küpers et al.19 8170 CpGs related to birth weight were identified, and of our top 66 CpG hits, there are 16 overlapping sites with the same directionality of methylation. In a meta-analysis assessing DNAm and gestational age in non-complicated births18 8899 CpGs were identified but only 10 CpGs overlapped with our study with the same directionality. They also report results for all births (complicated and non-complicated) with 17,095 CpGs related to gestational age, but there is considerable heterogeneity in this group. The overlap with the ”all births” model and our study is 25 CpGs. The meta-analyses mentioned here are mainly based on cord blood and may not be entirely representative of our study with Finnish adult participants.
Individuals born at VLBW have been shown to have several DMPs compared to term controls, but the results vary considerably across studies. A study of 12 individuals born at gestational age <31 weeks and 12 matched term controls identified 1,555 DMPs in cord blood at birth, most of which resolved by the age of 18.45 A study of 40 VLBW infants showed DNAm differences in buccal samples in IGF2 and FKBP5 at birth but they resolved after first year of life, which may reflect tissue-specific mechanisms and may not be comparable to studies based on blood.69 Another similar-sized study showed hypomethylation of PLAGL1 in VPT individuals at birth and at discharge.70 A small study comparing VLBW and term children at 12 months of age showed methylation differences in SLC6A3.71 IGF2 has been identified as a gene that is differentially methylated from VLBW infants up to 20-year-olds.20 A recent study by Cameron et al. compared DNAm between 109 VLBW individuals and 51 term controls using heel prick spot blood samples from birth, and venous blood at 28 years of age from 215 VLBW individuals and 96 term controls.22 They report 16,400 CpG-sites differing between the groups at birth, but at the age of 28 years, only twelve were differentially methylated after FDR-correction. It is of particular note, that of these twelve CpG-sites, eight were differentially methylated in our study as well. A study comparing ELBW individuals to normal birth weight individuals in adulthood found buccal cell DNAm differences in 1,354 loci in 694 genes in men, but only two loci in women.21 Epigenome-wide differences have also been studied in individuals born with a less severe exposure of LBW. Fewer DMPs tend to be identified when comparing LBW individuals to term controls than when studying VLBW individuals, which suggests that there might be a threshold- or dose-effect phenomenon connecting the degree of prematurity and methylation. Simpkin et al.72 aimed to see whether long-term changes to the epigenome could be identified by sampling 1018 mostly term-born children at birth, and at ages 7 and 17, but they could not detect persistent changes in the epigenome associated with birth weight. A study by Madden et al. reports only one significant DNAm CpG-site (cg00966482) associated with birth weight (n of LBW = 84) in adults with a cohort mean age of 29.5.73 Aside from the CpGs associating with GLI2, EBF4, and HIF3A reported by Cameron et al., none of the previously reported CpGs or genes from studies with VLBW or LBW participants were significant in our study, which underlines the heterogeneity of the results and the need for large-scale meta-analyses. Methylation patterns tend to change during early development, and reported methylation differences between preterm and term individuals tend to largely resolve, which may reflect metabolic adjustments during maturation. Persisting epigenetic differences may reflect long-lasting differences in metabolism, but this requires more study.
We aimed to explore whether DNAm could be linked to SGA or prematurity, as well as examining gestational age and birth weight as continuous variables. Based on our findings, the epigenetic signatures of these phenomena overlap, which is to be expected, as the clinical phenotypes themselves overlap. However, being SGA could associate with a separate DNAm identifier, as some adverse adult health outcomes of preterm birth may be more apparent in individuals born SGA. Our meta-analysis of preterm participants in NFBC1986 and NFBC1966 had too few SGA participants (n = 0 for NFBC1986 and n = 25 for NFBC1966) to reach conclusions. No definite conclusions about the other secondary exposures could be drawn from these results either. Therefore, our results need to be replicated in a larger sample of preterm and preterm at VLBW subjects. Furthermore, when considering the effect of prematurity on DNAm, the nature of the exposure should be considered carefully as both gestational age and birth weight may have separate, yet often overlapping effects. Thus, we explored both gestational length and size at birth as exposures.
A major strength of our discovery cohort is the large number of VLBW participants in adulthood, as much of the past research is conducted in either infants or individuals with a less severe exposure. We also have access to four extensively phenotyped and characterized independent follow-up cohorts of varying ages, which we combined into a meta-analysis, to validate our findings. The age of our cohorts spans over decades of life with PREDO starting from birth and finishing with NFBC1966 in adulthood. Methylation patterns change rapidly during early life, which may explain why only few of the differentially methylated CpGs in the discovery cohort could be identified from newborn samples.
The age of our discovery cohort subjects at the time of measurement was relatively early in adulthood and it remains to be seen whether the epigenetic differences persist further into adulthood and what their relationship with future cardiovascular risk is. It should also be noted that contrary to the other birth cohorts, PREDO is built around examining risk factors for pre-eclampsia, which may introduce confounding. Even though our study spans a wide age range of participants, no longitudinal analysis was possible. Thus, DNAm differences seen in adulthood between VLBW and term individuals could be due to other biological differences that have accumulated during the participants’ life. Causality cannot be shown in this setting. Our results showed some inflation, which is often observed in EWAS.37
We aimed to investigate the DNAm differences between young adults born at VLBW and their term controls. We found 66 differentially methylated CpG-sites, which suggests a marker of preterm birth at VLBW present at young adulthood. We further saw several of these sites when examining birth weight in independent cohorts of varying ages validating our findings. While there is heterogeneity and noise between published studies, consistent and replicable gene regions where epigenetic signatures of early life events persist are starting to emerge. We believe this highlights the importance of collaboration and wider meta-analyses.
Conclusions
In comparing young adults born preterm at low birth weight with controls born at term, we have discovered differentially methylated CpG-sites that are associated with birth weight in cohorts ranging from newborns to adulthood.
Our results support earlier findings and provide more robust evidence that epigenetic differences with regard to birth weight may remain even decades after preterm birth. Future studies should investigate whether our findings can be replicated in longitudinal cohorts and also continue the follow-up to see if the epigenetic differences show persistence in later life, and how these differences manifest phenotypically.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Chawanpaiboon, S. et al. Global, regional, and national estimates of levels of preterm birth in 2014: a systematic review and modelling analysis. Lancet Glob. Health 7, e37–e46 (2019).
Hovi, P. et al. Glucose regulation in young adults with very low birth weight. N. Engl. J. Med. 356, 2053–2063 (2007).
Hovi, P. et al. Decreased bone mineral density in adults born with very low birth weight: a cohort study. PLoS Med. 6, e1000135 (2009).
Hovi, P. et al. Blood pressure in young adults born at very low birth weight: adults born preterm international collaboration. Hypertension 68, 880–887 (2016).
Saarenpaa, H. K. et al. Lung function in very low birth weight adults. Pediatrics 136, 642–650 (2015).
Eves, R. et al. Association of very preterm birth or very low birth weight with intelligence in adulthood: an individual participant data meta-analysis. JAMA Pediatr. 175, e211058 (2021).
Pyhälä, R. et al. Neurocognitive abilities in young adults with very low birth weight. Neurology 77, 2052–2060 (2011).
Pyhala, R. et al. Self-reported mental health problems among adults born preterm: a meta-analysis. Pediatrics 139, e20162690 (2017).
Wolke, D., Johnson, S. & Mendonça, M. The life course consequences of very preterm birth. Annu. Rev. Dev. Psychol. 1, 69–92 (2019).
Smith, Z. D. & Meissner, A. DNA Methylation: Roles in Mammalian Development. Nat. Rev. Genet. 14, 204–220 (2013).
Bavineni, M. et al. Mechanisms linking preterm birth to onset of cardiovascular disease later in adulthood. Eur. Heart J. 40, 1107–1112 (2019).
Breton, C. V. et al. Prenatal tobacco smoke exposure is associated with childhood DNA Cpg methylation. PLoS One 9, e99716 (2014).
Hannon, E. et al. Variable DNA methylation in neonates mediates the association between prenatal smoking and birth weight. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374, 20180120 (2019).
Palma-Gudiel, H., Córdova-Palomera, A., Eixarch, E., Deuschle, M. & Fañanás, L. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: a meta-analysis. Epigenetics 10, 893–902 (2015).
Cao-Lei, L. et al. DNA Methylation mediates the impact of exposure to prenatal maternal stress on BMI and central adiposity in children at age 13½ years: project ice storm. Epigenetics 10, 749–761 (2015).
Williams, T. C. & Drake, A. J. Preterm birth in evolutionary context: a predictive adaptive response? Philos. Trans. R. Soc. Lond. B Biol. Sci. 374, 20180121 (2019).
Agha, G. et al. Birth weight-for-gestational age is associated with DNA methylation at birth and in childhood. Clin. Epigenetics 8, 118 (2016).
Merid, S. K. et al. Epigenome-wide meta-analysis of blood DNA methylation in newborns and children identifies numerous loci related to gestational age.Genome Med. 12, 25 (2020).
Küpers, L. K. et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat. Commun. 10, 1893 (2019).
Wehkalampi, K. et al. Altered methylation of IGF2 Locus 20 years after preterm birth at very low birth weight. PLoS One 8, e67379 (2013).
Mathewson, K. J. et al. DNA methylation profiles in adults born at extremely low birth weight. Dev. Psychopathol. 34, 19–36 (2022).
Cameron, V. A. et al. DNA methylation patterns at birth predict health outcomes in young adults born very low birthweight. Clin. Epigenetics 15, 47 (2023).
Hovi, P. et al. Ambulatory blood pressure in young adults with very low birth weight. J. Pediatr. 156, 54–59.e51 (2010).
Girchenko, P. et al. Cohort profile: prediction and prevention of preeclampsia and intrauterine growth restriction (Predo) study. Int. J. Epidemiol. 46, 1380–1381g (2016).
Suarez, A. et al. The epigenetic clock and pubertal, neuroendocrine, psychiatric, and cognitive outcomes in adolescents. Clin. Epigenetics 10, 96 (2018).
Strandberg, T. E., Järvenpää, A. L., Vanhanen, H. & McKeigue, P. M. Birth outcome in relation to licorice consumption during pregnancy. Am. J. Epidemiol. 153, 1085–1088 (2001).
Järvelin, M. R., Hartikainen-Sorri, A. L. & Rantakallio, P. Labour induction policy in hospitals of different levels of specialisation. Br. J. Obstet. Gynaecol. 100, 310–315 (1993).
Järvelin, M. R. et al. Ecological and individual predictors of birthweight in a northern Finland birth cohort 1986. Paediatr. Perinat. Epidemiol. 11, 298–312 (1997).
Nordström, T. et al. Cohort profile: 46 years of follow-up of the northern Finland birth cohort 1966 (Nfbc1966). Int J. Epidemiol. 50, 1786–1787j (2022).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical bayes methods. Biostatistics 8, 118–127 (2007).
Czamara, D. et al. Integrated analysis of environmental and genetic influences on cord blood DNA methylation in new-borns. Nat. Commun. 10, 2548 (2019).
Wiklund, P. et al. DNA methylation links prenatal smoking exposure to later life health outcomes in offspring. Clin. Epigenetics 11, 97 (2019).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinforma. 13, 86 (2012).
Bakulski, K. M. et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics 11, 354–362 (2016).
Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 36, 1–48 (2010).
van Iterson, M. et al. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 18, 19 (2017).
Saffari, A. et al. Estimation of a significance threshold for epigenome-wide association studies. Genet Epidemiol. 42, 20–33 (2018).
Mansell, G. et al. Guidance for DNA methylation studies: statistical insights from the Illumina epic array. BMC Genom. 20, 366 (2019).
Marine, J. C. et al. Socs1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell 98, 609–616 (1999).
Seufert, D. W., Hegde, R. S., Nekkalapudi, S., Kelly, L. E. & El-Hodiri, H. M. Expression of a novel Ski-like gene in Xenopus development. Gene Expr. Patterns 6, 22–28 (2005).
Shen, J. et al. Hif3a DNA methylation, obesity and weight gain, and breast cancer risk among Mexican American women. Obes. Res. Clin. Pr. 14, 548–553 (2020).
Cao, V. T. et al. A genome-wide methylation study of body fat traits in the Norfolk Island isolate. Nutr. Metab. Cardiovasc. Dis. 31, 1556–1563 (2021).
Alisch, R. S. et al. Age-associated DNA methylation in pediatric populations. Genome Res. 22, 623–632 (2012).
Cruickshank, M. N. et al. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Med. 5, 96 (2013).
Tan, Q. et al. Epigenetic signature of preterm birth in adult twins. Clin. Epigenetics 10, 87 (2018).
Mulligan, C. J., D’Errico, N. C., Stees, J. & Hughes, D. A. Methylation changes at Nr3c1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics 7, 853–857 (2012).
van Dijk, S. J. et al. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int. J. Obes. 42, 28–35 (2018).
Haworth, K. E. et al. Methylation of the Fgfr2 gene is associated with high birth weight centile in humans. Epigenomics 6, 477–491 (2014).
Jacob, J. & Briscoe, J. Gli proteins and the control of spinal-cord patterning. EMBO Rep. 4, 761–765 (2003).
Visel, A., Thaller, C. & Eichele, G. Genepaint.Org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 32, D552–D556 (2004).
Girgis, C. M., Cheng, K., Scott, C. H. & Gunton, J. E. Novel links between hifs, type 2 diabetes, and metabolic syndrome. Trends Endocrinol. Metab. 23, 372–380 (2012).
Wang, S. S., Betz, A. G. & Reed, R. R. Cloning of a novel Olf-1/Ebf-like gene, O/E-4, by degenerate oligo-based direct selection. Mol. Cell Neurosci. 20, 404–414 (2002).
Tolosa, E. J. et al. Gli1/Gli2 functional interplay is required to control Hedgehog/GLI targets gene expression. Biochem. J. 477, 3131–3145 (2020).
Kashima, K. et al. Identification of epigenetic memory candidates associated with gestational age at birth through analysis of methylome and transcriptional data. Sci. Rep. 11, 3381 (2021).
Julià, A. et al. Epigenome-wide association study of rheumatoid arthritis identifies differentially methylated loci in B cells. Hum. Mol. Genet. 26, 2803–2811 (2017).
Dickinson, M. E. et al. High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514 (2016).
Bohlin, J. et al. Prediction of gestational age based on genome-wide differentially methylated regions. Genome Biol. 17, 207 (2016).
Tanaka, T., Wiesener, M., Bernhardt, W., Eckardt, K. U. & Warnecke, C. The human Hif (Hypoxia-Inducible Factor)-3alpha gene is a Hif-1 target gene and may modulate hypoxic gene induction. Biochem J. 424, 143–151 (2009).
Koivunen, P., Serpi, R. & Dimova, E. Y. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition in cardiometabolic diseases. Pharm. Res 114, 265–273 (2016).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 13, 397–406 (2014).
Dick, K. J. et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet 383, 1990–1998 (2014).
Pfeiffer, S. et al. Hypoxia-inducible factor 3A gene expression and methylation in adipose tissue is related to adipose tissue dysfunction. Sci. Rep. 6, 27969 (2016).
Mansell, T. et al. Early-life determinants of hypoxia-inducible factor 3A gene (Hif3a) methylation: a birth cohort study. Clin. Epigenetics 11, 96 (2019).
Mansell, T. et al. Hif3a cord blood methylation and systolic blood pressure at 4 years—a population-based cohort study. Epigenetics 15, 1361–1369 (2020).
Morrison, K. M. et al. Cardiometabolic health in adults born premature with extremely low birth weight. Pediatrics 138, e20160515 (2016).
Tamiya, T., Kashiwagi, I., Takahashi, R., Yasukawa, H. & Yoshimura, A. Suppressors of cytokine signaling (Socs) proteins and Jak/Stat pathways: regulation of T-cell inflammation by Socs1 and Socs3. Arterioscler. Thromb. Vasc. Biol. 31, 980–985 (2011).
Knight, A. K. et al. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 17, 206 (2016).
Piyasena, C. et al. Dynamic changes in DNA methylation occur during the first year of life in preterm infants. Front Endocrinol. 7, 158 (2016).
Provenzi, L. et al. Very preterm birth is associated with Plagl1 gene hypomethylation at birth and discharge. Epigenomics 10, 1121–1130 (2018).
Arpon, A. et al. Methylation changes and pathways affected in preterm birth: a role for Slc6a3 in neurodevelopment. Epigenomics 10, 91–103 (2018).
Simpkin, A. J. et al. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum. Mol. Genet. 24, 3752–3763 (2015).
Madden, R. A. et al. Birth weight associations with DNA methylation differences in an adult population. Epigenetics 16, 783–796 (2021).
Funding
No funder was directly or indirectly involved with data collection, analysis, interpretation or reporting of results. No funder has reviewed the manuscript, and it is the sole work of the authors. This study was supported by the following institutions: the Doctoral Programme in Clinical Research, University of Helsinki; the Finnish Foundation for Pediatric Research; Finska Läkaresällskapet; the Juho Vainio Foundation; the Paulo Foundation; the Päivikki and Sakari Sohlberg Foundation; the Jalmari and Rauha Ahokas Foundation; the Novo Nordisk Foundation; the Signe and Ane Gyllenberg Foundation; the Sigrid Jusélius Foundation; and the Yrjö Jahnsson Foundation. The PREDO Study has been funded by the Academy of Finland (J.L.: 311617 and 269925, K.R.: 1312670, 128789, 1287891, 1324596), EraNet Neuron, EVO (a special state subsidy for health science research), University of Helsinki Research Funds, the Signe and Ane Gyllenberg Foundation, the Emil Aaltonen Foundation, the Finnish Medical Foundation, the Jane and Aatos Erkko Foundation, the Novo Nordisk Foundation, the Päivikki, and Sakari Sohlberg Foundation, the Sigrid Juselius Foundation granted to members of the Predo study board. Methylation assays were funded by the Academy of Finland (269925). GLAKU cohort has been supported by the Academy of Finland, Hope and Optimism Initiative, the Signe and Ane Gyllenberg Foundation, the Emil Aaltonen Foundation, the Foundation for Pediatric Research, the Foundation for Cardiovascular Research, the Juho Vainio Foundation, the Sigrid Jusélius Foundation, the Yrjö Jahnsson Foundation, and the University of Helsinki Research Funds. NFBC1986 was funded by EU QLG1-CT-2000-01643 (EUROBLCS) Grant no. E51560, NorFA Grant no. 731, 20056, 30167, USA/NIH 2000 G DF682 Grant no. 50945. NFBC1966 31 yr follow-up received financial support from University of Oulu Grant no. 65354, Oulu University Hospital Grant no. 2/97, 8/97, Ministry of Health and Social Affairs Grant no. 23/251/97, 160/97, 190/97, National Institute for Health and Welfare, Helsinki Grant no. 54121, Regional Institute of Occupational Health, Oulu, Finland Grant no. 50621, 54231. J.R., M.R.J and S.S. were supported by European Union’s Horizon 2020 research and innovation program under grant agreements No 874739 (LongITools), 733206 (LifeCycle) and 633595 (DynaHEALTH) and Academy of Finland under grant agreement No 285547 (EGEA). NFBC1966 (31 years study) received financial support from University of Oulu Grant No. 65354, Oulu University Hospital Grant No. 2/97, 8/97, Ministry of Health and Social Affairs Grant No. 23/251/97, 160/97, 190/97, National Public Health Institute, Helsinki Grant No. 54121, Regional Institute of Occupational Health, Oulu, Finland Grant No. 50621, 54231, NHLBI Grant 5R01HL087679-02 through the STAMPEED program (1RL1MH083268-01), NIH/NIMH (5R01MH63706:02). NFBC1966 (46 years study) received financial support from University of Oulu Grant No. 24000692, Oulu University Hospital Grant No. 24301140, ERDF European Regional Development Fund Grant No. 539/2010 A31592. Open Access funding provided by Finnish Institute for Health and Welfare.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. J.K. wrote the manuscript. D.C., H.H., J.L., P.H., M.M., J.R., J.E., S.A., M.J., S.S., K.R., E.B. and E.K. revised the manuscript. J.K., D.C., H.H., J.R., and E.B. contributed to the analyses. J.L., D.C., P.H., J.E., S.A., M.J., S.S., K.R., E.B., and E.K. contributed to study design.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study protocol was approved by the Northern Ostrobothnia Hospital District Ethical Committee (94/2011 (12.12.2011) for NFBC1966 and 108/2017 (15.1.2018) for NFBC1986), the Ethics Committee of Children’s and Adolescent’s Diseases and Psychiatry at Helsinki and Uusimaa Hospital District (HeSVA and PREDO), and the City of Helsinki and Helsinki and Uusimaa Hospital District (GLAKU). Written informed consent was given by all adult participants and parent/guardian by the minors. No individual-level data is presented anywhere in the manuscript.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kuula, J., Czamara, D., Hauta-alus, H. et al. Epigenetic signature of very low birth weight in young adult life. Pediatr Res (2024). https://doi.org/10.1038/s41390-024-03354-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41390-024-03354-6
- Springer Nature America, Inc.